75 Hereditary Myopathies
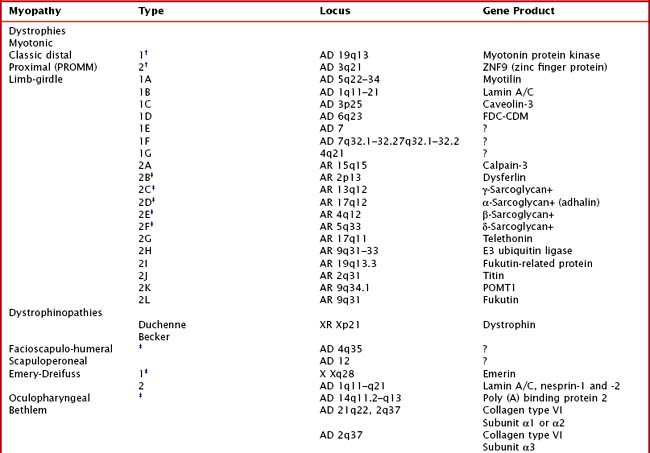
Hereditary muscle disorders are usually generalized muscle disorders that are progressive and are variously categorized as channelopathies, metabolic and mitochondrial myopathies, muscular dystrophies, and congenital myopathies (Table 75-1).


Channelopathies
Periodic Paralysis and Congenital Myotonic Disorders
Clinical Vignette
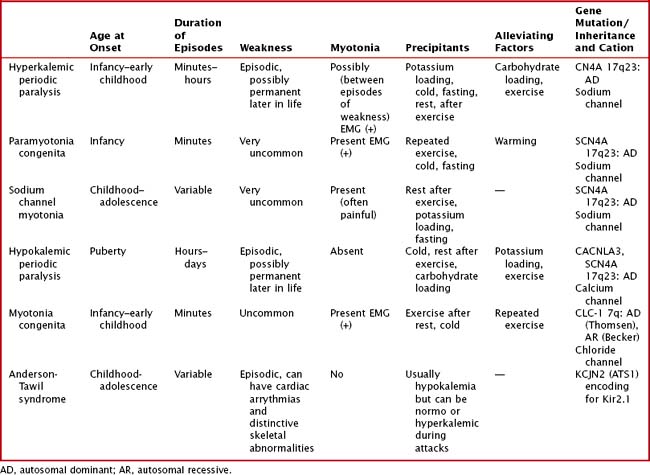
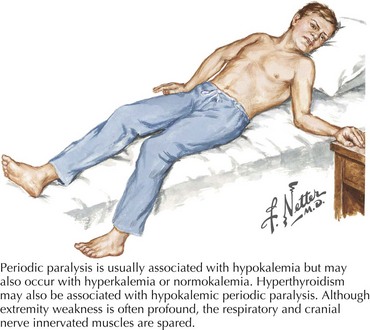
Variable mutations within genes encoding muscle membrane ion channels are responsible for the different forms of periodic paralyses as well as other myotonic disorders (Table 75-2). Most of these patients have an autosomal dominant inheritance. During the episodic paralyses, the skeletal muscle membrane excitability transiently disappears. The degree of weakness may vary from one family member to another; boys and men are more often significantly affected.
Clinical Presentation
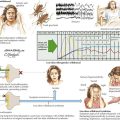
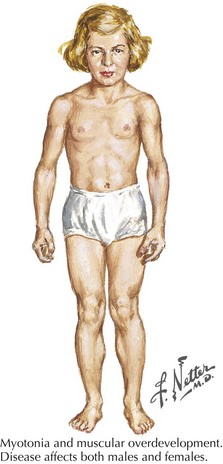
Myotonia congenita, a chloride channelopathy, is especially aggravated by immobility and ameliorated by exercise and warming; here the myotonia per se is easily elicited on examination. Fixed weakness is not usually present in dominant myotonia congenita. A rather unique characteristic of Thomsen disease variant is the pseudo-hypertrophy of the skeletal muscles providing the patient with a rather pseudo-herculean habitus (Fig. 75-2). It may be so profound that athletic coaches enthusiastically encourage these individuals to participate in sports activities. Unfortunately, some of these individuals may develop a mild progressive weakness. Transient episodes of true weakness precipitated by sudden movements after rest that are relieved by exercise are characteristic of myotonia congenita. Interesting examples include a baseball player who cannot run after hitting the ball or a subway rider who wishes to get off when the train stops but is frozen in place or falls when he arises to leave the train.
