Chapter 697 Hyperphosphatasia
Hyperphosphatasia is characterized by excessive elevation of the bone isoenzyme of alkaline phosphatase in serum and significant growth failure. Osteoid proliferation in the subperiosteal portion of bone results in separation of the periosteum from the bone cortex. Bowing and thickening of the diaphyses are common, along with osteopenia (see Fig. 697-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com  ). The disease usually has its onset by 2-3 yr of age, when painful deformity developing in the extremities leads to abnormal gait and sometimes fractures. Other common findings include pectus carinatum, kyphoscoliosis, and rib fraying. The skull is large, and the cranium is thickened (widened diploë) and may be deformed. Skull involvement can lead to progressive and profound hearing loss. Radiographically, the bony texture is variable; dense areas (showing a teased cotton-wool appearance) are interspersed with radiolucent areas and general demineralization. Long bones appear cylindrical, lose metaphyseal modeling, and contain pseudocysts that show a dense, bony halo.
). The disease usually has its onset by 2-3 yr of age, when painful deformity developing in the extremities leads to abnormal gait and sometimes fractures. Other common findings include pectus carinatum, kyphoscoliosis, and rib fraying. The skull is large, and the cranium is thickened (widened diploë) and may be deformed. Skull involvement can lead to progressive and profound hearing loss. Radiographically, the bony texture is variable; dense areas (showing a teased cotton-wool appearance) are interspersed with radiolucent areas and general demineralization. Long bones appear cylindrical, lose metaphyseal modeling, and contain pseudocysts that show a dense, bony halo.
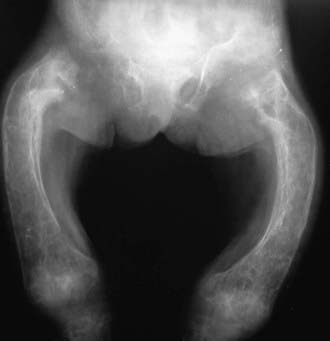
Figure 697-1 Hyperphosphatasia showing bowing and thickening of the diaphyses and osteopenia.
(From Slovis TL, editor: Caffey’s pediatric diagnostic imaging, ed 11, vol 2, Philadelphia, 2008, Mosby.)
Cundy T, Wheadon L, King A. Treatment of idiopathic hyperphosphatasia with intensive bisphosphonate therapy. J Bone Miner Res. 2004;19:703-711.
Whyte MP, Hughes AE. Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RANK and is allelic to familial expansile osteolysis. J Bone Miner Res. 2002;17:26-29.
Whyte MP. Enzyme defects and the skeleton. In: Rosen CJ, Compston JE, Lian JB, editors. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society for Bone and Mineral Research; 2008:454-458.





