Hypercalcemia
1. What is hypercalcemia? How does protein binding affect the calcium level?
Hypercalcemia is a corrected total serum calcium value above the upper limit of the normal range or an elevated ionized calcium value. Calcium is 50% free (ionized), 40% protein-bound, and 10% complexed to phosphate, citrate, bicarbonate, sulfate, and lactate. Only elevations in the free calcium are associated with symptoms and signs. Of the protein-bound calcium, about 80% is bound to albumin and 20% to globulins. A decrease or increase in serum albumin of 1 g/dL from 4 g/dL decreases or increases the serum calcium by 0.8 mg/dL. An increase or decrease in serum globulin by 1 g/dL increases or decreases serum calcium by 0.16 mg/dL. Such protein changes do not affect free calcium and do not cause calcium-related symptoms.
2. How common are hypercalcemia and its main associated conditions?
Hypercalcemia affects 0.5% to 1% of the general population. The incidence may increase to 3% among postmenopausal women. Primary hyperparathyroidism (PHPT) causes 70% of outpatient and 20% of inpatient cases of hypercalcemia. Cancer causes 50% of inpatient cases of hypercalcemia. Ten percent of patients with malignancy experience hypercalcemia. Hyperparathyroidism and cancer cause 90% of all hypercalcemia. About 10% of patients with hyperparathyroidism experience nephrolithiasis. Although calcium oxalate stones are usually the most common stone type, calcium phosphate stones are more common in hyperparathyroidism.
3. How would you classify mild, moderate, and severe hypercalcemia?
First, consider the patient’s general health, hypercalcemic symptoms, and the normal upper limit for calcium in your laboratory. For example, a patient with renal failure and a serum phosphorus value of 8.5 mg/dL may have metastatic calcification with a serum calcium level of 10.5 mg/dL. Then the serum calcium (Ca) is corrected for the albumin concentration, as follows:
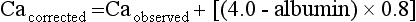
With this in mind, a serum calcium value 1.5 to 3.5 mg/dL above the upper normal limit defines moderate hypercalcemia. Mild hypercalcemia occurs below this range and severe hypercalcemia above. Thus if the upper normal limit for calcium is 10.5 mg/dL, a serum calcium value of 12 to 14 mg/dL indicates moderate hypercalcemia. A serum calcium value less than 12 mg/dL indicates mild hypercalcemia and a level greater than 14 mg/dL severe hypercalcemia.
4. Discuss the signs and symptoms of hypercalcemia.
No symptoms are usually present with mild hypercalcemia (< 12 mg/dL). Moderate or severe hypercalcemia and rapidly developing mild hypercalcemia may cause symptoms and signs. Common symptoms and signs involve (1) the central nervous system (lethargy, stupor, coma, mental changes, psychosis), (2) the gastrointestinal tract (anorexia, nausea, constipation, acid peptic disease, pancreatitis), (3) the kidneys (polyuria, nephrolithiasis), (4) the musculoskeletal system (arthralgias, myalgias, weakness), and (5) the vascular system (hypertension). The classic electrocardiographic (ECG) change associated with hypercalcemia is a short QT interval. Occasionally, severe hypercalcemia also causes dysrhythmias, sinus arrest, disturbances in atrioventricular (AV) conduction, and ST segment elevation mimicking myocardial infarction.
5. What are the sources of serum calcium?
Bone calcium approximates 1 kg and 99% of body calcium. A normal serum calcium level is maintained by integrated regulation of calcium absorption, resorption, and reabsorption; these processes occur, respectively, in the gut, bone, and kidney. Of the 1000 mg/day of dietary calcium intake, the gut absorbs 300 mg/day, secretes 100 mg/day, and excretes 800 mg/day. Net absorption averages 200 mg/day. Calcium absorption is usually about 30% of dietary calcium; however, absorption may increase to more than 50% when people also take large doses of 1,25(OH)2D. The kidney reabsorbs 98% of filtered calcium and excretes 200 mg/day. Bone exchanges about 500 mg of calcium per day with serum (Fig. 13-1).
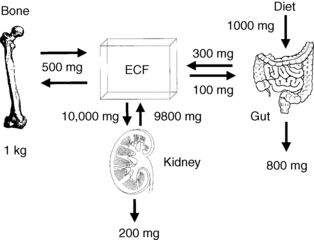
Figure 13-1. Normal calcium balance. ECF, extracellular fluid.