Differential Diagnosis
Myasthenia gravis (MG) classically has a remitting relapsing course predominantly confined to ocular as well as bulbar muscles and to a lesser extent proximal extremity musculature. MG is typified by fluctuating, rarely acute weakness with a very definitive diurnal variation (see Chapter 73).
Glycogen and Lipid Storage Disorders
Clinical Vignette
Comment: This is a classic example of muscle phosphorylase deficiency (Fig. 75-3) with onset in adolescence when individuals for the first time have the muscle power to allow them to stress their metabolic system to the point of actual muscle necrosis and subsequent myoglobinuria, the feature that most commonly brings them to medical attention.
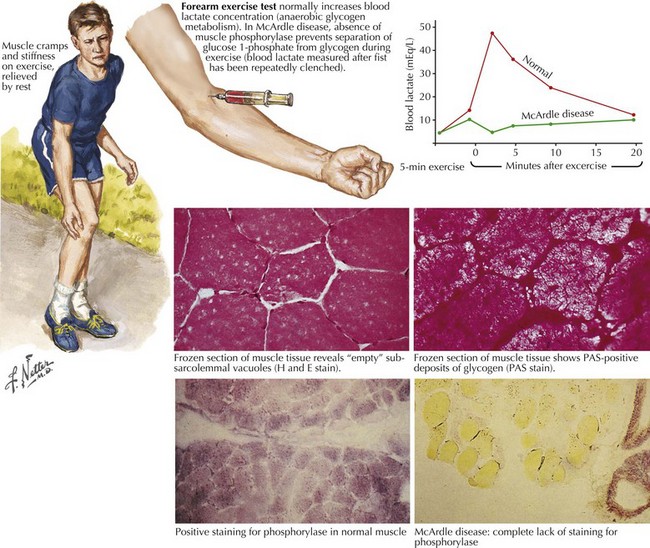
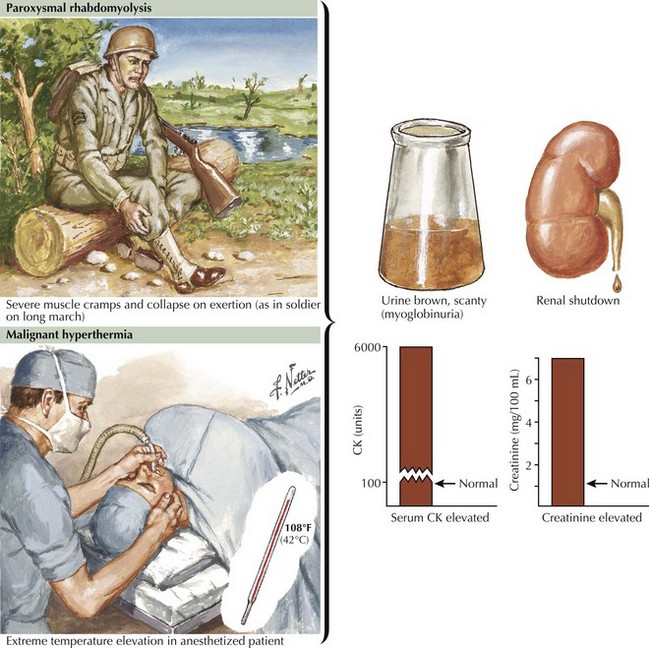
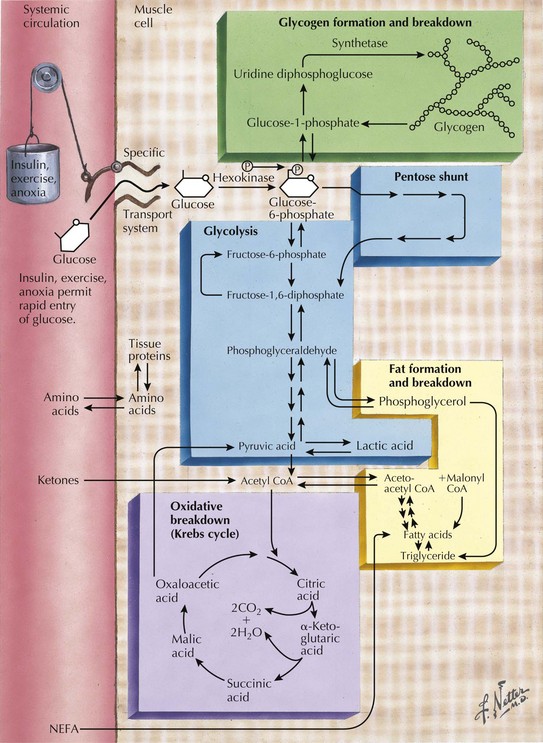
Glycogen storage disorders (GSDs) are very uncommon clinical entities. The classic picture is one of exercise-induced painful muscle cramps, associated with myoglobinuria. The concomitant laboratory documentation of profound elevated serum CK levels and myoglobinuria strongly implicates either a carbohydrate or lipid enzymatic deficiency of inborn metabolism (Fig. 75-4; Table 75-3). Very rarely prolonged use of one extremity, in isolation, will uncover the presence of a previously unsuspected glycogen storage disease (GSD). Muscle phosphorylase deficiency, an inborn error of glycogen metabolism, is the most common of these GSD myopathies.
