[level-membership-for-basic-science-category]
41 HIV infection
Pathogenesis
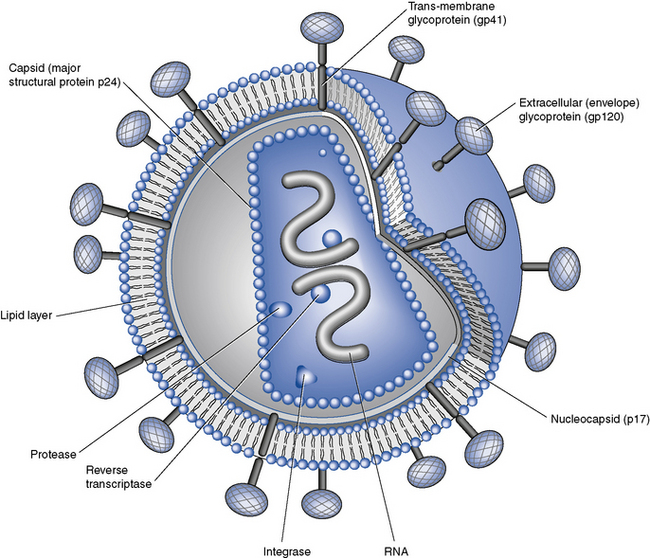
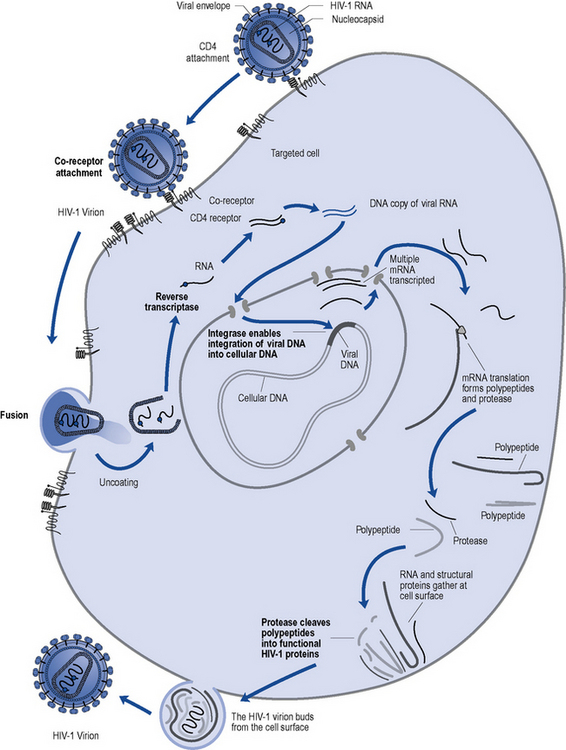
HIV, in common with other retroviruses, possesses the enzyme reverse transcriptase and consists of a lipid bilayer membrane surrounding the capsid (Fig. 41.1). Its surface glycoprotein molecule (gp120) has a strong affinity for the CD4 receptor protein found predominantly on the T-helper/inducer lymphocytes. Monocytes and macrophages may also possess CD4 receptors in low densities and can therefore also be infected. The process of HIV entry is more complex than originally thought, and in addition to CD4 attachment, subsequent binding to co-receptors such as CCR-5 or CXCR-4 and membrane fusion also occur (Fig. 41.2).
Clinical manifestations
The sequelae of untreated HIV infection can be broadly considered in five categories:
Opportunistic infections generally fall into two categories:
Investigations and monitoring
CD4 count
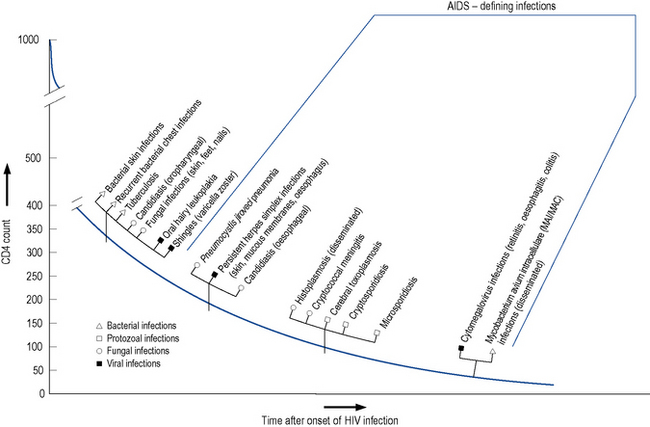
The level of immunosuppression is most easily estimated by monitoring a patient’s CD4 count. This measures the number of CD4-positive T-lymphocytes in a sample of peripheral blood. The normal range can vary between 500 and 1500 cells/mm3. As HIV disease progresses, the number of cells falls. Particular complications of HIV infection usually begin to occur at similar CD4 counts (Fig. 41.3) which can assist in differential diagnoses and enable the use of prophylactic therapies. For example, patients with a CD4 count of less than 200 cells/mm3 should always be offered prophylaxis against P. jiroveci pneumonia. Similarly, both patient and clinician are likely to use the CD4 count as the major indicator of when to consider starting antiretroviral therapy.
Viral load
The measurement of plasma HIV RNA (viral load) estimates the amount of circulating virus in the blood. This has been proven to correlate with prognosis, with a high viral load predicting faster disease progression (Mellors et al., 1997). Conversely, a reduction in viral load after commencement of antiviral therapy is associated with clinical benefit. This measure, in combination with the CD4 count, allows patients and clinicians to make informed decisions regarding when to start and when to change antiviral therapies, enabling the more effective use of such agents. There are on-line calculators utilising viral load and CD4 count to model risk of disease progression or death based on large cohort studies.
Drug treatment
The goals of therapy in HIV-positive individuals are to:
Antiretroviral therapy
Many organisations, such as the British HIV Association (BHIVA), the European AIDS Clinical Society (EACS) and the International AIDS Society (IAS), produce regularly updated guidelines on the use of antiretroviral therapy, for example, Gazzard et al. (2008). These guidelines include the most up-to-date considerations of:
Most studies evaluating triple combinations of antiretrovirals have been designed with so-called surrogate marker endpoints, measuring the effect on laboratory parameters such as CD4 count and HIV viral load. These trials are generally smaller and shorter in duration than clinical endpoint studies that are powered to measure the impact on survival and disease progression. The first large clinical endpoint trial that demonstrated the superiority of a triple combination over dual therapy was undertaken by Hammer et al. (1997). Following the results of this trial, the standard approach, where treatment is indicated, has been to use a combination of at least three agents. The reduction in morbidity and mortality associated with HAART has been confirmed in routine clinical practice, as well as in other trials (e.g. Palella et al., 1998; Smit et al., 2006). Subsequent clinical trials have largely been for licensing purposes and/or have served to refine therapeutic choices rather than to change the paradigm of treatment. The concept of intermittent rather than continuous therapy was evaluated in the SMART study but shown to be linked with an increased risk of co-morbidities not previously thought to be associated with HIV (such as cardiovascular disease, hepatic and renal failure) as well as HIV disease progression (El-Sadr et al., 2006). The use of protease inhibitor (PI) ‘monotherapy’ compared to conventional triple therapy has been evaluated in a number of small studies, for example, Arribas et al. (2009), and is being investigated in longer-term strategic studies. A large international study of early versus deferred treatment, to attempt to address the question of when to initiate treatment, is ongoing.