6. What are the major anatomic and physiologic determinants of vitamin D?
Diet, skin, liver, and kidney control the amount, synthesis, and secretion of vitamin D. Dietary sources of vitamin D include liver, fish oils, egg yolks, vitamin D–fortified foods, and vitamin D supplements. Skin exposure to ultraviolet sunlight activates 7-dehydrocholesterol to pre–vitamin D, which subsequently rearranges to form vitamin D. Hepatic 25-hydroxylase then converts vitamin D to 25-hydroxyvitamin D (25-OHD). 25-OHD circulates and interacts with two renal mitochondrial hydroxylases. High parathyroid hormone (PTH), low phosphate, and low calcium levels stimulate 1α-hydroxylase activity to increase conversion of 25-OHD to 1,25(OH)2D (calcitriol)—the most potent metabolite of vitamin D. Low PTH, high phosphate, and high calcium levels suppress 1α-hydroxylase activity and stimulate 24-hydroxylase activity. This process inhibits calcitriol production and, through 24-hydroxylase, converts 25-OHD to 24,25-dihydroxyvitamin D [24,25(OH)2D], which promotes antiresorptive effects on bone and positive calcium balance. This same sequence occurs less intensely with normal levels of PTH, PO4 (phosphate), and calcium. Calcitriol feeds back negatively on its own synthesis by suppressing 1α-hydroxylase activity, stimulating 24-hydroxylase activity, decreasing PTH, and increasing calcium and phosphate. Calcitriol is also degraded primarily through the enzyme 24-hydroxylase. The activity of 1α-hydroxylase is classically thought of as occurring only in the kidneys, but this enzyme is also present in bone, brain, pancreas, heart, intestines, lymph nodes, adrenal gland, prostate, and other tissues (Fig. 13-2). Fibroblast growth factor-23 (FGF-23) also has an important role in calcium and vitamin D metabolism.
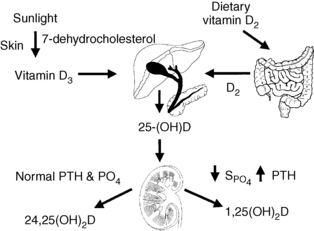
7. What is fibroblast growth factor-23 and what role does it play in calcium, phosphate, and vitamin D metabolism?
FGF-23 is a newly discovered phosphaturic hormone that is produced by osteocytes in response to increases in serum levels of phosphate, 1,25(OH)2D, and PTH. FGF-23 then decreases phosphate, 1,25(OH)2D, and PTH by reducing proximal renal tubular phosphate reabsorption, stimulating 24-hydroxylase, inhibiting 1α-hydroxylase, and variably inhibiting PTH synthesis and secretion by the parathyroid glands. FGF-23 accumulates to very high levels in chronic kidney disease (CKD) and is associated with increased mortality in CKD.
8. What are the classical and nonclassical effects of vitamin D and what is the role of the vitamin D receptor?
Calcitriol acts classically on intestine, bone, kidneys, and parathyroid glands to help regulate calcium and phosphate metabolism. When calcitriol activates the parathyroid vitamin D receptor (VDR), it decreases PTH messenger RNA (mRNA) synthesis by inhibiting the pre-pro-PTH gene at the vitamin D response element. This inhibition decreases PTH synthesis within the chief cell of the parathyroid gland and ultimately lowers PTH levels. Additionally, calcitriol increases intestinal calcium and phosphate absorption, increases bone calcium and phosphate resorption, enhances bone turnover, and enhances renal calcium and phosphate reabsorption. The VDR is a nuclear hormone receptor that is also regulated by calcium and PTH. Many proteins are downregulated and upregulated by the activated VDR. Downregulated proteins include PTH, 1α-hydroxylase, bone matrix protein, bone sialoprotein, type I collagen, interferons, interleukins, tumor necrosis factor (TNF), epidermal growth factor receptors, renin, and peroxisome proliferator-activated receptor (PPAR) gamma-2. Upregulated proteins include osteopontin, matrix Gla protein, type IV collagen, interleukins, VDR, calcium-sensing receptor (CaSR), and 24-hydroxylase. Through activation of the VDR, calcitriol has many (nonclassical) effects other than those related to calcium and phosphorus metabolism. VDR activation may ameliorate arterial calcification, retard neuronal degeneration, enhance host defenses against bacterial infection and tumor growth, enhance Sertoli cell function and spermatogenesis, enhance insulin synthesis and secretion from pancreatic beta cells, and assist with glycogen and transferrin synthesis in liver parenchymal cells. Additionally, calcitriol has antiproliferative and prodifferentiating effects on myeloid cell precursors, cardiac and smooth muscle cells, and a variety of skin cells, including keratinocytes, fibroblasts, hair follicles, and melanocytes.
