[level-membership-for-surgery-category]Chapter 38
Hypercoagulable States
Howard A. Liebman, Ilene Ceil Weitz
Thromboembolism is an enormous problem across the spectrum of medicine. Approximately 10 million individuals in the United States have symptomatic peripheral artery disease, and approximately 100,000 arterial reconstructive procedures are performed annually.1 In the United States and Europe, more than 600,000 people will experience a venous thromboembolism (VTE), and nearly 20% will die as a result.2 The incidence of arterial embolism and VTE is highly age dependent, contributing significant morbidity and mortality to an aging population.3–5
More than one hundred fifty years ago, the great German pathologist, Rudolf Virchow proposed the concept known as Virchow’s triad. He observed that thrombosis occurred when the following three situations were present; vascular wall (endothelial) injury, stasis of blood, and changes in the consistency of blood that enhance coagulation (hypercoagulability). An understanding of the latter, now termed the hypercoagulable state, was least apparent and could not be further characterized until there was a greater understanding of the normal mechanisms of hemostatic regulation (see Chapter 34). In the last 40 years, a number of inherited and acquired hemostatic disorders have been characterized that are associated with an increased risk of venous and/or arterial thrombosis. In most circumstances, the relationship between specific hemostatic defect and the hypercoagulable state is readily apparent by the respective role in normal hemostasis. However, the mechanism(s) by which other well-documented hypercoagulable defects, such as the antiphospholipid syndrome or elevated homocysteine, contribute to an increased risk of venous and arterial thrombosis remain poorly understood. The interactions of these hypercoagulable defects, alone or in combination, or when combined with other transient factors, such as vascular injury from surgery or trauma, can result is a significant increased risk of thrombosis. The interaction between inherited hypercoagulable defects and acquired additional factors, such as age or acute illness, can result in what has been termed an increased “thrombosis potential,” which upon reaching a “thrombosis threshold,” can result in symptomatic thromboembolism.6
Pathophysiology
Hemostasis is a highly organized series of reactions involving platelet adhesion and activation to form the platelet plug, followed by activation of coagulation proteins in a series of controlled enzymatic reactions to generate thrombin. Thrombin potentiates platelet aggregation and activation and acts on fibrinogen to generate the insoluble fibrin clot. Under normal conditions, these reactions occur on the endothelial surface only at sites of endothelial injury. This limits the size of the clot and allows blood to remain liquid and flowing. When this balance is altered, excess thrombin is generated, and abnormal thrombosis may occur. Therefore, thrombosis can result from defects in normal hemostasis that increase either the procoagulant activity or decrease the naturally occurring anticoagulants. These hypercoagulable defects have been termed thrombophilic syndromes and have been further classified as either congenital or acquired thrombophilias.6,7
In addition, there are a group of miscellaneous disorders whose prothrombotic mechanisms are poorly understood. These include elevations of homocysteine (hyperhomocysteinemia) and lipoprotein (a) (Lp[a]). The management of patients with thrombosis with these disorders remains controversial.8–13
Arterial thrombosis, unlike VTE, is associated with conditions that primarily affect the vascular wall and endothelium. Endothelial disruption, which occurs with atherosclerosis, vasculitis (vascular inflammation), infection, trauma, or surgery, is most often associated with an arterial thrombotic event. In well-controlled population studies, the majority of congenital hypercoagulable defects associated with venous thrombosis are not associated with a statistical increased risk of arterial thromboembolism.14–17 The few congenital hypercoagulable defects that may be associated with an increased risk of both venous and arterial thrombosis, such as hyperhomocysteinemia or increased Lp(a), are also associated with an increased risk of arterial atherosclerosis.8,9,18–20 Other defects associated with an increased risk of arterial thromboembolism, such as those observed with increased levels of fibrinogen and von Willebrand factor, are known to enhance platelet function.21,22
Congenital Hypercoagulability
Although the thrombosis potential or risk of thrombosis for inherited prothrombotic defects have been well characterized in a number of prospective and retrospective population studies, it is important to understand that thrombosis is a multigenetic and multifactor disorder. The thrombosis potential for specific congenital thrombophilic abnormalities is classically defined as a relative risk of thrombosis compared with a patient population without these abnormalities.6,7,14–16 In most circumstances, patients who inherit more than one abnormality have a significantly higher risk of thrombosis potential or relative risk of thrombosis.6,7,14–16,23 However, patients may have additional risk factors, such as aging, oral contraceptives (OCs) use, hormone replacement therapy (HRT) use, pregnancy, cancer, infection, trauma, or surgery. In these circumstances, a patient’s individual risk can also be increased by a factor that is more than the sum of each individual risk factor.23
Classification
Crowther and Kelton7 proposed a simple classification system that divides the congenital hypercoagulable (thrombophilic) states associated with VTE into two broad groups (Table 38-1). The first group constitutes defects associated with the reduced levels of the natural anticoagulants, such as antithrombin, protein C, and protein S.7 The defects in this “loss of inhibition” group are much less common, but are associated with a significantly higher risk of thrombosis.7 The second group is associated with defects that result in a “gain in procoagulant function” due to increased levels or function of coagulation factors.7 These include factor V Leiden (activated protein C resistance [APCR]) and prothrombin G20210 mutations, increases in coagulation factors VIII, IX, and XI, and the dysfibrinogenemias. Although these defects have a lower thrombosis potential, they are more frequently found in the general population, and therefore, more commonly associated with clinical thrombosis. The understanding that certain defects alone or in combination can be associated with a significantly higher risk of thrombosis may be important in determining the physician’s approach to antithrombotic prophylaxis, duration of anticoagulation after venous thrombosis, and family screening for patients with these defects.
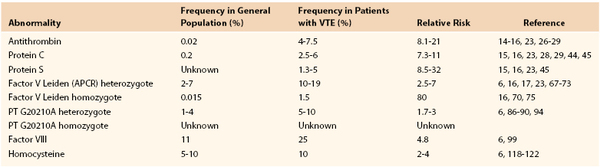
Group 1 Thrombophilia
Group 1 includes deficiencies of the naturally occurring anticoagulant factors antithrombin (AT-III), protein C, and protein S. All are rare, representing less than 1% of the population.7 However, they are highly prothrombotic, with 30% to 50% of carriers (heterozygote) having a symptomatic thrombotic event before they reach 60 years of age.7 A significant number of carriers will have had a spontaneous thromboembolic event before the age of 40 years. Frequently, there is a strong family history of venous thrombosis. Although the risk of thrombosis is high, routine prophylactic anticoagulation in ambulatory healthy individuals has not been demonstrated to be of benefit and should be reserved for high-risk situations, such as surgery, sepsis, pregnancy, and immobilization.7 Because of the high risk of recurrence, with group 1 deficiencies and patients homozygous for certain group 2 abnormalities, lifelong anticoagulation may be recommended after a spontaneous thrombotic event.7,24
Antithrombin Deficiency
Pathogenesis and Incidence.
Physiologically, AT-III is the most important inhibitor of thrombin and other activated clotting factors (e.g., factors Xa, IXa, and VIIa). AT-III physiologic activity is enhanced 1000-fold by the binding of naturally occurring or administered heparin or heparin sulfates.25 AT-III deficiency is rare, reportedly occurring in an estimated 0.02% of the general population in a study that screened samples from healthy blood donors.26,27 It has been reported in 4% to 7.5% of patients with VTE.14–16,28,29 AT-III inheritance is autosomal dominant.30 More than 250 mutations within the molecule have been described.31 Homozygosity, particularly for AT-III deficiency types I and II, is extremely rare and appears to be incompatible with life. Homozygosity for type III AT-III deficiency results in a severe thrombophilic phenotype.32
Types.
There are three general subtypes of AT-III deficiency.31 Type I is characterized by decreased AT-III functional activity and antigen. Type II defects are AT-III mutations that have reduced functional activity, but normal antigen levels resulting from a mutation in the active inhibitory site on the protein. Type III AT-III mutations are characterized by moderate decreased activity due to impaired interaction with heparin. Screening for AT-III deficiency should always be undertaken using a functional assay, because screening with an antigen assay may fail to diagnose type II and III defects. In the presence of low AT-III activity, further characterization can be made with an antigen assay.
Patients with AT-III deficiencies are at a significantly higher risk of thrombosis than patients with other congenital deficiencies. Approximately 60% of carriers of type I and II deficiencies will have a thrombotic event by the age of 60 years. The risk for type III deficiency may be lower.33,34 A strong family history of thrombosis is usually present. A Spanish Multi-Center study reported the relative risk of thrombosis in these thrombophilic families as 21-fold.29 A multicenter, multinational European Prospective Cohort on Thrombophilia (EPCOT) study reported an adjusted relative thrombotic risk of 17.5 (95% confidence interval [CI], 9.1-33.8).23 After the diagnosis of AT-III deficiency is made in an individual with thrombosis, screening of family members is recommended.
Clinical Presentation and Management.
The clinical presentation of AT-III deficiency is predominantly lower extremity thrombosis with or without pulmonary embolism.30,33,35 Recurrent events are common, and atypical thrombotic events involving the portal, mesenteric, and hepatic venous system, or cerebral veins have been reported.29,40,41 Arterial events are rare, and patients are not at increased risk more than the unaffected adult population.14–16,23 Pregnancy is a particularly high-risk situation, and prophylaxis with heparin is indicated throughout pregnancy and in the immediate postpartum period.35,36 Patients with breakthrough events can receive AT-III concentrates.37 An acquired form of AT-III deficiency has been reported in pregnant women with the fatty liver syndrome with disseminated intravascular coagulation.38 Treatment with plasma or AT-III concentrates rapidly reverses the coagulopathy.38
Protein C Deficiency
Pathogenesis and Incidence.
Protein C is a vitamin K-dependent anticoagulation protein that is activated by thrombin to activated protein C (APC).42 When thrombin levels are high, thrombin binds to the endothelial protein receptor, thrombomodulin (TM), which changes the specificity of thrombin from cleaving fibrinogen or activating platelets to activating protein C. Protein C binds to its specific endothelial receptor, termed the endothelial protein C receptor, which enhances its activation. APC is a potent serine protease anticoagulant that cleaves the coagulant cofactors VIIIa and Va, thus modulating thrombin generation and subsequent clot formation.42,43 Deficiency of protein C is found in 0.2% of the general population, and in 2.5% to 6% of patients with VTE.28,29,44,45
Types.
Similar to AT-III deficiency, multiple mutations resulting in protein C deficiency have been reported. These mutations have been classified into two general subtypes: type I mutations have reduced functional and antigenic protein levels; and type II mutations have reduced functional levels but preserved antigen levels of the protein.46 Adult heterozygous patients with protein C deficiency usually have activity levels of less than 60%.
Clinical Presentation and Management.
Patients who have VTE usually present in the lower extremities. Unusual sites of thrombosis have been reported and are similar to those seen with AT-III deficiency.42,43,45,46 Although rare arterial events have been reported, large cohort studies do not support an increased risk of arterial events.47 In protein C deficient patients from thrombophilic families who present with an unprovoked (idiopathic) thrombosis, recurrent thrombotic events are frequent. Life-long anticoagulation should be considered in these patients, particularly those who present before the age of 40 years.
Homozygosity for protein C deficiency, with absent protein C activity, may present at birth as a neonatal disorder termed purpura fulminans.48 This disorder is characterized by diffuse microvascular thrombosis of the skin and systemic organs. Immediate treatment with heparin, plasma, or protein C concentrates can be life saving.48 The majority of homozygous neonates with protein C deficiency will have functional levels less than 20% of normal.48
A similar severe disseminated thrombotic disorder, characterized by skin necrosis, can occur in heterozygous patients with protein C deficiency treated with higher doses of warfarin, usually without concomitant anticoagulation with heparin.49,50 The syndrome, termed warfarin necrosis, results from the disproportional decrease in protein C in comparison to other procoagulant vitamin K-dependent coagulation factors. Patients presenting with this disorder should be treated with fresh frozen plasma, vitamin K, and heparin.49,50 The syndrome can be prevented by initiating oral anticoagulation with lower doses of warfarin and concomitant use of heparin.
Protein S Deficiency
Pathogenesis and Incidence.
Protein S is the vitamin K-dependent cofactor necessary for the inactivation of factors Va and VIIIa by APC. A deficiency in protein S is phenotypically similar to protein C deficiency. Protein S exists in two forms: the functionally active free form that usually constitutes 20% to 40% of the total protein, and the remaining 60% to 80% that is active and bound to complement binding protein C4b.51,52 Most patients with protein S deficiency will have activity levels between 50% and 75% of normal.51–53
Types.
Similar to the other natural anticoagulant proteins, protein S deficiency can be classified into 3 subtypes: type I is characterized by reduced functional and antigen protein levels; type II has reduced functional activity, but normal antigen levels; and type III has normal antigen levels, but reduced free active protein S due to enhanced C4b binding.54 Type I and type III protein S deficiencies are the most common forms of the deficiency encountered and do not appear to differ in their risk of venous thrombosis.55
The measurement of protein S activity and antigen can be confounded by a number of physiologic and clinical conditions. In pregnancy, protein S levels fall in the second and third trimester.56,57 Reduced protein S levels have also been reported in patients with active cancer, lupus erythematosus, antiphospholipid antibody syndrome, sepsis, chronic inflammatory disorders (e.g., inflammatory bowel disease) and in advanced HIV disease.58–60
Clinical Presentation and Management.
Protein S deficiency is clinically characterized by venous thrombosis and has been frequently reported in association with venous thrombosis in atypical sites.51–53,61,62 These reports may be confounded by other comorbidities in these patients that can be associated with acquired protein S deficiency. In addition, several reports have suggested a relationship between low protein S levels and arterial thrombosis, including a reported association between low free protein S and cardiolipin antibodies in stroke patients.63
Like protein C deficiency, homozygous deficiency can be associated with neonatal purpura fulminans. Approach to management is similar to that of homozygous protein C deficiency.64 Warfarin necrosis can also develop in heterozygote protein S patients.64
Group 2 Thrombophilia
Although associated with a lower thrombosis potential than Group 1 thrombophilia, Group 2 hypercoagulable disorders are more frequently found in the general population and are found in a greater proportion of patients with VTE. They represent gain of function mutations resulting in increased thrombin generation. They are typified by either increased synthesis of specific coagulation factors or as observed with factor V Leiden, resistance to the inactivation of this cofactor by APC.
Factor V Leiden (Activated Protein C Resistance)
Pathogenesis and Incidence.
Factor V is a cofactor that accelerates the conversion of factor II (prothrombin) to thrombin by factor Xa. Under normal circumstances, factor V is degraded by the serine protease, APC, which cleaves the protein at two sites. Factor V Leiden has a mutation in the 506 position that results in a substitution of glycine for arginine. This renders one of the factor V cleavage sites resistant to the action of the APC, which, like all coagulation serine proteases, cleaves arginine bonds.65–67 This results in a slowing of the inactivation of the cofactor, which leads to increased thrombin generation.
Factor V Leiden is a common mutation, occurring in approximately 2% to 7% of individuals of European ancestry.67–69 It can be found in nearly 10% of people with VTE and in 30% to 50% of individuals being evaluated for thrombophilia.67–70 The mutation is very rare in native Asians and Africans.69,70 The mutation is also rare in African Americans (0.6%) and has not been reported in Native Americans.69,70 Compared with patients with group 1 thrombophilia, patients with factor V Leiden have a lower relative risk of thrombosis, estimated between 2.5- and 7-fold.16,17,23,71–74 A cross-sectional study from Italy found that only 6% of carriers of the mutation developed a thromboembolic event by the age of 65 years.75 Homozygotes are at much higher risk then heterozygotes, with an estimated relative risk of 80-fold.16,71,76 However, heterozygotes with combined defects have a significantly increased risk of VTE.16,23,75 A second, very rare, mutation of the second factor Va cleavage site (Arg306Thr) has also been reported to be associated with increased risk of thrombosis, whereas other studies have failed to find a relationship of this mutation with the development of VTE.77
Clinical Presentation and Management.