Choosing and monitoring therapy
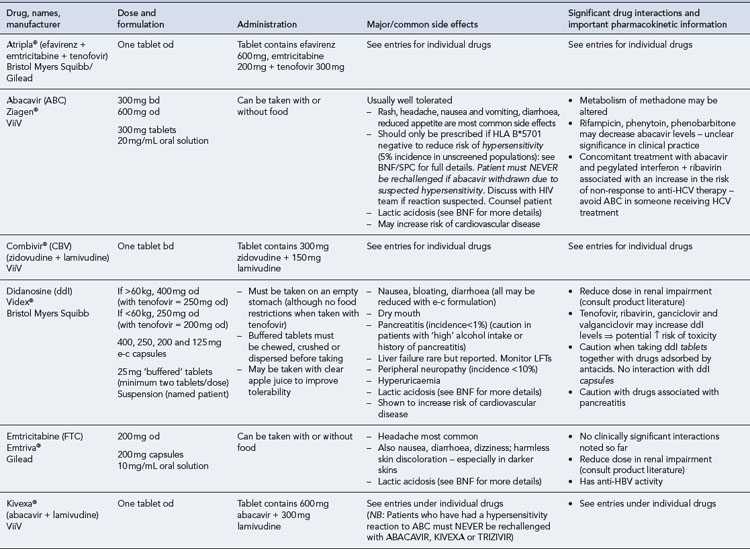
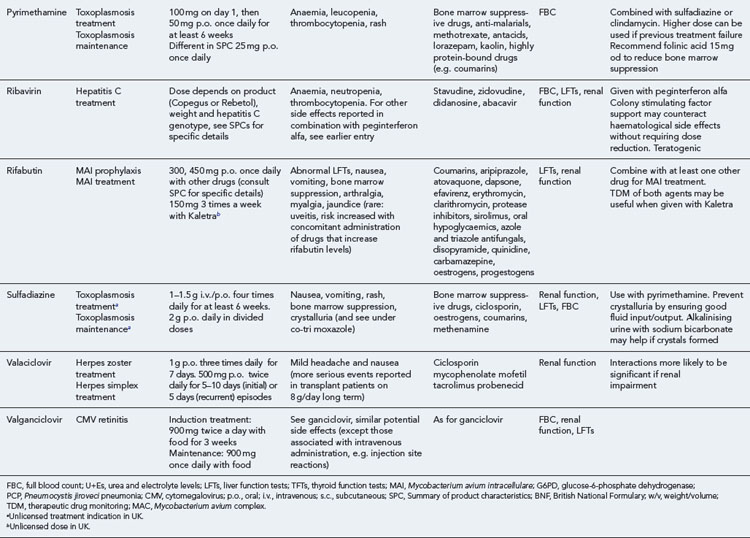
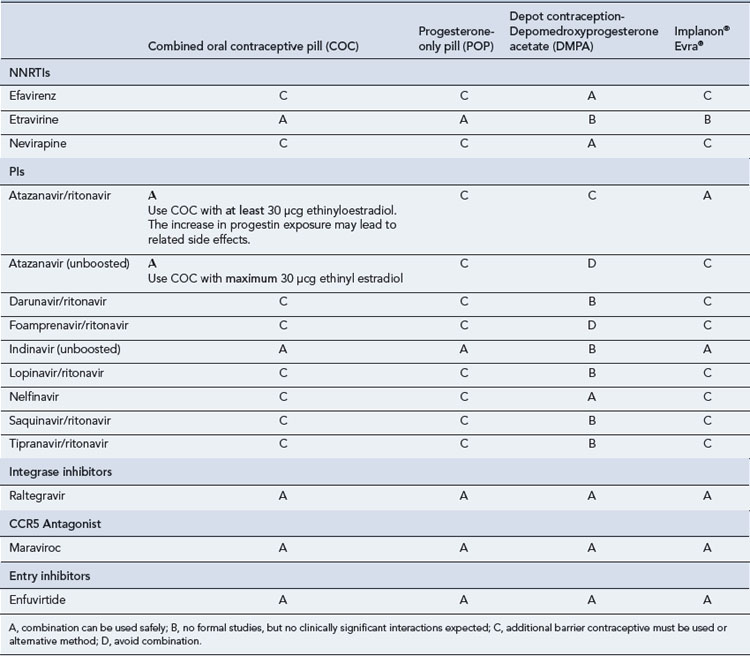
Many of the antiretrovirals, particularly the PIs and NNRTIs, exhibit a wide range of interactions, especially with other drugs that are metabolised by the cytochrome P450 enzyme system, including prescribed, ‘over-the-counter’, herbal and recreational drugs. HAART failure (detectable viral load and drug resistance) has been documented following co-administration of hepatic enzyme inducers, including non-prescribed agents such as St John’s Wort. Conversely, serious and even fatal toxicities due to enzyme inhibition by the PIs continue to be reported. These include Cushing’s syndrome following concomitant use of fluticasone or budesonide inhaler or nasal spray with a PI. This highlights the necessity of taking a comprehensive drug history prior to starting or switching HAART and ensuring patients and prescribers are aware of the need to check the interaction potential of new medicines. General prescribing guidelines for antiretrovirals are presented in Box 41.1 whilst details of common side effects and interactions of the currently available agents are summarised in Tables 41.1 to 41.5.