| Glycogenoses | Respiratory Chain Defects | Lipid Metabolism Disorders |
|---|---|---|
| Myophosphorylase deficiency (McArdle disease)—Type V | Complex 1 deficiency | Carnitine deficiency |
| Phosphofructokinase deficiency (Tauri disease)—Type VII | Coenzyme Q10 deficiency | Carnitine palmitoyltransferase deficiency |
| Phosphorylase B kinase deficiency—Type VIII | Complex III deficiency | Very long chain, long chain, medium chain, or short chain acyl CoA dehydrogenase deficiency |
| Phosphoglycerate kinase deficiency—Type IX | Complex IV deficiency | 3-Hydroxy Acyl-CoA dehydrogenase deficiency protein deficiency |
| Phosphoglycerate mutase deficiency—Type X | Complex V deficiency | Glutaric aciduria type II (electron-transferring flavoprotein and CoQ oxidoreductase deficiencies) |
| Lactate dehydrogenase deficiency—Type XI | Combination of I to V | Neutral lipid storage disease with myopathy; neutral lipid storage disease with ichthyosis |
| Beta enolase deficiency—Type XII |
Pathophysiology
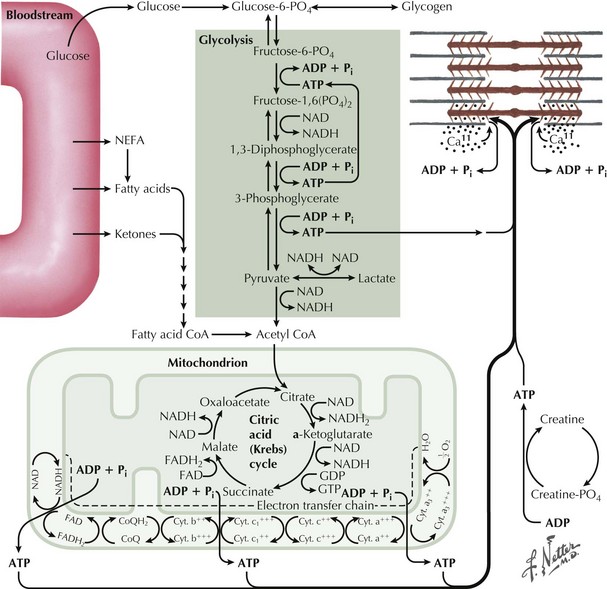
Skeletal muscle function is extremely energy dependent. Normal muscle metabolism requires the presence of both circulating glucose and free fatty acids (Figs. 75-5 and 75-6). At rest, muscles use fatty acids for basal metabolic demands. When one first begins to vigorously exercise, usually within the first 10 minutes, the glycolysis of glycogen, already stored within muscle tissues, is the primary energy source as its breakdown produces glucose but for a relatively short time period. However, when the vigorous exercise is prolonged past these first few minutes, the body shifts to anaerobic glycolysis. This is manifested clinically by the second wind phenomenon. Here lipid stores, in the form of free fatty acids, are mobilized as the primary source of energy. Effective glycolysis is blocked in the various muscle glycogenoses. This essentially deprives muscle of the initial need for glucose, and consequently an accumulation of underutilized glycogen occurs within muscle. In essence, that is, these muscles are inappropriately stressed by what for most healthy persons is no more than strenuous exercise.
