[level-membership-for-neurology-category]
CHAPTER 69 HEREDITARY SPASTIC PARAPLEGIAS*
The hereditary spastic paraplegias (HSPs), also known as familial spastic paraplegia and Strümpell-Lorrain disease (per Dorland’s),1 constitute a group of more than 30 inherited neurological disorders in which the predominant symptom is bilateral lower extremity spastic weakness. Previous reviews of HSP are available in articles by Fink and colleagues,2–4 in the Gene Reviews website (http://www.geneclinics.org/profiles/hsp/), in the University of Michigan’s Hereditary Spastic Paraplegia website (http://www.med.umich.edu/hsp), and in the website for the Spastic Paraplegia Foundation (http://www.sp-foundation.org).
EPIDEMIOLOGY
HSP affects individuals of all ethnic groups and ancestries without particular ethnic predilection. The prevalence of HSP has been estimated in Ireland (1.27 per 100,000),5 Italy (2.7 per 100,000),6 and Spain (9.6 per 100,000).5
GENETIC CLASSIFICATION: MODE OF INHERITANCE AND HSP LOCUS
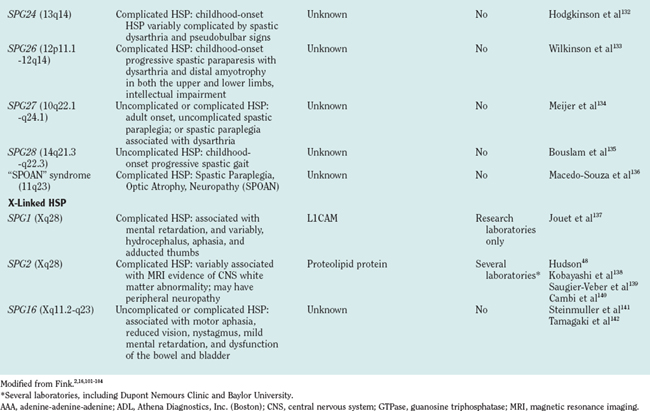
There are autosomal dominant, autosomal recessive, and X-linked forms of HSP, each of which is genetically heterogeneous: Many different, separate gene mutations cause clinically similar, often indistinguishable syndromes of lower extremity spastic weakness). HSP genetic loci are designated SPG (spastic gait) and numbered 1 through 28 in order of their discovery (Table 69-1).

CLINICAL CLASSIFICATION: “UNCOMPLICATED” AND “COMPLICATED” HSP
Classifying HSP as uncomplicated or complicated is also important for prognosis. Families with “uncomplicated” HSP (e.g., HSP caused by SPG4/spastin gene mutation) are not at risk of having offspring with “complicated” HSP. The converse is not always true, however. Families with some forms of “complicated” HSP (e.g., SPG7 or SPG10 HSP caused by paraplegin or kinesin heavy chain [KIF5A] gene mutations, respectively)7,8 may have offspring affected with either “uncomplicated” or “complicated” HSP.
Controversies in HSP classification arise as the knowledge of the HSPs expands. For example, seizures, cognitive impairment, and ataxia9 have been reported in patients with the most common form of dominantly inherited HSP (SPG4, caused by SPG4/spastin gene mutation), generally considered prototypical of uncomplicated HSP.10–14 Further studies to determine the frequency of “extraspinal” features in otherwise uncomplicated HSP are needed.
NEUROPATHOLOGY: DISTAL AXON DEGENERATION INVOLVING LONGEST MOTOR AND SENSORY FIBERS IN THE CENTRAL NERVOUS SYSTEM
Postmortem studies of uncomplicated HSP reveal relatively selective axonal degeneration involving terminal portions of corticospinal tracts (maximal in the thoracolumbar region) and dorsal column fibers (maximal in cervicomedullary region).15–20 Spinocerebellar fibers are involved to a lesser extent. Myelin loss is considered secondary to primary axonal degeneration. Decreased numbers of cortical motor neurons and anterior horn cells have been reported.18,19 Peripheral nerves and dorsal root ganglia are normal in uncomplicated HSP.19
Axonal degeneration in uncomplicated HSP thus involves the distal ends of the longest motor (corticospinal tracts) and sensory (dorsal column) fibers in the central nervous system (CNS). In this regard, uncomplicated HSP may be considered a “CNS homologue” of Charcot-Marie-Tooth disease type 2, in which distal motor and sensory axon degeneration is limited to the peripheral nervous system. To extend this analogy, it is noteworthy that one type of HSP (SPG10) is caused by mutation in KIF5A,8 whereas mutations in another kinesin (KIF1B) cause Charcot-Marie-Tooth disease type 2A1.21
EMERGING CONCEPTS OF HSP PATHOGENESIS
The molecular mechanisms underlying axon degeneration for most types of HSP are poorly understood. Nonetheless, the discovery of many HSP genes is generating new concepts about the pathophysiology of HSPs.16 Thus far, 28 HSP loci and 11 HSP genes have been discovered (see Table 69-1). The functions of most of these HSP proteins have not been fully elaborated; however, as a group, HSP genes (and their respective proteins) appear highly diverse. This diversity of HSP genes (and their proteins) suggests that axonal degeneration in various genetic types of HSP may be caused by diverse, primary biochemical disturbances.22 A tentative biochemical classification of HSP is emerging. It is likely that these disparate biochemical disturbances converge into one or more common pathways.