9. What is the CaSR, and what role does it play in calcium metabolism?
The CaSR is a membrane-bound calcium sensor-receptor. The most important locations of the CaSRs are the parathyroid glands and the renal tubular cells but the receptors are located in many other tissues, including at low levels in pancreatic beta cells and thyroid C cells. The major function of the CaSR is to maintain extracellular calcium concentrations in the normal range and prevent hypercalcemia. In the parathyroid chief cells, the CaSR has a large extracellular domain of 700 amino acids (primary calcium binding site), a seven-segment transmembrane portion (primary calcimimetic binding site), and a cytoplasmic carboxyl-terminal component of about 200 amino acids (primary effector site for metabolic changes). The CaSR belongs to subfamily C of the G protein–coupled receptor family. The CaSR senses the minutest change in ionized calcium (0.1 mg/dL) and regulates PTH secretion in order to maintain steady-state calcium levels within a narrow optimal range. These changes center on a set point for calcium-regulated PTH release that is unique for each individual. Cinacalcet, a calcimimetic drug, binds to the transmembrane portion of the CaSR, making it markedly more responsive to any level of ambient calcium. After activation by calcium, the CaSR activates phospholipase C, inhibits adenylate cyclase, and opens nonselective cation channels. This effect increases cytoplasmic calcium by mobilizing calcium from thapsigargin-sensitive intracellular stores and enhancing calcium influx through voltage-insensitive cation channels. These CaSR-induced changes in intracellular calcium act on the calcium response element of the pre-pro-PTH gene to decrease chief cell PTH messenger RNA synthesis, reduce PTH secretion, and decrease parathyroid gland hyperplasia. The parathyroid glands secrete both intact PTH (iPTH) and carboxy-terminal PTH fragments (CPTH). Intact PTH acts directly on bone PTH receptors. CPTH remains in the circulation much longer and at higher concentrations than iPTH and, although it was previously thought to be inactive, data now suggest that CPTH fragments can exert direct effects on bone cells through a novel class of CPTH receptors. CPTH fragments accumulate in renal failure. PTH functions to keep calcium in the normal range and helps prevent hypocalcemia.
10. What is the function of the CaSR in the kidneys?
In the kidney, as in the parathyroid glands, the CaSR functions to prevent hypercalcemia. Activation of the CaSR located on the basolateral membrane in the thick ascending limb of Henle’s loop decreases tubular reabsorption of calcium and increases excretion. Activation of the renal CaSR generates an arachidonic acid metabolite that inhibits the luminal potassium channel and the sodium-potassium adenosine triphosphatase (ATPase) pump on the basolateral membrane. This diminishes the lumen-positive electrical gradient needed for passive calcium and magnesium reabsorption. Thus there is less reabsorption and more excretion of calcium. Because PTH is decreased by the CaSR activated in the parathyroid gland, there is less PTH-mediated distal tubular reabsorption of calcium, net calcium loss, and lower plasma calcium.
