[level-membership-for-pediatrics-category]
Chapter 644 Hyperpigmented Lesions
Lentigines
Lentiginosis profusa involves innumerable small, pigmented macules that are present at birth or appear during childhood. There are no associated abnormalities, and mucous membranes are spared. LAMB syndrome (Carney complex), a multiple endocrine neoplasia syndrome, consists of lentigines of the face and vulva, atrial myxoma, mucocutaneous myxomas, and blue nevi (type 1, PRKAR1 gene; type 2, gene map locus 2p16-gene as yet to be identified). The Carney complex variant is associated with distal arthrogryposis (MYH8 gene). The multiple lentigines (LEOPARD) syndrome is an autosomal dominant entity consisting of a generalized, symmetric distribution of lentigines (Fig. 644-1) in association with electrocardiogram abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitals (cryptorchidism, hypogonadism, hypospadias), growth retardation, and sensorineural deafness (type 1, PTPN11 gene; type 2, RAF1 gene). Other features include hypertrophic obstructive cardiomyopathy and pectus excavatum or carinatum.
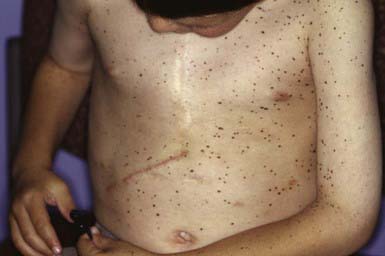
Polyposis usually involves the jejunum and ileum but may also occur in the stomach, duodenum, colon, and rectum (Chapter 337). Episodic abdominal pain, diarrhea, melena, and intussusception are frequent complications. Patients have a significantly increased risk of GI tract and non–GI tract tumors at a young age. GI cancer has been reported in ≈ 2-3% of patients; the lifetime relative risk for GI malignancy is 13. The relative risk of non–GI tract malignancies, including ovarian, cervical, and testicular tumors, is 9. Peutz-Jeghers syndrome must be differentiated from other syndromes associated with multiple lentigines (Laugier-Hunziker syndrome), from ordinary freckling, from Gardner syndrome, and from Cronkhite-Canada syndrome, a disorder characterized by GI polyposis, alopecia, onychodystrophy, and diffuse pigmentation of the palms, volar aspects of the fingers, and dorsal hands. Treatment of Peutz-Jeghers melanotic macules with multiple different lasers has been successful, in some cases.
Café-Au-Lait Spots
Café-au-lait spots are uniformly hyperpigmented, sharply demarcated macular lesions, the hues of which vary with the normal degree of pigmentation of the individual: They are tan or light brown in white individuals and may be dark brown in black children (Figs. 644-2 and 644-3). Café-au-lait spots vary tremendously in size and may be large, covering a significant portion of the trunk or limb. Generally the borders are smooth, but some have exceedingly irregular borders. The lesions are characterized by increased numbers of melanocytes and melanin in the epidermis but lack the clubbed rete ridges that typify lentigines. One to 3 café-au-lait spots are common in normal children; ≈ 10% of normal children have café-au-lait macules. The spots may be present at birth or may develop during childhood.
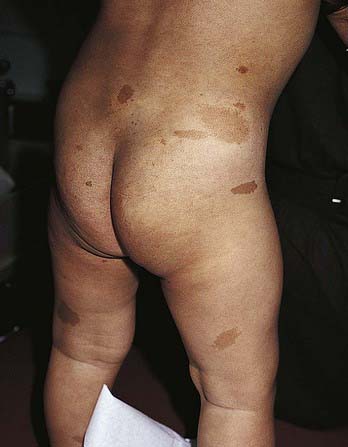
Figure 644-2 Multiple café au lait macules on a child with neurofibromatosis type 1.
(From Eichenfield LF, Frieden IJ, Esterly NB: Textbook of neonatal dermatology, Philadelphia, 2001, WB Saunders, p 372.)
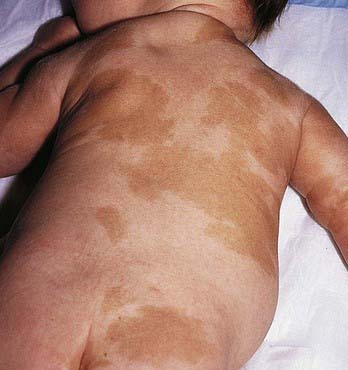
Figure 644-3 Multiple patterned café-au-lait spots in a child with McCune-Albright syndrome.
(From Eichenfield LF, Frieden IJ, Esterly NB: Textbook of neonatal dermatology, Philadelphia, 2001, WB Saunders, p 373.)
Large, often asymmetric café-au-lait spots with irregular borders are characteristic of patients with McCune-Albright syndrome (GNAS1 gene) (Chapter 556.6). This disorder includes polyostotic fibrous dysplasia of bone, leading to pathologic fractures; precocious puberty; and numerous hyperfunctional endocrinopathies. The macular hyperpigmentation may be present at birth or may develop late in childhood (see Fig. 644-3). Cutaneous pigmentation is typically most extensive on the side showing the most severe bone involvement.
Neurofibromatosis Type 1 (Von Recklinghausen Disease)
The café-au-lait spot is the most familiar cutaneous hallmark of the autosomal dominant neurocutaneous syndrome known as neurofibromatosis type 1 (neurofibromin gene) (see Fig. 644-2 and Chapter 589.1). The lesions also occur with certain other disorders, including other types of neurofibromatosis, but in these disorders the café-au-lait spots are not a major feature of the disorder and do not aid in diagnosis (Table 644-1). Included in the criteria for this diagnosis is the presence of 5 or more café-au-lait spots >5 mm in diameter in prepubertal patients or 6 or more café-au-lait spots >15 mm in diameter in postpubertal patients. Multiple café-au-lait macules commonly produce a freckled appearance of non–sun-exposed areas such as the axillae (Crowe sign), the inguinal and inframammary regions, and under the chin.
Incontinentia Pigmenti (Bloch-Sulzberger Disease)
See Chapter 589.7 for a full discussion of this condition.
Boikos SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Oncol. 2007;19:24-29.
McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci. 2006;63:2135-2144.
Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
Williams VC, Lucas J, Babcock, et al. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124-133.
Zacharin M. The spectrum of McCune Albright syndrome. Pediatr Endocrinol Rev. 2007;Suppl 4:412-418..
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 644 Hyperpigmented Lesions
Lentigines
Lentiginosis profusa involves innumerable small, pigmented macules that are present at birth or appear during childhood. There are no associated abnormalities, and mucous membranes are spared. LAMB syndrome (Carney complex), a multiple endocrine neoplasia syndrome, consists of lentigines of the face and vulva, atrial myxoma, mucocutaneous myxomas, and blue nevi (type 1, PRKAR1 gene; type 2, gene map locus 2p16-gene as yet to be identified). The Carney complex variant is associated with distal arthrogryposis (MYH8 gene). The multiple lentigines (LEOPARD) syndrome is an autosomal dominant entity consisting of a generalized, symmetric distribution of lentigines (Fig. 644-1) in association with electrocardiogram abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitals (cryptorchidism, hypogonadism, hypospadias), growth retardation, and sensorineural deafness (type 1, PTPN11 gene; type 2, RAF1 gene). Other features include hypertrophic obstructive cardiomyopathy and pectus excavatum or carinatum.
[/not-level-membership-for-pediatrics-category]