Box 41.1 General prescribing and monitoring information for antiretroviral agents
Table 41.1 General prescribing points for nucleoside/nucleotide analogue reverse transcriptase inhibitors (NRTIs)
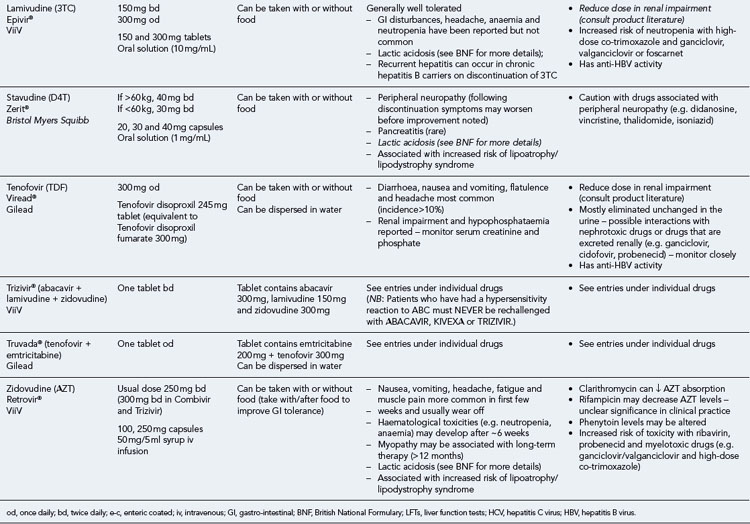
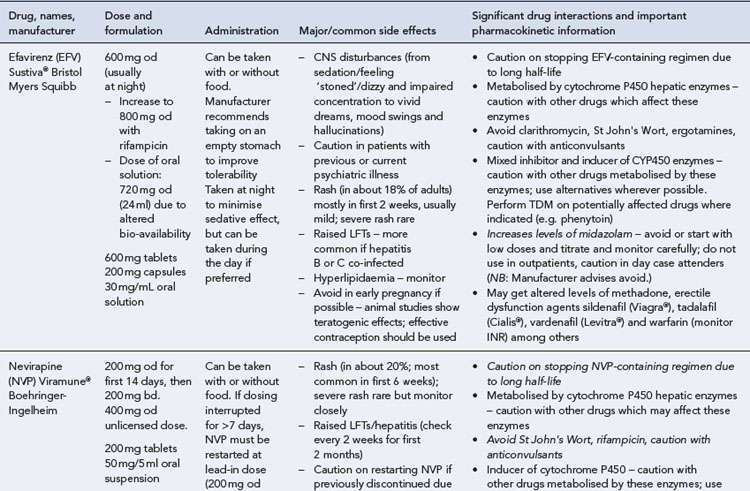
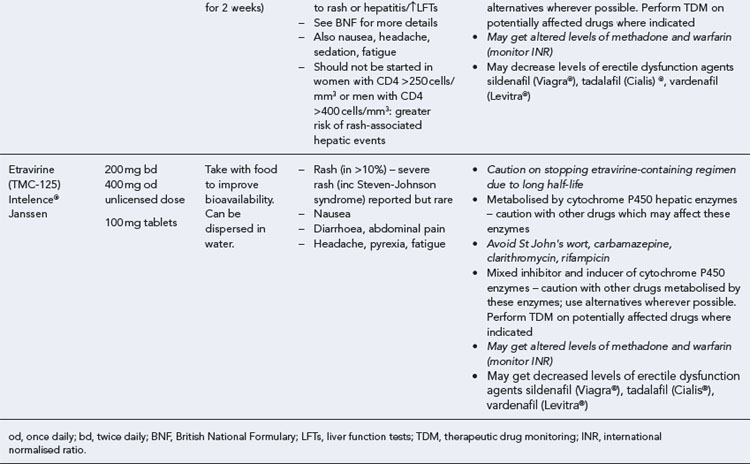
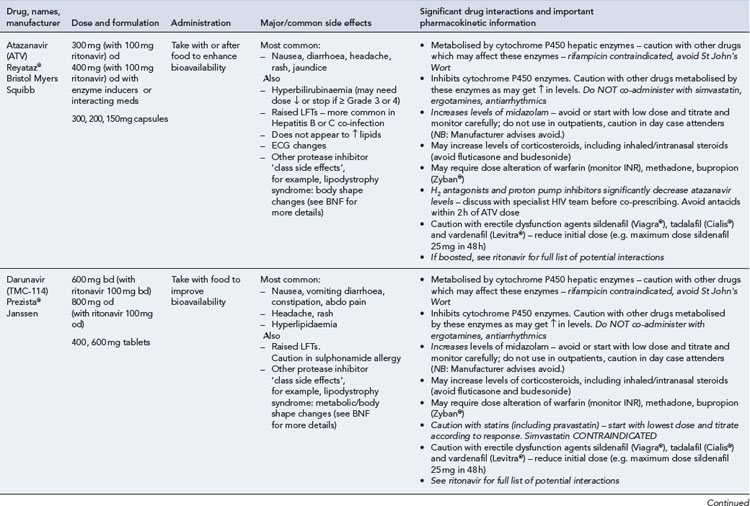
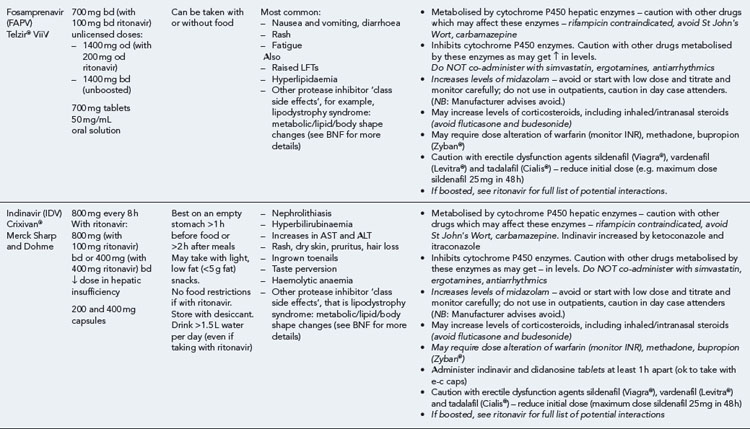
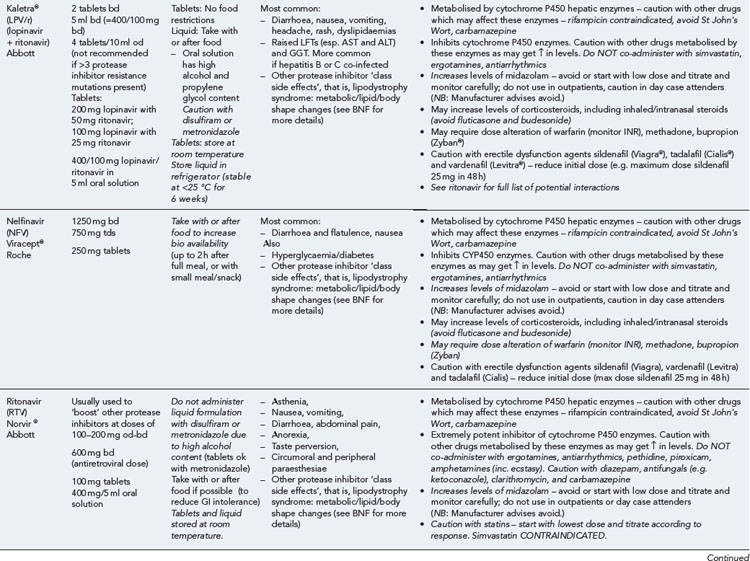
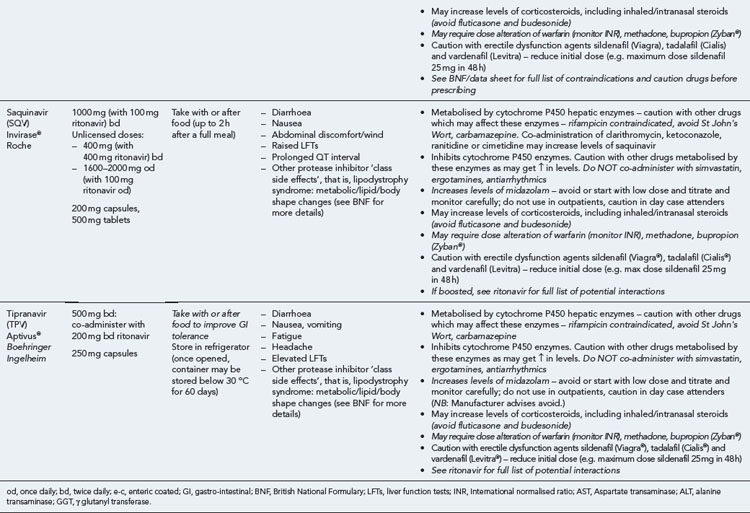
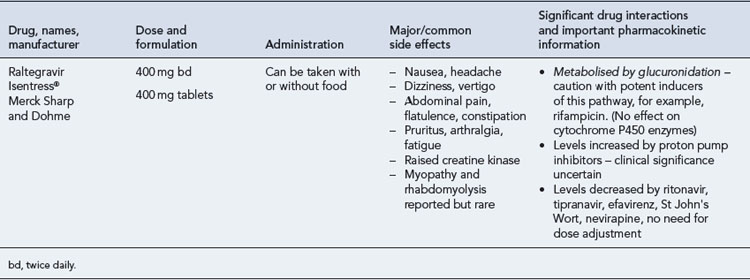
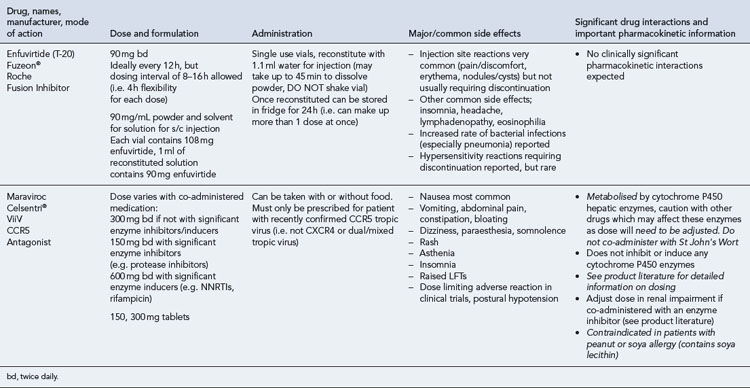
The routine use of therapeutic drug monitoring is not recommended but blood levels of PIs and NNRTIs should be measured in selected patients, for example, during pregnancy, where there is liver impairment and where there are concerns regarding potentially interacting drugs (Gazzard et al., 2008).