11. What are the overall effects of PTH, vitamin D, and FGF-23 on calcium metabolism?
Plasma calcium must be maintained within a narrow concentration range because of the key role it plays in a diverse array of physiologic processes, including intracellular signal transduction, muscle contraction, blood clotting, and neuronal transmission. Indeed, calcium in a given person is so tightly regulated that the average daily ionized calcium level does not deviate more than 0.3 mg/dL. Regulation of plasma calcium depends on normal amounts of PTH and calcitriol. Both hormones are also necessary for normal bone health. PTH and calcitriol provide the main control of serum calcium. Both PTH and calcitriol increase bone resorption by increasing osteoclast activity. At physiologic levels, PTH and calcitriol also increase bone formation. Because osteoclasts have no known receptors for either hormone, PTH and calcitriol stimulate osteoclast activity indirectly. Both hormones promote normal bone formation by direct action on the osteoblast line of cells. PTH enhances the activity of osteoblasts, which secrete factors such as interleukin-6 (IL-6) that stimulate osteoclastic bone resorption. PTH and calcitriol promote osteoclast differentiation from promonocytes to monocytes to macrophages to pre-osteoclasts and finally to osteoclasts. This is accompanied by an increase in osteoclast number and activity and decreased collagen synthesis. In addition, calcitriol increases calcium transport from bone to blood and maintains a favorable calcium-phosphate product necessary for normal bone mineralization. Both PTH and calcitriol stimulate osteoblast production of receptor activator of nuclear factor kappa B ligand (RANKL). RANKL binds to its membrane-bound receptor (RANK) on pre-osteoclasts and osteoclasts. This action stimulates osteoclast differentiation and osteoclast attachment to the bone via integrins and, ultimately, bone resorption. Both PTH and calcitriol also control osteoblast production and secretion of osteoprotegerin (OPG), which blocks the effects of excess RANKL and promotes normal bone metabolism. This process depends on the concentration of PTH and calcitriol. Higher PTH and calcitriol levels increase bone resorption abnormally and may cause hypercalcemia and loss of bone mass. Denosumab is a monoclonal antibody to RANKL that in clinical trials has decreased hypercalcemia but is not yet approved for hypercalcemia treatment. Bone resorption is the major mechanism of most occurrences of hypercalcemia (see Table 13-3). However, PTH and calcitriol also act on the kidney to increase Ca reabsorption. PTH increases renal phosphate excretion, and calcitriol increases its reabsorption. PTH has no direct effect on the intestine, but calcitriol increases absorption of both calcium and phosphate. The higher calcium and calcitriol levels provide negative feedback on PTH secretion, whereas higher phosphate levels provide positive feedback. The net effect is normal bone function and plasma calcium at physiologic levels of PTH and calcitriol and loss of bone mineral and hypercalcemia at high levels. FGF-23 counters the effects of excess PTH and calcitriol by inhibiting synthesis of both hormones and increasing renal phosphate excretion.
12. How do calcium and phosphate interact with calcium-regulating hormones?
Table 13-1 summarizes the main factors controlling serum calcium. The arrows show direct actions of factors in the left column on factors in the top row, whereas the plus (+) and minus (−) signs show indirect actions. As a rule, the direct effects predominate as the net effect. Table 13-2 outlines the specific effects of each of these factors.
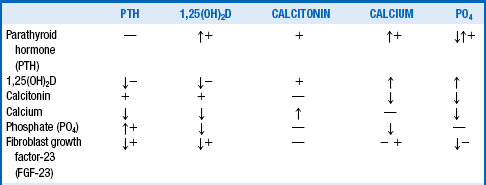
TABLE 13-2.
SUMMARY OF CALCIUM AND PHOSPHATE CONTROL
| VARIABLE | DIRECT ACTION(S) |
| Parathyroid hormone (PTH) | Increased bone resorption of calcium and phosphate Increased distal renal tubular calcium reabsorption Decreased renal tubular phosphate reabsorption Increased renal production of 1,25(OH)2D Net effect: increased serum calcium and decreased phosphate |
| 1,25(OH)2D | Increased bone resorption of calcium and phosphate Increased renal reabsorption of calcium and phosphate Increased gut absorption of calcium and phosphate Decreased parathyroid production of PTH Decreased renal production of 1,25(OH)2D Net effect: increased serum calcium and phosphate |
| Calcitonin | Decreased bone resorption of calcium and phosphate Decreased renal reabsorption of calcium and phosphate Decreased gut absorption of calcium and phosphate Net effect: decreased serum calcium and phosphate |
| Calcium | Decreased PTH synthesis and secretion Decreased 1,25(OH)2D production in the kidney Increased calcitonin release from the thyroid C cells Decreased phosphate |
| Phosphate | Decreased 1,25(OH)2D production in the kidney Decreased calcium Increased PTH synthesis in parathyroid chief cells |
| Fibroblast growth factor-23 (FGF-23) | Decreased 1,25(OH)2D production in the kidney Decreased renal tubular phosphate reabsorption Decreased parathyroid production of PTH |
13. List the main causes of hypercalcemia.
The mnemonic VITAMINS TRAP (see Pont, 1989) includes most causes of hypercalcemia (Box 13-1).