HSP Caused by Axonal Transport Abnormality
There is increasing evidence that disturbance of axonal transport occurs in a variety of motor neuron disorders, including HSP.23,24 The clearest example of axonal transport involvement in HSP is autosomal dominant SPG10 HSP caused by KIF5A mutation. KIF5A is a molecular motor component involved in axonal transport of organelles and macromolecules. SPG4, the most common cause of dominantly inherited HSP, resulting from spastin gene mutation, may be another example of axonal transport or cytoskeletal involvement in HSP. There is accumulating evidence that spastin interacts with microtubules and may be involved in microtubule severing.25–34
HSP Caused by Golgi Abnormalities
Atlastin mutations cause approximately 25% of childhood-onset dominantly inherited HSP.35–37 Spartin mutations cause autosomal recessive HSP associated with distal muscle atrophy (Troyer’s syndrome).38 Although the functions of both atlastin and spartin have not been elucidated, it is known that both of these proteins are localized to the Golgi apparatus.39,40
HSP Caused by Mitochondrial Abnormality
Two HSP genes encode integral mitochondrial proteins: chaperonin 60/heat shock protein 60, mutations of which cause autosomal dominant uncomplicated SPG13 HSP,41 and paraplegin, mutations of which cause autosomal, complicated recessive SPG7 HSP.42–45 Some but not all patients with paraplegin mutation have evidence of mitochondrial abnormalities in skeletal muscle biopsy.7
HSP Caused by Primary Myelin Disturbance
It is important to note that axon degeneration in at least one form of HSP arises not from an intrinsic axon or neuron abnormality but rather from glial abnormality. X-linked SPG2 HSP is caused by proteolipid protein gene mutation. Proteolipid protein is an intrinsic myelin protein, and mutations in the gene cause both Pelizaeus-Merzbacher disease46 (an X-linked infantile-onset dysmyelination disorder) and X-linked HSP (a childhood-onset slowly progressive spastic gait disorder).47,48 Patients and gene-targeted mice lacking proteolipid protein develop length-dependent axon degeneration in the absence of demyelination.49
HSP Caused by Embryonic Development of Corticospinal Tracts
Mutations in the neuronal cell adhesion molecule L1 (L1CAM) gene cause a variety of X-linked neurological disorders, including X-linked hydrocephalus; the syndrome of mental retardation, aphasia, shuffling gait, and adducted thumbs; and complicated X-linked spastic paraplegia.50–52 L1CAM knockout mice53 exhibit weak hind limbs and reduced size of corticospinal tracts.
NEUROLOGICAL EXAMINATION: UPPER MOTOR NEURON SIGNS IN THE LEGS; IMPAIRED VIBRATION SENSATION IN THE TOES
Although all patients with HSP have lower extremity spasticity, the degree of weakness is variable. Some patients have lower extremity spasticity but normal muscle strength. Although Harding19,54 used the variable proportion of weakness and spasticity (along with age at symptom onset) to classify HSP as type I (greater spasticity than weakness) and type II (significant weakness), this classification is not widely used because estimates of the relative contributions of weakness versus spasticity are qualitative and because some genetic types of HSP may manifest as both type I and type II.
Vibration sensation in the toes is often mildly impaired in uncomplicated HSP. Distal lower extremity vibratory impairment may not manifest for several years, but when present, this is a helpful diagnostic sign. When not attributed to other disorders (such as peripheral neuropathy or cervical spondylosis), impaired vibration sensation in the toes helps to distinguish HSP’s motor (corticospinal tract) and sensory (dorsal column) patterns of involvement from primary lateral sclerosis (involvement of upper motor neurons with sparing of dorsal columns).55 Although vibratory sense may be mildly impaired in HSP, severe dorsal column disturbance is not typical of uncomplicated HSP and would prompt a search for other disorders (including Friedreich’s ataxia, subacute combined degeneration, and tertiary syphilis).
Spastic Gait
Patients with uncomplicated HSP generally exhibit bilaterally symmetrical gait disturbance,56 including short stride length (because of difficulty flexing the thighs and dorsiflexing the feet), circumduction, anterior-foot strike (tendency to walk on the balls of the feet or on the toes), scissoring, hyperlordosis, and sometimes hyperextension at the knee. The ability to walk on the heels is generally compromised. The abnormality of gait varies between individuals. Careful analysis of each affected individual’s gait is necessary to provide specific exercise recommendations, to determine whether the patient would benefit from spasticity-reducing medication, and to determine whether the patient would benefit from ankle-foot orthotic devices.
SYNDROME VARIABILITY
Within a given genetic type of HSP (such as SPG4 HSP caused by spastin gene mutation), there may be significant clinical variability. Part of this variability may result from the effects of different mutations.57 For example, whereas SPG4 HSP (caused by spastin mutation) is usually uncomplicated, ataxia in addition to spastic paraplegia has been reported in a family with SPG4 mutation GLN490Stop.9
Significant variability between affected patients who share the same HSP gene mutation reflects the influence of modifying genetic and possibly environmental factors. One source of modifying genes is polymorphisms in HSP genes themselves. Recently, Svenson and associates analyzed benign SPG4/spastin polymorphisms (S44L and P45Q) and showed that L44 and Q45 are each associated with a striking decrease in age at onset in the presence of the pathogenic mutations in SPG4/spastin’s adenine-adenine-adenine domain.58
Age at Symptom Onset
Age at symptom onset may be quite variable between different genetic types of HSP, between patients with one particular genetic type, and even within a family in which affected patients share the same HSP gene mutation. Although the average age at symptom onset is earlier for some types of HSP (SPG10, SPG3A, and SPG12)4 than for other types of HSP (SPG4, SPG13, SPG8, and SPG6), there is significant overlap in the range of ages at which symptoms begin. For SPG4 HSP, meta-analysis of 75 families did not reveal a correlation between spastin mutation class (missense, aberrant splicing, frameshift, premature truncation mutations) and age at symptom onset.59
Genetic Anticipation
Genetic anticipation has been reported in SPG4 HSP,60 including patients later shown to have point mutations (not trinucleotide repeat expansions) in the SPG4/spastin gene. The author and colleagues have observed apparent genetic anticipation in SPG3A HSP. For example, they identified the SPG3A mutation V253I in a 70-year-old patient who was asymptomatic and had normal neurological examination findings; his mutation-bearing son developed HSP in his 20s; and his mutation-bearing grandson developed HSP before age 7 (J. K. Fink, 2005 unpublished observation).
