[level-membership-for-pediatrics-category]
Chapter 605 Hereditary Motor-Sensory Neuropathies
605.1 Peroneal Muscular Atrophy (Charcot-Marie-Tooth Disease; HMSN Type I)
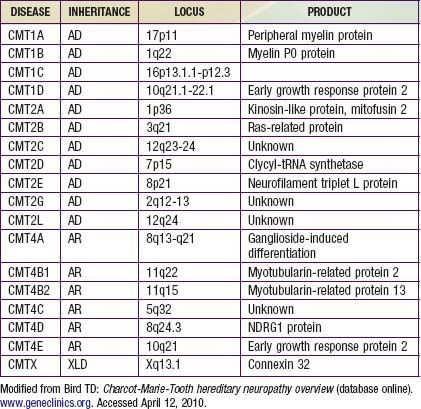
Charcot-Marie-Tooth disease is the most common genetically determined neuropathy and has an overall prevalence of 3.8/100,000. It is transmitted as an autosomal dominant trait with 83% expressivity; the 17p11.2 locus is the site of the abnormal gene. Autosomal recessive transmission also is described but is rarer. The gene product is peripheral myelin protein 22 (PMP22). A much rarer X-linked HMSN type I results from a defect at the Xq13.l locus, causing mutations in the gap junction protein connexin-32. Other forms have been reported (Table 605-1).
Clinical Manifestations
Davidenkow syndrome is a variant of HMSN type I with a scapuloperoneal distribution.
605.6 Fabry Disease
Treatment
(See Chapter 80.4 for specific therapy of Fabry disease, including enzyme replacement.)
Medical therapy of painful neuropathies includes management of the initiating disease and therapy directed to the neuropathic pain independent of etiology. Pain may be burning or associated with paresthesias, hyperalgesia (abnormal response to noxious stimuli), or allodynia (induced by non-noxious stimuli; Chapter 71). Neuropathic pain is often successfully managed by tricyclic antidepressants; selective serotonin reuptake inhibitors are less effective. Anticonvulsants (carbamazepine, phenytoin, gabapentin, lamotrigine) are effective, as are narcotic and non-narcotic analgesics. Enzyme replacement therapy has improved the short and long term prognosis.
605.9 Tomaculous (Hypermyelinating) Neuropathy; Hereditary Neuropathy with Liability to Pressure Palsies
605.10 Leukodystrophies
Several hereditary degenerative diseases of white matter of the CNS also cause peripheral neuropathy. The most important are Krabbe disease (globoid cell leukodystrophy), metachromatic leukodystrophy, and adrenoleukodystrophy (see Chapters 80 and 592).
Bruno C, Bertini E, Federico A, et al. Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology. 2004;62:13-16.
Chance PF. Inherited focal, episodic neuropathies: hereditary neuropathy with liability to pressure palsies and hereditary neuralgic amyotrophy. Neuromolec Med. 2006;8:159-174.
Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143-151.
Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602-606.
Gordon N. Giant axonal neuropathy. Dev Med Child Neurol. 2004;46:717-719.
Houlden H, Blake J, Reilly MM. Hereditary sensory neuropathies. Curr Opin Neurol. 2004;17:569-577.
Kochanski A, Drac H, Kabzinska D, et al. A novel MPZ gene mutation in congenital neuropathy with hypomyelination. Neurology. 2004;62:2122-2123.
Kovach MJ, Lin JP, Boyadjiev S, et al. A unique point mutation in the PMP22 gene is associated with Charcot-Marie-Tooth disease and deafness. Am J Hum Genet. 1999;64:1580-1593.
Landouré G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-tooth disease type 2C. Nat Genet. 2010;42(2):170-174.
Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marine-tooth neuropathy. N Engl J Med. 2010;362(13):1181-1190.
Mendell JR, Sahenk Z. Painful sensory neuropathy. N Engl J Med. 2003;348:1243-1255.
Pleasure D. New treatments for denervating diseases. J Child Neurol. 2005;20:258-262.
Ries M, Gupta S, Moore DF, et al. Pediatric Fabry disease. Pediatrics. 2005;115:344-355.
Seithi PK, Khandelwal D, Sethi NK, et al. Hereditary infantile motor axonal neuropathy mimicking SMA: a case report. J Pediatr Neurol. 2009;7:175-179.
Shy ME. Charcot-Marie-Tooth disease: An update. Curr Opin Neurol. 2004;17:579-585.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 605 Hereditary Motor-Sensory Neuropathies
605.1 Peroneal Muscular Atrophy (Charcot-Marie-Tooth Disease; HMSN Type I)
Charcot-Marie-Tooth disease is the most common genetically determined neuropathy and has an overall prevalence of 3.8/100,000. It is transmitted as an autosomal dominant trait with 83% expressivity; the 17p11.2 locus is the site of the abnormal gene. Autosomal recessive transmission also is described but is rarer. The gene product is peripheral myelin protein 22 (PMP22). A much rarer X-linked HMSN type I results from a defect at the Xq13.l locus, causing mutations in the gap junction protein connexin-32. Other forms have been reported (Table 605-1).
Clinical Manifestations
Davidenkow syndrome is a variant of HMSN type I with a scapuloperoneal distribution.