14. How do various causes of hypercalcemia increase the serum calcium level?
True hypercalcemia results from altered bone resorption, renal tubular reabsorption, and gut absorption of calcium. Although the bone (resorption and formation), kidney (reabsorption and excretion), and gut (absorption and secretion) each have two major processes involved with mineral metabolism, only resorption, reabsorption, and absorption play a significant role in hypercalcemia. An exception to this rule occurs when decreased renal function from renal or prerenal disease impairs calcium filtration and excretion. In Figure 13-3, solid arrows represent potential causes of increased calcium, and dashed arrows represent potential causes of decreased calcium.
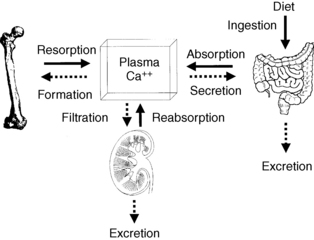
15. What are the mechanisms and causes of hypercalcemia?
From the preceding discussions, one can appreciate that mechanisms of hypercalcemia are usually multifactorial. However, most hypercalcemic syndromes have a primary or predominant mechanism, as outlined in Table 13-3. Most resorptive hypercalcemia is humoral (PTH, PTH-related peptide [PTHrP], transforming growth factor-α [TGF-α], TNF) or local osteolytic hypercalcemia (PTHrP, interleukins, prostaglandins). Increased calcium absorption usually occurs because of high 1,25(OH)2D levels either from excess vitamin D ingestion or produced by tumors or granulomas. Ninety percent of hypercalcemia cases result from hyperparathyroidism or cancer.
TABLE 13-3.
MECHANISMS AND CAUSES OF HYPERCALCEMIA
| PRIMARY MECHANISM | CAUSE(S) OF HYPERCALCEMIA |
| Increased bone resorption | Hyperparathyroidism Local osteolytic hypercalcemia Humoral hypercalcemia of malignancy Thyrotoxicosis Pheochromocytoma Excessive vitamin A (usually > 25,000 IU/day) Lithium carbonate Immobilization Addison’s disease |
| Increased renal reabsorption or decreased excretion | Milk-alkali syndrome Rhabdomyolysis Thiazide diuretics Familial hypocalciuric hypercalcemia Renal failure Lithium carbonate Addison’s disease |
| Increased gut absorption | Excessive vitamin D (usually > 10,000 IU/day) Berylliosis Candidiasis, coccidioidomycosis Eosinophilic granuloma Histoplasmosis Sarcoidosis Silicone implants Tuberculosis Inflammatory disorders Acquired immunodeficiency syndrome Lymphomas |
 KEY POINTS 1: HYPERCALCEMIA
KEY POINTS 1: HYPERCALCEMIA
1. Therapy for hypercalcemia should be directed at the underlying etiology, including excess bone resorption, renal tubular reabsorption, and gut absorption.
2. Although there are more than 30 different causes of hypercalcemia, hyperparathyroidism and hypercalcemia of malignancy account for more than 90% of cases.
3. Most patients with severe hypercalcemia require normal saline hydration and multiple-drug therapy, but most therapies for hypercalcemia inhibit bone resorption.
4. Zoledronic acid is the most potent bisphosphonate approved for treatment of hypercalcemia and has the advantage over pamidronate of a shorter infusion time and a longer duration of action.
5. Cinacalcet is a calcimimetic that lowers parathyroid hormone (PTH), calcium, and phosphorus levels. It is approved for treatment of secondary hyperparathyroidism (HPT) and parathyroid carcinoma. It is now approved for treating primary HPT in patients with clinically significant hypercalcemia when parathyroidectomy is not clinically appropriate or is contraindicated. Although not approved for such situations, cinacalcet also lowers PTH and calcium in kidney transplant recipients who have persistently elevated PTH.
16. What is the relative frequency of skeletal lesions in patients with advanced cancer?
The relative frequency is as follows: myeloma 95% to 100%, breast and prostate 70%, thyroid 60%, bladder 40%, lung 35%, renal 25%, and melanoma 14% to 45%. Common sites of bone metastases are ribs, spine, pelvis, and proximal extremities.
