[level-membership-for-pediatrics-category]
Chapter 452 Hereditary Spherocytosis
Etiology
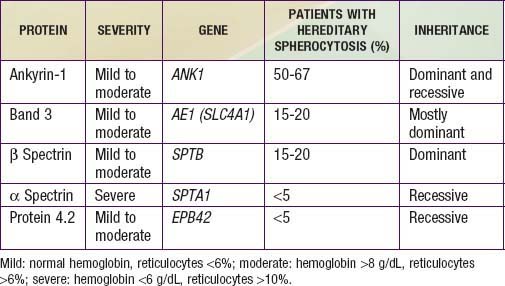
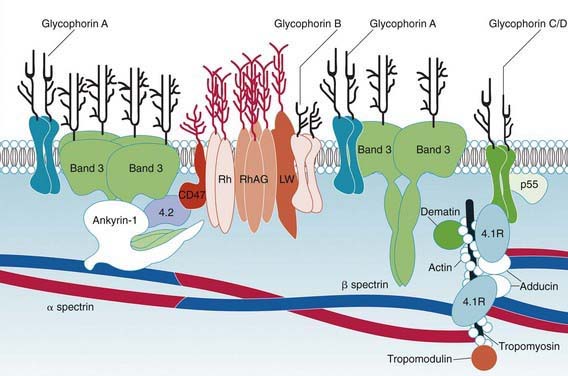
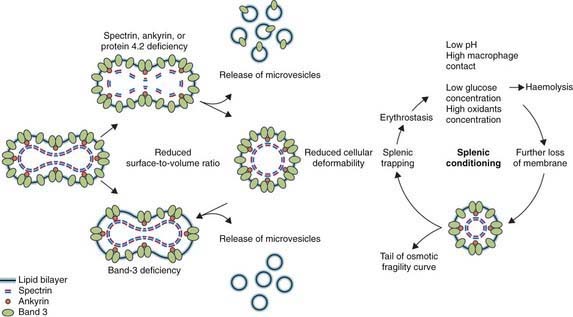
Hereditary spherocytosis usually is transmitted as an autosomal dominant or, less commonly, as an autosomal recessive disorder. As many as 25% of patients have no previous family history. Of these patients, most represent new mutations, and a few cases result from recessive inheritance or represent nonpaternity. The most common molecular defects are abnormalities of spectrin or ankyrin, which are major components of the cytoskeleton responsible for RBC shape. A recessive defect has been described in α-spectrin. Dominant defects have been described in β-spectrin and protein 3. Dominant and recessive defects have been described in ankyrin (Table 452-1). A deficiency in spectrin, protein 3, or ankyrin results in uncoupling in the “vertical” interactions of the lipid bilayer skeleton and the loss of membrane microvesicles (Figs. 452-1 and 452-2). The loss of membrane surface area without a proportional loss of cell volume causes sphering of the RBCs and an associated increase in cation permeability, cation transport, adenosine triphosphate (ATP) use, and glycolysis. The decreased deformability of the spherocytic RBCs impairs cell passage from the splenic cords to the splenic sinuses, and the spherocytic RBCs are destroyed prematurely in the spleen. Splenectomy markedly improves RBC life span and cures the anemia.
Clinical Manifestations
Hereditary spherocytosis may be a cause of hemolytic disease in the newborn and can manifest as anemia and hyperbilirubinemia sufficiently severe to require phototherapy or exchange transfusions. Hemolysis may be more prominent in the newborn because hemoglobin F binds 2,3-diphosphoglycerate poorly, and the increased level of free 2,3-diphosphoglycerate destabilizes interactions among spectrin, actin, and protein 4.1 in the RBC membrane (see Fig. 452-1).