Genetics
Muscle phosphorylase deficiency usually has a dominant inheritance pattern. In contrast, the much more rare phosphoglycerate kinase deficiency is usually X-linked. The remaining other glycogenoses, as well as the various disorders of lipid metabolism, the respiratory chain defects, and muscle adenylate deaminase deficiency are usually recessively inherited. As based on the specific enzyme deficiency, there are 12 different types (I-XII) of GSDs. Type V, McArdle disease, is the most common, presenting with classic exercise-induced painful symptomatology (see Fig. 75-3). Similar phenotypes occur with types VII, IX, X, and XI GSDs.
Clinical Presentation
Severe painful muscle cramping and stiffness occurring after exertion are the hallmark of an enzymatic deficiency glycogen or lipid storage disorder. There may be a characteristic second-wind phenomenon, where brief periods of rest at the onset of myalgia alleviate symptoms and enable prolonged exercise. Symptomatic relief may come with rest (see Fig. 75-4). Recurrent myoglobinuria is common, and permanent weakness may develop in older patients. Patients with myalgias and exercise intolerance, with or without hyperCKemia, are commonly evaluated by neuromuscular specialists. The exercise-induced symptomatology with the subsequent myalgia (in contrast to joint or soft tissues), muscle stiffness, and myoglobinuria provide the primary diagnostic criteria for investigating the possibility of a metabolic myopathy. Although specific defects of muscle energy metabolism, as described in this chapter, are sometimes precisely defined, more commonly and very frustratingly, specific enzymatic defects cannot be identified in this setting.
Diagnostic Approach
Precise diagnosis of a GSD requires biochemical findings manifested by an abnormal FET. Baseline measurement of plasma lactate, pyruvate, and ammonia are obtained. The patients then vigorously exercise their hand for 1–2 minutes (see Fig. 75-6). Subsequently serial lactate and ammonia determinations are made immediately after exercise and 1, 3, 6, and 10 minutes thereafter. Normally there is a fivefold rise in serum lactate and a tenfold rise in the serum ammonia level. Glycogen metabolism storage disorders are suspected where there is failure to achieve the normal increase of serum lactate. Muscle adenylate deaminase deficiency is defined by a lack of the expected increase in plasma ammonia after exercise and the normal increase in serum lactate. The test sensitivity is dependent on patient effort. Permanent weakness or rhabdomyolysis, leading to renal impairment, either one precipitated by the ischemic testing format has been rarely reported. Therefore we no longer include an induced ischemic component to this study.
Muscle Biopsy
Abnormal accumulation of glycogen or lipid may be detected in a muscle biopsy specimen using periodic acid-Schiff or oil red O staining, respectively. Muscle phosphorylase, phosphofructokinase, and myoadenylate deaminase deficiency are the primary enzyme-specific stains available for metabolic myopathies. Lipid stains may be normal or show mild lipid accumulation in carnitine palmitoyltransferase II deficiency. Biochemical testing of muscle is available for some myopathies (Table 75-4).

Treatment and Prognosis
One crucial caveat is that patients with hyperCKemia, regardless of cause, have an increased risk for development of malignant hyperthermia (see Fig. 75-4, bottom). This is a potentially life-threatening anesthesia-induced complication of certain fluorinated hydrocarbon inhalation agents such as halothane. Thus, we suggest that any patient with hyperCKemia wear a MedicAlert bracelet at all times to alert the anesthesiologist to the potential for this life-threatening disorder.
Muscular Dystrophies
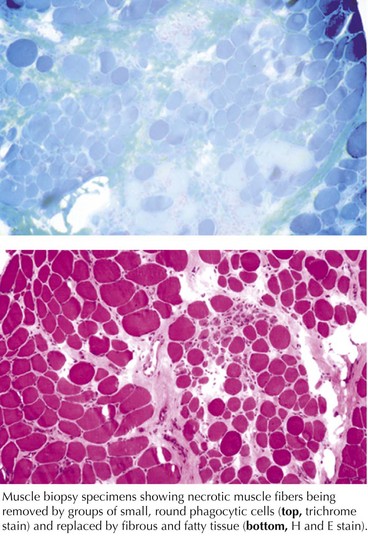
These are genetically determined myopathies distinguished from congenital myopathies by their generally progressive clinical course and characteristic dystrophic histology profile, that is, myofiber degeneration, regeneration, and fibrosis and fatty replacement (Fig. 75-7, bottom panel). The milder dystrophies, for example, oculopharyngeal muscular dystrophy, may lack some of these classic histologic features.