17. What is the incidence of hypercalcemia in patients with cancer?
Hypercalcemia affects 10% to 20% of patients with cancer. It is most common in squamous cell carcinoma of the lung, head, and neck, renal cell carcinoma, breast cancer, multiple myeloma, and lymphoma.
18. What are the multiple endocrine neoplasia syndromes?
Multiple endocrine neoplasia (MEN) is associated with three familial syndromes, two of which manifest as hypercalcemia due to hyperparathyroidism. MEN 1, or Wermer’s syndrome, includes the three Ps: pituitary, parathyroid, and pancreatic tumors. Hypercalcemia due to hyperparathyroidism is usually the first feature of this syndrome to appear. MEN 2 has two variants. Patients with MEN 2A, or Sipple’s syndrome, have medullary carcinoma of the thyroid (MCT), pheochromocytoma, and hyperparathyroidism. Patients with MEN 2B have MCT, pheochromocytoma, multiple mucosal neuromas, and marfanoid habitus; they usually do not have hyperparathyroidism. In comparison with sporadic hyperparathyroidism, parathyroid tumors in the MEN syndromes are more often bilateral, hyperplastic, and malignant.
19. How would you diagnose familial hypocalciuric hypercalcemia?
Familial hypocalciuric hypercalcemia (FHH), also called familial benign hypercalcemia, is due to autosomal dominant CaSR gene mutations, encoding inactive forms of the CaSR on parathyroid and renal tubular cell membranes. Important diagnostic features of FHH are the combination of no symptoms, a family history of benign hypercalcemia, mild hypercalcemia, normal to high serum PTH levels, and decreased renal calcium clearance (fractional excretion of calcium [FECa] < 1%). The clinical importance of FHH is to distinguish it from PHPT to avoid needless and ineffective parathyroidectomy. Patients with PHPT usually have an FECa value greater than 2%. In addition, patients with FHH usually have a 24-urinary calcium level lower than 100 mg, and those with HPT have values higher than 200 mg.
20. What is the likely cause of hypercalcemia in the following patient?
An 18-year-old man has had serum calcium values of 10.5 to 11.8 mg/dL for 2 years; his physical examination is normal, and he has a family history of hypercalcemia. Current laboratory values are as follows: calcium 11.5 mg/dL, intact PTH 70 pg/mL (normal NL < 65), plasma creatinine (PCr) 1.0 mg/dL, random urine calcium (UCa) 5 mg/dL, and urine creatinine (UCr) 90 mg/dL. Because circulating proteins bind 40% of the calcium, the kidney filters only 60%. Plasma calcium (PCa) available for filtration is 0.6 × 11.5, or 6.9 mg/dL, as shown by the following calculations:
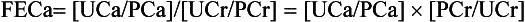
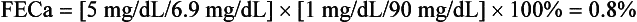
The history, physical findings, laboratory values, and FECa < 1% support the diagnosis of FHH.
21. What therapy is useful for hypercalcemia?
Most patients with severe hypercalcemia require treatment with multiple drugs. The lowest amount and least frequent dose that will achieve and maintain acceptable serum calcium levels should be given. The usual order of therapy is normal saline, calcitonin, zoledronic acid, and glucocorticoids, if indicated. Furosemide should be given only after good hydration, primarily to avoid volume overload and improve urinary volume. Gallium nitrate is rarely used for severe hypercalcemia of malignancy that is refractory to all other therapy. Consultation with nephrology should be sought, and dialysis considered, for acute management of severe and refractory hypercalcemia and hypercalcemic crisis (Table 13-4).
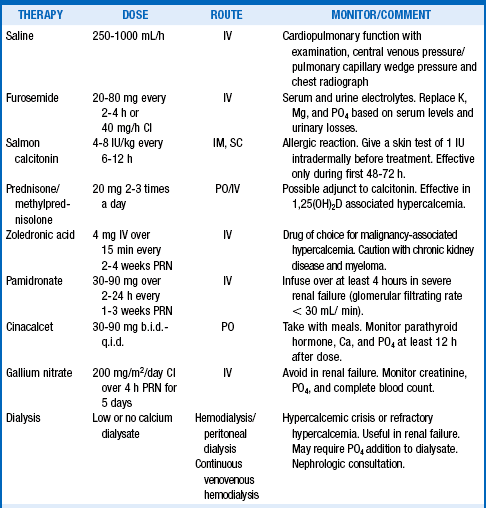
22. Describe the mechanisms of action of drug therapies for hypercalcemia.
TABLE 13-5.