HIV mutates readily and resistance to some antiretrovirals, particularly reverse transcriptase inhibitors and integrase inhibitors, develops rapidly in the face of suboptimal treatment, for example, monotherapy or subtherapeutic blood levels. A high level of adherence to treatment is crucial to the sustained, successful outcome of antiretroviral regimens and has been the subject of much research. For example, in one study of people taking their first regimen containing nelfinavir, it was found that at least 95% adherence was required to achieve a sustained response in the majority (78%) of patients. The chances of treatment success declined as the level of adherence dropped, such that 80% of patients whose adherence was below 80% experienced virological failure. Virological success was also found to correlate with a better clinical outcome in terms of fewer hospitalisations, opportunistic infections and deaths (Paterson et al., 2000). Such clinical trial data have also been supported by clinical experience in the UK and elsewhere, although it has yet to be established if the level of adherence required is the same for all regimens and every patient. In view of this, patients should be advised to take HAART as close as possible to the same time every day and certainly within 1 hour of the agreed time each day. If they forget a dose, it should be taken as soon as they remember and then return to the original schedule.
There has been significant progress over recent years in reducing some of the physical burden of therapy, through the development of combination tablets and the use of strategies such as ritonavir boosting to reduce dietary restrictions and dosing frequency. Adherence aids such as pill boxes, medication record cards and alarms (e.g. on mobile phone) can also help to support adherence. However, practical issues are not the only barriers to adherence and the individual’s health beliefs and motivation, particularly around HIV and antiretroviral therapy, should also be addressed before treatment is commenced, as these are likely to have a significant impact on outcome (Horne et al., 2004). Although there is little evidence to demonstrate what the optimal interventions to improve adherence are, multidisciplinary and multiagency approaches appear to be most useful (Poppa et al., 2003).
Treatment interruptions
For many reasons, including toxicity, cost and adherence, patients and clinicians have been interested in considering ‘drug holidays’ or treatment interruptions. However, this strategy is no longer recommended in routine practice (El-Sadr et al., 2006). It is now recognised that there are dangers associated with this approach because of CD4 decline, disease progression, mortality related to co-morbidities, for example, cardiovascular disease, and viral load rebound associated with increased transmission risk and a seroconversion-like syndrome. Further, as different anti-HIV medications have different half-lives, there may be a risk of functional monotherapy, particularly with NNRTIs, and the development of resistance if combinations are stopped abruptly in an unplanned fashion.
Post-exposure prophylaxis
Post-exposure prophylaxis (PEP) involves the use of antiretroviral drugs to prevent infection with HIV after possible exposure, which may be recommended after occupational injuries (DH, 2008) or sexual exposure (Fisher et al., 2006). Whilst PEP is a largely unproven and unlicensed indication for the drugs used, it is supported by animal model data and case–control studies. Where recommended in guidelines, PEP is usually commenced as a 3–5-day starter regimen of two NRTIs and a boosted PI, followed by an ongoing course for a total of 4-week post-exposure. It is believed this will reduce the likelihood of infection by at least 80%, although toxicity issues are not insignificant. Therefore, the decision to prescribe or take PEP must reflect a careful risk/benefit evaluation. Studies of pre-exposure prophylaxis (PREP), using one or two antiretrovirals (orally or topically) before potential exposure to HIV, have so far yielded mixed results, but may offer additional options to reduce transmission.
Nucleoside and nucleotide analogue reverse transcriptase inhibitors
There is also one formulation combining two NRTIs with an NNRTI:
Other triple and quadruple mixed class co-formulations are in development.
Toxicity of antiretroviral therapies
Whilst there are many individual drug toxicities (see Tables 41.1–41.5), there are also a number of class-specific or therapy-related toxicities (Carr and Cooper, 2000).
Mitochondrial toxicity
Mitochondrial toxicity is increasingly recognised in patients with prolonged exposure to nucleoside analogue antiretrovirals, particularly stavudine, didanosine and, to a lesser extent, zidovudine, and is thought to explain such side effects as peripheral neuropathy, myopathy, pancreatitis and lactic acidosis. If these problems should arise, management is to switch the likely causative agent, if possible (McComsey and Lonergan, 2004).
Rash and hepatitis
These are both recognised side effects of the NNRTI class, although the incidence and severity appear greatest with nevirapine, particularly in patients with a higher CD4 count (>250 cells/mm3 for women and >400 cells/mm3 for men). Management is either close observation (in mild-to-moderate cases) or withdrawal of the causative agent (in severe cases). An abacavir hypersensitivity reaction is well characterised and historically occurs in approximately 6–8% of individuals who receive abacavir. It typically presents in the first 6 weeks of therapy as a progressive illness with fevers, rash and flu-like symptoms. Fatalities have been reported where the drug has subsequently been reintroduced in patients with a prior history of hypersensitivity reaction. Management has traditionally been to withdraw the agent and never reintroduce. However, the use of newer pharmacogenomic techniques, for example, HLA B*5701 testing, and subsequent prescription of abacavir only to those who are B*5701 negative have dramatically reduced the incidence of hypersensitivity reaction (Mallal et al., 2008).
Lipodystrophy
Lipodystrophy has been well reported in individuals on HAART. This is characterised by one or both of lipoatrophy (fat loss, particularly from the face, upper limbs and buttocks) and lipohypertrophy (abnormal fat deposition, particularly affecting the abdomen and neck). Whilst these body shape changes are associated with drug therapy, predominantly stavudine and zidovudine for lipoatrophy and possibly PIs for lipohypertrophy, it is likely that host and disease factors also play a role in aetiology (Carr, 2003; Lichtenstein, 2005). Management at present is to avoid or switch away from the causative agent(s) and/or to use cosmetic approaches (fillers or liposuction). Since the decline in use of the older NRTIs, the incidence of lipodystrophy has decreased dramatically.