Relatively Nonprogressive versus Progressive Forms of HSP
When HSP begins after adolescence, symptoms usually progress steadily over many years. When symptoms begin in childhood, there may be very little progression even over 10 years.2
Syndromic Features
Although complicated forms of HSP have “syndromic” features (e.g., SPG9, SPG10, and SPG17 have motor neuropathy or distal wasting), such features may be present in only a minority of affected family members (e.g., some with SPG1061 and some with SPG745 had complicated HSP, whereas others had uncomplicated HSP).
Subclinical Cognitive Disturbance and Late-Onset Dementia
These features have been described in some but not all patients with the most common form of dominantly inherited HSP (caused by SPG4 mutations)10–14 and may be correlated with specific spastin mutations.62 Cognitive impairment is a feature of several forms (see Table 69-1) of complicated HSP, particularly SPG11, which appears to be the most common form of autosomal recessive HSP.
LABORATORY STUDIES, NEUROIMAGING, AND NEUROPHYSIOLOGICAL EVALUATION
Neuroimaging is important for ruling out alternative disorders, including multiple sclerosis, leukodystrophies, and structural abnormalities affecting the brain or spinal cord (see “Differential Diagnosis” section). Whereas brain magnetic resonance imaging in uncomplicated HSP is usually normal, those of complicated forms of HSP may reveal syndrome-specific abnormalities, such as thin corpus callosum in autosomal recessive SPG11 HSP (e.g., Casali et al63) and cerebral or cerebellar abnormalities in autosomal recessive SPG7 HSP. Magnetic resonance imaging of the thoracic spinal cord often demonstrates atrophy in uncomplicated HSP.9–11,64
Electromyographic nerve conduction studies usually yield normal results in uncomplicated HSP. Such studies are useful in ruling out other disorders (such as amyotrophic lateral sclerosis, Friedreich’s ataxia, Machado-Joseph disease). Such study results are usually normal in uncomplicated HSP.51,52,65 Exceptions have been reported, however, including peripheral neuropathy in some patients with SPG4 HSP caused by spastin frameshift mutation 906delT53 and axonal neuropathy associated with SPG3A HSP caused by atlastin mutation R495W.66 Subclinical sensory neuropathy in otherwise uncomplicated HSP has been described.67,68 Several types of complicated HSP are associated with peripheral neuropathy (see Table 69-1).
Somatosensory evoked potentials may demonstrate dorsal column impairment in uncomplicated HSP. Whereas somatosensory evoked potentials recorded from lower extremities often show delayed conduction, somatosensory evoked potentials recorded from the upper extremities are usually normal.52,69–72 This finding may help distinguish patients with autosomal recessive, uncomplicated HSP (and those with uncomplicated spastic paraplegia who do not have a family history of the disorder) from patients in an early stage of primary lateral sclerosis (in whom vibration sensation and dorsal column function are normal).55
Cortical evoked potentials provide a useful measurement of corticospinal tract conduction velocity. Studies of patients with uncomplicated HSP often reveal reduced conduction velocity and amplitude when cortical evoked potentials are recorded from the lower extremities.73–76 In contrast, cortical evoked potentials recorded from cervical spinal segments are usually normal or show only mildly reduced conduction velocity.75
Muscle Biopsy
Paraplegin, mutations of which cause autosomal recessive SPG7 HSP, is mitochondrial protein. Muscle biopsies from some patients with SPG7 HSP reveal ragged red fibers and cytochrome c oxidase–negative fibers.7 These do not appear to be general phenomena for uncomplicated HSP, however. Muscle biopsies, including analysis of enzymes of oxidation-phosphorylation, yielded normal results in patients with autosomal dominant uncomplicated SPG3A, SPG4, SPG6, and SPG8 HSP. Together, these represent the majority of cases of uncomplicated autosomal dominant HSP.77,78 There is some controversy, however, because McDermott and associates79 reported decreases in mitochondrial respiratory chain complexes I and IV in patients with HSP for whom SPG4 HSP (spastin mutation) and SPG7 HSP (paraplegin mutation) were ruled out.
DIAGNOSTIC CRITERIA
Role of Gene Testing in the Diagnosis of HSP
Identifying HSP gene mutations (available through Athena Diagnostics, Inc., Boston, for SPG3A/atlastin,35 SPG4/spastin,80 and SPG6/NIPA181 genes, and through DuPont Nemours Clinic for SPG2/proteolipid protein gene) can be used to confirm the clinical diagnosis. When a mutation is identified in an affected patient, this information can be applied to prenatal genetic testing.82,83
Differential Diagnosis
The differential diagnosis of HSP (reviewed previously2,3) includes treatable disorders (e.g., vitamin B12 deficiency and central folate deficiency84) and conditions whose prognoses differ significantly from those of HSP. Such conditions include structural disorders of the brain and spine (e.g., tethered cord syndrome, spinal cord compression, cervical spondylosis), disorders of central white matter (including vitamin B12 deficiency, multiple sclerosis, adrenomyeloneuropathy85 and other leukodystrophies); infectious diseases (e.g., tropical spastic paraplegia caused by human T cell leukocyte virus type 1 infection86 and tertiary syphilis); and other degenerative disorders (e.g., spinal cord arteriovenous malformation, Machado-Joseph disease [spinocerebellar ataxia type 3], Friedreich’s ataxia,86 primary lateral sclerosis,55 amyotrophic lateral sclerosis, and lathyrism). It is always important to consider the possibility of dopamine-responsive dystonia,87 particularly in children.88
Diagnostic “Red Flags”
Muscle bulk is usually preserved in uncomplicated HSP. Although some patients have mildly decreased muscle bulk in their shins, lower motor neuron disturbance is not a feature of uncomplicated HSP. Although distal muscle wasting is a feature of SPG17 complicated autosomal dominant HSP (Silver’s syndrome), SPG20 recessive HSP (Troyer’s syndrome), and HSP syndromes associated with peripheral neuropathy or motor neuronopathy,89,90–92 muscle wasting and fasciculations are not consistent with uncomplicated HSP and should prompt consideration of an alternative or coexisting diagnosis (such as generalized motor neuron disease or amyotrophic lateral sclerosis).
