CHAPTER 57 Hereditary, Familial, and Genetic Disorders of the Pancreas and Pancreatic Disorders in Childhood
DEFINITIONS AND TERMINOLOGY
There are a growing number of pancreatic disorders with a known genetic basis. However, there is no consensus terminology to distinguish disorders of different etiology that have identical end-stage pathology. Acute pancreatitis represents an event triggered by sudden pancreatic injury that is followed by sequential inflammatory responses (see Chapter 58). Chronic pancreatitis, on the other hand, is a process that usually begins with recurrent acute pancreatitis and ends with immune-mediated destruction of the pancreas and widespread glandular fibrosis (see Chapter 59).1 Therefore, the terms acute pancreatitis and chronic pancreatitis describe syndromes with similar clinical and pathologic characteristics caused by multiple etiologies and varying mechanistic pathways.2
Hereditary pancreatitis refers to recurrent acute or chronic pancreatitis in an individual from a family in which the pancreatitis phenotype appears to be inherited through a disease-causing gene mutation expressed in an autosomal dominant pattern. Individuals with pancreatitis who carry a gene mutation that causes autosomal dominant pancreatitis have hereditary pancreatitis. Familial pancreatitis refers to pancreatitis from any cause that occurs in a family with an incidence that is greater than would be expected by chance alone, given the size of the family and incidence of pancreatitis within a defined population. Familial pancreatitis may or may not be caused by a genetic defect. Tropical pancreatitis (TP) is a form of early age-onset, nonalcoholic chronic pancreatitis occurring in tropical regions3 that is often clustered among family members, and that has a complex genetic basis. TP is further subdivided into fibrocalculous pancreatic diabetes (FCPD) and tropical calcific pancreatitis (TCP) based on the presenting feature of diabetes with fibrosis (FCPD) or severe pain with fibrosis and calcifications (TCP).4 The majority of children previously classified as having early onset idiopathic chronic pancreatitis have identifiable genetic etiologies, but there is no consensus terminology to distinguish the clinical diagnosis from the underlying etiology, especially when there is significant overlap.
MODELS OF PANCREATITIS AS A COMPLEX DISORDER
New models are necessary to understand the relationship among risk factors, etiologies, and the pathology of pancreatic disorders.5,6 Chronic pancreatitis, for example, can no longer be approached from an allopathic perspective (germ theory of disease in which one etiologic agent is responsible for a specific disease), but from the perspective of personalized medicine in which multiple factors must be considered in designing patient-specific treatments. For historical reasons most knowledge about chronic pancreatitis was derived from linking clinical symptoms in patients with abdominal pain and loss of pancreatic exocrine and endocrine function to pathologic changes in the pancreas observed at autopsy. The goal was to find the agent that caused chronic pancreatitis. Because of this approach the major advances in the late 20th century were technical in nature, allowing physicians to obtain biopsies (or high-resolution images) without an autopsies.7 Therapeutic interventions were directed at replacing lost function and attempting to control pain. However, the factors that determine which patient with known environmental risk factors (e.g., alcoholism and tobacco smoking) would progress to chronic pancreatitis whereas others did not, or why other patients without identifiable environmental risk factors progressed from a normal pancreas to chronic pancreatitis remained obscure. Resolving the multiple etiologies and mechanistic pathways could not be accomplished by examining end-stage pathology or by comparative gene expression profiles among patients with a normal pancreas and those with chronic pancreatitis because gene profiling defines abnormalities at a molecular level without discerning the proximal causes of the disease process.