The clinical presentation of thrombosis with factor V Leiden is overwhelmingly venous. Although rare, unusual sites, such as cerebral vein and retinal vein thrombosis, have been reported.78,79 The onset of thrombosis is frequently at an older age than seen in patients with type 1 thrombophilia.17 In the Physicians Health Study, the risk of VTE in men with the factor V Leiden mutation did not become statistically significant until after the age of 50 years.17 The study also found no association with the factor V Leiden and an increased risk of stroke or myocardial infarction.17 Thrombosis is frequently triggered by transient risk factors, such as a prolonged plane flight, oral contraceptive (OC) use, or pregnancy.80–83 In women carriers of the mutation, the reported thrombosis risk associated with the use of second-generation OCs varies widely, ranging from 6- to 50-fold.80–83 The wide variation reported in the thrombotic risk of OCs may be explained, in part, by a higher risk of thrombosis in women more than 30 years old who are taking OCs and some variation in the specific OCs used in the European studies, some of which contain prothrombotic progesterone substitutes.83
There is no excess mortality in carriers of the factor V Leiden mutation. No prophylaxis is recommended for carriers except what is recommended by guidelines for surgical interventions. Antepartum prophylaxis is not recommended for women who have not had a previous thrombotic event or a history of recurrent fetal loss.84 The risk of recurrent VTE after cessation of oral anticoagulation is not greater than that observed in other patients with unprovoked VTE.85
Prothrombin Gene Mutation G20210A
Pathogenesis and Incidence.
The prothrombin G20210A mutation is an abnormality located at the untranslated 3′ end of the prothrombin gene that results in increased plasma levels of prothrombin. The mutation affects the 5′ end cleavage signal, leading to increased prothrombin mRNA stability.86 The thrombotic risk is relatively low.87–89 The gene frequency in the European population is about 1% to 4%, with the greatest frequency in southern Europe and Spain.90–91 It is present in 5% to 10% of patients with VTE, and 15% of patients with thrombophilia.87–91 Similar to factor V Leiden, the prothrombin G20210A mutation is very rare in native Asians, Africans, African Americans, and Native Americans.92 Many carriers with a history of VTE have co-inheritance of the factor V Leiden mutation.23,93 The relative risk of thrombosis is low in carriers of the prothrombin mutation, ranging from two- to threefold higher in carriers compared with noncarriers of the mutation.94,95
Clinical Presentation and Management.
The clinical presentation is predominantly venous thrombosis of the lower extremities, and unusual sites are rare.87–89,94,95 Some controversy exists as to whether the prothrombin mutation is associated with an increased risk of arterial thrombosis.95–97 In carriers of the mutation who develop thrombosis, studies to date have shown either no increase in risk or only a slight increased risk of recurrent VTE after discontinuation of anticoagulation.95,98
The risk of VTE in women on OCs has been reported to be significantly increased, similar to that reported for factor V Leiden.94 Women who are carriers of both the prothrombin G20210A mutation and the factor V Leiden mutation have a markedly high risk of VTE.94 An association between the development of cerebral venous thrombosis in women carriers of the prothrombin mutation taking OCs has been reported, in which 20% of patients carried the mutation versus only 3% of controls.99
Elevated Factors VII, XI, and IX
Pathogenesis and Incidence.
Koster et al.100 first reported an increased risk of VTE in patients with an elevated factor VIII coagulant protein of more than the 90th percentile (>150%). Factor VIII activity levels more than 150% were associated with an adjusted relative risk of 4.8% compared with factor VIII levels 100% or less.100 These elevations appear to be independent of ABO blood type and were measured in patients without evidence of inflammation confirmed by a normal erythrocyte sedimentation rate (ESR) and C-reactive protein.101,102
Subsequent studies have supported this observation and found both a family and racial clustering suggestive of an inherited propensity for increased factor VIII levels.101,103 A study of African-American women found a statistically higher level of factor VIII compared with the white and Asian population in the United States.104 Unlike female carriers of the factor V Leiden and prothrombin mutations, the use of OCs did not appear to significantly increase the risk of VTE.105 Elevations of factor VIII activity have also been associated with an increased risk of recurrent VTE.106
The measurement of factor VIII activity is confounded by the fact that factor VIII, along with its carrier protein, von Willebrand factor, is an acute phase reactant and increases with bleeding and inflammation.101,102 Therefore, measurement of factor VIII activity should be performed with a simultaneous acute phase marker, such as the ESR or C-reactive protein. The assay should also be repeated at least twice at distant time intervals.100–102 Each laboratory must define the 90th percentile range for its own population to determine the patient population at risk.
Clinical Presentation and Management.
The clinical presentation of patients with elevations in factor VIII activity is predominantly VTE of the lower extremities.100 Because factor VIII levels increase with inflammation, elevation of this coagulation protein is an important cofactor in the development of thrombosis associated with infection, inflammatory bowel disease, and cancer. There is controversy as to whether increased levels of factor VIII are associated with arterial thrombosis, because elevations in factor VIII activity are associated with increases in von Willebrand factor. Increased levels of von Willebrand factor have been shown in population-based studies to be associated with an increased risk of arterial thrombosis.107
Increased levels of factors IX and XI have been associated with a twofold increase in risk of VTE.108,109 These are relatively weak risk factors for thrombosis, but if combined with other defects, they may become significant. To date, no molecular markers have been reported to characterize these elevations in factors VIII, IX, and XI.110
Hyperhomocysteinemia
An association was made between markedly elevated levels of homocysteine and arterial vascular disease in individuals with homocysteinuria.111,112 Additional studies confirmed this association and found additional evidence that individuals with homocysteinuria also experience an increased incidence of VTE.111 Subsequent studies found an association between elevations of plasma homocysteine and an increased risk of atherosclerosis and arterial thrombosis in apparently healthy individuals.113–121 Additional studies that focused on risk factors for venous thrombosis reported a similar association.121–124
Homocysteine metabolism involves two enzymatic pathways that require essential vitamin cofactors: folate, vitamin B12, and vitamin B6 (pyridoxine).125 The vitamine B6-dependent pathway involves the enzymatic conversion of homocysteine to cystathionine by the enzyme cystathionine-β-synthase (CBS).112,125 The second pathway involves re-methylation of homocysteine back to methionine requiring folate, vitamin B12, and the two enzymes 5,10-methylenetetrahydrofolatate reductase (MTHFR) and methionine synthetase.125 The primary mutations responsible for homocysteinuria are in the CBS gene.112 However, in the general population, independent of deficiencies in the vitamin cofactors, a common mutation in the MTHFR gene at position 677 is most frequently associated with elevations in homocysteine.125 This C to T mutation results in a 50% reduction in the enzymatic activity of the gene.125 However, even in MTFR C677T homozygotes, folate supplementation or a diet intrinsically rich in folate can reduce homocysteine levels into the normal range.125,126
The reported mechanisms by which homocysteine increases the risk of both arterial and venous thrombosis include a direct toxic effect on endothelium, enhanced platelet activation, oxidation of low-density lipoprotein cholesterol, an inflammatory decrease in endothelial TM, and an increase in von Willebrand factor and factor VIII.125 However, no single reported effect of homocysteine adequately explains its prothrombotic effect in both the arterial and venous vasculature.
A number of cohort studies and some prospective studies have found a statistically significant association between elevated homocysteine and an increased risk for myocardial infarction and stroke with estimated risks of 2- to 7.8-fold for individuals in the highest quartile.113–115 However, several prospective studies have failed to confirm this association.116–118 Data from the Physicians’ Health Study,118 the Atherosclerosis Risk in Communities Study,119 and the Women’s Health Study116,117 suggest a gender-defined risk for elevated homocysteine in which the risk is most significant in women. Elevations of homocysteine have also been found in case-controlled studies to be a weak risk factor for VTE, with a relative risk of only 2 to 4.120–124 Meta-analyses further confirmed an increased risk in patients with homocysteine levels more than the 95th percentile.124
Complicating the issue of elevated homocysteine as a risk factor for both arterial and VTE disease is the lack of response, in randomized controlled trials, to lowering homocysteine in reducing the risk of recurrent arterial10–12 and VTE13 disease in patients treated with vitamin therapy.
The measurement of homocysteine in patients can be complicated by comorbid conditions, such as vitamin deficiency, renal insufficiency, and improper plasma collection and handling. Measurements are best performed using freshly collected plasma, preferentially with patients in the fasting state. It is reasonable to repeat the assay on at least two separate occasions. Studies to date do not support an advantage for determination of the MTHFR genotype over direct measurement of homocysteine.126
Lipoprotein(a)
Lp(a) is a serum lipoprotein composed of a low-density lipid particle with a disulfide link to a long polypeptide chain, apolipoprotein (a).20,127 The protein has “kringle-like” domains that compete with tissue plasminogen activator, plasminogen, and plasmin for binding to fibrin and endothelial annexin II, thereby inhibiting fibrinolysis.127,128 The plasma levels of Lp(a) vary greatly within and between different racial populations, with African Americans having higher levels.129 Also, Lp(a) may increase as an acute phase reactant130,131 and has been reported to be elevated in certain inflammatory rheumatologic disorders.132
Early retrospective studies suggested a relationship between elevated levels of Lp(a) and the risk of premature atherosclerosis and arterial thrombosis.133–135 However, prospective studies have been less supportive and at the most suggest only a small increase in risk for individuals with elevated plasma Lp(a).136–142 Additional reports have suggested a role for elevated Lp(a) as an independent risk factor for VTE,143,144 although a failure to find an association has been reported by other investigators.145
Sticky Platelet Syndrome
Sticky platelet syndrome (SPS) is an inherited, autosomal dominant disorder associated with early-onset myocardial infarction and peripheral vascular disease.146–148 Although most often associated with arterial vasculopathy, VTE has been reported.146–148 A causal relationship to early miscarriage has also been reported. Patients usually present with a history of myocardial infarction or thromboembolism as young adults. The events often are precipitated by stress.146–148 The defect appears to be common in patients with unexplained thromboembolic events. In an analysis of 200 patients and family members identified with SPS, 21% had arterial thromboembolic events and 13.2% had VTE events.147 SPS platelets demonstrate hyperreactivity to epinephrine and adenosine diphosphate (ADP) even with dilution, but have normal responses to thrombin, collagen, and arachadonic acid.146–148 Three types of platelet responses have been noted: type I is hyperreactive to both ADP and epinephrine; type II is hyperreactive to epinephrine only; and type III is hyperreactive to ADP only. Increased arterial events may be mediated through polymorphisms of glycoprotein IIIa allele PLA-A2.149 Although other investigators have reported similar hyperreactivity of platelets in patients with ischemic stroke,150 there remains controversy as to whether SPS represents a true congenital prothrombotic syndrome. In patients with documented platelet reactivity and thrombosis, low-dose acetylsalicylic acid (81 mg) appears to be effective in inhibiting platelet hyperreactivity.148
Idiopathic (Unprovoked) Venous Thrombosis
Unprovoked or idiopathic venous thrombosis is defined as the development of deep vein thrombosis and/or pulmonary embolism in the absence of known genetic prothrombotic mutations, permanent factors, or acquired risk factors. Patients who develop VTE provoked by surgery, trauma, immobilization, pregnancy, or female hormone intake are at low risk of recurrent thrombosis.151,152 Patients with unprovoked VTE have a recurrence risk of 5% to 10% per year, with nearly 50% developing a recurrent event by 10 years.151,152 A prospective study that screened 66 patients with idiopathic venous thrombosis found abnormalities in 26 (39.3%) patients when they were screened for thrombophilia (antithrombin, protein C, protein S, factor V Leiden, prothrombin G20210A, antiphospholipid antibodies).153 Therefore, 60% of patients with unprovoked VTE have no underlying known major inherited or acquired thrombophilia and still remain at high risk of recurrent VTE. It has been proposed that an underlying inflammatory state may be responsible for the increased risk of VTE in patients with unprovoked events.154,155 Laboratory assessment of patients with idiopathic VTE have demonstrated higher levels of interleukin-6 and -8 with low levels of interleukin-10 compared with age- and sex-matched controls.156 It is notable that the same inflammatory markers are linked to an increased risk of atherothrombotic disease.154–157
Acquired Hypercoagulability
An increased risk of thrombosis can be associated with a variety of acquired abnormalities. Disorders such as the antiphospholipid antibody syndrome (APLAS) and cancer are significant prothrombotic syndromes.158,159 Common clinical situations such as cancer, pregnancy, infection, and estrogen use are transient causes of hypercoagulability.160–162 Surgery is a well-documented transient cause of hypercoagulability in which the risk of thrombosis is related to a number of factors, such as the type of surgery, duration of surgery, age of the patient, and other patient comorbidities.162 A significant iatrogenic thrombotic disorder is heparin-induced thrombocytopenia (HIT), in which early diagnosis and treatment may be life-saving.163 Chronic inflammatory and autoimmune disorders, such as inflammatory bowel disease, Behcet’s syndrome, or lupus erythematosus, with and without antiphospholipid antibodies, are also associated with a significant risk of thrombosis.164–167
Acquired elevations in factor VIII levels or depressions in protein S may be an important contributing factor to a number of prothrombotic disorders, including cancer, pregnancy, infection, and chronic inflammatory disorders. In patients with cancer, the expression of tissue factor by the malignant cell further increases the thrombotic risk.168,169 Paroxysmal nocturnal hemoglobinuria, a rare hemolytic disorder in which nearly half of the patients develop symptomatic thrombosis, has been shown to be associated with both increased platelet activation and increased leukocyte tissue factor expression.170,171 Thrombosis in patients with myeloproliferative syndromes frequently involves unusual sites, such as the portal and hepatic veins, and is more often associated with syndromes associated with the Janus kinase-2 (JAK-2) mutation.172–175 The presence of JAK-2 mutation has also been shown to be associated with thrombosis in the splanchnic, portal, and hepatic veins, even in the presence of normal blood counts.175
Antiphospholipid Antibody Syndrome
The APLAS is one of the more common causes of acquired hypercoagulability.158,159 The syndrome is associated with an increased risk of both arterial and venous thrombosis.158,159 Although the APLAS can occur with systemic lupus erythematosus (SLE), 50% of the patients do not fulfill criteria for lupus or other autoimmune disorders.176 The syndrome is characterized by the presence of antibodies that inhibit in vitro coagulation reactions, the lupus anticoagulant (LA), and antibodies that bind to anticardiolipin and β2-glycoprotein.158,159 Clinical criteria include a history of either thrombosis and/or pregnancy complications with fetal loss.177
Pathogenesis and Incidence
There is strong epidemiologic evidence to support a relationship between the presence of these antiphospholipid antibodies and thrombosis.158,159,178 There appears to be a differential risk with the highest risk of thrombosis associated with the presence of an LA and high titer immunoglobulin-G (IgG) anticardiolipin antibody.158,178 The Leiden Thrombophilia Study, a large population-based, case-control study of unselected patients with a first episode of venous thrombosis, found a 3.6-fold increased risk for deep venous thrombosis for individuals positive for LA (odds ratio [OR], 3.6; 95% confidence interval [CI], 1.2-10.9).178 However, patients who were positive for both the LA and either antiprothrombin or anti-β2-glycoprotein-1 antibodies had an estimated 10-fold increased risk of VTE (OR, 10.1; 95% CI, 1.3-79.8).