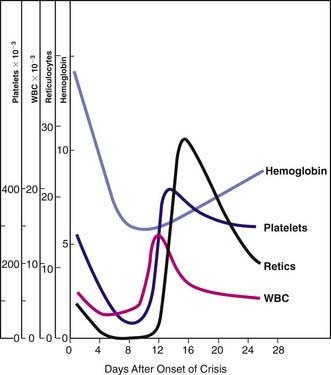
Because of the high RBC turnover and heightened erythroid marrow activity, children with hereditary spherocytosis are susceptible to aplastic crisis, primarily as a result of parvovirus B19 infection, and to hypoplastic crises associated with various other infections (Fig. 452-3). The erythroid marrow failure can rapidly result in profound anemia (hematocrit <10%), high-output heart failure, hypoxia, cardiovascular collapse, and death. White blood cell and platelet counts can also fall (see Fig. 452-3).
Long-term complications include gout, myopathy, and spinocerebellar degenerations.
Laboratory Findings
Evidence of hemolysis includes reticulocytosis and indirect hyperbilirubinemia. The hemoglobin level usually is 6-10 g/dL, but it can be in the normal range. The reticulocyte percentage often is increased to 6-20%, with a mean of approximately 10%. The mean corpuscular volume (MCV) is normal, although the mean corpuscular hemoglobin concentration often is increased (36-38 g/dL RBCs). The RBCs on the blood film vary in size and include polychromatophilic reticulocytes and spherocytes (Fig. 452-4). The spherocytes are smaller in diameter and appear hyperchromic on the blood film as a result of the high hemoglobin concentration. The central pallor is less conspicuous than in normal cells. Spherocytes may be the predominant cells or may be relatively sparse, depending on the severity of the disease, but they usually account for >15-20% of the cells when hemolytic anemia is present. Erythroid hyperplasia is evident in the marrow aspirate or biopsy. Marrow expansion may be evident on routine roentgenographic examination. Other evidence of hemolysis can include decreased haptoglobin and the presence of gallstones on ultrasonography.
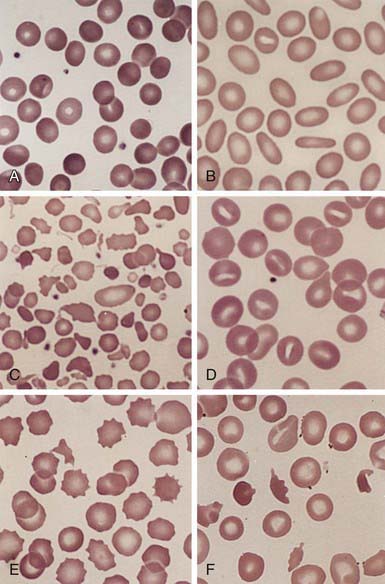
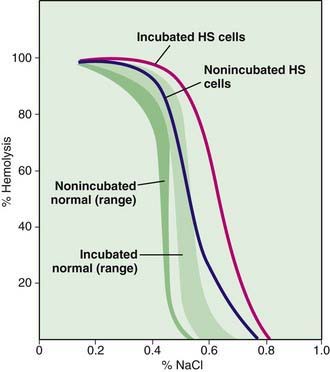
The diagnosis of hereditary spherocytosis usually is established clinically from the blood film, which shows many spherocytes and reticulocytes, from the family history, and from splenomegaly. The presence of spherocytes in the blood can be confirmed with an osmotic fragility test (Fig. 452-5). The RBCs are incubated in progressive dilutions of an iso-osmotic buffered salt solution. Exposure to hypotonic saline causes the RBCs to swell, and the spherocytes lyse more readily than biconcave cells in hypotonic solutions. This feature is accentuated by depriving the cells of glucose overnight at 37°C, known as the incubated osmotic fragility test. Unfortunately, this test is not specific for hereditary spherocytosis, and results may be abnormal in immune and other hemolytic anemias. A normal test result also may be found in 10-20% of patients. Other tests, such as the cryohemolysis test, osmotic gradient ektacytometry, and the eocin-5-maleimide test, may be more sensitive but are not readily available. Detection of a population of hyperdense RBCs using a laser-based instrument or a Coulter counter may prove more convenient as an approach to diagnosis.