Metabolic disturbances
Hypercholesterolaemia and hypertriglyceridaemia, in particular, are frequently seen in patients receiving HAART. Again, the aetiology of these toxicities is likely to represent a combination of drug and host factors. However, they are particularly associated with PIs, although the incidence appears lower with atazanavir. Whilst it is likely that this hyperlipidaemia contributes to an increased cardiovascular disease risk, this needs to be considered in the context of traditional risk factors, for example, smoking, which may be present in this patient group. Management is to reduce all modifiable risk factors and either switch away from the likely causative agent to one more metabolically favourable or consider adjunctive lipid-lowering therapy, taking into consideration potential drug–drug interactions (Schambelan et al., 2002).
Cardiovascular disease
Cohort studies have suggested an increased risk of cardiovascular disease with some PIs (Kaletra® and indinavir) and with the NRTI, abacavir (Sabin et al., 2008; Worm et al., 2010). This risk is independent of the effect on lipids and the mechanism has yet to be determined. As with lipid disturbances, the decision to start or continue these agents needs to be considered as part of a holistic approach to cardiovascular risk.
Opportunistic infections and malignancies
Detailed information on treatment guidelines can be obtained by consulting the relevant national guidelines, for example, British HIV Association guidelines for the management of opportunistic infections (Nelson et al., 2010), tuberculosis (Pozniak et al., 2010), malignancies (Bower et al., 2008). Dosing schedules for the most commonly used drugs used to treat opportunistic infections are summarised in Table 41.6.
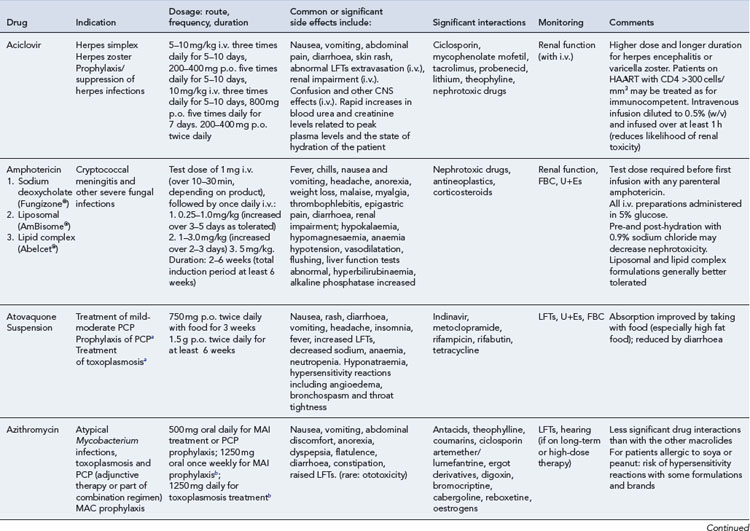
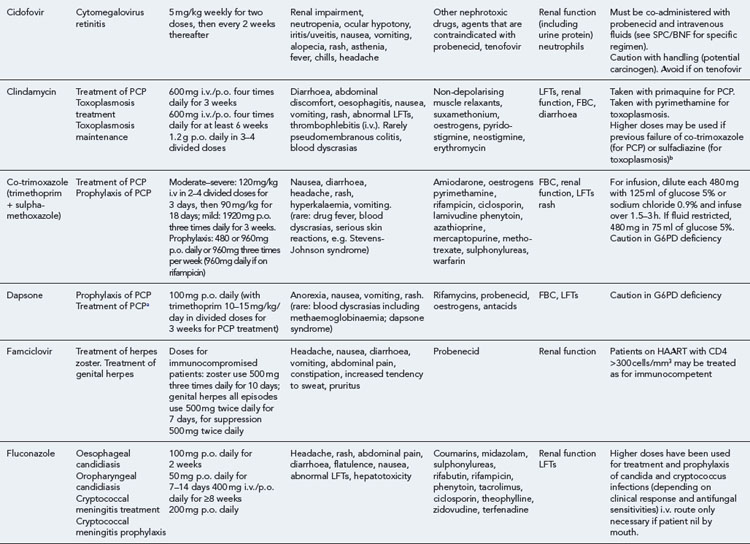
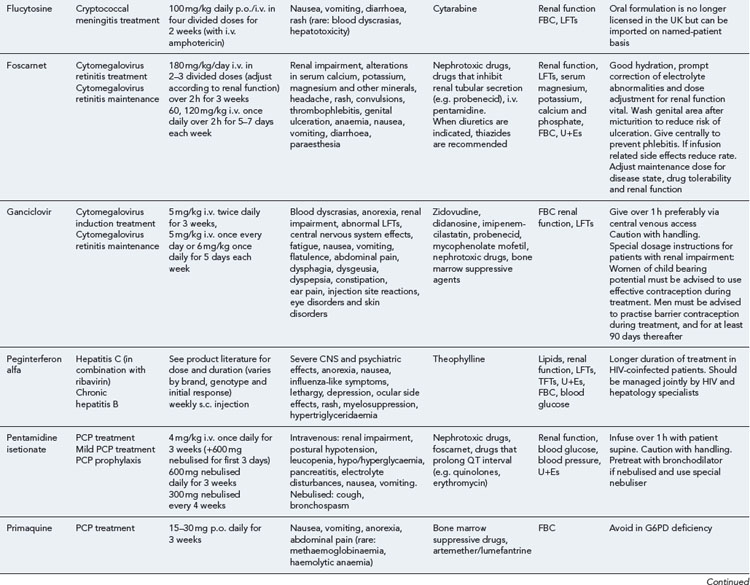
Bacterial infections
Mycobacteria
Treatment for pulmonary and extrapulmonary tuberculosis should follow conventional guidelines for immunocompetent individuals. Meningitis is an unusual but significant complication of tuberculosis. Treatment for all forms of CNS tuberculosis should consist of four drugs (isoniazid, rifampicin, pyrazinamide, ethambutol) for 2 months followed by two drugs (isoniazid, rifampicin) for at least 10 months. Adjunctive corticosteroids (either dexamethasone or prednisolone) should be given to all patients with tuberculous meningitis, regardless of disease severity (Thwaites et al., 2009).
Managing tuberculosis and HIV co-infection is further complicated by drug–drug interactions between anti-tuberculous and antiretroviral agents, overlapping toxicities and the risk of development of immune reconstitution disease (see later). This is a complex area and treatment should be guided by clinicians with the relevant specialist expertise (Pozniak et al., 2010). One of the most frequent concerns is when to initiate therapy for HIV in an individual receiving TB treatment. This decision is based upon the risk of developing other opportunistic infections in the medium term. HAART is usually started after 2 weeks of tuberculosis treatment in those with severe immunosuppression (CD4 <100 cells/mm3), after 2 months if the CD4 is between 100 and 200 cells/mm3, and on completion of tuberculosis treatment or at 6 months if the CD4 is greater than 200 cells/mm3.