The syndrome is something of a paradox in that this thrombotic disorder is characterized by antibodies that prolong in vitro coagulation tests, most commonly, the activated partial thromboplastin time (aPTT). The mechanism(s) by which these antibodies result in thrombosis remains unclear.179 Activation of monocytes, platelets, and endothelial cells by antibody/β2-glycoprotein-1 complexes has been implicated in the etiology of thrombotic events.180–183 Antibodies to annexin II on endothelial cells, tissue plasminogen activator, and plasmin have been proposed as additional antigenic targets.182 Complement (C5a) mediated inflammation has been demonstrated to be associated with increased thrombogenicity and recurrent fetal loss.179,183
Diagnosis
Laboratory diagnosis of the APLAS should include screening for LA with two different assay systems.177 Assessment of anticardiolipin antibodies should include screening for both IgG and IgM antibodies.177 Anti-β2-glycoprotein-1 screening can provide further confirmatory evidence for the diagnosis.177 Positive tests should be confirmed 12 weeks after the initial screen, because transiently positive values may be seen during infections, autoimmune hemolytic anemia, and with malignancy.177
Clinical Presentation and Management
APLAS has been associated with both arterial and VTE.158,159,176 No laboratory test can distinguish patients at risk for either an arterial or venous event. However, patients who have recurrent events most often recur in the vascular sites of previous thrombosis.184 Thrombosis at unusual sites is well described with the APLAS, including multiple arterial emboli due to nonbacterial thrombotic endocarditis.184 Retrospective studies have reported a risk of recurrence as high as 50%.185,186 However, recent prospective treatment trials have estimated the risk to be somewhat lower, but still significant, with a relative risk of recurrence after stopping oral anticoagulants of 4- to 7.7-fold.187–189 This has led to consensus recommendations to consider extended or indefinite anticoagulation for patients with the APLAS and thromboembolism.24
Two prospective randomized trials in patients with APLAS associated thrombosis compared moderate- to high-intensity warfarin (prothrombin time [PT] international normalized ratio [INR] 2.0-3.0 versus 3.0-4.0) for secondary antithrombotic prophylaxis. Both trials demonstrated that moderate-intensity warfarin was equivalent to high-intensity treatment with a lower risk of bleeding complications.188,189 Assessing adequate anticoagulation with the PT INR may, at times, be difficult in the rare patient with an LA that affects the PT test.
A catastrophic form of APLAS designated as CAPS is a life-threatening variant of the disorder characterized by diffuse macro- and microvascular thrombosis with multiple organ involvement.190 Prompt recognition and treatment with corticosteroids, heparin, and in refractory cases, plasmapheresis, can be life-saving.190
Cancer
Pathogenesis and Incidence
The etiology of thrombosis in patients with cancer is complex, including expression of tissue factor by tumor cells and malignancy-induced inflammation resulting in prothrombotic hemostatic changes in blood, such as increases in fibrinogen, factor VIII, and platelet count.160,191,192 Additional prothrombotic risk factors frequently encountered by the cancer patient include compression and invasion of vessels by tumors, surgical procedures, indwelling catheters, and systemic chemotherapy.160,191–193
The association of cancer with an increased propensity for thrombosis has been recognized since 1861, when Armand Trousseau made his seminal presentation.194 According to various studies, the incidence of clinically significant VTE ranges from 1% to 43% among cancer patients, depending on the type and stage of the tumor, the modality of cancer treatment, contributing risk factors, and comorbidities.160,195–198 Cancer patients receiving chemotherapy are at a significantly higher risk of VTE.161,195,196 In comparison to patients without cancer, cancer patients have an approximately 7-fold increased risk of VTE, and with metastatic disease, the risk may be as high as 20-fold.161 Furthermore, the identification of unsuspected pulmonary emboli among cancer patients at autopsy and by highly sensitive computed tomography scanning now used for routine staging suggests that VTE remains an underdiagnosed phenomenon.198,199
Clinical Presentation and Management
Venous.
Cancer patients with VTE have a three-fold higher incidence of recurrent thrombosis and a higher risk of treatment-related bleeding than patients without cancer.200,201 In a prospective study comparing patients with and without cancer who were newly diagnosed with VTE and treated with unfractionated heparin (UFH) or low-molecular-weight heparin (LMWH) followed by oral warfarin, the 12-month cumulative incidence of recurrent VTE among the cancer patients was 20.7% versus 6.8% among patients without cancer, although the degree of anticoagulation achieved was superior among the cancer patients.201 Recurrences were more likely in patients with more extensive disease. The 1-year cumulative incidence of major bleeding was 12.4% in cancer patients, as opposed to 4.9% in patients without cancer, with no difference in the number of patients who had levels of anticoagulation above the therapeutic range.201 The results of these retrospective studies resulted in a reassessment in the use of warfarin for secondary antithrombotic prophylaxis in cancer patients with VTE. Subsequent clinical trials202,203 and resulting guidelines204,205 now recommend against the use of warfarin in favor of long-term LMWH for the management of cancer-associated VTE.
Arterial.
Arterial thromboembolic events have been reported to account for 10% to 30% of the thrombotic complications in cancer patients.160 Cancer patients, who in general, represent an older age population, have the same risk factors for arterial thrombotic events, such as stroke or myocardial infarction, as older patients without cancer. However, the impression emerges of a greater risk imposed by the presence of a malignancy. As with VTE, systemic therapy of cancer may increase the risk of arterial thrombosis. In a meta-analysis of five completed cancer trials using chemotherapy plus the antiangiogenic agent, bevacizumab, the incidence of arterial thromboembolic events was 3.8%, twice that seen in patients who received chemotherapy alone.205 The mortality associated with these events was 0.8%, again twice that seen with chemotherapy alone.
Cerebral emboli resulting in infarction may result from underlying nonbacterial thrombotic endocarditis (NBTE). NBTE may be the most common cause of stroke in patients with solid tumors.206,207 NBTE is the cardiac manifestation of systemic hemostatic activation in the cancer patient, resulting in the formation of platelet and fibrin vegetations on cardiac valves. In one study, atherosclerosis accounted for 14.5% of symptomatic cerebral infarcts compared with 18.5% of thrombotic events associated with NBTE.208 Thirty percent to 70% of patients with NBTE have laboratory evidence of overt disseminated intravascular coagulation (DIC).206,207,210 Although NBTE is more frequently diagnosed with advanced malignancy, patients with localized or occult malignancy have been reported.206,207 In autopsy series, NBTE has been detected in 0.9% to 1.3% of patients dying of cancer.209,210 Antemortem diagnosis of NBTE with transesophageal echocardiography (TEE) has the greatest diagnostic yield.207 However, a negative TEE cannot completely exclude the diagnosis of NBTE in cancer patients with new-onset stroke or other arterial thrombotic event. Hemostatic studies to look for DIC should be performed before NBTE is ruled out.207–210
Pregnancy and Oral Contraceptives
Pregnancy
Pathogenesis and Incidence.
Pregnancy induces an acquired prothrombic state due to hormone-related elevations of fibrinogen, factor VIII, reduced protein S, and depressed fibrinolysis.56,57 Therefore, it is not surprising that the incidence of VTE significantly increases for patients who have underlying inherited thrombophilia.211 Patients with congenital deficiencies of AT-III and proteins C or S are eight times more likely to have thromboembolic complications during pregnancy compared with controls and are more likely to have antepartum thromboembolism.35,81,84,212 Heligren et al81 reported, in a noncontrolled study of northern European pregnant patients with VTE, that 60% had the factor V Leiden mutation. In a case-controlled study of 119 women with a history of VTE during pregnancy and postpartum, the prevalence of factor V Leiden was 43.7% compared with 7.7% among normal women.212 The prothrombin G20210A mutation was detected in 16.9% of women with VTE compared with only 1.3% in the nonpregnant controls.212 In an Italian case-controlled study, 39% of women presenting with their first venous thrombotic event during a pregnancy had a thrombophilic defect.213 Factor V Leiden was the most commonly associated thrombophilia.221
The risk of VTE associated with pregnancy is increased four to sixfold compared with comparable age-matched nonpregnant patients.211–215 The overall incidence of VTE, as estimated by the Rochester Epidemiology Project (Olmsted County, Minnesota) from 1966 to 1995, was 199.7 per 100,000 woman-years.211 Greer214 estimated the acute antepartum risk of deep venous thrombosis to be 0.6 per 1000 in women younger than 35 years and 1.2 per 1000 in women older than 35 years. VTE is a leading cause of maternal mortality in the United States, representing 11% of maternal deaths.215 Thromboembolism can develop any time during pregnancy; the lowest risk occurs in the first trimester, but the risk does not increase during the last 20 weeks of gestation.212 The postpartum period accounts for greater than 50% of pregnancy-related VTEs.212,215–217 The incidence is higher after cesarean section than vaginal delivery.216 The left leg is affected in 90% of cases, which is felt to be due to a compressive effect on the left iliac vein by the right iliac artery where the two blood vessels cross.218–221
Clinical Presentation and Management.
Women with a history of pregnancy-associated thrombosis without documented thrombophilia can have antepartum prophylactic anticoagulation withheld, but should be followed closely and receive prophylaxis in the immediate postpartum period.84 Patients with congenital deficiencies, such as AT-III, protein C, protein S, and antiphospholipid syndrome should be considered for both ante- and postpartum prophylaxis, even if they have not had a previous thrombembolic event. Patients with the factor V Leiden mutation should receive prophylaxis in the peri- and postpartum periods (6 weeks), unless they have had an antepartum thrombosis with a previous pregnancy. Pregnant patients with active thrombosis should receive a full dose of LMWH.
Oral Contraceptives
Pathogenesis and Incidence.
Estrogen alone as HRT or when used in combination with progestin in OCs is a well-documented risk factor for venous thrombosis.222–225 A reduction in the estrogen content of OCs from 150 mcg to the present preparations containing 15 to 20 mcg resulted in a reduction of arterial thrombotic events in American studies, but not European studies.226–229 The progestagen component of OCs was changed from the second-generation compounds (levonorgestrel, norgestrione, norgestrel) to the third-generation agents (desgestrel, gestodene) in hope of lower androgenic effects and beneficial effects on high-density lipoprotein cholesterol levels. However, these third-generation progestagen compounds were associated with a doubling of the risk of VTE compared with second-generation agents.230–232
The first-generation OCs were associated with a 10-fold increased risk of VTE that decreased to a 4-fold risk with the second-generation OCs.232–234 The third-generation OCs containing the newer progestagen compounds have a two to threefold increased risk of VTE over second-generation OCs.232–234 A meta-analysis including case-controlled studies and cohorts performed before October 1995 estimated the overall adjusted ORs for third- versus second-generation OCs to be 1.7 (95% CI, 1.4-2.0).234 The role of OCs in the development of venous thrombosis in unusual sites remains controversial. A meta-analysis of studies on risk factors associated with cerebral venous thrombosis estimated a sixfold increased risk for users of OCs,235 whereas a study of risk factors for the development of portal vein thrombosis failed to document an increased risk for users of OCs.236 Retrospective studies have demonstrated that the combination of OC use and inherited thrombophilia have a multiplicative effect on the risk of VTE.82,83,94,237–239
Case-controlled studies240,241 and randomized clinical trials242–244 of HRT in postmenopausal women demonstrated a two to threefold increased risk of VTE without demonstrable benefit in reduction of cardiovascular disease. The risk appeared to be the highest in the first year and significantly higher in women with thrombophilia.245,246 A recent case-controlled study suggested that transdermal estrogen replacement might be associated with a lower risk of VTE, even in women with inherited thrombophilia.247,248
Heparin-Induced Thrombocytopenia
Heparin use in cardiac and vascular surgery patients is ubiquitous, therefore, the differential diagnosis of thrombocytopenia in these patients must include consideration of HIT. HIT is the most significant adverse event associated with heparin use. HIT is an immunopathologic disorder that places patients at a significant higher risk of developing venous and arterial thrombotic complications and is associated with a substantial mortality.163,249–255 Although HIT is associated with thrombocytopenia, HIT is not a bleeding disorder.
Pathogenesis and Incidence
HIT is an immunologic disorder in which there is a polyclonal antibody response against neoantigens expressed on platelet factor 4 (PF4) upon binding to heparin.256,265 Platelet activation is mediated solely by IgG antibodies directed against the heparin–PF4 complex.264–266 The larger heparin molecules contained in unfractionated heparin induce larger PF4 bundles, resulting in greater antibody binding. This may explain the greater incidence of HIT observed with unfractionated heparin as opposed to LMWH or fondaparinux.
Upon binding to the heparin-PF4 complex, the Fc domain of the HIT IgG antibodies activate platelets through the platelet Fc receptors.267,268 Upon activation, platelets release significant membrane microparticles,269 which express P-selectin capable of activating monocytes and inducing tissue factor expression, the initiator of the coagulation cascade.270 However, heparin-PF4/HIT IgG complexes have also been reported to independently induce tissue factor expression and cytokine release from monocytes and macrophages.271 The resulting activation of coagulation leads to significant thrombin generation, which, in turn, results in further platelet activation and thrombocytopenia. In many ways, clinical HIT can be viewed as similar to disseminated intravascular coagulation because both syndromes are mediated by excessive thrombin generation.272 Although venous thrombosis predominates over arterial thrombotic events at a rate of 4 : 1, the targeting of specific thrombotic events in patients with HIT can be explained in part by the clinical circumstances associated with the development of HIT.251 Patients who develop HIT during treatment of venous thrombosis are most likely to develop new or progressive VTE. Antecedent arterial events may precipitate additional arterial thromboses. If HIT is not diagnosed promptly and appropriately treated, thrombotic events at multiple sites can occur, as seen in patients with DIC.273 The incidence of HIT varies depending upon the patient population, duration and type of heparin utilized, and medical or surgical procedure.249–253,257,259–263
Diagnosis and Management
The diagnosis of HIT is based upon clinical findings, including the development of thrombocytopenia and/or thrombosis during or immediately following heparin use.163,250,251,256,257 Rapid-onset HIT can occur as early as 24 hours of heparin treatment in patients who have been exposed to heparin within the previous 100 days and have developed HIT antibodies.258 Although the development of thrombocytopenia or an unexplained decrease in the platelet count (>50% of pretreatment baseline) during heparin exposure is the clinical hallmark of this disorder, thrombotic sequelae can develop in 30% to 70% of HIT patients, and this accounts for much of the morbidity associated with this diagnosis.252,253,256,257 The thrombotic death rates in these retrospective studies are between 4% and 5%.252,253,256,257 Thrombotic complications may occur even when the platelet count nadir remains greater than 150 × 109/L.256 Severe thrombocytopenia (platelet count <20 K) is rare with HIT. In large clinical studies of HIT, the median platelet count nadir has been in the range of 50 to 60 × 109/L.252,253,257 The development of new thrombosis or progression of an existing venous or arterial thrombosis in a patient receiving heparin, particularly if accompanied by a drop in the platelet count, is highly suggestive of HIT.