Differential Diagnosis
The major alternative considerations when large numbers of spherocytes are seen on the blood film are isoimmune and autoimmune hemolysis. Isoimmune hemolytic disease of the newborn, particularly due to ABO incompatibility, mimics hereditary spherocytosis. The detection of antibody on an infant’s RBCs using a direct antiglobulin (Coombs) test should establish the diagnosis of immune hemolysis. Autoimmune hemolytic anemias also are characterized by spherocytes, and there may be evidence of previously normal values for hemoglobin, hematocrit, and reticulocyte count. Rare causes of spherocytosis include thermal injury, clostridial septicemia with exotoxemia, and Wilson disease, each of which can manifest as transient hemolytic anemia (see Table 451-1).
Alizai NK, Richards EM, Stringer MD. Is cholecystectomy really an indication for concomitant splenectomy in mild hereditary spherocytosis? Arch Dis Child. 2010;95:596-599.
Bolton-Maggs PBH, Stevens RF, Dodd NJ, et al. Guidelines for the diagnosis and management of hereditary spherocytosis. Br J Haematol. 2004;126:455-474.
Delhommeau F, Cynober T, Schischmanoff PO, et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95:393-397.
Diesen DL, Zimmerman SA, Thornburg CD, et al. Partial splenectomy for children with congenital hemolytic anemia and massive splenomegaly. J Pediatr Surg. 2008;43:466-472.
Eber S, Lux SE. Hereditary spherocytosis: defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118-141.
Iolascon A, Perrotta S, Stewart GW. Red cell membrane defects. Rev Clin Exp Hematol. 2003;7:22-56.
Minkes RK, Lagzdins M, Langer JC, et al. Laparoscopic versus open splenectomy in children. J Pediatr Surg. 2000;35:699-700.
Perrota S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
Shah S, Vega R. Hereditary spherocytosis. Pediatr Rev. 2004;25:168-172.
Tracy ET, Rice HE. Partial splenectomy for hereditary spherocytosis. Pediatr Clin North Am. 2008;55:503-519.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 452 Hereditary Spherocytosis
Etiology
Hereditary spherocytosis usually is transmitted as an autosomal dominant or, less commonly, as an autosomal recessive disorder. As many as 25% of patients have no previous family history. Of these patients, most represent new mutations, and a few cases result from recessive inheritance or represent nonpaternity. The most common molecular defects are abnormalities of spectrin or ankyrin, which are major components of the cytoskeleton responsible for RBC shape. A recessive defect has been described in α-spectrin. Dominant defects have been described in β-spectrin and protein 3. Dominant and recessive defects have been described in ankyrin (Table 452-1). A deficiency in spectrin, protein 3, or ankyrin results in uncoupling in the “vertical” interactions of the lipid bilayer skeleton and the loss of membrane microvesicles (Figs. 452-1 and 452-2). The loss of membrane surface area without a proportional loss of cell volume causes sphering of the RBCs and an associated increase in cation permeability, cation transport, adenosine triphosphate (ATP) use, and glycolysis. The decreased deformability of the spherocytic RBCs impairs cell passage from the splenic cords to the splenic sinuses, and the spherocytic RBCs are destroyed prematurely in the spleen. Splenectomy markedly improves RBC life span and cures the anemia.
Clinical Manifestations
Hereditary spherocytosis may be a cause of hemolytic disease in the newborn and can manifest as anemia and hyperbilirubinemia sufficiently severe to require phototherapy or exchange transfusions. Hemolysis may be more prominent in the newborn because hemoglobin F binds 2,3-diphosphoglycerate poorly, and the increased level of free 2,3-diphosphoglycerate destabilizes interactions among spectrin, actin, and protein 4.1 in the RBC membrane (see Fig. 452-1).
Because of the high RBC turnover and heightened erythroid marrow activity, children with hereditary spherocytosis are susceptible to aplastic crisis, primarily as a result of parvovirus B19 infection, and to hypoplastic crises associated with various other infections (Fig. 452-3
[/not-level-membership-for-pediatrics-category]