A new etiologic model of pancreatitis became necessary when it was discovered that the major genetic susceptibility factors for chronic pancreatitis actually caused acute pancreatitis through genetic defects that affected the ability of the pancreas to protect itself from premature trypsin activation.8 Trypsin was recognized as the critical molecule in pancreatitis because it is the master enzyme that controls activation of the other digestive enzymes inside the pancreas, and these enzymes cause tissue injury that triggers an inflammatory response. Most factors that increase susceptibility to pancreatitis disrupt a mechanism protecting the pancreas from trypsin-associated injury. Because trypsinogen is synthesized, stored, and transported almost exclusively by the pancreas, loss of mechanisms that protect the body from trypsin-associated injury specifically target the pancreas for recurrent injury. The classic example is hereditary chronic pancreatitis in which it has been observed that affected individuals experience recurrent acute pancreatitis that precedes chronic pancreatitis by a number of years.9–11 It is now recognized that a variety of genetic mutations exists that each increase the risk of pancreatitis through inadequate injury protection from trypsin. Furthermore, individuals with pancreatitis susceptibility genes only have occasional episodes of acute pancreatitis that are often triggered by identifiable environmental factors such as alcohol consumption or cigarette smoking. Because any number of stressors can trigger pancreatitis in genetically susceptible individuals, these factors can be categorized as metabolic and environmental stressors. Some individuals who inherit pancreatitis susceptibility genes and other genetically intact individuals exposed to strong environmental stimuli never develop recognizable acute pancreatitis, suggesting that triggering recurrent acute pancreatitis requires at least one factor in both domains.
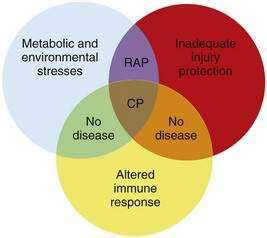
Another observation is that not all patients carrying the hereditary pancreatitis gene who have recurrent acute pancreatitis develop chronic pancreatitis.11 Chronic pancreatitis is characterized as chronic inflammation and fibrosis, which are immune-mediated processes.1 Fibrosis is the product of activated pancreatic stellate cells that are driven by anti-inflammatory cytokines including transforming growth factor-β (TGF-β) and interleukin-10 (IL-10).12–14 The inflammatory process is independent of the mechanism of injury and can be modified by genetic and environmental factors that influence the severity of fibrosis.8,15 If the normal response to pancreatic injury is recovery, then a complication of recurrent acute pancreatitis is extensive fibrosis (i.e., chronic pancreatitis). Factors that affect the immune system by accelerating fibrosis can be grouped together in the category of altered immune response to pancreatic injury. Figure 57-1 illustrates the complexity of chronic pancreatitis and the typical interaction of at least three different categories of risk factors before chronic pancreatitis develops. The study of pancreatic diseases, including chronic pancreatitis, must advance to predictive mathematical models that include all of the relevant risk factors and variables that are common in the human population, so that patient-specific treatments can be designed. This chapter focuses on the major genetic variables that will necessarily become part of the eventual predictive models, but that provide insight into specific disease syndromes that are already recognized.
MAJOR SUSCEPTIBILITY GENES CAUSING PANCREATIC DISEASE
CATIONIC TRYPSINOGEN GENE MUTATIONS
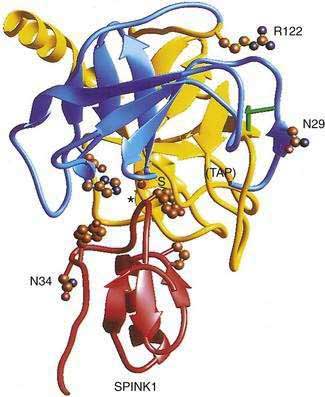
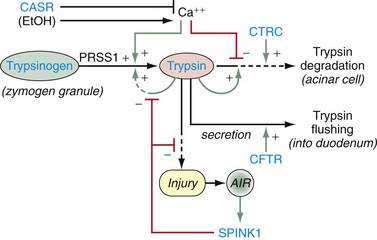
The cationic trypsinogen gene (UniGene name: protease, serine 1; PRSS1) was identified as the first pancreatitis-specific susceptibility gene through genetic linkage studies in families with hereditary pancreatitis.9 Cationic trypsinogen is the major form of trypsinogen (≈65%) followed by anionic trypsinogen (PRSS2, ≈30%), and mesotrypsin (PRSS3, ≈5%). The trypsin molecule is formed by a single peptide that folds into an enzyme with an active site between two globular domains linked by a single connecting chain. An eight amino acid extension of the enzyme, called the trypsinogen activation peptide (TAP), maintains the enzyme as inactive trypsinogen until it is cleaved by enterokinase or another trypsin molecule (autoactivation). Cleavage of TAP allows a conformation change that activates trypsin. Trypsin is also susceptible to trypsin-mediated autolysis beginning at the arginine 122 (R122) site of the connecting chain. The connecting chain is further degraded by chymotrypsin C (CTRC),16 which was biochemically characterized as enzyme Y.17 Finally, the trypsinogen molecule also has two separate calcium binding pockets that play key roles in trypsin regulation by exposing the activation site and blocking the autolysis site, respectively.