Appropriate platelet monitoring is essential to the early diagnosis of HIT. In heparin-naive patients, the platelet count should be monitored every other day, beginning 5 days after starting heparin. In patients with previous heparin exposure, the platelets should be monitored with the start of heparin use. A 50% or greater decrease in platelet count from the pretreatment platelet count should alert the clinician to development of HIT.163,250,274 The development of platelet activating, noncomplement fixing IgG antibodies directed against the complex of heparin and PF4 is the hallmark of this immunopathic disorder.258,264,265,274 Therefore, confirmation of a clinical suspicion of HIT requires serologic screening for HIT-associated heparin/PF4 antibodies.258,264,265 Prospective studies of heparin /PF4 antibody formation indicate that clinical HIT occurs in a minority of patients who form antibodies.275 Antibody detection can be performed by either the use of functional assays that detect antibodies capable of inducing platelet activation (serotonin release assay) or by a PF4/heparin immunoassay (enzyme-linked immunosorbent assay [ELISA]). The more readily available heparin/PF4 ELISA has significant sensitivity (>90%-95%) and specificity (80%-90%).274 Therefore, a negative ELISA has a strong negative predictive value of more than 95%, suggesting an alternate diagnosis for thrombocytopenia. A strongly positive ELISA is also more likely to be associated with a positive functional assay as determined by either a washed platelet C14-serotonin-release assay (SRA) or heparin-induced washed platelet aggregation assay.274 A positive platelet SRA or platelet aggregation assay has a reported sensitivity of greater than 90% and specificity for clinical HIT of greater than 95%.274 The combined use of both the PF4/heparin ELISA and a functional assay provide a diagnostic sensitivity approaching 100% for clinical HIT.274
Prompt recognition of the disorder, immediate cessation of all forms of heparin and rapid initiation of a direct thrombin inhibitor, such as argatroban, lepirudin, or bivalrudin, is essential in preserving life and limb. Approximately 50% of patients with HIT characterized by thrombocytopenia alone will develop thrombosis if managed with only discontinuation of heparin.251 The use of warfarin alone in these patients is not protective, and warfarin loading after a diagnosis of HIT can result in severe thrombotic complications, such as ischemic limb gangrene.251,276 Other thrombotic complications reported in patients with HIT include overt DIC with hypofibrinogenemia, adrenal necrosis, and cutaneous necrosis at the sites of heparin injections.163,256 HIT appears to be a unique event in many patients, without recurrence. However, patients within 100 days of diagnosis should not receive any form of heparin therapy.258,277 Patients beyond 100 days may receive heparin if indicated, but they should have daily platelet counts monitored. Antibody testing should be closely followed to make sure the patient does not have a recurrence.
Obesity
Obesity, as defined by a body mass index (BMI) greater than 25 kg/m2, has been shown to be a comorbidity and an independent risk for VTE. In a prospective study of risk factors, the Copenhagen City Heart Study, a BMI of more than 35 kg/m2 was found to be associated with a hazard ratio of 2.1 versus patients with a BMI less than 20 kg/m2.278 In the Tromsø study, the combination of tall stature and obesity (BMI >30 kg/m2) was associated with an increased risk of VTE in both men and women.279 Tall, obese men had a fivefold increase in VTE (hazard ratio 5.16) and tall, obese women had a threefold increase (hazard ratio 2.89). Moreover, in an analysis of patients with idiopathic VTE, the presence of the “metabolic syndrome,” defined as abdominal obesity, impaired glucose metabolism, arterial hypertension, and dyslipidemia, was detected in 23.5% of patients with idiopathic VTE (P = <0.001; OR, 2.99).280
In association with other risk factors such as family history, OCs, and surgery, the risk for VTE in obese patients increases significantly independent of age and sex.281 Data stratified by age, regardless of race and family history of VTE showed obesity to be a statistically significant risk factor for VTE (age 18-44 years: OR, 3.5, 95% CI 1.6-7.6; age 45-54 years: OR, 2.6, 95% CI 1.3-5.0; age ≥55 years: OR, 2.6, 95% CI 1.3-5.2). Further, when the association between family history and VTE was examined, among men and women, regardless of race, a family history of VTE was a statistically significant risk factor for VTE among both men and women who were obese (OR, 3.5, 95% CI 1.8-6.6 versus OR, 2.2, 95% CI 1.3-3.7, respectively).281 Among obese patients, however, a family history of VTE in a relative before age 50 years was a statistically significant risk factor for VTE only among men (OR, 6.9, 95% CI 2.2-21.6), although the OR was elevated among women (OR, 1.8, 95% CI 0.86-3.6), but the P value for interaction was not significant.281
Evaluation of the Patient with Suspected Thrombophilia
The purpose for testing for thrombophilia in patients with venous thrombosis is to determine the risk of recurrent thrombosis, and therefore. the duration of anticoagulation. Figure 38-1 provides a suggested outline for hypercoagulable testing. Patients with significant prothrombotic defects alone or in combination can be at a significantly higher risk of recurrent VTE, and therefore, extended or lifelong anticoagulation may be recommended. In patients in whom significant prothrombotic defects are detected, further testing of family members, who have not had a VTE, should be performed. This will allow asymptomatic carriers to be counseled regarding high-risk situations of increased hemostatic stress, such as surgery, pregnancy, oral contraceptive pills, HRT, or prolonged travel. Patients with VTE who should be tested include those younger than age of 50 years with unprovoked (idiopathic) VTE, patients with unprovoked recurrent VTE, and patients who develop thrombosis, including those associated with a transient risk factor, if there is a strong family history of VTE.
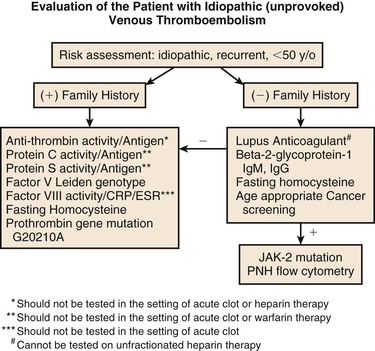
Figure 38-1 Indications for evaluation and laboratory testing for hypercoagulability in patients with venous thromboembolism. CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; Ig, immunoglobulin; JAK-2, Janus kinase-2; PNH, paroxysmal nocturnal hemoglobinuria.
In the younger patient (<50 years of age) without a history of diabetes, who has experienced an unprovoked arterial thrombotic event, a screening evaluation can lead to the identification of prothrombotic risk factors. This allows for effective treatment strategies to reduce the risk of a recurrent arterial event. Table 38-2 lists the recommended studies for evaluation of patients with VTE and patients with arterial thrombotic events.
Table 38-2
Recommended Tests for Evaluation of Congenital and Acquired Prothrombotic Defects in Patients With Venous and Arterial Thrombosis
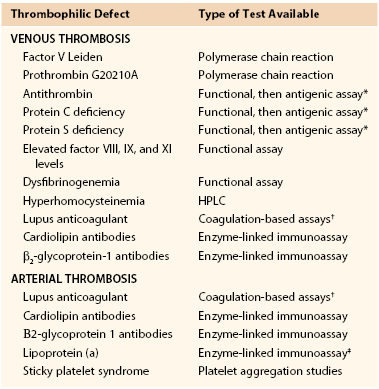
* Abnormal functional assays should be performed first, followed by antigen assays to subtype the defect.
† Two different coagulation-based assays should be performed in screening for a lupus anticoagulant.
‡ Screening for elevated lipoprotein (a) should be performed in conjunction with a full cholesterol and lipid panel.
HPLC, High-performance liquid chromatography.
Selected References
Crowther MA, Kelton JG. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138:128–134.
De Stefano V, Leone G, Mastrangelo S, Tripodi A, Rodeghiero F, Castaman G, Barbui T, Finazzi G, Bizzi B, Mannucci PM. Clinical manifestations and management of inherited thrombophilia: retrospective analysis and follow-up after diagnosis of 238 patients with congenital deficiency of antithrombin III, protein C and protein S. Thromb Haemost. 1994;72:352–358.
Large clinical multicenter study of the three major inherited thrombophilic states..
Goldhaber S. Thrombosis and thromboembolism, venous thromboembolism: clinical impact and multifactorial etiology. Marcel Dekker, Inc.: New York; 2002.
An excellent overview of venous thrombosis, pathophysiology, and epidemiology..
Holst AG, Jensen G, Prescott E. Risk Factors for Venous Thrombosis: Results of the Copenhagen City Heart Study. Circulation. 2010;109:1896–1903.
Lim W, Crowther MA. Antiphospholipid antibodies: a critical review of the literature. Curr Opin Hematol. 2007;14:494–499.
Excellent review and analysis of the medical literature on the antiphospholipid syndrome..
Lyman GH, Khorana AA, Falanga A, Clarke-Pearson D, Flowers C, Jahanzeb M, Kakkar A, Kuderer NM, Levine MN, Liebman H, Mendelson D, Raskob G, Somerfield MR, Thodiyil P, Trent D, Francis CW, American Society of Clinical Oncology. American Society of Clinical Oncology Guideline: Recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25:5490–5505.
Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia: recognition, treatment and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:311S–337S.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Rutkow IM, et al. An analysis of vascular surgical manpower requirements in the United States. J Vasc Surg. 1997;3:74.
2. Goldhaber S. Thrombosis and thromboembolism, venous thromboembolism: clinical impact and multifactorial etiology. Marcel Dekker, Inc.: New York; 2002.
3. Jackson E, et al. Epidemiology of arterial thrombosis. Colman RW, et al. Hemostasis and thrombosis; basic principles & clinical practice. ed 4. Lippincott, Williams & Wilkins: Philadelphia; 2000:1179–1196.
4. Anderson FA, et al. A population-based perspective of hospital incidence and case-fatality rates of deep vein thrombosis and pulmonary embolism. Arch Intern Med. 1991;151:933.
5. Coon WW, et al. Venous thromboembolism and other venous disease in the Tecumseh community health study. Circulation. 1973;48:839.
6. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1993;353:1167.
7. Crowther MA, et al. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138:128.
8. D’Angelo A, et al. Homocysteine and thrombotic disease. Blood. 1997;90:1.
9. Frohlich J. Lipoproteins and homocyt(e)ine as risk factors for atherosclerosis: assessment and treatment. Can J Cardiol. 1995;11(suppl C):18C.
10. Lange H, et al. Folate therapy and In-stent restenosis after coronary stenting. N Engl J Med. 2004;350:2673.
11. Jamison RL, et al. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease. JAMA. 2007;298:1163.
12. Toole JF, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction and death. The vitamin intervention for stroke prevention randomized controlled trial. JAMA. 2004;291:565.
13. den Heijer M, et al. Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: a randomized placebo-controlled, double-blind trial. Blood. 2007;109:139.
14. De Stefano V, et al. Clinical manifestations and management of inherited thrombophilia: retrospective analysis and follow-up after diagnosis of 238 patients with congenital deficiency of antithrombin III, protein C and protein S. Thromb Haemost. 1994;72:352.
15. Pabinger I, et al. Thrombotic risk in hereditary antithrombin III, protein C or protein S deficiency. Arteroscler Thromb Vasc Biol. 1996;16:742.
16. Bucciarelli P, et al. Risk of venous thromboembolism and clinical manifestations in carriers of antithrombin, protein C, protein S deficiency or activated protein C resistance. A multicenter collaborative study. Arteroscler Thromb Vasc Biol. 1999;19:1026.
17. Ridker PM, et al. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med. 1995;332:912.
18. McCully KS. Vascular pathology of homocysteinuria: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111.
19. Alfthan G, et al. Relation of serum homocysteine and lipoprotein (a) concentrations to atherosclerotic disease in a prospective Finnish population based study. Atherosclerosis. 1994;106:9–19.
20. Scanu AM. Lipoprotein (a): a genetic risk factor for premature coronary heart disease. JAMA. 1992;267:3326.
21. Wu KK, et al. Association of coagulation factors and inhibitors with carotid artery atherosclerosis. Early results of the Atherosclerosis Risk in Communities Study. Ann Epidemiol. 1992;2:471.
22. Tracy RP, et al. Fibrinogen and factor VIII, but not factor VII are associated with measurements of subclinical cardiovascular disease in the elderly. Results from the Cardiovascular Health Study. Arteroscler Thromb Vasc Biol. 1995;15:1269.
23. Vossen CY, et al. Familial thrombophilia and lifetime risk of venous thrombosis. J Thromb Haemost. 2004;2:1526–1532.
24. Buller HR, et al. Antithrombotic therapy for venous thromboembolic disease: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(suppl 3):401S.
25. Hirsh J. Heparin. N Engl J Med. 1991;324:1565.
26. Wells PS, et al. Prevalence of antithrombin deficiency in healthy blood donors: a cross-sectional study. Am J Hematol. 1994;45:321.
27. Tait RC, et al. Prevalence of antithrombin deficiency in the healthy population. Br J Haematol. 1994;87:106.
28. Ben-Tal O, et al. The relative frequency of hereditary thrombotic disorders among 107 patients with thrombophilia in Israel. Thromb Haemost. 1989;61:50.
29. Mateo J, et al. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism. Results of the Spanish Multicentric Study on Thrombophilia (EMET-study). Thromb Haemost. 1997;77:444.
30. Cosgriff TM, et al. Familial antithrombin III deficiency: its natural history, genetics, diagnosis, and treatment. Medicine. 1983;62:209.
31. Lane DA, et al. Antithrombin mutation database: 2nd (1997) update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1997;77:197.
32. Bauters A, et al. Homozygous variant of antithrombin with lack of affinity for heparin: management of severe thrombotic complications associated with intrauterine demise. Blood Coagul Fibrinolysis. 1996;7:705.
33. Winter JH, et al. Familial antithrombin III deficiency. Q J Med. 1982;51:373.
34. Finazzi G, et al. Different prevalence of thromboembolism in the subtypes of congenital antithrombin deficiency: review of 404 cases (letter). Thromb Haemost. 1987;58:1094.
35. Hellgren M, et al. Pregnancy in women with congenital antithrombin III deficiency: experience of treatment with heparin and antithrombin. Gynecol Obstet Invest. 1982;14:127.
36. Blondel-Hill E, et al. The pregnancy antithrombin III deficient patient: management without antithrombin III concentrate. Thromb Res. 1992;65:193.
37. Schwartz RS, et al. Antithrombin III Study Group. Clinical experience with antithrombin III concentrate in the treatment of congenital acquired deficiency of antithrombin. Am J Med. 1989;87:53S.
38. Liebman HA, et al. Severe depression of antithrombin III associated with disseminated intravascular coagulation of women with fatty liver of pregnancy. Ann Intern Med. 1983;98:330.
39. DeStefano V, et al. Mortality related to thrombosis in congenital antithrombin III deficiency. Lancet. 1991;337:847.
40. Wilson C, et al. Mesenteric venous thrombosis and antithrombin III deficiency. J Clin Pathol. 1987;40:906.
41. Tuite P, et al. Cerebral vein thrombosis due to antithrombin III deficiency. Can J Neurol Sci. 1993;20:158.
42. Griffin JH, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–1373.
43. Dahlback B. The protein C anticoagulant system: inherited defects as the basis of for venous thrombosis. Thrombosis Res. 1995;77:1.
44. Tait RC, et al. Prevalence of protein C deficiency in the healthy population. Thromb Haemost. 1995;73:87–93.
45. Pabinger I, et al. Hereditary deficiency of antithrombin III, protein C, and protein S: prevalence in patients with a history of venous thrombosis and criteria for rational patient screening. Blood Coagul Fibrinolysis. 1992;3:547.
46. Reitsma PH, et al. Protein C deficiency: a database of mutations 1995 update. On behalf of the Subcommittee on Plasma Coagulation Inhibitors of the Scientific and Standardization Committee of the ISTH. Thromb Haemost. 1995;73:876–889.