TREATMENT
There is no specific treatment to reverse, retard, or prevent progressive axonal degeneration in HSP. Symptomatic treatment includes efforts to reduce muscle spasticity through muscle-stretching exercises and medications such as oral or intrathecal baclofen (Lioresal), dantrolene, or tizanidine.93 Oxybutynin is useful in reducing urinary urgency.
GENETIC COUNSELING
Genetic counseling in HSP is guided by the mode of inheritance (X-linked, autosomal dominant, autosomal recessive), the frequency of spontaneous mutations, the extent of genetic penetrance, and the degree of phenotypic variability. Spontaneous mutation for autosomal dominant HSP has been reported in SPG3A uncomplicated HSP94 and, although it may occur,95 appears to be uncommon in dominantly inherited SPG3A, SPG4, and SPG6 HSP. Fewer than 10% of patients who have all signs and symptoms of HSP but do not have a family history have a mutation in the SPG3A, SPG4, and NIPA1 genes.34,96,97
Genetic penetrance in uncomplicated HSP is age-dependent and, although high (70% to 85% for SPG4 HSP, for example), may be incomplete. Incomplete penetrance implies that some patients may possess an HSP gene mutation, remain asymptomatic with normal neurological examinations, and yet transmit the condition to progeny. Incomplete genetic penetrance has been reported for SPG4,98 SPG8,77 and SPG3A HSP.37,99
Genetic counselors must recognize that the age at symptom onset and extent of disability may vary significantly98 between families with different genetic types of HSP, between families with the same genetic type of HSP, and between individuals from the same family who share the same HSP gene mutation. Often, the extent of clinical variability is a caution against assuming that disability will be similar in all affected relatives. The author have seen families in which some members had progressively disabling spastic paraparesis and others had mild, nondisabling spastic gait. Small families with few affected patients may not enable accurate assessment of the full range of phenotypic variation. As noted previously, a number of families with “complicated” forms of HSP have been described in which the “complicating features” (such as distal muscle atrophy)100 were not present in each patient with spastic paraplegia.7
The author and colleagues have encountered a number of families in which the condition was diagnosed in one or more children before a parent developed symptoms of HSP. Genetic anticipation has been reported in SPG460 and observed in SPG3A HSP (J. K. Fink, 2005 unpublished observation). The mechanism for genetic anticipation in these circumstances presumably involves a tandem repeat expansion in an HSP modifying gene (because the causative HSP gene mutations were missense mutations and not tandem repeat expansions).
CONCLUSIONS
Fink JK. The hereditary spastic paraplegias: nine genes and counting. Arch Neurol. 2003;60:1045-1049.
Fink JK. Hereditary spastic paraplegia. In: Rimoin DL, Pyeritz RE, Connor JM, et al, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. 4th ed. London: Churchill Livingston; 2001:3124-3145.
Fink JK. Progressive spastic paraparesis: hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin Neurol. 2001;21:199-208.
1 Strümpell A. Die primaere Seitenstrangsklerose (spastis-cheSpinalparalyse). Dtsch Z Nervenheilk. 1904;27:291-339.
2 Fink JK. Hereditary spastic paraplegia. In: Rimoin D, Pyeritz RE, Connor J, et al, editors. Emery & Rimoin’s Principles and Practice of Medical Genetics. 4th ed. London: Harcourt; 2002:3124-3145.
3 Fink JK, Heiman-Patterson T, Bird T, et al. Hereditary spastic paraplegia: advances in genetic research. Neurology. 1996;46:1507-1514.
4 Fink JK, Hedera P. Hereditary spastic paraplegia: genetic heterogeneity and genotype-phenotype correlation. Semin Neurol. 1999;19:301-310.
5 Polo AE, Calleja J, Combarros O, et al. Hereditary ataxias and paraplegias in Cantabria, Spain: an epidemiological and clinical study. Brain. 1991;114:855-856.
6 Filla A, DeMichele G, Marconi R, et al. Prevalence of hereditary ataxias and spastic paraplegias in Molise, a region of Italy [Abstract]. J Neurol. 1992;239:351-353.
7 DeMichele G, DeFusco M, Cavalcanti F, et al. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. Am J Hum Genet. 1998;63:135-139.
8 Reid E, Kloos M, Ashley-Koch A, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10).. Am J Hum Genet. 2002;71:1189-1194.
9 Nielsen JE, Johnsen B, Koefoed P, et al. Hereditary spastic paraplegia with cerebellar ataxia: a complex phenotype associated with a new SPG4 gene mutation. Eur J Neurol. 2004;11:817-824.
10 Heinzlef O, Paternotte C, Mahieux F, et al. Mapping of a complicated familial spastic paraplegia to locus SPG4 on chromosome 2p. J Med Genet. 1998;35:89-93.
11 McMonagle P, Byrne P, Hutchinson M. Further evidence of dementia in SPG4-linked autosomal dominant hereditary spastic paraplegia. Neurology. 2004;62:407-410.
12 Webb S, Coleman D, Byrne P, et al. Autosomal dominant hereditary spastic paraparesis with cognitive loss linked to chromosome 2p. Brain. 1998;121:601-609.
13 Byrne PC, Webb S, McSweeney F, et al. Linkage of AD HSP and cognitive impairment to chromosome 2p: haplotype and phenotype analysis indicates variable expression and low or delayed penetrance. Eur J Hum Genet. 1998;6:275-282.
