Chapter 554 Hyperpituitarism, Tall Stature, and Overgrowth Syndromes
Tall Stature
Differential Diagnosis
Table 554-1 lists the causes of tall stature in childhood and adolescence. Of those listed, the normal variant, familial or constitutional tall stature, is by far the most common cause. Almost invariably, a family history of tall stature can be obtained, and no organic pathology is present. The child is often taller than his or her peers throughout childhood and enjoys excellent health. The parent of the constitutionally tall adolescent might reflect unhappily upon his or her own adolescence as a tall teenager. There are no abnormalities in the physical examination, and the laboratory studies, if obtained, are negative. Additional features of overgrowth syndromes are noted in Table 554-2.
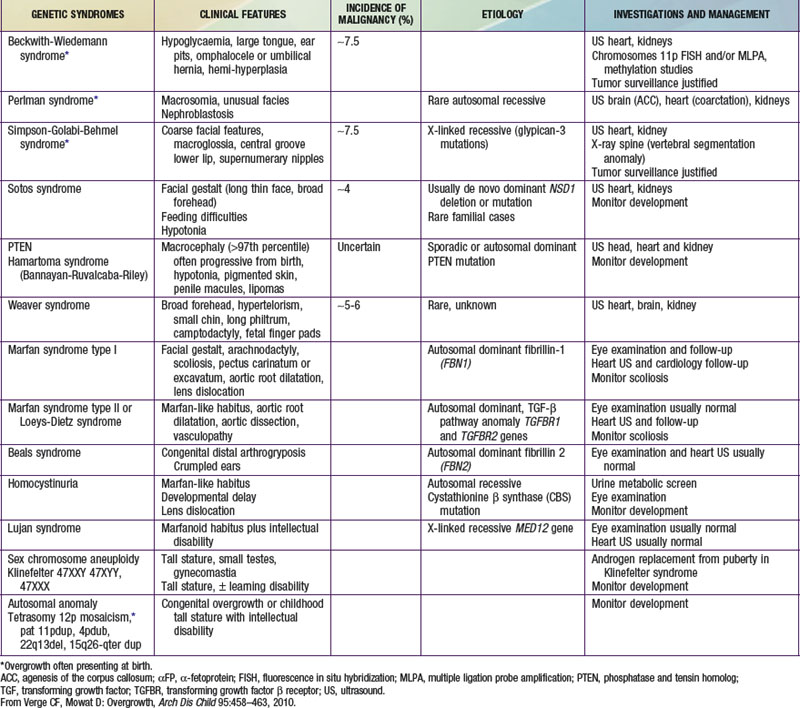
Table 554-1 DIFFERENTIAL DIAGNOSIS OF TALL STATURE AND OVERGROWTH SYNDROMES
FETAL OVERGROWTH
POSTNATAL OVERGROWTH LEADING TO CHILDHOOD TALL STATURE
POSTNATAL OVERGROWTH LEADING TO ADULT TALL STATURE
ACTH, adrenocorticotropic hormone; GH, growth hormone; IGF, insulin-like growth factor; MEN, multiple endocrine neoplasia.
Marfan syndrome is an autosomal dominant connective tissue disorder consisting of tall stature, arachnodactyly, thin extremities, increased arm span, and decreased upper to lower body segment ratio (Chapter 693). Additional abnormalities include ocular abnormalities (e.g., lens subluxation), hypotonia, kyphoscoliosis, cardiac valvular deformities, and aortic root dilatation.
Homocystinuria is an autosomal recessive inborn error of amino acid metabolism, caused by a deficiency of the enzyme cystathionine synthetase. It is characterized by mental retardation when untreated, and many of its clinical features resemble Marfan syndrome, particularly ocular manifestations (Chapter 79). Hyperthyroidism in adolescents is associated with rapid growth but normal adult height. It is almost always caused by Graves disease and is much more common in girls (Chapter 562).
Precocious puberty, whether mediated centrally (increased gonadotropin secretion) or peripherally (increased secretion of androgens, estrogens, or both), results in accelerated linear growth during childhood, mimicking the pubertal growth spurt. Because skeletal maturation is also advanced, adult height is often compromised. The diagnostic evaluation and management of precocious puberty are discussed in Chapter 556.
Prolactinoma
Prolactinomas should not be confused with the hyperprolactinemia and pituitary hyperplasia that can occur in patients with primary hypothyroidism, which is readily treated with thyroid hormone (Chapter 559). Moderate elevations (<200 ng/mL) of prolactin are also associated with a variety of medications (mainly antipsychotic), with pituitary stalk dysfunction such as can occur with craniopharyngioma, and with other benign conditions. In most patients this can be effectively treated with the dopamine agonist bromocriptine or long-acting cabergoline. When dopamine agonist treatment has been unsuccessful in lowering the serum prolactin concentration or size of the adenoma, and when symptoms or signs due to hyperprolactinemia or adenoma size persist during treatment, transsphenoidal surgery should be considered.