Trypsin plays a critical role in pancreatic physiology as the activator of the other pancreatic zymogens, a process that normally occurs within the duodenum where the zymogen activation cascade is initiated by the enterokinase-stimulated conversion of trypsinogen to trypsin. Trypsinogen activation and trypsin inactivation are primarily controlled by trypsin (autoactivation and autolysis), and the ambient calcium concentration serves as the switch between on and off (Fig. 57-2).8 Calcium binding to the first binding pocket that is formed by four aspartic acids within the TAP portion of trypsinogen facilitates trypsinogen activation by trypsin. Calcium binding to the second calcium binding pocket formed by a peptide loop in both trypsinogen and trypsin located adjacent to the autolysis loop, prevents exposure of the trypsin-sensitive R122 autolysis site and thereby prevents autolysis. Thus, physiologic regulation of trypsin activity is determined by cellular calcium, with increased cellular calcium facilitating activation and preventing inactivation and low cellular calcium levels limiting activation and permitting autolysis.
The maintenance of low calcium concentrations within the acinar cells is critical to protecting them from premature trypsinogen activation. However, other protective mechanisms are used to limit trypsinogen activation within the high calcium concentrations of the pancreatic duct fluid. Acinar cell calcium can rise through neurohormonal hyperstimulation (which opens basolateral calcium channels and is linked to calcium tunnels transporting calcium to the acinar pole)18,19; high extracellular calcium concentrations and submaximal pancreatic stimulation20; bile acid reflux, which opens apical membrane calcium pathways21; and prolonged, high-dose alcohol consumption, which lowers the threshold for stimulation-induced acute pancreatitis,22 possibly through mitochondrial damage23 and other factors that regulate intracellular calcium.24 Any process that increases acinar cell calcium will predispose to acute pancreatitis through a calcium-dependent trypsinogen activation and stabilization mechanism.24
More than 20 mutations have been identified in PRSS1 that increase susceptibility to recurrent acute pancreatitis,25 although the R122H and N29I mutations are most common. The locations of the R122H and N29I (N21I) mutations are shown in relationship to the active site in Figure 57-2 and in a mechanistic model in Figure 57-3. The mutations are clustered in regions associated with calcium-dependent trypsin regulation and may confer “gain-of-function” features by facilitating trypsinogen activation or retarding trypsinogen inactivation independent of cell calcium. Gain-of-function mutations often result in an autosomal dominant inheritance pattern; only one of the two trypsinogen alleles must code for a super-functional trypsin in order to prematurely trigger the zymogen activation cascade and cause pancreatitis, thus manifesting the phenotype. Other trypsinogen mutations that are unrelated to calcium-dependent trypsin regulation may predispose to recurrent pancreatitis by altering the activation or inactivation process normally regulated by pH or through interaction with other molecules,26 but the clinical relevance of these potential types of trypsinogen variants remains an area of investigation. The fact that the trypsinogen molecule has two calcium “switch” sites may explain why pancreatitis occurs only intermittently. The clinically important trypsinogen mutations and polymorphisms are discussed following.
The importance of maintaining a tight regulation of cationic trypsinogen is further highlighted by additional genetic findings. First, the R122H and N29I mutations appear to be gene conversions, in which a segment of deoxyribonucleic acid (DNA) from another similar gene replaces a similar segment of the gene of interest. There is now strong evidence that the “H” of PRSS1 R122H is a conversion mutation from Tryp 6,27 and the “I” of N29I is from PRSS2.28,29 These findings highlight the critical importance of the high-fidelity regulatory mechanism of PRSS1 in preventing recurrent, premature trypsinogen activation or trypsin survival in low-calcium environments. Secondly, it has been reported that gene copy number variants are also associated with a risk of chronic pancreatitis in France.30,31 In this case a segment of the genome containing the trypsinogen genes is duplicated on one chromosome, and when combined with the expression of trypsinogen for the opposite allele gives an extra “dose” of trypsinogen—enough to increase the risk of pancreatitis.