14 Reid E, Grayson C, Rubinsztein DC, et al. Subclinical cognitive impairment in autosomal dominant “pure” hereditary spastic paraplegia. J Med Genet. 1999;36:797-798.
15 Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576-584.
16 Fink JK. Hereditary spastic paraplegia: nine genes and counting. Arch Neurol. 2003;60:1045-1049.
17 Schwarz GA, Liu C-N. Hereditary (familial) spastic paraplegia. Further clinical and pathologic observations. AMA Arch Neurol Psychiatry. 1956;75:144-162.
18 Behan W, Maia M. Strümpell’s familial spastic paraplegia: genetics and neuropathology. J Neurol Neurosurg Psychiatry. 1974;37:8-20.
19 Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13:333-336.
20 Sack GH, Huether CA, Garg N. Familial spastic paraplegia: clinical and pathologic studies in a large kindred. Johns Hopkins Med J. 1978;143:117-121.
21 Zhao C, Takita J, Tanaka Y, et al. Charcot-Marie-Tooth disease type2A caused by mutation in a microtubule motor KIF1Bβ. Cell. 2001;105:587-597.
22 Gould RM, Brady ST. Neuropathology: many paths lead to hereditary spastic paraplegia. Curr Biol. 2004;14:P903-P904.
23 Holzbaur EL. Motor neurons rely on motor proteins. Trends Cell Biol. 2004;14:233-240.
24 Guzik BW, Goldstein LS. Microtubule-dependent transport in neurons; steps towards and understanding of regulation, function and dysfunction. Curr Opin Cell Biol. 2004;16:443-450.
25 Sherwood NT, Sun Q, Xue M, et al. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429.
26 Roll-Mecak A, Vale RD. The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr Biol. 2005;15:650-655.
27 Reid E, Connell J, Edwards TL, et al. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex–associated endosomal protein CHMP1B. Hum Mol Genet. 2005;14:19-38.
28 McDermott CJ, Grierson AJ, Wood JD, et al. Hereditary spastic paraparesis: disrupted intracellular transport associated with spastin mutation. Ann Neurol. 2003;54:748-759.
29 Wharton SB, McDermott CJ, Grierson AJ, et al. The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J Neuropathol Exp Neurol. 2003;62:1166-1177.
30 Molon A, DiGiovanni S, Chen YW, et al. Large-scale disruption of microtubule pathways in morphologically normal human spastin muscle. Neurology. 2004;62:97-104.
31 Trotta N, Orso G, Rossetto MG, et al. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol. 2004;14:1135-1147.
32 Errico A, Claudiani P, D’Addio M, et al. Spastin interacts with the centrosomal protein NA14, and is enriched in the spindle pole, the midbody, and the distal axon. Hum Mol Genet. 2004;13:2121-2132.
33 Evans KJ, Gomes ER, Reisenweber SM, et al. Linking axonal degeneration to microtubule remodeling by spastinmediated microtubule severing. J Cell Biol. 2005;168:599-606.
34 Proukakis C, Auer-Grumbach M, Wagner K, et al. Screening of patients with hereditary spatic paraplegia reveals seven novel mutations in the SPG4 (Spastin) gene. Hum Mutat. 2003;21:170.
35 Zhao X, Alvarado D, Rainier S, et al. Mutations in a novel GTPase cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326-331.
36 Alvarado DM, Ming L, Hedera P, et al. Atlastin gene analysis in early onset hereditary spastic paraplegia [Abstract]. Am J Hum Genet. 2001;69:597.
37 Durr A, Camuzat A, Colin E, et al. Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch Neurol. 2004;61:1867-1872.
38 Patel H, Cross H, Proukakis C, et al. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet. 2002;31:347-348.
39 Zhu PP, Patterson A, Lavoie B, et al. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063-49071.
40 Crosby AH, Patel H, Patton MA, et al. Spartin, the Troyer syndrome gene, suggests defective endosomal trafficking underlies some forms of hereditary spastic paraplegia [Abstract]. Am J Hum Genet. 2002;71:516.
41 Hansen JJ, Durr A, Cournu-Rebeix I, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70:1328-1332.
42 Atorino L, Silvestri L, Koppen M, et al. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol. 2003;163:777-787.
43 Ferreirinha F, Quattrini A, Pirozzi M, et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231-242.
44 Wilkinson PA, Crosby AH, Turner C, et al. A clinical and genetic study of SPG5A linked autosomal recessive hereditary spastic paraplegia. Neurology. 2003;61:235-238.
45 Casari G, Fusco M, Ciarmatori S, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973-983.
46 Hodes ME, Zimmerman AW, Aydanian A, et al. Different mutations in the same codon of the proteolipid protein gene, PLP, may help in correlating genotype with phenotype in Pelizaeus-Merzbacher disease/X-linked spastic paraplegia (PMD/SPG2). Am J Med Genet. 1999;82:132-139.
47 Willard HF, Riordan JR. Assignment of the gene for myelin proteolipid protein to the X chromosome: implications for X-linked myelin disorders. Science. 1985;230:940-942.
48 Hudson LD. Pelizaeus-Merzbacher disease and spastic paraplegia type 2: two faces of myelin loss from mutations in the same gene. J Child Neurol. 2003;18:616-624.
49 Garbern JY, Yool DA, Moore GJ, et al. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551-561.
50 Bateman A, Jouet M, MacFarlane J, et al. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 1996;15:6050-6059.
51 Owens LA, Peterson CR. Familial spastic paraplegia: a clinical and electrodiagnostic evaluation. Arch Phys Med Rehabil. 1982;63:357-361.
52 Dimitrijevic MR, Lenman JAR, Prevec T, et al. A study of posterior column function in familial spastic paraplegia. J Neurol Neurosurg Psychiatry. 1982;45:46-49.
53 Orlacchio A, Kawarai T, Gaudiello F, et al. Clinical and genetic study of a large SPG4 Italian family. Mov Disord. 2005;20:1055-1059.