47. Boekholdt SM, et al. Arterial thrombosis and the role of thrombophilia. Semin Thromb Hemost. 2007;33:588–595.
48. Estrelles A, et al. Severe “homozygous” protein C deficiency in a newborn infant. Thromb Haemost. 1984;52:53.
50. Broekman AW, et al. Protein C and the development of skin necrosis during anticoagulant therapy. Thromb Haemost. 1983;49:251.
51. Borgel D, et al. Protein S deficiency. Thromb Haemost. 1997;78:351.
52. Comp PC, et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest. 1984;74:2082.
53. Schwartz HP, et al. Plasma protein S deficiency in familial thrombotic disease. Blood. 1984;64:1297.
54. Comp PC. Laboratory evaluation of protein S status. Semin Thromb Hemost. 1990;16:177.
55. Castoldi E, et al. Similar hypercoagulable state and thrombosis risk in type I and type III protein S-deficient individuals from families with mixed type I/III deficiency. Haematologica. 2010;95:1563.
56. Faught W, et al. Changes in protein C and protein S levels in normal pregnancy. Am J Obstet Gynecol. 1995;172:147.
57. Comp PC, et al. Functional and immunologic protein S levels are decreased during pregnancy. Blood. 1986;68:881.
58. Kemkes-Matthes B. Acquired protein S deficiency. Clin Invest Med. 1992;70.
59. Vigano-D’Angelo S, et al. Protein S deficiency occurs in the nephrotic syndrome. Ann Intern Med. 1987;107:42.
60. Levine AM, et al. Advancing prothrombotic state in women with advancing HIV disease. J Acquir Immune Defici Syndr. 2006;42:572.
61. Clark DA, et al. Mesenteric vein thrombosis associated with familial deficiency of free protein S. Arch Pathol Lab Med. 1991;115:617.
62. Sas G, et al. A protein S deficient family with portal vein thrombosis. Thromb Haemost. 1985;54:724.
63. Vrethem M, et al. Association between deficiency of free protein S and anticardiolipin antibodies in patients ≤ 65 years of age with ischemic stroke and TIA. Eur J Neurol. 1998;5:491.
64. Anderson DR, et al. Warfarin induced skin necrosis in 2 patients with protein S deficiency: successful reinstatement of warfarin therapy. Haemostasis. 1992;22:124.
65. Dahlback B, et al. Familial thrombophilia due to a previously unrecognized mechanism due to poor anticoagulant response to activated protein: prediction of a cofactor to activated protein C. Proc Natl Acad Science USA. 1993;90:1004.
66. Bertina RM, et al. Mutation in blood coagulation Factor V associated with resistance to activated protein C. Nature. 1994;369:64.
67. Dahlback B, et al. Inherited resistance to activated protein C is corrected by anticoagulant cofactor activity found to be a property of factor V. Proc Natl Acad Sci USA. 1994;91:1396.
68. Lee DH, et al. Prevalence of factor V Leiden in a Canadian blood donor population. CMAJ. 1996;155:285.
69. Rees DC. The population genetics of factor V Leiden (Arg506Gln). Br J Haematol. 1996;95:579.
70. Ridiker PM, et al. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA. 1997;277:1305.
71. Dulicek P, et al. Risk of thrombosis in patients homozygous for and heterozygous for factor V Leiden in the East Bohemian region. Clin Appl Thromb Hemost. 2000;6:87.
72. Simioni P, et al. The risk of venous thromboembolism in patients with an Arg506Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med. 1997;336:399.
73. Price DT, et al. Factor V Leiden and the risk for thromboembolic disease: a clinical perspective. Ann Intern Med. 1997;127:895.
74. Vossen CY, et al. Risk of a first venous thrombotic event in carriers of a familial thrombophilic defect. The European Prospective Cohort on Thrombophilia (EPCOT). J Thromb Haemost. 2005;3:459.
75. Rodeghiero F, et al. Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med. 1999;130:643.
76. Rosendaal FR, et al. High risk of thrombosis in patients homozygous for factor V Leiden. Blood. 1995;85:1504.
77. Williamson D, et al. Factor V Cambridge: a new mutation (Arg306Thr) associated with resistance to activated protein C. Blood. 1998;91:1140.
78. Zuber M, et al. Factor V Leiden in cerebral venous thrombosis. Stroke. 1996;27:1721.
79. MA AD, et al. Activated protein C resistance and retinal vein occlusion. Retina. 1998;18:297.
80. Hirsch DR, et al. Pulmonary embolism and deep vein thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J. 1996;131:1145.
81. Heligren M, et al. Resistance to activated protein C as a basis for venous thromboembolism associated with pregnancy and oral contraceptives. Am J Obstet Gynecol. 1995;173:210.
82. Vandenbroucke JP, et al. Increased risk of venous thrombosis in oral contraceptive users who are carriers of factor V Leiden mutation. Lancet. 1994;344:1453.
83. Bloemenkamp KW, et al. Enhancement of factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing third generation progestagen. Lancet. 1995;346:1593.
84. Brill-Edwards P, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. N Engl J Med. 2000;343:1439.
85. Ridker PM, et al. Long-term, low intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med. 2003;348:1425.
86. Poort SR, et al. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased in venous thrombosis. Blood. 1996;88:3698.
87. Margaglione M, et al. Increased risk of venous thrombosis in carriers of the prothrombin G-A20210 gene variant. Ann Intern Med. 1998;129:89.
88. Leroyer C, et al. Prevalence of 20210 A allele of the prothrombin gene in venous thromboembolism patients. Thromb Haemost. 1998;80:49.
89. Brown K, et al. Risk of venous thromboembolism associated with the G to A transition at position 20210 in the 3′-untranslated region of the prothrombin gene. Br J Haematol. 1997;98:907.
90. Rosendaal FR, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost. 1998;79:706.
91. Franco RF, et al. The 20210 G to A mutation in the 3′-untranslated region of the prothrombin gene in different human populations. Acta Haematol. 1998;100:9.
92. Zivelin A, et al. Prothrombin 20210G>A is an ancestral prothrombotic mutation that occurred in whites approximately 24,000 years ago. Blood. 1998;92:1119.
93. Markis M, et al. Co-inheritance of the 20210A allele of the prothrombin gene increases the risk of thrombosis in subjects with familial thrombophilia. Thromb Haemost. 1997;78:1426.
94. Martinelli I, et al. Interaction between G20210A mutation of the prothrombin gene and oral contraceptive use in deep vein thrombosis. Arteroscler Thromb Vasc Biol. 1999;19:700.
95. Ridker PM, et al. G20210A mutation in prothrombin gene and risk of myocardial infarction, stroke, and venous thrombosis in a large cohort of US men. Circulation. 1999;99:999.
96. Ferraresi P, et al. The heterozygous 20210 G/A prothrombin genotype is associated with early venous thrombosis in inherited thrombophilias and is not increased in frequency in artery disease. Arterioscler Thromb Vasc Biol. 1997;17:2418.
97. Rosendaal FR, et al. A common prothrombin variant (20210 G toA) increases the risk of myocardial infarction in young women. Blood. 1997;90:1747.
98. Lindmarker P, et al. The risk of recurrent venous thromboembolism in carriers and non-carriers of the G1691A allele in the coagulation factor V gene and the G20210A allele in the prothrombin gene. DURAC Trial Study Group. Duration of Anticoagulation. Thromb Haemost. 1999;81:684.
99. Martinelli I, et al. High risk of cerebral-vein thrombosis in carriers of a prothrombin-gene mutation and in users of oral contraceptives. N Engl J Med. 1998;338:1793.
100. Koster T, et al. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet. 1995;345:152.
102. Kamphusien PW, et al. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:731.
103. Schambeck CM, et al. Familial clustering of high factor VIII levels in patients with venous thromboembolism. Arteroscler Thromb Vasc Biol. 2001;21:289.
104. Miller CH, et al. Population differences in von Willebrand factor levels affect the diagnosis of von Willebrand disease in African-American women. Am J Hematol. 2001;67:125.
105. Bloemenkamp KWM, et al. Venous thrombosis, oral contraceptives and high factor VIII levels. Thromb Haemost. 1999;82:1024.
106. Kyrle PA, et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med. 2000;343:457.
107. Campos M, et al. Influence of single nucleotide polymorphisms in factor VIII and von Willebrand factor genes on plams factor VIII activity: the ARIC study. Blood. 2012;119:1929.
108. van Hylckama Vlieg A, et al. High levels of factor IX increase the risk of venous thrombosis. Blood. 2000;95:3678.
109. Meijers JC, et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696.
110. Mansveld EPG, et al. Analysis of the F8 gene in individuals with high plasma factor VIII:C levels and associated venous thrombosis. Thromb Haemost. 1998;80:561.
111. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of atherosclerosis. Am J Pathol. 1969;56:111.
112. Mudd SH, et al. The natural history of homocysteinuria due to cystathionine-beta-synthase deficiency. Am J Hum Genet. 1985;37:1.
113. Arnesen E, et al. Serum total homocysteine and coronary heart disease. Int J Epidemiol. 1995;24:704–709.
114. Nygard O, et al. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med. 1997;337:230–236.
115. Wald NJ, et al. Homocysteine and ischemic heart disease: results of a prospective study with implications regarding prevention. Arch Intern Med. 1998;158:862–867.
116. Ridker PM, et al. Homocysteine and the risk of cardiovascular disease among postmenopausal women. JAMA. 1999;281:1817.
117. Ridker PM, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836.
118. Stampfer MJ, et al. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. 1992;268:877–881.
119. Folsom AR, et al. Prospective study of coronary heart disease incidence in relation to fasting total homocysteine, related genetic polymorphisms and B vitamins: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 1998;98:204–210.
120. Falcon C, et al. High incidence of hyperhomocysteinemia in juvenile venous thrombosis. Atheroscler Thromb. 1994;14:1080.
121. Fermo I, et al. Prevalence of moderate hyperhomocysteinemia in patients with early onset venous and arterial occlusive disease. Ann Intern Med. 1995;123:747.
122. Simioni P, et al. Hyperhomocysteinemia and deep-vein thrombosis: a case controlled study. Thromb Haemost. 1996;76:883.
123. den Heijer M, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl Med. 1996;334:759.
124. den Heijer M, et al. Hyperhomocysteinemia and venous thrombosis: a meta-analysis. Thromb Haemost. 1998;80:874.
125. D’Angelo A, et al. Homocysteine and thrombotic disease. Blood. 1997;90:1.
126. Bezemer ID, et al. No association between the common MTHFR 677 C to T polymorphism and venous thrombosis. Results from the MEGA study. Arch Intern Med. 2007;167:497.
127. Angles-Cano E, et al. Inhibition of fibrinolysis by lipoprotein (a). Ann New York Acad Sci. 2001;931:261.
128. Collet JP, et al. Altered fibrin architecture is associated with hypofibinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. 2006;2567.
129. Howard BV, et al. Concentrations of Lp(a) in black and white young adults: relations to risk factors for cardiovascular disease. Ann Epidemiol. 1994;341.
130. Slunga L, et al. Lipoprotein (a) and acute-phase proteins in myocardial infarction. Scand J Clin Lab Invest. 1992;52:95.
131. Rosenson RS. Myocardial injury: the acute phase response and lipoprotein metabolism. J Am Coll Cardiol. 1993;22:933.
132. Balik O, et al. Serum lipoprotein (a) levels and Behcet’s disease: is there an association? Int J Dermatol. 2007;46:827.
133. Rhoads GG, et al. Lp(a) lipoprotein as a risk factor for myocardial infarction. JAMA. 1986;256:2540.
134. Kostner GM, et al. Lipoprotein Lp(a) and the risk for myocardial infarction. Atherosclerosis. 1981;38:51.
135. Sandkamp M, et al. Lipoprotein (a) is an independent risk factor for myocardial infarction at a young age. Clin Chem. 1990;36:20–23.
136. Rosengren A, et al. Lipoprotein (a) and coronary heart disease: a prospective case-control study in a general population sample of middle aged men. BMJ. 1990;301:1248.
137. Jauhiainen M, et al. Lipoprotein (a) and coronary heart disease risk: a nested case-control study of the Helsinki Heart Study participants. Atherosclerosis. 1991;89:59.
138. Wald NJ, et al. Apolipoproteins and ischaemic heart disease: implications for screening. Lancet. 1994;343:75.
139. Schaefer EJ, et al. Lipoprotein (a) levels and the risk of coronary heart disease in men. The Lipid Research Clinics Coronary Primary Prevention Trial. JAMA. 1994;271:999.
140. Cremer P, et al. Lipoprotein Lp(a) as predictor of myocardial infarction in comparison to fibrinogen, LDL, cholesterol and other risk factors: results from the prospective Gottingen Risk Incidence and Prevalence Study. Eur J Clin Invest. 1994;24:444.
141. Ridker PM, et al. A prospective study of lipoprotein (a) and the risk of myocardial infarction. JAMA. 1993;270:2195.
142. Ridker PM, et al. Plasma concentration of lipoprotein (a) and the risk of future stroke. JAMA. 1995;273:1269.
143. Marcucci R, et al. Increased plasma levels of lipoprotein (a) and the risk of idiopathic and recurrent venous thromboembolism. Am J Med. 2003;115:601.
144. Ignatescu M, et al. Plasma Lp (a) levels are increased in patients with chronic thromboembolic pulmonary hypertension. Thromb Haemost. 1998;80:231.
145. Vormittag R, et al. Lipoprotein (a) in patients with spontaneous venous thromboembolism. Thromb Res. 2007;120:15.
146. Mammen EF, et al. “Sticky platelet syndrome”: a congenital platelet abnormality predisposing to thrombosis. Folia Haematologica. 1988;115:361.
147. Mammen EF. Ten years experience with the “sticky platelet syndrome”. Clin Appl Thromb Hemost. 1995;1:66.
148. Frenkel EP, et al. Sticky platelet syndrome and thrombocythemia. Hematol/Oncol Clin N Am. 2003;17:63.
149. Kubisz P, et al. The glycoprotein IIIa PL(A1/A2) polymorphism–a defect responsible for the sticky platelet syndrome? Clin Appl Thromb Hemost. 2006;12:117.
150. Fateh-Moghadam S, et al. Hyperresponsiveness of platelets in ischemic stroke. Thromb Haemost. 2007;97:974.
151. Prandoni P, et al. The risk of recurrent venous thromboembolism after discontinuing anticoagulation in patients with acute proximal deep vein thrombosis or pulmonary embolism. A prospective cohort study in 1,626 patients. Haematologica. 2007;92:199.
152. Baglin T, et al. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet. 2003;362:523.
153. Pottier P, et al. Efficiency of systematic thrombophilia screening in idiopathic venous thrombosis: a prospective study in internal medicine. Clin Appl Thrombosis/Hemostasis. 2005;11:243.
154. Anderson FA, et al. Risk factors for venous thromboembolism. Circulation. 2003;107:19.
155. Conde I, et al. Classification of venous thromboembolism (VTE). Role of acute inflammatory stress in venous thromboembolism. J Thromb Haemost. 2005;3:2573.
156. Poredos P, et al. In patients with idiopathic venous thrombosis, interleukin-10 is decreased and related to endothelial dysfunction. Heart Vessels. 2011;26:596.
157. Prandoni P, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med. 2003;348:1435.
158. Galli M, et al. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid antibody syndrome: a systematic review of the literature. Blood. 2003;101:1827.