ANIONIC TRYPSINOGEN GENE MUTATIONS
Anionic trypsinogen (UniGene name: protease, serine 2; PRSS2) is a form of pancreatic trypsinogen that is usually expressed at about half the amount as cationic trypsinogen, although this ratio may change in some cases.32,33 To date, no gain-of-function mutations have been identified. However, a loss-of-function mutation, PRSS2 G191R, is associated with protection from pancreatitis.34,35 The mutation introduces an arginine (“R”) into a surface loop of PRSS2, making it a target for trypsin-mediated degradation.
CALCIUM-SENSING RECEPTOR GENE POLYMORPHISMS
Regulation of intra-acinar cell calcium is critical for the prevention of pancreatic injury.24,36 The calcium-sensing receptor (CASR) is a membrane-bound member of the G-protein–coupled receptor superfamily.37 CASR plays an important role in calcium homeostasis, as is reflected in its expression by cells of the parathyroid gland and renal tubules that are involved in the calcium metabolism. CASR has been identified in human pancreatic acinar and ductal cells, as well as in various nonexocrine tissues,38 although its functional significance in the pancreas has not yet been determined. More than 100 functional mutations (40 activating and 72 inactivating) have been described in the CASR mutation database related to familial hypocalcuric hypercalcemia (FHH), neonatal severe primary hyperparathyroidism (NSPHT), autosomal dominant hypocalcaemia (ADH) and related hypercalcemic or hypocalcemic disorders.39
In 2003 Felderbauer40 investigated a kindred with familial pancreatitis and a serine protease inhibitor Kazal type 1 (SPINK1) gene mutation (see following for discussion of SPINK1 mutations). However, only two of these family members had chronic pancreatitis, and both were found to have a novel CASR 518T>C mutation that was linked to hypercalcemia. An association between additional CASR variants, with or without SPINK1 mutations, was subsequently identified in patients in India with TP,41 as well as in the United States in sporadic and alcoholic chronic pancreatitis in which the CASR mutation doubles and triples the relative risk, respectively.42 The finding of different CASR polymorphisms in different populations is intriguing, but it appears that the presumed mild hypercalcemia is a cofactor rather than an independent risk factor, as seen in animal models of hypercalcemia.20,43 At the present time, CASR mutations analysis is not offered for clinical decision making.
CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR GENE MUTATIONS
The cystic fibrosis transmembrane conductance regulator (CFTR) gene is the most important molecule for regulating pancreatic duct cell function. This includes generating the bicarbonate-rich pancreatic juice that helps neutralize the acidic chyme coming from the stomach and flushing digestive enzymes out of the pancreas and into the duodenum. Both functions are accomplished by anion secretion through the CFTR located on the apical side of the pancreatic duct cells (see Chapter 56). The CFTR molecule becomes relevant to pancreatic diseases when its function, or regulation of its function, is altered by various gene mutations.
The CFTR molecule forms a regulated ion channel expressed on epithelial cells in the respiratory system, sweat glands, the digestive tract mucosa, biliary epithelium, pancreatic duct cells, and other locations. The primary anions conducted through CFTR under physiologic conditions are chloride and, under some conditions, bicarbonate. The CFTR gene contains more than 4300 nucleotides, with 24 exons and three splice variants that code for a single protein of 1480 amino acids.44 The CFTR molecule has 12 membrane spanning domains, 2 nucleotide-binding domains (NBD1 and NBD2), and a regulatory domain (R domain) with multiple phosphorylation sites (Fig. 57-4).