Impact of HAART on opportunistic infections
Cancers
Kaposi’s sarcoma
This is the most common malignancy in people with HIV infection and may be triggered by infection with human herpes virus 8 (HHV-8). The majority of lesions affect the skin and appear as raised purple papules. These may be single or multiple and in severe cases may result in oedema, ulceration and infection. Visceral involvement is not uncommon but rarely causes clinically significant disease. In some cases, no therapeutic intervention is necessary and cosmetic camouflage may be sufficient. Indeed, treatment of HIV with antiretroviral therapy usually results in improvement, and in most cases, complete resolution, of Kaposi’s sarcoma. When individual lesions are troublesome, local radiotherapy or intralesional vinblastine can be beneficial. Alitretinoin gel (0.1%) (9-cis-retinoic acid) is a topical, self-administered therapy approved for the treatment of KS in the USA but not licensed in Europe. In cases of widespread cutaneous disease or significant visceral involvement, systemic chemotherapy is used, though this is often withheld until the potential benefits of HAART have been established. The liposomal anthracyclines and taxanes are now the mainstay of systemic chemotherapy for Kaposi’s sarcoma (Bower et al., 2008).
Hepatitis B co-infection
There are a number of ways in which HIV can impact on hepatitis B (HBV) infection:
Management requires an understanding of both viruses and is facilitated by the availability of drugs with dual HIV and hepatitis B activity (Brook et al., 2010). The HAART regimen should include two agents with anti-hepatitis B activity, usually tenofovir with either emtricitabine or lamivudine. If HIV develops resistance to these agents, they can still be continued for their anti-hepatitis B activity.
Hepatitis C co-infection
HIV also impacts on hepatitis C infection in a number of ways:
The management of hepatitis C/HIV co-infection is complicated (Brook et al., 2010). If treatment for hepatitis C is needed for someone on HAART, the antiretrovirals used must be compatible with hepatitis C therapy (pegylated α interferon and ribavirin). Didanosine, abacavir, stavudine and zidovudine should be avoided. It is commonplace to treat individuals with co-infection for a longer duration, for example, 48 weeks rather than 24 weeks for individuals with genotype 2 and 3 hepatitis C, and possibly 72 weeks for genotypes 1 and 4 – dependent upon early virological responses. Side effects of hepatitis C therapy tend to be more frequent and severe in the co-infected population. The proactive use of the colony-stimulating factors erythropoietin and G-CSF may enable optimal dosing of hepatitis C therapy and thereby improve outcome. The management of HCV is likely to be revolutionised by the availability of increasing numbers of Directly Acting Antivirals (DAAs) of which a large number are currently in development. The optimal way to use these and the potential drug-drug interactions with antiretrovirals have yet to be fully elucidated.
Women with HIV
Guidelines are available that set out the management of HIV infection in pregnant women and the prevention of mother-to-child transmission (de Ruiter et al., 2008). In general, intervention reflects a risk/benefit evaluation between the efficacy of reducing transmission and the potential harmful effects to the mother and fetus. Where HAART is clinically indicated for the mother herself, this should utilise a regimen of optimal efficacy with a favourable safety profile. Where therapy is initiated to reduce transmission, this is usually a HAART regimen but could be zidovudine monotherapy if the viral load is low (consistently <10,000 copies/mL). In the latter situation, the time of therapy initiation is guided by the viral load (Read et al., 2010):
Ethnicity
Questions
Answers
Case 41.2
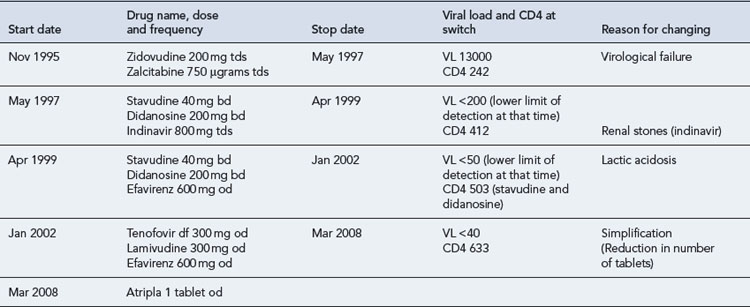
In 1995, Mr B, a 47-year-old man, presented with P. jiroveci pneumonia. He had a CD4 count of 123 cells/mm3, and plasma HIV RNA was unknown, as viral load testing was not routinely available in clinics at the time. He made a good response to treatment with high-dose co-trimoxazole and was subsequently commenced on dual combination therapy with zidovudine and zalcitabine (ddC, an NRTI, no longer marketed). He remained on this regimen for 18 months, until HIV RNA testing became available and he was found to have a viral load of 13,000 copies/mL and CD4 242 cells/mm3. His full antiretroviral therapy history, with reasons for switching, is detailed as follows (Table 41.8).
Question
Answer
Questions
Answers
Answers
Acknowledgements
The authors would like to acknowledge the contribution of Claire Richardson for assistance in updating Tables 41.2–41.5, of David Annandale for assistance in updating Table 41.6 and Laura Baber for permission to use Table 41.7.
Arribas J.R., Delgado R., Arranz A., et al. Lopinavir-ritonavir monotherapy versus lopinavir-ritonavir and 2 nucleosides for maintenance therapy of HIV: 96-week analysis. J. AIDS. 2009;51:147-152.
Arribas J.R., Horban A., Gerstoft J., et al. The MONET trial: darunavir/ritonavir with or without nucleoside analogues, for patients with HIV RNA below 50 copies/ml. AIDS. 2010;24:223-230.
Bower M., Collins S., Cottrill C., on behalf of the AIDS Malignancy Subcommittee. British HIV Association guidelines for HIV-associated malignancies 2008. HIV Med.. 2008;9:336-388. Available at http://www.bhiva.org/documents/Guidelines/Malignancy/080627MaligFinal.pdf. Accessed Nov. 2010
Brook G., Main J., Nelson M., et al. British HIV Association guidelines for the management of coinfection with HIV-1 and hepatitis B or C virus 2010. HIV Med.. 2010;11:1-30. Available at http://www.bhiva.org/documents/Guidelines/HepBC/2010/hiv_781.pdf. Accessed Nov. 2010
Carr A. HIV lipodystrophy: risk factors, pathogenesis, diagnosis and management. AIDS. 2003;17(Suppl. 1):S141-S148.
Carr A., Cooper D.A. Adverse effects of antiretroviral therapy. Lancet. 2000;356:1423-1430.
de Ruiter A., Mercey D., Anderson J., et al. British HIV Association and Children’s HIV Association guidelines for the management of HIV infection in pregnant women 2008. HIV Med.. 2008;9:452-502. Available at http://www.bhiva.org/documents/Guidelines/Pregnancy/2008/PregnancyPub.pdf. Accessed Nov. 2010
Department of Health. HIV post-exposure prophylaxis: guidance from the UK Chief Medical Officers’ Expert Advisory Group on AIDS. London: Department of Health, 2008. Available at http://www.dh.gov.uk/ab/EAGA/index.htm Accessed Nov. 2010
El-Sadr W.M., Lundgren J.D., Neaton J.D., et al. CD4+ count-guided interruption of antiretroviral treatment. N. Engl. J. Med.. 2006;355:2283-2296.
Fisher M., Benn P., Evans B., et al. UK guideline for the use of post-exposure prophylaxis for HIV following sexual exposure. Int. J. STD AIDS. 2006;17:81-92.
Gazzard B., on behalf of the BHIVA Treatment Guidelines Writing Group. British HIV Association guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy. HIV Med.. 2008;9:563-608. Available at http://onlinelibrary.wiley.com/doi/10.1111/j.1468–1293.2008.00636.x/full Accessed Nov. 2010
Hammer S.M., Squires K.E., Hughes M.D., et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med.. 1997;337:725-733.