159. Lim W, et al. Antiphospholipid antibodies: a critical review of the literature. Curr Opin Hematol. 2007;14:494.
160. Lyman GH, et al. American Society of Clinical Oncology Guideline: Recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25:5490.
161. Blom JW, et al. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–722.
162. Geerts WH, et al. Prevention of venous thromboembolism: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(suppl 3):338S.
163. Warkentin TE, et al. Heparin-induced thrombocytopenia: recognition, treatment and prevention. Chest. 2004;126:311S.
164. Liebman HA, et al. Increased risk of venous thrombosis with inflammatory bowel disease who are carriers of the factor V Leiden mutation. Gastroenterol. 1998;115:830.
165. Sakane T, et al. Behcet’s disease. N Engl J Med. 1999;341:1284.
166. Cervera R, et al. Morbidity and mortality in systemic lupus erythematosus during a 5-year period: a multicenter prospective study of 1000 patients. Medicine. 1999;78:167.
167. Long AA, et al. The relationship of antiphospholipid antibodies to thromboembolic disease in systemic lupus erythematosus: a cross-sectional study. Thromb Haemost. 1991;66:520.
168. Silberberg JM, et al. Identification of tissue factor in two human pancreatic tumor cell lines. Cancer Res. 1989;49:5443.
169. Rao LV. Tissue factor as a tumor procoagulant. Cancer Metastasis Rev. 1992;11:249–266.
170. Weidmer T, et al. Complement-induced vesciculation and exposure of membrane prothrombinase sites in platelets of paroxysmal noctural hemoglobinuria. Blood. 1993;91:1192.
171. Liebman HA, et al. Thrombosis in patients with paroxysmal nocturnal hemoglobinuria is associated with markedly elevated plasma levels of leukocyte tissue factor. Thrombosis Research. 2003;111:235.
172. Gruppo Italiano Studie Policitemia. Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studie Policitemia. Ann Intern Med. 1995;123:656.
173. Cortelazzo S, et al. Incidence and risk factors for thrombotic complications in historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol. 1990;8:556.
174. Hexner EO. JAK2 V617F: Implications for thrombosis in myeloproliferative diseases. Curr Opin Hematol. 2007;14:450.
175. DeStefano V, et al. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemost. 2007;5:708.
176. Vianna JL, et al. Comparison of primary and secondary antiphospholipid syndrome: a European multicenter study of 114 patients. Am J Med. 1994;96:3.
177. Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295.
178. de Groot PG, et al. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost. 2005;3:1993.
179. Giannakopoulis B, et al. Current concepts on the pathogenesis of the antiphospholipid syndrome. Blood. 2007;109:422.
180. Zhou H, et al. Characterization of monocytes tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood. 2004;104:2352.
181. Dignat-George F, et al. Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost. 2004;91:667.
182. Meroni P, et al. Beta-2-glycoprotein I as a “cofactor” for anti-phospholipid reactivity with endothelial cells. Lupus. 1998;7(suppl 2):S44.
183. Pierangeli SS, et al. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum. 2005;52:2120.
184. Turiel M, et al. Thrombotic risk factors in primary antiphospholipid syndrome. A 5-year prospective study. Stroke. 2005;36:1490.
185. Rosove MH, et al. Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med. 1992;117:303.
186. Khamashta MA, et al. The management of thrombosis in the antiphospholipid antibody syndrome. N Engl J Med. 1995;332:993.
187. Schulman S, et al. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Am J Med. 1998;104:332.
188. Crowther MA, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med. 2003;349:1133.
189. Finazzi G, et al. A randomized clinical trial of high intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome. J Thromb Haemost. 2005;3:848.
190. Cervera R, et al. Catastrophic antiphospholipid syndrome: lesions from the “CAPS” registry. A tribute to the late Josep Font. Ann NY Acad Sci. 2007;1108:448.
191. Rickles FR, et al. Epidemiology of thrombosis in cancer. Acta Haematol. 2001;106:6.
192. Taubman MB. Tissue factor in cancer angiogenesis and coagulopathy. Khorana AA, et al. Cancer-associated thrombosis. Informa Healthcare: New York; 2007:35–50.
193. Weitz I, et al. Chemotherapy-induced hemostatic activation and thrombosis in cancer. Khorana AA, et al. Cancer-associated thrombosis. Informa Healthcare: New York; 2008:65–76.
194. Trousseau A. Phlegmasia alba dolen. Clinique médicale de l’Hôtel-Dieu de Paris. JB Bailliere: Paris; 1865:654.
195. Heit JA, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160:809.
196. Levitan N, et al. Rates of initial and recurrent thromboembolic disease among patients with malignancy versus those without malignancy: risk analysis using Medicare claims data. Medicine. 1999;78:285.
197. Sallah S, et al. Venous thrombosis in patients with solid tumors. Determination of frequency and characteristics. Thromb Haemost. 2002;87:575.
198. Stein PD, et al. Prevalence of acute pulmonary embolism among patients in a general hospital and at autopsy. Chest. 1995;108:978.
199. O’Connell C, et al. Unsuspected pulmonary emboli in cancer patients: clinical correlates and significance. J Clin Oncol. 2006;24:4928.
200. Hutten BA, et al. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol. 2000;18:3078.
201. Prandoni P, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood. 2002;100:3484.
202. Lee AYY, et al. Long-term treatment with dalteparin low-molecular weight heparin (LMWH) may improve survival in patients with non-metastatic malignancy and venous thromboembolism. J Clin Oncol. 2005;23:2123.
204. Lyman GH, et al. American Society of Clinical Oncology Guideline: Recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25:5490.
205. Scappaticci F, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. 2007;99:1232.
206. Rosen P, et al. Nonbacterial thrombotic endocarditis in patients with malignant neoplastic diseases. Am J Med. 1973;54:23.
207. Edoute Y, et al. Cardiac valvular vegetations in cancer patients: a prospective echocardiographic study of 200 patients. Am J Med. 1997;102:252.
208. Graus F, et al. Cerebrovascular complications in patients with cancer. Medicine. 1985;64:16.
209. Bedikian A, et al. Nonbacterial thrombotic endocarditis: comparison of characteristics of patients with and without concomitant disseminated intravascular coagulation. Med Pediatr Oncol. 1978;4:149.
210. Rogers LR, et al. Cerebral infarction from non-bacterial thrombotic endocarditis. Clinical and pathological study including the effects of anticoagulation. Am J Med. 1987;746.
211. Heit JA, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30 year population-based study. Ann Intern Med. 2005;143:697.
212. Eldor A. Thrombophilia, thrombosis and pregnancy. Thromb Haemost. 2001;86:104.
213. Andersen BS, et al. The cumulative incidence of venous thromboembolism during pregnancy and the puerperium. Acta Obstet Gynecol Scand. 1998;77:170.
214. Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet. 1999;353:1258.
215. Andres RL, et al. Venous thromboembolism and pregnancy. Obstet Gynecol Clin North Am. 2001;28:613.
216. James AH, et al. Thrombosis during pregnancy and the postpartum period. Am J Obstet Gynecol. 2005;193:216.
217. Hull RD, et al. Serial IPG in pregnant patients with clinically suspected DVT: clinical validity of negative findings. Ann Intern Med. 1990;112:663.
218. Ginsberg J, et al. DVT during pregnancy: leg and trimester presentation. Thromb Haemost. 1992;67:519.
219. Cockett FB, et al. Iliac vein compression: its relation to iliofemoral thrombosis and the post-thrombotic syndrome. BMJ. 1967;2:14.
220. Gerhardt A, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and puerperium. N Engl J Med. 2000;342:374.
221. Martinelli I, et al. Inherited thrombophilia and the first thromboembolism during pregnancy and the puerperium. Thromb Haemost. 2002;87:791.
222. Inman WHW, et al. Thromboembolic disease and the steroidal content of oral contraceptives: a report to the committee on safety of drugs. BMJ. 1970;2:203.
223. Meirik O. World Health Organization Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception: effect of different progestogens in low oestrogen oral contraceptive on venous thromboembolic disease. Lancet. 1995;346:1582.
224. Poulter NR. World Health Organization Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception venous thromboembolic disease and combined oral contraceptives: results of international multicentre case-control study. Lancet. 1995;346:1575.
225. Rosendaal FR, et al. Female hormones and thrombosis. Arteroscler Thromb Vasc. 2002;22:201.
226. Lewis MA. Myocardial infarction and stroke in young women: what is the impact of oral contraceptive? Am J Obstet Gynecol. 1996;179:S66–S77.
227. Schwartz SM, et al. Use of low-dose oral contraceptives and stroke in young women. Ann Intern Med. 1997;127:596–603.
228. Petiti DB, et al. Stroke in users of low-dose oral contraceptives. N Engl J Med. 1996;335:8–15.
229. Heinemann LA, et al. Case-control study of oral contraceptives and risk of thromboembolic stroke: results from international study on oral contraceptives and health of young women. BMJ. 1997;315:1502–1504.
230. Hannaford P. Health consequences of oral combination contraceptives. Br Med Bull. 2000;56:749–760.
231. Rosendaal FR, et al. Oral contraceptives, hormone replacement therapy and thrombosis. Thromb Haemost. 2001;86:112–123.
232. WHO. Effect of different progestagens in low oestrogen contraceptives on venous thromboembolic disease. World Health Organization Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Lancet. 1995;346:1582–1588.
233. Jick H, et al. Risk of idiopathic cardiovascular death and nonfatal venous thromboembolism in women using oral contraceptives with differing progestagens components. Lancet. 1995;346:1589–1593.
234. Kemmeren J, et al. Third generation oral contraceptives and the risk of venous thrombosis: meta-analysis. BMJ. 2001;323:131–134.
235. Dentali F, et al. Thrombophilic abnormalities, oral contraceptives and the risk of cerebral venous thrombosis: a meta-analysis. Blood. 2006;107:2766–2773.
236. Primignani M, et al. Risk factors for thrombophilia in extrahepatic portal vein obstruction. Hepatology. 2005;41(3):603–608.
237. Pabinger I, et al. Thrombotic risk of women with hereditary antithrombin III, protein C and protein S deficiency taking oral contraceptive medication. [and the GTH study group] Thromb Haemost. 1994;71:548–552.
238. Van Vijimen EFW, et al. Oral contraceptives and absolute risk of venous thromboembolism in women with single or multiple thrombophilic defects: results from a retrospective family cohort study. Arch Intern Med. 2007;167:282–289.
239. Martinelli I. Risk factors in venous thromboembolism. Thromb Haemost. 2001;86:395–403.
240. Daly E, et al. Risk of venous thromboembolism in users of hormone replacement therapy. Lancet. 1996;348:977–980.
241. Jick H, et al. Risk of hospital admission for idiopathic venous thromboembolism among users of postmenopausal oestrogens. Lancet. 1996;348:981–983.
242. Grady D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease: the Heart and Estrogen/Progestin Replacement Study. Ann Intern Med. 2000;132:689–696.
243. Herrington D, et al. Effect of estrogen replacement on the progression of coronary-artery atherosclerosis. N Engl J Med. 2000;343:522–529.
244. Lowe G. Hormone replacement therapy and cardiovascular disease: increased risk of venous thromboembolism and stroke, and no protection from coronary heart disease. J Intern Med. 2004;256:361–374.
245. Miller J, et al. Postmenopausal estrogen replacement and risk for venous thromboembolism: a systematic review and meta-analysis for the US Preventive Services Task Force. Ann Intern Med. 2002;136:680–690.
246. Herrington D, et al. Factor V Leiden, hormone replacement therapy, and risk of venous thrombosis. Arteroscler Thromb Vasc Biol. 2002;22:1012–1017.
247. Canonico M, et al. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER Study. Circulation. 2007;115:840–845.
248. Straczek C, et al. Prothrombotic mutations, hormone therapy, and venous thromboembolism among postmenopausal women: impact of route of estrogen administration. Circulation. 2005;112:3495–3500.
249. Kelton JG. The pathophysiology of heparin-induced thrombocytopenia. Biological basis for treatment. Chest. 2005;127:9S.
250. Warkentin TE. New approaches to the diagnosis of heparin-induced thrombocytopenia. Chest. 2005;127:35S.
251. Warkentin TE, et al. A 14 year study of heparin-induced thrombocytopenia. Am J Med. 1996;101:502.
253. Wallis DE, et al. Failure of early heparin cessation as treatment for heparin-induced thrombocytopenia. Am J Med. 1999;106:629.
254. Weismann RE, et al. Arterial embolism occurring during systemic heparin therapy. AMA Arch Surg. 1958;76:219.
255. Rhodes GR, et al. Heparin induced thrombocytopenia with thrombotic and hemorrhagic manifestations. Surg Gynecol Obstet. 1973;136:409.
256. Warkentin TE. Clinical presentation of heparin-induced thrombocytopenia. Semin Hematol. 1998;35(4 suppl 5):9.
257. Warkentin TE. Management of heparin-induced thrombocytopenia: a critical comparison of lepirudin and argatroban. Thromb Res. 2003;110:73.
258. Warkentin TE, et al. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med. 2001;344:1286.
259. Hong AP, et al. Central venous catheters and upper extremity deep-vein thrombosis complicating immune heparin-induced thrombocytopenia. Blood. 2003;101:3049.
260. Girolami B, et al. The incidence of heparin-induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study. Blood. 2003;101:2955.
261. Warkentin TE, et al. An improved definition of immune heparin-induced thrombocytopenia in postoperative orthopedic patients. Arch Intern Med. 2003;163:2514.
262. Jackson MR, et al. The incidence of heparin-induced antibodies in patients undergoing vascular surgery: a prospective study. J Vasc Surg. 1998;28:439.
263. Lindhoff-Last E, et al. A prospective study on the incidence and clinical relevance of heparin-induced antibodies in patients after vascular surgery. Thromb Res. 2000;97:387.
264. Amiral J, et al. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost. 1992;68:95.
265. Kelton JG, et al. Immunoglobulin G from patients with heparin-induced thrombocytopenia binds to a complex of heparin and platelet factor 4. Blood. 1994;83:3232.
266. Horsewood P, et al. The epitope specificity of heparin-induced thrombocytopenia. Br J Haematol. 1996;95:161.
267. Kelton JG, et al. Heparin-induced thrombocytopenia: laboratory studies. Blood. 1988;72:925.
268. Nieswandt B, et al. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcR-gamma chain. J Biol Chem. 2000;275:23998.
269. Warkentin TE, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood. 1994;84:3691.
270. Furie B, et al. P-selectin induction of tissue factor biosynthesis. Haemostasis. 1996;26(suppl 1):60.
271. Pouplard C, et al. Induction of monocytes tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood. 2001;3300.
272. Liebman HA, et al. Disseminated intravascular coagulation. [Chapter 125] Hoffman R, et al. Hematology: basic principles and practice. ed 4. McGraw-Hill, Inc.: New York, NY; 2004:2169–2182.
273. Klein HG, et al. Disseminated intravascular coagulation during heparin therapy. Ann Intern Med. 1974;80:477.
274. Warkentin TE. Platelet count monitoring and laboratory testing for heparin-induced thrombocytopenia. Recommendations of the College of American Pathologists. Arch Pathol Lab Med. 2002;126:1415.
275. Lee DH, et al. Frequency of heparin-induced thrombocytopenia. Warkentin TE, et al. Heparin-induced thrombocytopenia. ed 3. Marcel Dekker: New York, NY; 2004:107–148.