54 Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151-1155.
55 Fink JK. Progressive spastic paraparesis: hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin Neurol. 2001;21:199-208.
56 Klebe S, Stolze H, Kopper F, et al. Gait analysis of sporadic and hereditary spastic paraplegia. J Neurol. 2004;251:571-578.
57 Bonsch D, Schwindt A, Navratil P, et al. Motor system abnormalities in hereditary spastic paraparesis type 4 (SPG4) depend on the type of mutation in the spastin gene. J Neurol Neurosurg Psychiatry. 2003;74:1109-1112.
58 Svenson IK, Kloos M, Gaskell PC, et al. Intragenic modifiers of hereditary spastic paraplegia due to spastin gene mutations. Neurogenetics. 2004;5:157-164.
59 Yip AG, Durr A, Marchuk DA, et al. Meta-analysis of age at onset in spastin-associated hereditary spastic paraplegia provides no evidence for a correlation with mutational class [Abstract]. J Med Genet. 2003;40:e106.
60 Nielsen JE, Koefoed P, Abell K, et al. CAG repeat expansion in autosomal dominant pure spastic paraplegia linked to chromosome 2p21-p24. Hum Mol Genet. 1997;6:1811-1816.
61 Pericak-Vance MA, Kloos MT, Reid E, et al. A kinesin heavy chain (K1F5A) mutation in Hereditary Spastic Paraplegia (SPG10) [Abstract]. Am J Hum Genet. 2002;71:165.
62 Tallaksen CME, Gomez EG, Verpillat P, et al. Subtle cognitive impairment but no dementia in patients with spastin mutations [Abstract]. Arch Neurol. 2003;60:1113-1118.
63 Stevanin G, Montagna G, Azzedine H, et al. Spastic paraplegia with thin corpus callosum: description of 20 new families, refinement of the SPG11 locus, candidate gene analysis and evidence of genetic heterogeneity. Neurogenetics. 2006;3:149-156.
64 Hedera P, Eldevik OP, Maly P, et al. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology. 2005;47:730-734.
65 Mcleod JG, Morgan JA, Reye C. Electrophysiological studies in familial spastic paraplegia. Neurol Neurosurg Psychiatry. 1993;40:611-615.
66 Scarano V, Mancini P, Criscuolo C, et al. The R495W mutation in SPG3A causes spastic paraplegia associated with axonal neuropathy. J Neurol. 2005;252:901-903.
67 Schady W, Scheard A. A qualitative study of sensory functions in hereditary spastic paraplegia. Brain. 1990;113:709-720.
68 Schady W, Smith DI. Sensory neuropathy in hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. 1994;57:693-698.
69 Pelosi L, Lanzillo B, Perretti A. Motor and somatosensory evoked potentials in hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. 1991;54:1099-1102.
70 Pedersen L, Trojaborg W. Visual, auditory and somatosensory pathway involvement in hereditary cerebellar ataxia, Friedreich’s ataxia and familial spastic paraplegia. Electroencephalogr Clin Neurophys. 1981;52:283-297.
71 Uncini A, Treviso M, Basciani M, et al. Strümpell’s familial spastic paraplegia: an electrophysiological demonstration of selective central distal axonopathy. Electroencephalogr Clin Neurophys. 1987;66:132-136.
72 Battistella PA, Suppiej A, Mandara V. Evoked potentials in familial spastic paraplegia: description of three brothers and review of the literature. Giorn Neuropsi Evol. 1997;17:201-212.
73 Schulte T, Miterski B, Bornke C, et al. Neurophysiological findings in SPG4 patients differ from other types of spastic paraplegia. Neurology. 2003;60:1529-1532.
74 Nardone R, Tezzon F. Transcranial magnetic stimulation study in hereditary spastic paraparesis. Eur Neurol. 2003;49:234-237.
75 Claus D, Waddy HM, Harding AE. Hereditary motor and sensory neuropathies and hereditary spastic paraplegia: a magnetic stimulation study. Ann Neurol. 1990;28:43-49.
76 Claus D, Jaspert A. Central motor conduction in hereditary spastic paraparesis (Strümpell’s disease) and tropical spastic paraparesis. Neurol Croatica. 1995;44:23-31.
77 Hedera P, DiMauro S, Bonilla E, et al. Phenotypic analysis of autosomal dominant hereditary spastic paraplegia linked to chromosome 8q. Neurology. 1999;53:44-50.
78 Hazan J, Fonknechten N, Mavel D, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23:296-303.
79 McDermott CJ, Taylor RW, Hayes C, et al. Investigation of mitochondrial function in hereditary spastic paraparesis. Genet Nerv Syst Dis. 2003;14:485-488.
80 Hazan J, Fontaine B, Bruyn RPM, et al. Linkage of a new locus for autosomal dominant familial spastic paraplegia to chromosome 2p. Hum Mol Genet. 1994;3:1569-1573.
81 Rainier S, Chai J-H, Tokarz D, et al. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6).. Am J Hum Genet. 2003;73:967-971.
82 Nielsen JE, Koefoed P, Kjaergaard S, et al. Prenatal diagnosis of autosomal dominant hereditary spastic paraplegia (SPG4) using direct mutation detection. Prenat Diagn. 2004;24:363-366.
83 Hedera P, Williamson J, Alvarado D, et al. Prenatal diagnosis of hereditary spastic paraplegia. Prenat Diagn. 2001;21:202-206.
84 Hansen FJ, Blau N. Cerebral folate deficiency: life-changing supplementation with folinic acid. Mol Genet Metab. 2005;84:371-373.
85 Shaw-Smith CJ, Lewis SJ, Reid E. X-linked adrenoleukodystrophy presenting as autosomal dominant pure hereditary spastic paraparesis. J Neurol Neurosurg Psychiatry. 2004;75:686-688.