Horne R., Buick D., Fisher M., et al. Doubts about necessity and concerns about adverse effects: identifying the types of beliefs that are associated with non-adherence to HAART. Int. J. STD AIDS. 2004;15:38-44.
Lichtenstein K.A. Redefining lipodystrophy syndrome: risks and impact on clinical decision making. J. AIDS. 2005;39:395-400.
Mallal S., Phillips E., Carosi G., et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med.. 2008;358:568-579.
McComsey G., Lonergan J.T. Mitochondrial dysfunction: patient monitoring and toxicity management. J. AIDS. 2004;37:S30-S35.
Mellors J.W., Munoz A., Giorgi J.V., et al. Plasma viral load and CD4 lymphocytes as prognostic markers of HIV-1 infection. Ann. Intern. Med.. 1997;126:946-954.
Nelson M., Dockrell D., Edwards S., on behalf of the BHIVA Guidelines Subcommittee. British HIV Association guidelines for the treatment of opportunistic infection in HIV-positive individuals 2010. London: British HIV Association, 2010. Available at http://www.bhiva.org/PublishedandApproved.aspx
Paterson D.L., Swindells S., Mohr J., et al. Adherence to protease inhibitor therapy and outcome in patients with HIV infection. Ann. Intern. Med.. 2000;133:21-30.
Palella F.J.Jr., Delaney K.M., Moorman A.C. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med.. 1998;338:853-860.
Poppa Poppa A., Davidson O., Deutsch J., et al. British HIV Association (BHIVA)/British Association for Sexual Health & HIV (BASHH) guidelines on provision of adherence support to individuals receiving antiretroviral therapy. British HIV Association; 2003. Available online at www.bhiva.org Accessed Nov. 2010
Pozniak A.L., Collins S., Coyne C.M., on behalf of BHIVA Guidelines Writing Committee. British HIV Association guidelines for the treatment of TB/HIV co-infection 2010. Available at http://www.bhiva.org/PublishedandApproved.aspx, 2010.
Read P., Khan P., Mandalia S., et al. When Should HAART Be Initiated in Pregnancy to Achieve an Undetectable Viral Load?. 17th Conference on Retroviruses and Opportunistic Infections, February 2010. Poster 896. 2010. Available at www.retroconference.org/2010/PDFs/896.pdf Accessed Nov. 2010
Sabin C.A., Worm S.W., Weber R., et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet. 2008;371:1417-1426.
Schambelan M., Benson C.A., Carr A., et al. Management of metabolic complications associated with antiretroviral therapy for HIV-1 infection: recommendations of an International AIDS Society-USA Panel. J. AIDS. 2002;31:257-275.
Smit C., Geskus R., Walker S., et al. Effective therapy has altered the spectrum of cause-specific mortality following HIV seroconversion. AIDS. 2006;20:741-749.
Thwaites G., Fisher M., Hemingway C., et al. British Infection Society guidelines for the diagnosis and treatment of tuberculosis of the central nervous system in adults and children. J. Infect.. 2009;59:167-187.
Worm S.W., Sabin C., Weber R., et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the data collection on adverse events of anti HIV drugs (D:A:D) study. J. Infect. Dis.. 2010;201:318-330.
Bhagani S., Sweny P., Brook G. Guidelines for kidney transplantation in patients with HIV disease. HIV Med., 7. 2006:133-139. 10.1111/j.1468–1293.2006.00367.x. Available at http://onlinelibrary.wiley.com/doi/10.1111/j.1468–1293.2006.00367.x/full Accessed Nov. 2010.
British HIV Association, British Association of Sexual Health and HIV, British Infection Society. UK national guidelines for HIV testing. Available at www.bhiva.org/HIVTesting2008.aspx, 2008. Accessed Nov. 2010
Department of Health. HIV Infected Health Care Workers: Guidance on Management and Patient Notification. London: Department of Health; 2005. Available at http://www.clinical-virology.org/pdfs/DH_4116416.pdf Accessed Nov. 2010
Geretti A.M., on behalf of the BHIVA Immunization Writing Committee. British HIV Association Guidelines for immunization of HIV-infected adults 2008. HIV Med.. 2008;9:795-848. Available at http://onlinelibrary.wiley.com/doi/10.1111/j.1468–1293.2008.00637.x/full Accessed Nov. 2010
Gupta S.K., Eustace J.A., Winston J.A., et al. Guidelines for the management of chronic kidney disease in HIV-infected patients: recommendations of the HIV Medicine Association of the Infectious Diseases Society of America. Clin. Infect. Dis.. 2005;40:1559-1585.
O’Grady J., Taylor C., Brook G., et al. Guidelines for liver transplantation in patients with HIV infection. British HIV Association A. HIV Med.. 2005;6(Suppl. 2):149-153. Available at http://onlinelibrary.wiley.com/doi/10.1111/j.1468–1293.2005.00303.x/pdf Accessed Nov. 2010
British Association for Sexual Health and HIV. www.bashh.org.
British HIV Association. www.bhiva.org.
Clinical Care Options (US-based website focusing on HIV, hepatitis and oncology). www.clinicaloptions.com.
European AIDS Clinical Society Guidelines. www.europeanaidsclinicalsociety.org/guidelines.asp.
HIV i-Base HIV i-Base (UK-based community provider of wide range of HIV-related information, including drug/treatment updates, conference reports and daily news items). i-base.info/home/.
Johns Hopkins University HIV Guide. www.hopkins-aids.edu.
Medscape Medscape (US-based medical website with HIV specialty home page). www.medscape.com.
NAM NAM (UK-based community provider of wide range of HIV-related information, including drug/treatment updates, conference reports and daily news items). www.aidsmap.com.
Toronto General Hospital Immunodeficiency Clinic. www.hivclinic.ca/main/home.html.
United States National HIV Guidelines (antiretroviral treatment, management of opportunistic infections, co-infections and post-exposure prophylaxis). www.aidsinfo.nih.gov/guidelines/.
University of California San Francisco HIV Educational Resource. www.hivinsite.ucsf.edu.
University of Liverpool HIV Pharmacology Group (includes drug interaction charts). www.hiv-druginteractions.org.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
41 HIV infection
Pathogenesis
HIV, in common with other retroviruses, possesses the enzyme reverse transcriptase and consists of a lipid bilayer membrane surrounding the capsid (Fig. 41.1). Its surface glycoprotein molecule (gp120) has a strong affinity for the CD4 receptor protein found predominantly on the T-helper/inducer lymphocytes. Monocytes and macrophages may also possess CD4 receptors in low densities and can therefore also be infected. The process of HIV entry is more complex than originally thought, and in addition to CD4 attachment, subsequent binding to co-receptors such as CCR-5 or CXCR-4 and membrane fusion also occur (Fig. 41.2).
Clinical manifestations
The sequelae of untreated HIV infection can be broadly considered in five categories:
Opportunistic infections generally fall into two categories:
Investigations and monitoring
CD4 count
The level of immunosuppression is most easily estimated by monitoring a patient’s CD4 count. This measures the number of CD4-positive T-lymphocytes in a sample of peripheral blood. The normal range can vary between 500 and 1500 cells/mm3. As HIV disease progresses, the number of cells falls. Particular complications of HIV infection usually begin to occur at similar CD4 counts (Fig. 41.3) which can assist in differential diagnoses and enable the use of prophylactic therapies. For example, patients with a CD4 count of less than 200 cells/mm3 should always be offered prophylaxis against P. jiroveci pneumonia. Similarly, both patient and clinician are likely to use the CD4 count as the major indicator of when to consider starting antiretroviral therapy.