Clinical Vignette
Myotonic Muscular Dystrophy, Type 1 (DM1)
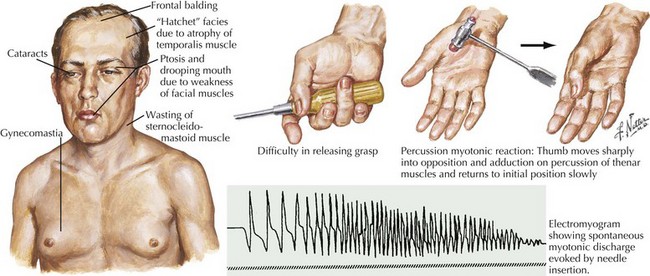
The classic autosomal dominant form usually presents in early adulthood but may be recognized from the neonatal period presenting as a floppy infant similar to some of the congenital myopathies and congenital dystrophies (Fig. 75-8). This condition presents with distal weakness that progresses to involve proximal muscles. Myotonia is delayed skeletal muscle relaxation and is best demonstrated with a forceful handgrip or by thenar muscle eminence percussion. Temporalis, masseter, and sternocleidomastoid wasting, frontal balding, and ptosis contribute to the characteristic myotonic facies (Fig. 75-9). Facial, pharynx, tongue, and neck muscles are also weak. Limb weakness predominantly affects distal extensor muscle groups and then progresses proximally.
Limb-Girdle Muscular Dystrophies
Limb-girdle muscular dystrophies (LGMDs) are a genetically heterogeneous group of disorders wherein the precise classification is rendered more complicated by the frequency of variable phenotypes resulting from mutations of the same gene. It is difficult to distinguish clinically the various subtypes of LGMD, although patterns of weakness or other clinical features may suggest various genotypes. Classification of LGMDs is by number (1 for dominant inheritance, 2 for recessive) and letter (usually in the order of discovery of the chromosomal locus) (see Table 75-1).
Dystrophinopathies
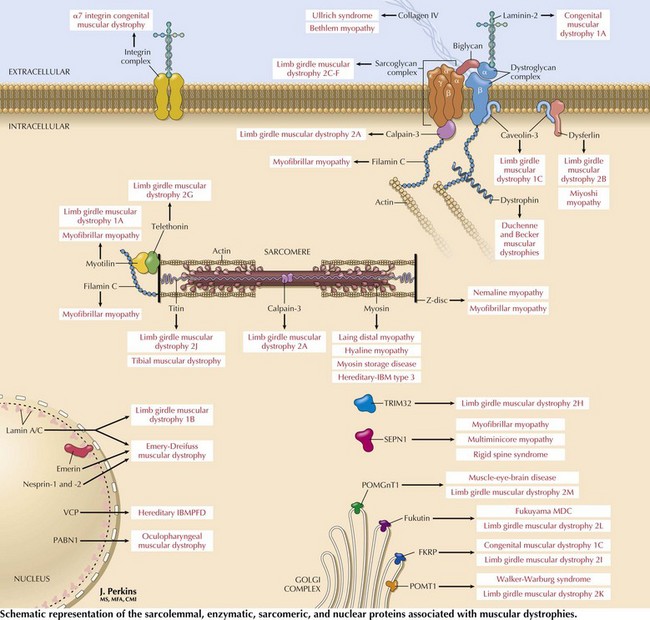
These dystrophinopathies are the most common muscular dystrophies occurring during childhood and in some adults. Dystrophin is a subsarcolemmal protein present in skeletal and cardiac muscle. Dystrophin along with the sarcolemmal proteins forms the dystrophin-sarcoglycan complex (Fig. 75-10).