276. Warkentin TE, et al. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Intern Med. 1997;127:804.
277. Lubenow N, et al. Heparin-induced thrombocytopenia: temporal pattern of thrombocytopenia in relation to initial use or reexposure to heparin. Chest. 2002;122:37.
278. Holst AG Jensen G, et al. Risk factors for venous thrombosis: results of the Copenhagen City Heart Study. Circulation. 2010;109:1896.
279. Braekkan SK, et al. Body height and the risk of thromboembolism: the Tromso Study. Am J Epidemiol. 2010;171:1109–1115.
280. Di Minno MN, et al. Abnormally high prevalence of major components of the metabolic syndrome in subjects with early-onset idiopathic venous thromboembolism. Thromb Res. 2011;127:193–197.
281. Cushman M, et al. Impact of other risk factors on the association of obesity with venous thrombosis: the longitudinal investigation of thromboembolism etiology (LITE) study. J Thromb Haemostas. 2011;9(Suppl 2):872.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 38
Hypercoagulable States
Howard A. Liebman, Ilene Ceil Weitz
Thromboembolism is an enormous problem across the spectrum of medicine. Approximately 10 million individuals in the United States have symptomatic peripheral artery disease, and approximately 100,000 arterial reconstructive procedures are performed annually.1 In the United States and Europe, more than 600,000 people will experience a venous thromboembolism (VTE), and nearly 20% will die as a result.2 The incidence of arterial embolism and VTE is highly age dependent, contributing significant morbidity and mortality to an aging population.3–5
More than one hundred fifty years ago, the great German pathologist, Rudolf Virchow proposed the concept known as Virchow’s triad. He observed that thrombosis occurred when the following three situations were present; vascular wall (endothelial) injury, stasis of blood, and changes in the consistency of blood that enhance coagulation (hypercoagulability). An understanding of the latter, now termed the hypercoagulable state, was least apparent and could not be further characterized until there was a greater understanding of the normal mechanisms of hemostatic regulation (see Chapter 34). In the last 40 years, a number of inherited and acquired hemostatic disorders have been characterized that are associated with an increased risk of venous and/or arterial thrombosis. In most circumstances, the relationship between specific hemostatic defect and the hypercoagulable state is readily apparent by the respective role in normal hemostasis. However, the mechanism(s) by which other well-documented hypercoagulable defects, such as the antiphospholipid syndrome or elevated homocysteine, contribute to an increased risk of venous and arterial thrombosis remain poorly understood. The interactions of these hypercoagulable defects, alone or in combination, or when combined with other transient factors, such as vascular injury from surgery or trauma, can result is a significant increased risk of thrombosis. The interaction between inherited hypercoagulable defects and acquired additional factors, such as age or acute illness, can result in what has been termed an increased “thrombosis potential,” which upon reaching a “thrombosis threshold,” can result in symptomatic thromboembolism.6
Pathophysiology
Hemostasis is a highly organized series of reactions involving platelet adhesion and activation to form the platelet plug, followed by activation of coagulation proteins in a series of controlled enzymatic reactions to generate thrombin. Thrombin potentiates platelet aggregation and activation and acts on fibrinogen to generate the insoluble fibrin clot. Under normal conditions, these reactions occur on the endothelial surface only at sites of endothelial injury. This limits the size of the clot and allows blood to remain liquid and flowing. When this balance is altered, excess thrombin is generated, and abnormal thrombosis may occur. Therefore, thrombosis can result from defects in normal hemostasis that increase either the procoagulant activity or decrease the naturally occurring anticoagulants. These hypercoagulable defects have been termed thrombophilic syndromes and have been further classified as either congenital or acquired thrombophilias.6,7
In addition, there are a group of miscellaneous disorders whose prothrombotic mechanisms are poorly understood. These include elevations of homocysteine (hyperhomocysteinemia) and lipoprotein (a) (Lp[a]). The management of patients with thrombosis with these disorders remains controversial.8–13
Arterial thrombosis, unlike VTE, is associated with conditions that primarily affect the vascular wall and endothelium. Endothelial disruption, which occurs with atherosclerosis, vasculitis (vascular inflammation), infection, trauma, or surgery, is most often associated with an arterial thrombotic event. In well-controlled population studies, the majority of congenital hypercoagulable defects associated with venous thrombosis are not associated with a statistical increased risk of arterial thromboembolism.14–17 The few congenital hypercoagulable defects that may be associated with an increased risk of both venous and arterial thrombosis, such as hyperhomocysteinemia or increased Lp(a), are also associated with an increased risk of arterial atherosclerosis.8,9,18–20 Other defects associated with an increased risk of arterial thromboembolism, such as those observed with increased levels of fibrinogen and von Willebrand factor, are known to enhance platelet function.21,22
Congenital Hypercoagulability
Although the thrombosis potential or risk of thrombosis for inherited prothrombotic defects have been well characterized in a number of prospective and retrospective population studies, it is important to understand that thrombosis is a multigenetic and multifactor disorder. The thrombosis potential for specific congenital thrombophilic abnormalities is classically defined as a relative risk of thrombosis compared with a patient population without these abnormalities.6,7,14–16 In most circumstances, patients who inherit more than one abnormality have a significantly higher risk of thrombosis potential or relative risk of thrombosis.6,7,14–16,23 However, patients may have additional risk factors, such as aging, oral contraceptives (OCs) use, hormone replacement therapy (HRT) use, pregnancy, cancer, infection, trauma, or surgery. In these circumstances, a patient’s individual risk can also be increased by a factor that is more than the sum of each individual risk factor.23
Classification
Crowther and Kelton7 proposed a simple classification system that divides the congenital hypercoagulable (thrombophilic) states associated with VTE into two broad groups (Table 38-1). The first group constitutes defects associated with the reduced levels of the natural anticoagulants, such as antithrombin, protein C, and protein S.7 The defects in this “loss of inhibition” group are much less common, but are associated with a significantly higher risk of thrombosis.7 The second group is associated with defects that result in a “gain in procoagulant function” due to increased levels or function of coagulation factors.7 These include factor V Leiden (activated protein C resistance [APCR]) and prothrombin G20210 mutations, increases in coagulation factors VIII, IX, and XI, and the dysfibrinogenemias. Although these defects have a lower thrombosis potential, they are more frequently found in the general population, and therefore, more commonly associated with clinical thrombosis. The understanding that certain defects alone or in combination can be associated with a significantly higher risk of thrombosis may be important in determining the physician’s approach to antithrombotic prophylaxis, duration of anticoagulation after venous thrombosis, and family screening for patients with these defects.
Group 1 Thrombophilia
Group 1 includes deficiencies of the naturally occurring anticoagulant factors antithrombin (AT-III), protein C, and protein S. All are rare, representing less than 1% of the population.7 However, they are highly prothrombotic, with 30% to 50% of carriers (heterozygote) having a symptomatic thrombotic event before they reach 60 years of age.7 A significant number of carriers will have had a spontaneous thromboembolic event before the age of 40 years. Frequently, there is a strong family history of venous thrombosis. Although the risk of thrombosis is high, routine prophylactic anticoagulation in ambulatory healthy individuals has not been demonstrated to be of benefit and should be reserved for high-risk situations, such as surgery, sepsis, pregnancy, and immobilization.7 Because of the high risk of recurrence, with group 1 deficiencies and patients homozygous for certain group 2 abnormalities, lifelong anticoagulation may be recommended after a spontaneous thrombotic event.7,24
Antithrombin Deficiency
Pathogenesis and Incidence.
Physiologically, AT-III is the most important inhibitor of thrombin and other activated clotting factors (e.g., factors Xa, IXa, and VIIa). AT-III physiologic activity is enhanced 1000-fold by the binding of naturally occurring or administered heparin or heparin sulfates.25 AT-III deficiency is rare, reportedly occurring in an estimated 0.02% of the general population in a study that screened samples from healthy blood donors.26,27 It has been reported in 4% to 7.5% of patients with VTE.14–16,28,29 AT-III inheritance is autosomal dominant.30 More than 250 mutations within the molecule have been described.31 Homozygosity, particularly for AT-III deficiency types I and II, is extremely rare and appears to be incompatible with life. Homozygosity for type III AT-III deficiency results in a severe thrombophilic phenotype.32
Types.
There are three general subtypes of AT-III deficiency.31 Type I is characterized by decreased AT-III functional activity and antigen. Type II defects are AT-III mutations that have reduced functional activity, but normal antigen levels resulting from a mutation in the active inhibitory site on the protein. Type III AT-III mutations are characterized by moderate decreased activity due to impaired interaction with heparin. Screening for AT-III deficiency should always be undertaken using a functional assay, because screening with an antigen assay may fail to diagnose type II and III defects. In the presence of low AT-III activity, further characterization can be made with an antigen assay.
Patients with AT-III deficiencies are at a significantly higher risk of thrombosis than patients with other congenital deficiencies. Approximately 60% of carriers of type I and II deficiencies will have a thrombotic event by the age of 60 years. The risk for type III deficiency may be lower.33,34 A strong family history of thrombosis is usually present. A Spanish Multi-Center study reported the relative risk of thrombosis in these thrombophilic families as 21-fold.29 A multicenter, multinational European Prospective Cohort on Thrombophilia (EPCOT) study reported an adjusted relative thrombotic risk of 17.5 (95% confidence interval [CI], 9.1-33.8).23 After the diagnosis of AT-III deficiency is made in an individual with thrombosis, screening of family members is recommended.
Clinical Presentation and Management.
The clinical presentation of AT-III deficiency is predominantly lower extremity thrombosis with or without pulmonary embolism.30,33,35 Recurrent events are common, and atypical thrombotic events involving the portal, mesenteric, and hepatic venous system, or cerebral veins have been reported.29,40,41 Arterial events are rare, and patients are not at increased risk more than the unaffected adult population.14–16,23 Pregnancy is a particularly high-risk situation, and prophylaxis with heparin is indicated throughout pregnancy and in the immediate postpartum period.35,36 Patients with breakthrough events can receive AT-III concentrates.37 An acquired form of AT-III deficiency has been reported in pregnant women with the fatty liver syndrome with disseminated intravascular coagulation.38 Treatment with plasma or AT-III concentrates rapidly reverses the coagulopathy.38
Protein C Deficiency
Pathogenesis and Incidence.
Protein C is a vitamin K-dependent anticoagulation protein that is activated by thrombin to activated protein C (APC).42 When thrombin levels are high, thrombin binds to the endothelial protein receptor, thrombomodulin (TM), which changes the specificity of thrombin from cleaving fibrinogen or activating platelets to activating protein C. Protein C binds to its specific endothelial receptor, termed the endothelial protein C receptor, which enhances its activation. APC is a potent serine protease anticoagulant that cleaves the coagulant cofactors VIIIa and Va, thus modulating thrombin generation and subsequent clot formation.42,43 Deficiency of protein C is found in 0.2% of the general population, and in 2.5% to 6% of patients with VTE.28,29,44,45
Types.
Similar to AT-III deficiency, multiple mutations resulting in protein C deficiency have been reported. These mutations have been classified into two general subtypes: type I mutations have reduced functional and antigenic protein levels; and type II mutations have reduced functional levels but preserved antigen levels of the protein.46 Adult heterozygous patients with protein C deficiency usually have activity levels of less than 60%.
Clinical Presentation and Management.
Patients who have VTE usually present in the lower extremities. Unusual sites of thrombosis have been reported and are similar to those seen with AT-III deficiency.42,43,45,46 Although rare arterial events have been reported, large cohort studies do not support an increased risk of arterial events.47 In protein C deficient patients from thrombophilic families who present with an unprovoked (idiopathic) thrombosis, recurrent thrombotic events are frequent. Life-long anticoagulation should be considered in these patients, particularly those who present before the age of 40 years.
Homozygosity for protein C deficiency, with absent protein C activity, may present at birth as a neonatal disorder termed purpura fulminans.48 This disorder is characterized by diffuse microvascular thrombosis of the skin and systemic organs. Immediate treatment with heparin, plasma, or protein C concentrates can be life saving.48 The majority of homozygous neonates with protein C deficiency will have functional levels less than 20% of normal.48
A similar severe disseminated thrombotic disorder, characterized by skin necrosis, can occur in heterozygous patients with protein C deficiency treated with higher doses of warfarin, usually without concomitant anticoagulation with heparin.49,50 The syndrome, termed warfarin necrosis, results from the disproportional decrease in protein C in comparison to other procoagulant vitamin K-dependent coagulation factors. Patients presenting with this disorder should be treated with fresh frozen plasma, vitamin K, and heparin.49,50 The syndrome can be prevented by initiating oral anticoagulation with lower doses of warfarin and concomitant use of heparin.
Protein S Deficiency
Pathogenesis and Incidence.
Protein S is the vitamin K-dependent cofactor necessary for the inactivation of factors Va and VIIIa by APC. A deficiency in protein S is phenotypically similar to protein C deficiency. Protein S exists in two forms: the functionally active free form that usually constitutes 20% to 40% of the total protein, and the remaining 60% to 80% that is active and bound to complement binding protein C4b.51,52 Most patients with protein S deficiency will have activity levels between 50% and 75% of normal.51–53
Types.
Similar to the other natural anticoagulant proteins, protein S deficiency can be classified into 3 subtypes: type I is characterized by reduced functional and antigen protein levels; type II has reduced functional activity, but normal antigen levels; and type III has normal antigen levels, but reduced free active protein S due to enhanced C4b binding.54 Type I and type III protein S deficiencies are the most common forms of the deficiency encountered and do not appear to differ in their risk of venous thrombosis.55
The measurement of protein S activity and antigen can be confounded by a number of physiologic and clinical conditions. In pregnancy, protein S levels fall in the second and third trimester.56,57 Reduced protein S levels have also been reported in patients with active cancer, lupus erythematosus, antiphospholipid antibody syndrome, sepsis, chronic inflammatory disorders (e.g., inflammatory bowel disease) and in advanced HIV disease.58–60
Clinical Presentation and Management.
Protein S deficiency is clinically characterized by venous thrombosis and has been frequently reported in association with venous thrombosis in atypical sites.51–53,61,62 These reports may be confounded by other comorbidities in these patients that can be associated with acquired protein S deficiency. In addition, several reports have suggested a relationship between low protein S levels and arterial thrombosis, including a reported association between low free protein S and cardiolipin antibodies in stroke patients.63
Like protein C deficiency, homozygous deficiency can be associated with neonatal purpura fulminans. Approach to management is similar to that of homozygous protein C deficiency.64 Warfarin necrosis can also develop in heterozygote protein S patients.64
Group 2 Thrombophilia
Although associated with a lower thrombosis potential than Group 1 thrombophilia, Group 2 hypercoagulable disorders are more frequently found in the general population and are found in a greater proportion of patients with VTE. They represent gain of function mutations resulting in increased thrombin generation. They are typified by either increased synthesis of specific coagulation factors or as observed with factor V Leiden, resistance to the inactivation of this cofactor by APC.
Factor V Leiden (Activated Protein C Resistance)
Pathogenesis and Incidence.
Factor V is a cofactor that accelerates the conversion of factor II (prothrombin) to thrombin by factor Xa. Under normal circumstances, factor V is degraded by the serine protease, APC, which cleaves the protein at two sites. Factor V Leiden has a mutation in the 506 position that results in a substitution of glycine for arginine. This renders one of the factor V cleavage sites resistant to the action of the APC, which, like all coagulation serine proteases, cleaves arginine bonds.65–67 This results in a slowing of the inactivation of the cofactor, which leads to increased thrombin generation.