Viral load
The measurement of plasma HIV RNA (viral load) estimates the amount of circulating virus in the blood. This has been proven to correlate with prognosis, with a high viral load predicting faster disease progression (Mellors et al., 1997). Conversely, a reduction in viral load after commencement of antiviral therapy is associated with clinical benefit. This measure, in combination with the CD4 count, allows patients and clinicians to make informed decisions regarding when to start and when to change antiviral therapies, enabling the more effective use of such agents. There are on-line calculators utilising viral load and CD4 count to model risk of disease progression or death based on large cohort studies.
Drug treatment
The goals of therapy in HIV-positive individuals are to:
Antiretroviral therapy
Many organisations, such as the British HIV Association (BHIVA), the European AIDS Clinical Society (EACS) and the International AIDS Society (IAS), produce regularly updated guidelines on the use of antiretroviral therapy, for example, Gazzard et al. (2008). These guidelines include the most up-to-date considerations of:
Most studies evaluating triple combinations of antiretrovirals have been designed with so-called surrogate marker endpoints, measuring the effect on laboratory parameters such as CD4 count and HIV viral load. These trials are generally smaller and shorter in duration than clinical endpoint studies that are powered to measure the impact on survival and disease progression. The first large clinical endpoint trial that demonstrated the superiority of a triple combination over dual therapy was undertaken by Hammer et al. (1997). Following the results of this trial, the standard approach, where treatment is indicated, has been to use a combination of at least three agents. The reduction in morbidity and mortality associated with HAART has been confirmed in routine clinical practice, as well as in other trials (e.g. Palella et al., 1998; Smit et al., 2006). Subsequent clinical trials have largely been for licensing purposes and/or have served to refine therapeutic choices rather than to change the paradigm of treatment. The concept of intermittent rather than continuous therapy was evaluated in the SMART study but shown to be linked with an increased risk of co-morbidities not previously thought to be associated with HIV (such as cardiovascular disease, hepatic and renal failure) as well as HIV disease progression (El-Sadr et al., 2006). The use of protease inhibitor (PI) ‘monotherapy’ compared to conventional triple therapy has been evaluated in a number of small studies, for example, Arribas et al. (2009), and is being investigated in longer-term strategic studies. A large international study of early versus deferred treatment, to attempt to address the question of when to initiate treatment, is ongoing.
Choosing and monitoring therapy
Many of the antiretrovirals, particularly the PIs and NNRTIs, exhibit a wide range of interactions, especially with other drugs that are metabolised by the cytochrome P450 enzyme system, including prescribed, ‘over-the-counter’, herbal and recreational drugs. HAART failure (detectable viral load and drug resistance) has been documented following co-administration of hepatic enzyme inducers, including non-prescribed agents such as St John’s Wort. Conversely, serious and even fatal toxicities due to enzyme inhibition by the PIs continue to be reported. These include Cushing’s syndrome following concomitant use of fluticasone or budesonide inhaler or nasal spray with a PI. This highlights the necessity of taking a comprehensive drug history prior to starting or switching HAART and ensuring patients and prescribers are aware of the need to check the interaction potential of new medicines. General prescribing guidelines for antiretrovirals are presented in Box 41.1 whilst details of common side effects and interactions of the currently available agents are summarised in Tables 41.1 to 41.5.
Box 41.1 General prescribing and monitoring information for antiretroviral agents
Table 41.1 General prescribing points for nucleoside/nucleotide analogue reverse transcriptase inhibitors (NRTIs)
The routine use of therapeutic drug monitoring is not recommended but blood levels of PIs and NNRTIs should be measured in selected patients, for example, during pregnancy, where there is liver impairment and where there are concerns regarding potentially interacting drugs (Gazzard et al., 2008).
HIV mutates readily and resistance to some antiretrovirals, particularly reverse transcriptase inhibitors and integrase inhibitors, develops rapidly in the face of suboptimal treatment, for example, monotherapy or subtherapeutic blood levels. A high level of adherence to treatment is crucial to the sustained, successful outcome of antiretroviral regimens and has been the subject of much research. For example, in one study of people taking their first regimen containing nelfinavir, it was found that at least 95% adherence was required to achieve a sustained response in the majority (78%) of patients. The chances of treatment success declined as the level of adherence dropped, such that 80% of patients whose adherence was below 80% experienced virological failure. Virological success was also found to correlate with a better clinical outcome in terms of fewer hospitalisations, opportunistic infections and deaths (Paterson et al., 2000). Such clinical trial data have also been supported by clinical experience in the UK and elsewhere, although it has yet to be established if the level of adherence required is the same for all regimens and every patient. In view of this, patients should be advised to take HAART as close as possible to the same time every day and certainly within 1 hour of the agreed time each day. If they forget a dose, it should be taken as soon as they remember and then return to the original schedule.
There has been significant progress over recent years in reducing some of the physical burden of therapy, through the development of combination tablets and the use of strategies such as ritonavir boosting to reduce dietary restrictions and dosing frequency. Adherence aids such as pill boxes, medication record cards and alarms (e.g. on mobile phone) can also help to support adherence. However, practical issues are not the only barriers to adherence and the individual’s health beliefs and motivation, particularly around HIV and antiretroviral therapy, should also be addressed before treatment is commenced, as these are likely to have a significant impact on outcome (Horne et al., 2004). Although there is little evidence to demonstrate what the optimal interventions to improve adherence are, multidisciplinary and multiagency approaches appear to be most useful (Poppa et al., 2003).
Treatment interruptions
For many reasons, including toxicity, cost and adherence, patients and clinicians have been interested in considering ‘drug holidays’ or treatment interruptions. However, this strategy is no longer recommended in routine practice (El-Sadr et al., 2006). It is now recognised that there are dangers associated with this approach because of CD4 decline, disease progression, mortality related to co-morbidities, for example, cardiovascular disease, and viral load rebound associated with increased transmission risk and a seroconversion-like syndrome. Further, as different anti-HIV medications have different half-lives, there may be a risk of functional monotherapy, particularly with NNRTIs, and the development of resistance if combinations are stopped abruptly in an unplanned fashion.
Post-exposure prophylaxis
Post-exposure prophylaxis (PEP) involves the use of antiretroviral drugs to prevent infection with HIV after possible exposure, which may be recommended after occupational injuries (DH, 2008) or sexual exposure (Fisher et al., 2006). Whilst PEP is a largely unproven and unlicensed indication for the drugs used, it is supported by animal model data and case–control studies. Where recommended in guidelines, PEP is usually commenced as a 3–5-day starter regimen of two NRTIs and a boosted PI, followed by an ongoing course for a total of 4-week post-exposure. It is believed this will reduce the likelihood of infection by at least 80%, although toxicity issues are not insignificant. Therefore, the decision to prescribe or take PEP must reflect a careful risk/benefit evaluation. Studies of pre-exposure prophylaxis (PREP), using one or two antiretrovirals (orally or topically) before potential exposure to HIV, have so far yielded mixed results, but may offer additional options to reduce transmission.
Nucleoside and nucleotide analogue reverse transcriptase inhibitors
There is also one formulation combining two NRTIs with an NNRTI:
Other triple and quadruple mixed class co-formulations are in development.