MECHANISMS OF ACTION OF HYPERCALCEMIC THERAPY
| DRUG | MECHANISM(S) OF ACTION |
| Saline | Dilutes serum calcium by volume expansion and increases urinary flow and calcium excretion |
| Furosemide | Impairs renal sodium and calcium reabsorption in Henle’s loop, increasing urinary flow and calcium excretion |
| Calcitonin | Binds to receptors on osteoclasts, inhibiting osteoclast activity and decreasing bone resorption; also decreases renal reabsorption |
| Glucocorticoids | Antagonism of vitamin D, causing decreased calcium absorption and reabsorption; in tumoral states, may be tumor lytic and may decrease production of osteoclast-activating factors and vitamin D |
| Bisphosphonates | Impair osteoclast differentiation, recruitment, motility, and attachment; incorporate into bone matrix, making the matrix resistant to hydrolysis; overall effect is decreased bone resorption |
| Cinacalcet | Calcimimetic that binds to the calcium sensing receptor, making it markedly more responsive to calcium activation |
| Gallium nitrate | Adsorbs to and decreases solubility of hydroxyapatite crystals, decreasing bone resorption |
| Dialysis | Direct removal of calcium from blood |
23. How might calcimimetic drugs be useful in therapy for hypercalcemia?
On March 8, 2004, the U.S. Food and Drug Administration (FDA) approved cinacalcet, the first-in-class oral calcimimetic for clinical use. Calcimimetics are potentially the most useful drugs for the treatment of hypercalcemia caused by hyperparathyroidism. Cinacalcet remains the only calcimimetic available for patient care. It acts by increasing the sensitivity of the CaSR to calcium activation (see questions 8 and 9). The increased sensitivity shifts the PTH-calcium curve to the left, increasing parathyroid cell sensitivity to the PTH-suppressive effects of high extracellular calcium and decreasing its responsiveness to the PTH-stimulatory effects of low calcium. By increasing CaSR calcium sensitivity in Henle’s loop, cinacalcet also increases renal calcium excretion. The net effect is a dose-dependent marked reduction in PTH secretion, an increase in urinary calcium excretion, and a drop in serum calcium. Cinacalcet is FDA approved for treatment of secondary HPT in patients with chronic kidney disease undergoing dialysis, of parathyroid carcinoma, and of severe hypercalcemia in patients with primary HPT who are unable to undergo parathyroidectomy. Although not approved for the following uses, cinacalcet has successfully improved persistent HPT after kidney transplantation, hypercalcemia due to FHH, and lithium-induced hypercalcemia (see question 24).
24. How does lithium cause hypercalcemia?
Lithium decreases urinary calcium through competitive inhibition of the CaSR in the thick ascending limb of Henle’s loop, causing increased calcium reabsorption, decreased calcium excretion, and hypercalcemia. Urinary calcium may be low in lithium-treated patients as it is in patients with FHH. Lithium also decreases the sensitivity of the parathyroid CaSR to calcium and shifts the PTH-calcium curve in the parathyroids to the right. Thus, for any given calcium level, there is less suppression of PTH secretion and synthesis and higher PTH levels. Unlike in hyperparathyroidism, serum phosphate tends to be normal and magnesium higher in lithium-treated patients. Because hypercalcemia and elevated PTH may persist after lithium is discontinued, therapy other than just discontinuing lithium may be indicated if the hypercalcemia is symptomatic. Cinacalcet has successfully corrected or ameliorated PTH and serum Ca levels in such patients. This effect is expected because cinacalcet sensitizes the CaSR to calcium and shifts the PTH-calcium curve to the left.