Duchenne Muscular Dystrophy
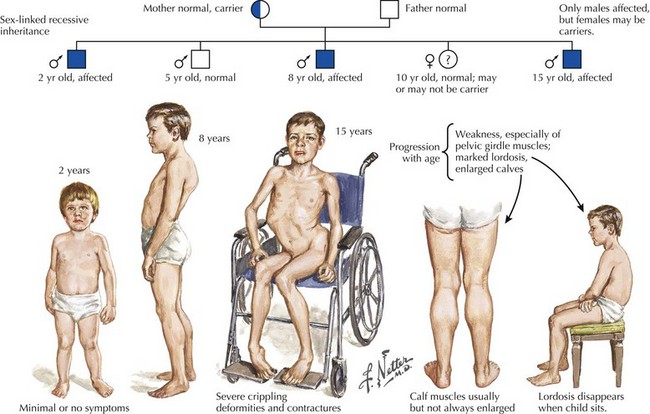
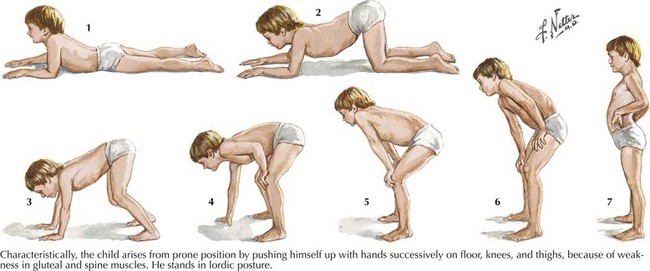
Duchenne muscular dystrophy (DMD) is a primarily X-linked recessive dystrophinopathy; however, in a third of patients this occurs sporadically, presenting in early childhood with proximal weakness and difficulty walking (Fig. 75-11). Untreated patients, usually boys, become wheelchair dependent by midadolescence. Calf hypertrophy, heel cord shortening, and blunted intellect help to differentiate this disorder from other myopathies. Common initial signs are a clumsy gait, frequent falls, and proximal lower extremity weakness. Children cannot rise from a squatting position on the floor and use their hands to push off on their legs (Fig. 75-12). Most patients are wheelchair bound by 12 years of age.
Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral (FSH) muscular dystrophy is a dominantly inherited disorder with variable penetrance. Patients may present with facial weakness and scapular winging in the second to fifth decade of life (Fig. 75-13). Atrophy and weakness of biceps and triceps muscles typically occur; paradoxically, there is relative sparing of deltoid and forearm strength. Ankle dorsiflexors are usually first affected in the lower extremity. An asymmetric pattern of weakness is a common feature. Abdominal muscle weakness and a Beevor sign (caudal or rostral movement of the umbilicus with head flexion) may be present. Variations in phenotype include the rare absence of any demonstrable facial weakness. An infantile form presents with a rapidly progressive course leading to wheelchair dependency by the age of 10 years.
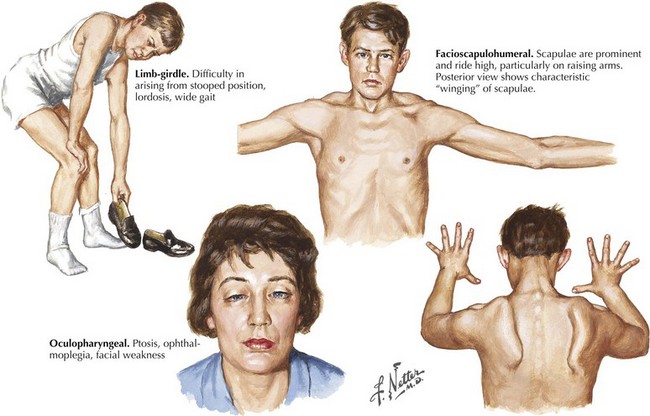
Oculopharyngeal Muscular Dystrophy
This autosomal dominant myopathy is particularly prevalent in North America among individuals of French Canadian ancestry. Most patients present in mid- to late adulthood with ptosis and dysphagia. Mild proximal weakness and ophthalmoparesis may develop. Genetic testing for the presence of a GCG repeat in the PABN1 gene on chromosome 14q11.1 if positive will confirm the diagnosis (see Fig. 75-13, bottom right panel).