86 Matsumura R, Takayanagi T, Fujimoto Y, et al. The relationship between trinucleotide repeat length and phenotypic variation in Machado-Joseph disease. J Neurol Sci. 1996;139:52-57.
87 Nygaard TG. Dopa-responsive dystonia: clinical, pathological, and genetic distinction from juvenile parkinsonism. Segawa M, Nomura Y, editors. Age-Related Dopamine-Dependent Disorders: Monographs in Neural Sciences. vol 14. Basel: Karger; 1995:109-119.
88 Fink JK, Filling-Katz M, Barton NW, et al. Treatable dystonia presenting as spastic cerebral palsy. Pediatrics. 1988;82:138.
89 McKusick VA. Mendelian Inheritance in Man: Catalogs of Autosomal Dominant, Autosomal Recessive, and X-Linked Phenotypes, 8th ed. Baltimore: Johns Hopkins University Press, 1994.
90 Cavanagh NPC, Eames RA, Galvin RJ, et al. Hereditary sensory neuropathy with spastic paraplegia. Brain. 1979;102:79-84.
91 Stewart RM, Tunell G, Ehle A. Familial spastic paraplegia, peroneal neuropathy, and crural hypopigmentation: a new neurocutaneous syndrome. Neurology. 1981;31:754-757.
92 Vazza GZM, Boaretto F, Micaglio GF, et al. A new locus for autosomal recessive spastic paraplegia associated with mental retardation and distal motor neuropathy, SPG14, maps to chromosome 3q27-q28. Am J Hum Genet. 2000;67:504-509.
93 Van Schaeybroeck P, Nuttin B, Lagae L, et al. Intrathecal baclofen for intractable cerebral spasticity: a prospective placebo-controlled, double-blind study. Neurosurgery. 2000;46:603-612.
94 Rainier S, Sher C, Reish O, et al. De novo occurrence of novel SPG3A/atlastin mutation presenting as cerebral palsy. Arch Neurol. 2006;63:445-447.
95 Alber B, Rothmund G, Ludolph AC, et al: Two novel mutations in the spastin gene in a family with hereditary spastic paraparesis and in one patient with apparently sporadic spastic paraplegia [Abstract]. Presented at the 13th Annual International Symposium on ALS/MND, Melbourne, Australia, 2002.
96 Patrono C, Scarano V, Cricchi F, et al. Autosomal dominant hereditary spastic paraplegia: DHPLC-based mutation analysis of SPG4 reveals eleven novel mutations. Hum Mutat. 2005;25:506.
97 Sauter S, Miterski B, Klimpe S, et al. Mutation analysis of the spastin gene (SPG4) in patients in Germany with autosomal dominant hereditary spastic paraplegia. Hum Mutat. 2002;20:127-132.
98 Fonknecten N, Mavel D, Byrne P, et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet. 2000;9:637-644.
99 D’Amico A, Tessa A, Sabino A, et al. Incomplete penetrance in an SPG3A-linked family with a new mutation in the atlastin gene. Neurology. 2004;62:2138-2139.
100 Reid E, Dearlove AM, Rhodes M, et al. A new locus for autosomal dominant “pure” hereditary spastic paraplegia mapping to chromosome 12q13 and evidence for further genetic heterogeneity. Am J Hum Genet. 1999;65:757-763.
101 Fink JK. Hereditary spastic paraplegia. In: Kaminski H, editor. Neuromuscular Disorders in Clinical Practice. London: Butterworth-Heinemann; 2002:1290-1297.
102 Fink JK. Hereditary spastic paraplegia. In: Noseworthy J, Rowland LP, editors. Neurological Therapeutics: Principles and Practice. London: Taylor & Francis; 2003:2705-2713.
103 Fink JK. Hereditary spastic paraplegia. In: Lynch DR, Farmer JM, editors. Neurogenetics. Philadelphia: WB Saunders; 2002:711-726.
104 Fink JK. Hereditary spastic paraplegia. In: Beal F, Lang A, Ludolph A, editors. Neurodegenerative Disease: Neurobiology, Pathogenesis, and Treatment. Cambridge, UK: Cambridge University Press; 2005:794-802.
105 Hazan J, Lamy C, Melki J, et al. Autosomal dominant familial spastic paraplegia is genetically heterogeneous and one locus maps to chromosome 14q. Nat Genet. 1993;5:163-167.
106 Sauter SM, Engel W, Neumann LM, et al. Novel mutations in the Atlastin gene (SPG3A) in families with autosomal dominant hereditary spastic paraplegia and evidence for late onset forms of HSP linked to the SPG3A locus. Hum Mutat. 2004;23:98.
107 Paternotte C, Rudnicki D, Fizames C, et al. Quality assessment of whole genome mapping data in the refined familial spastic paraplegia interval on chromosome 14q. Genome Res. 1998;8:1216-1227.
108 Charvin D, Fonknechten N, Cifuentes-Diaz C, et al. Mutations in SPG4 are responsible for a loss of function of spastin, an abundant neuronal protein localized to the nucleus [Abstract]. Am J Hum Genet. 2002;71:516.
109 Hentati A, Pericak-Vance MA, Lennon F, et al. Linkage of the late onset autosomal dominant familial spastic paraplegia to chromosome 2p markers. Hum Mol Genet. 1994;3:1867-1871.
110 Fink JK, Wu C-TB, Jones SM, et al. Autosomal dominant familial spastic paraplegia: tight linkage to chromosome 15q. Am J Hum Genet. 1995;56:188-192.
111 Fink JK, Sharp G, Lange B, et al. Autosomal dominant hereditary spastic paraparesis, type I: clinical and genetic analysis of a large North American family. Neurology. 1995;45:325-331.
112 Chen S, Song C, Guo H, et al. Distinct novel mutations affecting the same base in the NIPA1 gene cause autosomal dominant hereditary spastic paraplegia in two Chinese families. Hum Mutat. 2005;25:135-141.