 WEBSITES
WEBSITES
1. American Academy of Family Physicians: Available at http://www.aafp.org/afp/20030501/1959.html.
2. National Cancer Institute: Available at http://www.meb.uni-bonn.de/cancer.gov/CDR0000062737.html.
3. Zometa International: Zoledronic acid review: Available at http://www.zometa.com/monographs.html.
4. Endoctext.org: http://www.endotext.org/parathyroid/parathyroid2/parathyroidframe2.htm.
Bergua, C, Torregrosa, JV, Cofán, F, et al. Cinacalcet for the treatment of hypercalcemia in renal transplanted patients with secondary hyperparathyroidism. Transplant Proc. 2007;39:2254–2255.
Body, JJ. Current and future directions in medical therapy, hypercalcemia. Cancer. 2000;88(Suppl 12):3054–3058.
Bringhurst, FR, Demay, MB, Kronenberg, HM, et al. Hormones and disorders of mineral metabolism. In: Melmed S, Polonsky KS, Larsen PR, eds. Williams Textbook of Endocrinology. ed 12. Philadelphia: Elsevier Saunders; 2011:1237.
Carroll, MF, Schade, DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67:1959–1966.
Carroll, R, Matfin, G, Review. Endocrine and metabolic emergencieshypercalcaemia. Ther Adv Endocrinol Metab. 2010;1(5):225–234.
Festen-Spanjer, B, Haring, CM, Koster, JB, et al. Correction of hypercalcaemia by cinacalcet in familial hypocalciuric hypercalcaemia. Clin Endocrinol. 2008;68:324–325.
Jacobs, TP, Bilezikian, JP, Clinical review. rare causes of hypercalcemia. J Clin Endocrinol Metab 2005;90:6316–6322.
Kallas, M, Green, F, Hewison, M, et al, Rare causes of calcitriol-mediated hypercalcemia. a case report and literature review. J Clin Endocrinol Metab. 2010;95(7):3111–3117.
Kaye, TB, Hypercalcemia. how to pinpoint the cause and customize treatment. Postgrad Med 1995;97:153–160.
Maalouf, NM. Calcium homeostasis. In: Kovacs WJ, Ojeda SR, eds. Textbook of Endocrine Physiology. ed 6. New York: Oxford University Press; 2012:381.
Major, P. The use of zoledronic acid, a novel, highly potent bisphosphonate, for the treatment of hypercalcemia of malignancy. Oncologist. 2002;7:481–491.
Major, PP, Coleman, RE, Zoledronic acid in the treatment of hypercalcemia of malignancy. results of the International Clinical Development Program. Semin Oncol 2001;28:17–24.
Martin, A, David, V, Quarles, LD. Regulation and function of the FGF23/Klotho endocrine pathways. Physiol Rev. 2012;92(1):131–155.
Mihai, R, Wass, JAH, Sadler, GP. Asymptomatic hyperparathyroidism—need for multicentre studies. Clin Endocrinol. 2008;68:155–164.
Mundy, GR, Edward, JR. PTH-Related peptide in hypercalcemia. J Am Soc Nephrol. 2008;19:672–675.
Nishi, SPE, Barbagelata, NA, Atar, S, et al. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol. 2006;39:298–300.
Peacock, M, Bilezikian, JP, Bolognese, MA, et al. Cinacalcet HCL reduces hypercalcemia in primary hyperparathyroidism across a wide spectrum of disease severity. J Clin Endocrinol Metab. 2011;96(1):E9–E18.
Pont, A. Unusual causes of hypercalcemia. Endocrinol Metab Clin North Am. 1989;18:753–764.
Popovtzer, MM. Disorders of calcium, phosphorus, vitamin D, and parathyroid hormone activity. In: Schrier RW, ed. Renal and Electrolytes Disorders. ed 7. Philadelphia: Lippincott Williams & Wilkins; 2010:216.
Rabbani, SA, Molecular mechanism of action of parathyroid hormone related peptide in hypercalcemia of malignancy. therapeutic strategies [review]. Int J Oncol 2000;16:197–206.
Silverberg, SH, Rubin, MR, Faiman, C, et al. Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3803–3808.
Sloand, JA, Shelly, MA. Normalization of lithium-induced hypercalcemia and hyperparathyroidism with cinacalcet hydrochloride. Am J Kidney Dis. 2006;48:832–837.
Thakker, RV, Newey, PJ, Walls, GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990–3011.
Varghese, J, Rich, T, Jimenez, C. Benign familial hypocalciuric hypercalcemia. Endocr Pract. 2011;17(Suppl 1):13–17.