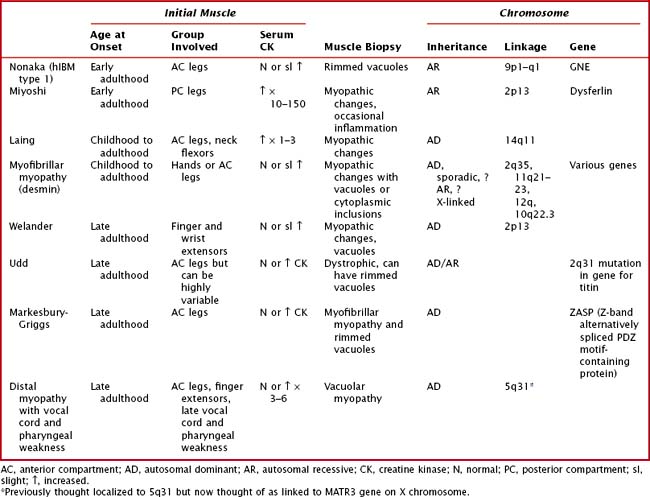
Distal Myopathies or Muscular Dystrophies
The distal myopathies are rare and present with progressive, distal weakness and may be sporadic or inherited. The distal myopathies are classified based on the inheritance, pattern of weakness, and histopathologic findings (Table 75-5). It is important to remember that other myopathies can present with a distal pattern of weakness, for example, DM1, EDMD, FSHD, scapuloperoneal, nemaline myopathy, and centronuclear myopathy due to dynamin-2 mutation.
Congenital Myopathies
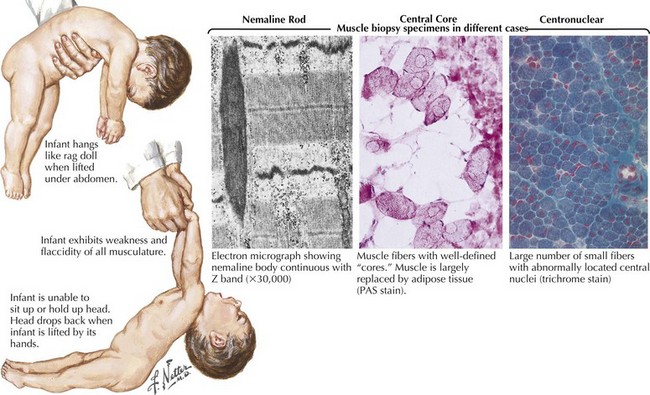
The most common congenital myopathies are centronuclear (myotubular) myopathy, central core disease, and nemaline myopathy (see Fig. 75-8, bottom panel). These are well-defined clinically and genetically heterogeneous disorders. Concomitant congenital skeletal changes such as high arched palates and kyphoscoliosis are commonplace and are suggestive of these genetically determined disorders. Although the presentation is usually that of a floppy infant, some individuals are mildly affected and may not present until early to midadulthood. Babies surviving infancy tend to have minimal progression and reasonable life expectancy.
Diagnostic Approach to Genetic Myopathies
The diagnostic testing required in patients with generalized chronic myopathies varies depending on clinical presentation (Table 75-6), as provided in guidelines for routinely and selectively ordered tests. Many diagnostic tools are available for evaluating a patient with an apparent myopathy. Extensive analyses of muscle should be undertaken only when warranted by a reasonable index of clinical suspicion.
Table 75-6 Evaluation of a Patient with Suspected Muscle Disease*
| Primary Studies | Studies That May Be Indicated in Some Patients |
|---|---|
Treatment
General goals for the management of chronic myopathies are largely supportive rather than disease specific and do not usually affect the natural disease course (Box 75-1).
Amato AA, Russell JA. Neuromuscular Disorders. New York: McGraw-Hill; 2008.
Jones HR. Case presentation and discussion. June 5, 1997. Case records of the Massachusetts General Hospital. N Engl J Med. 1998;339:182-191.
Miller A. Muscle disease. American Academy of Neurology. Continuum. Philadelphia, Pa: Lippincott Williams & Wilkins; 2006:12. No 3
Moxley R. Channelopathies affecting skeletal muscle in childhood. In: Jones HR, De Vivo D, Darras. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Philadelphia, Pa: Butterworth-Heinemann; 2003:783-812.
Neuromuscular disease center—Washington University, St. Louis www.neuro.wustl.edu/neuromuscular.
North K, Goebel HH. Congenital myopathies. In: Jones HR, De Vivo D, Darras BT, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Philadelphia, Pa: Butterworth-Heinemann; 2003:601-632.
North K, Jones K. Congenital muscular dystrophies. In: Jones HR, De Vivo D, Darras BT, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Philadelphia, Pa: Butterworth-Heinemann; 2003:633-648.
Olafsson E, Jones HR, Guay AT, et al. Myopathy of endogenous Cushing’s syndrome: a review of the clinical and electromyographic features in eight patients. Muscle Nerve. 1994;17:692-693.
Snyder R. Bioethical issues. In: Jones HR, De Vivo D Darras, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Philadelphia, Pa: Butterworth-Heinemann; 2003:1279-1286.