Factor V Leiden is a common mutation, occurring in approximately 2% to 7% of individuals of European ancestry.67–69 It can be found in nearly 10% of people with VTE and in 30% to 50% of individuals being evaluated for thrombophilia.67–70 The mutation is very rare in native Asians and Africans.69,70 The mutation is also rare in African Americans (0.6%) and has not been reported in Native Americans.69,70 Compared with patients with group 1 thrombophilia, patients with factor V Leiden have a lower relative risk of thrombosis, estimated between 2.5- and 7-fold.16,17,23,71–74 A cross-sectional study from Italy found that only 6% of carriers of the mutation developed a thromboembolic event by the age of 65 years.75 Homozygotes are at much higher risk then heterozygotes, with an estimated relative risk of 80-fold.16,71,76 However, heterozygotes with combined defects have a significantly increased risk of VTE.16,23,75 A second, very rare, mutation of the second factor Va cleavage site (Arg306Thr) has also been reported to be associated with increased risk of thrombosis, whereas other studies have failed to find a relationship of this mutation with the development of VTE.77
Clinical Presentation and Management.
The clinical presentation of thrombosis with factor V Leiden is overwhelmingly venous. Although rare, unusual sites, such as cerebral vein and retinal vein thrombosis, have been reported.78,79 The onset of thrombosis is frequently at an older age than seen in patients with type 1 thrombophilia.17 In the Physicians Health Study, the risk of VTE in men with the factor V Leiden mutation did not become statistically significant until after the age of 50 years.17 The study also found no association with the factor V Leiden and an increased risk of stroke or myocardial infarction.17 Thrombosis is frequently triggered by transient risk factors, such as a prolonged plane flight, oral contraceptive (OC) use, or pregnancy.80–83 In women carriers of the mutation, the reported thrombosis risk associated with the use of second-generation OCs varies widely, ranging from 6- to 50-fold.80–83 The wide variation reported in the thrombotic risk of OCs may be explained, in part, by a higher risk of thrombosis in women more than 30 years old who are taking OCs and some variation in the specific OCs used in the European studies, some of which contain prothrombotic progesterone substitutes.83
There is no excess mortality in carriers of the factor V Leiden mutation. No prophylaxis is recommended for carriers except what is recommended by guidelines for surgical interventions. Antepartum prophylaxis is not recommended for women who have not had a previous thrombotic event or a history of recurrent fetal loss.84 The risk of recurrent VTE after cessation of oral anticoagulation is not greater than that observed in other patients with unprovoked VTE.85
Prothrombin Gene Mutation G20210A
Pathogenesis and Incidence.
The prothrombin G20210A mutation is an abnormality located at the untranslated 3′ end of the prothrombin gene that results in increased plasma levels of prothrombin. The mutation affects the 5′ end cleavage signal, leading to increased prothrombin mRNA stability.86 The thrombotic risk is relatively low.87–89 The gene frequency in the European population is about 1% to 4%, with the greatest frequency in southern Europe and Spain.90–91 It is present in 5% to 10% of patients with VTE, and 15% of patients with thrombophilia.87–91 Similar to factor V Leiden, the prothrombin G20210A mutation is very rare in native Asians, Africans, African Americans, and Native Americans.92 Many carriers with a history of VTE have co-inheritance of the factor V Leiden mutation.23,93 The relative risk of thrombosis is low in carriers of the prothrombin mutation, ranging from two- to threefold higher in carriers compared with noncarriers of the mutation.94,95
Clinical Presentation and Management.
The clinical presentation is predominantly venous thrombosis of the lower extremities, and unusual sites are rare.87–89,94,95 Some controversy exists as to whether the prothrombin mutation is associated with an increased risk of arterial thrombosis.95–97 In carriers of the mutation who develop thrombosis, studies to date have shown either no increase in risk or only a slight increased risk of recurrent VTE after discontinuation of anticoagulation.95,98
The risk of VTE in women on OCs has been reported to be significantly increased, similar to that reported for factor V Leiden.94 Women who are carriers of both the prothrombin G20210A mutation and the factor V Leiden mutation have a markedly high risk of VTE.94 An association between the development of cerebral venous thrombosis in women carriers of the prothrombin mutation taking OCs has been reported, in which 20% of patients carried the mutation versus only 3% of controls.99
Elevated Factors VII, XI, and IX
Pathogenesis and Incidence.
Koster et al.100 first reported an increased risk of VTE in patients with an elevated factor VIII coagulant protein of more than the 90th percentile (>150%). Factor VIII activity levels more than 150% were associated with an adjusted relative risk of 4.8% compared with factor VIII levels 100% or less.100 These elevations appear to be independent of ABO blood type and were measured in patients without evidence of inflammation confirmed by a normal erythrocyte sedimentation rate (ESR) and C-reactive protein.101,102
Subsequent studies have supported this observation and found both a family and racial clustering suggestive of an inherited propensity for increased factor VIII levels.101,103 A study of African-American women found a statistically higher level of factor VIII compared with the white and Asian population in the United States.104 Unlike female carriers of the factor V Leiden and prothrombin mutations, the use of OCs did not appear to significantly increase the risk of VTE.105 Elevations of factor VIII activity have also been associated with an increased risk of recurrent VTE.106
The measurement of factor VIII activity is confounded by the fact that factor VIII, along with its carrier protein, von Willebrand factor, is an acute phase reactant and increases with bleeding and inflammation.101,102 Therefore, measurement of factor VIII activity should be performed with a simultaneous acute phase marker, such as the ESR or C-reactive protein. The assay should also be repeated at least twice at distant time intervals.100–102 Each laboratory must define the 90th percentile range for its own population to determine the patient population at risk.
Clinical Presentation and Management.
The clinical presentation of patients with elevations in factor VIII activity is predominantly VTE of the lower extremities.100 Because factor VIII levels increase with inflammation, elevation of this coagulation protein is an important cofactor in the development of thrombosis associated with infection, inflammatory bowel disease, and cancer. There is controversy as to whether increased levels of factor VIII are associated with arterial thrombosis, because elevations in factor VIII activity are associated with increases in von Willebrand factor. Increased levels of von Willebrand factor have been shown in population-based studies to be associated with an increased risk of arterial thrombosis.107
Increased levels of factors IX and XI have been associated with a twofold increase in risk of VTE.108,109 These are relatively weak risk factors for thrombosis, but if combined with other defects, they may become significant. To date, no molecular markers have been reported to characterize these elevations in factors VIII, IX, and XI.110
Hyperhomocysteinemia
An association was made between markedly elevated levels of homocysteine and arterial vascular disease in individuals with homocysteinuria.111,112 Additional studies confirmed this association and found additional evidence that individuals with homocysteinuria also experience an increased incidence of VTE.111 Subsequent studies found an association between elevations of plasma homocysteine and an increased risk of atherosclerosis and arterial thrombosis in apparently healthy individuals.113–121 Additional studies that focused on risk factors for venous thrombosis reported a similar association.121–124
Homocysteine metabolism involves two enzymatic pathways that require essential vitamin cofactors: folate, vitamin B12, and vitamin B6 (pyridoxine).125 The vitamine B6-dependent pathway involves the enzymatic conversion of homocysteine to cystathionine by the enzyme cystathionine-β-synthase (CBS).112,125 The second pathway involves re-methylation of homocysteine back to methionine requiring folate, vitamin B12, and the two enzymes 5,10-methylenetetrahydrofolatate reductase (MTHFR) and methionine synthetase.125 The primary mutations responsible for homocysteinuria are in the CBS gene.112 However, in the general population, independent of deficiencies in the vitamin cofactors, a common mutation in the MTHFR gene at position 677 is most frequently associated with elevations in homocysteine.125 This C to T mutation results in a 50% reduction in the enzymatic activity of the gene.125 However, even in MTFR C677T homozygotes, folate supplementation or a diet intrinsically rich in folate can reduce homocysteine levels into the normal range.125,126
The reported mechanisms by which homocysteine increases the risk of both arterial and venous thrombosis include a direct toxic effect on endothelium, enhanced platelet activation, oxidation of low-density lipoprotein cholesterol, an inflammatory decrease in endothelial TM, and an increase in von Willebrand factor and factor VIII.125 However, no single reported effect of homocysteine adequately explains its prothrombotic effect in both the arterial and venous vasculature.
A number of cohort studies and some prospective studies have found a statistically significant association between elevated homocysteine and an increased risk for myocardial infarction and stroke with estimated risks of 2- to 7.8-fold for individuals in the highest quartile.113–115 However, several prospective studies have failed to confirm this association.116–118 Data from the Physicians’ Health Study,118 the Atherosclerosis Risk in Communities Study,119 and the Women’s Health Study116,117 suggest a gender-defined risk for elevated homocysteine in which the risk is most significant in women. Elevations of homocysteine have also been found in case-controlled studies to be a weak risk factor for VTE, with a relative risk of only 2 to 4.120–124 Meta-analyses further confirmed an increased risk in patients with homocysteine levels more than the 95th percentile.124
Complicating the issue of elevated homocysteine as a risk factor for both arterial and VTE disease is the lack of response, in randomized controlled trials, to lowering homocysteine in reducing the risk of recurrent arterial10–12 and VTE13 disease in patients treated with vitamin therapy.
The measurement of homocysteine in patients can be complicated by comorbid conditions, such as vitamin deficiency, renal insufficiency, and improper plasma collection and handling. Measurements are best performed using freshly collected plasma, preferentially with patients in the fasting state. It is reasonable to repeat the assay on at least two separate occasions. Studies to date do not support an advantage for determination of the MTHFR genotype over direct measurement of homocysteine.126
Lipoprotein(a)
Lp(a) is a serum lipoprotein composed of a low-density lipid particle with a disulfide link to a long polypeptide chain, apolipoprotein (a).20,127 The protein has “kringle-like” domains that compete with tissue plasminogen activator, plasminogen, and plasmin for binding to fibrin and endothelial annexin II, thereby inhibiting fibrinolysis.127,128 The plasma levels of Lp(a) vary greatly within and between different racial populations, with African Americans having higher levels.129 Also, Lp(a) may increase as an acute phase reactant130,131 and has been reported to be elevated in certain inflammatory rheumatologic disorders.132
Early retrospective studies suggested a relationship between elevated levels of Lp(a) and the risk of premature atherosclerosis and arterial thrombosis.133–135 However, prospective studies have been less supportive and at the most suggest only a small increase in risk for individuals with elevated plasma Lp(a).136–142 Additional reports have suggested a role for elevated Lp(a) as an independent risk factor for VTE,143,144 although a failure to find an association has been reported by other investigators.145
Sticky Platelet Syndrome
Sticky platelet syndrome (SPS) is an inherited, autosomal dominant disorder associated with early-onset myocardial infarction and peripheral vascular disease.146–148 Although most often associated with arterial vasculopathy, VTE has been reported.146–148 A causal relationship to early miscarriage has also been reported. Patients usually present with a history of myocardial infarction or thromboembolism as young adults. The events often are precipitated by stress.146–148 The defect appears to be common in patients with unexplained thromboembolic events. In an analysis of 200 patients and family members identified with SPS, 21% had arterial thromboembolic events and 13.2% had VTE events.147 SPS platelets demonstrate hyperreactivity to epinephrine and adenosine diphosphate (ADP) even with dilution, but have normal responses to thrombin, collagen, and arachadonic acid.146–148 Three types of platelet responses have been noted: type I is hyperreactive to both ADP and epinephrine; type II is hyperreactive to epinephrine only; and type III is hyperreactive to ADP only. Increased arterial events may be mediated through polymorphisms of glycoprotein IIIa allele PLA-A2.149 Although other investigators have reported similar hyperreactivity of platelets in patients with ischemic stroke,150 there remains controversy as to whether SPS represents a true congenital prothrombotic syndrome. In patients with documented platelet reactivity and thrombosis, low-dose acetylsalicylic acid (81 mg) appears to be effective in inhibiting platelet hyperreactivity.148
Idiopathic (Unprovoked) Venous Thrombosis
Unprovoked or idiopathic venous thrombosis is defined as the development of deep vein thrombosis and/or pulmonary embolism in the absence of known genetic prothrombotic mutations, permanent factors, or acquired risk factors. Patients who develop VTE provoked by surgery, trauma, immobilization, pregnancy, or female hormone intake are at low risk of recurrent thrombosis.151,152 Patients with unprovoked VTE have a recurrence risk of 5% to 10% per year, with nearly 50% developing a recurrent event by 10 years.151,152 A prospective study that screened 66 patients with idiopathic venous thrombosis found abnormalities in 26 (39.3%) patients when they were screened for thrombophilia (antithrombin, protein C, protein S, factor V Leiden, prothrombin G20210A, antiphospholipid antibodies).153 Therefore, 60% of patients with unprovoked VTE have no underlying known major inherited or acquired thrombophilia and still remain at high risk of recurrent VTE. It has been proposed that an underlying inflammatory state may be responsible for the increased risk of VTE in patients with unprovoked events.154,155 Laboratory assessment of patients with idiopathic VTE have demonstrated higher levels of interleukin-6 and -8 with low levels of interleukin-10 compared with age- and sex-matched controls.156 It is notable that the same inflammatory markers are linked to an increased risk of atherothrombotic disease.154–157
Acquired Hypercoagulability
An increased risk of thrombosis can be associated with a variety of acquired abnormalities. Disorders such as the antiphospholipid antibody syndrome (APLAS) and cancer are significant prothrombotic syndromes.158,159 Common clinical situations such as cancer, pregnancy, infection, and estrogen use are transient causes of hypercoagulability.160–162 Surgery is a well-documented transient cause of hypercoagulability in which the risk of thrombosis is related to a number of factors, such as the type of surgery, duration of surgery, age of the patient, and other patient comorbidities.162 A significant iatrogenic thrombotic disorder is heparin-induced thrombocytopenia (HIT), in which early diagnosis and treatment may be life-saving.163 Chronic inflammatory and autoimmune disorders, such as inflammatory bowel disease, Behcet’s syndrome, or lupus erythematosus, with and without antiphospholipid antibodies, are also associated with a significant risk of thrombosis.164–167
Acquired elevations in factor VIII levels or depressions in protein S may be an important contributing factor to a number of prothrombotic disorders, including cancer, pregnancy, infection, and chronic inflammatory disorders. In patients with cancer, the expression of tissue factor by the malignant cell further increases the thrombotic risk.168,169 Paroxysmal nocturnal hemoglobinuria, a rare hemolytic disorder in which nearly half of the patients develop symptomatic thrombosis, has been shown to be associated with both increased platelet activation and increased leukocyte tissue factor expression.170,171 Thrombosis in patients with myeloproliferative syndromes frequently involves unusual sites, such as the portal and hepatic veins, and is more often associated with syndromes associated with the Janus kinase-2 (JAK-2) mutation.172–175 The presence of JAK-2 mutation has also been shown to be associated with thrombosis in the splanchnic, portal, and hepatic veins, even in the presence of normal blood counts.175
Antiphospholipid Antibody Syndrome
The APLAS is one of the more common causes of acquired hypercoagulability.158,159 The syndrome is associated with an increased risk of both arterial and venous thrombosis.158,159 Although the APLAS can occur with systemic lupus erythematosus (SLE), 50% of the patients do not fulfill criteria for lupus or other autoimmune disorders.176 The syndrome is characterized by the presence of antibodies that inhibit in vitro coagulation reactions, the lupus anticoagulant (LA), and antibodies that bind to anticardiolipin and β2-glycoprotein.158
[/not-level-membership-for-surgery-category]