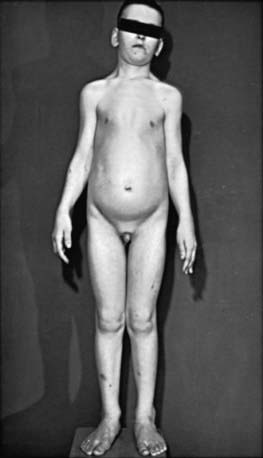
Sotos Syndrome (Cerebral Gigantism)
Children with cerebral gigantism (also known as Sotos syndrome) are above the 90th percentile for both length and weight at birth; they can also have macrocrania at that time. The NSD1 (nuclear receptor SET domain-containing protein 1) gene is responsible for this syndrome. Although it is characterized by rapid growth, there is no evidence that Sotos syndrome is caused by endocrine dysregulation. A hypothalamic defect has been suggested as a cause, but none has been demonstrated functionally or at necropsy. Growth is markedly rapid; by 1 yr of age, affected infants are greater than the 97th percentile in height. Accelerated growth continues for the first 4-5 yr and then returns to a normal rate (Fig. 554-1). Puberty usually occurs at the expected time but may occur slightly early. Adult height is usually in the upper normal range.
Acharya SV, Gopal RA, Bandgar TR, et al. Clinical profile and long term follow up of children and adolescents with prolactinomas. Pituitary. 2009;12:186-189.
Bademci G. Pitfalls in the management of Cushing’s disease. J Clin Neurosci. 2007;14:401-408.
Bulun SE. Aromatase deficiency and estrogen resistance: from molecular genetics to clinic. Semin Reprod Med. 2000;18:31-39.
Carel JC, Blumberg J, Bougeard-Julien M, et al. Long-acting lanreotide in adolescent girls with constitutional tall stature. Horm Res. 2009;71:228-236.
Chahal HS, Stals K, Unterlander M, et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med. 2011;364(l):43-50.
Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocr Rev. 1998;19:540-558.
Faravelli F. NSD1 mutations in Sotos syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:24-31.
Feenstra J, de Herder WW, ten Have SM, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365:1644-1646.
Horvath A, Stratakis CA. Clinical and molecular genetics of acromegaly: MEN1, Carney complex, McCune-Albright syndrome, familial acromegaly and genetic defects in sporadic tumors. Rev Endocr Metab Disord. 2008;9:1-11.
Lania A, Spada A. G-protein and signalling in pituitary tumours. Horm Res. 2009;71(Suppl 2):95-100.
Leventopoulos G, Kitsiou-Tzeli S, et al. A clinical study of Sotos syndrome patients with review of the literature. Pediatr Neurol. 2009;40:357-364.
Melmed S, Colao A, Barkan A, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94:1509-1517.
Melmed S, Cook D, Schopohl J, et al. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13:18-28.
Misra M, Cord J, Prabhakaran R, et al. Growth hormone suppression after an oral glucose load in children. J Clin Endocrinol Metab. 2007;92:4623-4629.
Morison IM, Becroft DM, Taniguchi T, et al. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat Med. 1996;2:311-316.
Narayanaswamy V, Rettig KR, Bhowmick SK. Excessive growth. Clin Pediatr (Phila). 2008;47:705-708.
Prevedello DM, Pouratian N, Sherman J, et al. Management of Cushing’s disease: outcome in patients with microadenoma detected on pituitary magnetic resonance imaging. J Neurosurg. 2008;109:751-759.
Reddy R, Hope S, Wass J. Acromegaly. BMJ. 2010;341:c4189.
Reeve AE. Role of genomic imprinting in Wilms’ tumour and overgrowth disorders. Med Pediatr Oncol. 1996;27:470-475.
Riccio A, Sparago A, Verde G, et al. Inherited and sporadic epimutations at the IGF2-H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumor. Endocr Dev. 2009;14:1-9.
Rosenfield RL, Lipton RB, Drum ML. Thelarche, pubarche, and menarche attainment in children with normal and elevated body mass index. Pediatrics. 2009;123:84-88.
Sacks DA. Etiology, detection, and management of fetal macrosomia in pregnancies complicated by diabetes mellitus. Clin Obstet Gynecol. 2007;50:980-989.
Simm PJ, Bajpai A, Russo VC, et al. Estrogens and growth. Pediatr Endocrinol Rev. 2008;6:32-41.
Socin HV, Chanson P, Delemer B, et al. The changing spectrum of TSH-secreting pituitary adenomas: diagnosis and management in 43 patients. Eur J Endocrinol. 2003;148:433-442.
Sotos JF. Overgrowth: genetic syndromes and other disorders associated with overgrowth. Clin Pediatr. 1997;36:157-170.
Sotos JF, Argente J. Overgrowth disorders associated with tall stature. Adv Pediatr. 2008;55:213-254.
Tajima T, Tsubaki J, Ishizu K, et al. Case study of a 15-year-old boy with McCune-Albright syndrome combined with pituitary gigantism: effect of octreotide-long acting release (LAR) and cabergoline therapy. Endocr J. 2008;55:595-599.
Topaloglu AK, Sansaricq C, Snyderman SE. Influence of metabolic control on growth in homocystinuria due to cystathionine B-synthase deficiency. Pediatr Res. 2001;49:796-798.
Venn A, Bruinsma F, Werther G, et al. Oestrogen treatment to reduce the adult height of tall girls: long-term effects on fertility. Lancet. 2004;364:1513-1518.
Verge CF, Mowat D. Overgrowth. Arch Dis Child. 2010;95:458-463.
Zeger MP, Zinn AR, Lahlou N, et al. Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome. J Pediatr. 2008;152:716-722.