113 Hedera P, Rainier S, Alvarado D, et al. Novel locus for autosomal dominant hereditary spastic paraplegia on chromosome 8q. Am J Hum Genet. 1999;64:563-569.
114 Reid E, Dearlove AM, Osborn M, et al. A locus for autosomal dominant “pure” hereditary spastic paraplegia maps to chromosome 19q13. Am J Hum Genet. 2000;66:728-732.
115 Seri M, Cusano R, Forabosco P, et al. Genetic mapping to 10q23.3-q24.2, in a large Italian pedigree, of a new syndrome showing bilateral cataracts, gastroesophageal reflux, and spastic paraparesis with amyotrophy. Am J Hum Genet. 1999;64:586-593.
116 Fichera M, Lo Giudice M, Falco M, et al. Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology. 2004;63:1108-1110.
117 Fontaine B, Davoine C-S, Durr A, et al. A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q34. Am J Hum Genet. 2000;66:702-707.
118 Patel H, Hart PE, Warner TT, et al. The Silver syndrome variant of hereditary spastic paraplegia maps to chromosome 11q12-q14, with evidence for genetic heterogeneity within this subtype. Am J Hum Genet. 2001;69:209-215.
119 Auer-Grumbach M, Schlotter-Weigel B, Lochmuller H, et al. Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol. 2005;57:415-424.
120 Windpassinger C, Auer-Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271-276.
121 Hentati A, Pericack-Vance MA, Hung W-Y, et al. Linkage of the “pure” recessive familial spastic paraplegia to chromosome 8 markers and evidence of genetic locus heterogeneity [Abstract]. Hum Genet. 1993;53:1013.
122 Muglia M, Criscuolo C, Magariello A, et al. Narrowing of the critical region in autosomal recessive spastic paraplegia linked to the SPG5 locus. Neurogenetics. 2004;5:49-54.
123 Tang BS, Chen X, Zhao GH, et al. Clinical features of hereditary spastic paraplegia with thin corpus callosum: report of 5 Chinese cases. Chin Med J (Engl). 2004;117:1002-1005.
124 Garner CC, Garner A, Huber G, et al. Molecular cloning of microtubule-associated protein 1 (MAP1A) and microtubule-associated protein 5 (MAP1B): identification of distinct genes and their differential expression in developing brain. J Neurochem. 1990;55:146-154.
125 Martinez-Murillo F, Kobayashi H, Pegoraro E, et al. Genetic localization of a new locus for recessive spastic paraplegia to 15q13–15. Neurology. 1999;53:50-56.
126 Winner B, Uyanik G, Gross C, et al. Clinical progression and genetic analysis in hereditary spastic paraplegia with thin corpus callosum in spastic gait gene 11 (SPG11).. Arch Neurol. 2004;61:117-121.
127 Hughes CA, Byrne PC, Webb S, et al. SPG15, a new locus for autosomal recessive complicated HSP on chromosome 14q. Neurology. 2001;56:1230-1233.
128 Cross HE, McKusick VA. The Troyer syndrome. A recessive form of spastic paraplegia with distal muscle wasting. Arch Neurol. 1967;16:473-485.
129 Proukakis C, Cross H, Patel H, et al. Troyer syndrome revisited. A clinical and radiological study of a complicated hereditary spastic paraplegia. J Neurol. 2004;251:1105-1110.
130 Simpson MA, Cross H, Proukakis C, et al. Maspardin is mutated in Mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet. 2003;73:1147-1156.
131 Blumen SC, Bevan S, Abu-Mouch S, et al. A locus for complicated hereditary spastic paraplegia maps to chromosome 1q24-q32. Ann Neurol. 2004;54:796-803.
132 Hodgkinson CA, Bohlega S, Abu-Amero SN, et al. A novel form of autosomal recessive pure hereditary spastic paraplegia maps to chromosome 13q14. Neurology. 2002;59:1905-1909.
133 Wilkinson PA, Simpson MA, Bastaki L, et al. A new locus for autosomal recessive complicated hereditary spastic paraplegia (SPG26) maps to chromosome 12p11.1*12q14. J Med Genet. 2005;42:80-82.
134 Meijer IA, Cossette P, Roussel J, et al. A novel locus for pure recessive hereditary spastic paraplegia maps to 10q22.110q24.1. Ann Neurol. 2004;56:579-582.
135 Bouslam N, Benomar A, Azzedine H, et al. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28).. Ann Neurol. 2005;57:567-571.
136 Macedo-Souza LI, Kok F, Santos S, et al. Spastic paraplegia, optic atrophy, and neuropathy is linked to chromosome 11q13. Ann Neurol. 2005;57:730-737.
137 Jouet M, Rosenthal A, Armstrong G, et al. X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet. 1994;7:402-407.
138 Kobayashi H, Hoffman EP, Marks HG. The rumpshaker mutation in spastic paraplegia. Nat Genet. 1994;7:351-352.
139 Saugier-Veber P, Munnich A, Bonneau D, et al. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet. 1994;6:257-262.
140 Cambi F, Tang XM, Cordray P, et al. Refined genetic mapping and proteolipid protein mutation analysis in X-linked pure hereditary spastic paraplegia. Neurology. 1996;46:1112-1117.
141 Steinmuller R, Lantingua-Cruz A, Carcia-Garcia R, et al. Evidence of a third locus in X-linked recessive spastic paraplegia [Letter]. Hum Genet. 1997;100:287-289.
142 Tamagaki A, Shima M, Tomita R, et al. Segregation of a pure form of spastic paraplegia and NOR insertion into Xq11.2. Am J Med Genet. 2000;94:5-8.
* This research is supported by grants from the Veterans Affairs Merit Review and the National Institutes of Health (NINDS R01NS33645, R01NS36177 and R01NS38713) to John K. Fink. The expert secretarial assistance of Ms. Lynette Girbach is gratefully acknowledged.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]<
[/not-level-membership-for-neurology-category]