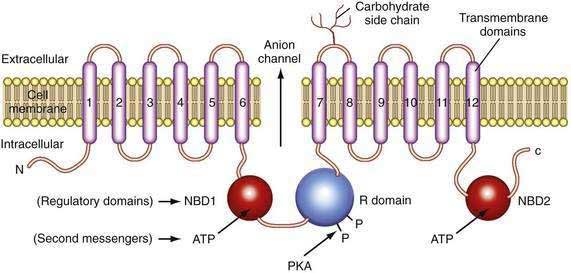
Although the regulation of CFTR is complex, many of the components that are relevant to the pancreas are now understood. CFTR-associated secretion is stimulated when the duct cell is stimulated by secretin or vasoactive intestinal peptide (VIP) acting on receptors that increase intracellular cyclic adenosine monophosphate (cAMP). The cAMP activates protein kinase A–mediated phosphorylation of various sites in the R domain, followed by increased anion conductance (e.g., chloride, bicarbonate) through the CFTR channel. The function associated with the individual phosphorylation sites on the R domain differs.45 Possible consequences of R domain phosphorylation include movement and insertion into the apical membrane, increased or decreased channel activity or specificity, or stabilization of other parts of the molecule such as NBD1. Duct cell stimulation by cholinergic agents or other agonists that increase intracellular calcium also potentiate anion secretion. Recent studies demonstrate that chloride conductance is regulated by cytoplasmic glutamate, while bicarbonate conductance is regulated by binding of ATP to NBD1 and NBD2,46 which may form a heterodimer to maximize adenosine triphosphatase (ATPase) activity.47 Structural studies of the CFTR protein suggest that the molecule exists in two different conformations depending on the presence or absence of ATP binding to NBD1 and NBD2.48 If the basolateral membrane of the duct cell is nearly impermeable to chloride during bicarbonate secretion and if bicarbonate conductance through CFTR is limited because of an unfavorable CFTR conformation, then net ion transport across the basolateral and apical membranes would markedly be reduced49 and the duct cells would not be able to help flush digestive enzymes out of the duct. This would put the pancreas at risk of recurrent acute pancreatitis because prematurely activated digestive enzymes would not be removed from the pancreas.
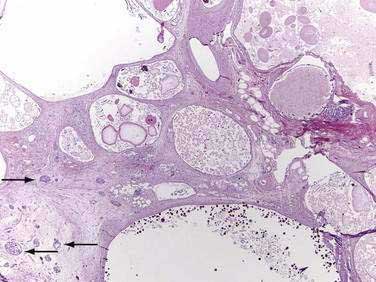
Major mutations in both CFTR alleles result in loss of CFTR function. The consequences include inability to adequately hydrate mucus and other macromolecules, leading to accumulation of viscid material and inspissated glands. This condition results in progressive organ destruction of the pancreas and respiratory system, and dysfunction of the liver, intestine, sweat glands, and other sites where epithelial cell secretion plays an important physiologic role. The pancreas incurs a double risk because much of its proteins are zymogens and trypsin activation will lead to recurrent injury and eventually destruction of the pancreas through progressive fibrosis. Trypsin-mediated injury and destruction of the pancreas in children with CF is consistent with this model because the pancreatic pathology in CF is pseudocyst formation and fibrosis rather than atrophy (as expected with duct obstruction).50 It appears that pancreatic gland injury in CF children roughly parallels the expression of trypsinogen in the developing acinar cells, which begins at 16 weeks’ gestation and gradually increases in concentration until birth and through the first six months of life when levels markedly rise.51,52 The resulting histology has many of the features of end-stage chronic pancreatitis that develops in children and adults, but also has striking expanded ducts that appear as multiple protein filled cysts (Fig. 57-5).
The overall clinical picture in an individual case depends on the nature of the combined CFTR mutations, the genetic background in which the defective genes operates (e.g., modifier genes), and environmental factors.50,53 About 70% of white patients with CF have a three-base pair deletion of the phenylalanine-coding codon 508 (?F508), although 1600 other mutations have been reported. Distinct mutations are common to certain ethnic groups, including African Americans who carry the 3120+1G>A mutation at a frequency of 12.3%54 and the R334W mutation in Hispanics, which is associated with CF with pancreatic sufficiency (PS) but recurrent acute pancreatitis.55 Patients with one severe CFTR mutation and one mild CFTR mutation (e.g., R117H or R334W) often have CF with pancreatic sufficiency.56,57 The reason may be that mutations such as R117H markedly reduce chloride conductance without affecting bicarbonate secretion.46,58 Reduction of chloride conductance would affect all of the epithelial cells in organs that use CFTR to transport chloride, but would have much less effect on the pancreatic duct because it uses CFTR to transport bicarbonate.49 On the other hand, CFTR mutations that specifically inhibit bicarbonate secretion put the pancreas at risk of recurrent pancreatitis without affecting the organs that use CFTR to transport chloride.
The functional consequence of CFTR mutations depends on the combined effects of both CFTR alleles with the severity of the phenotype dependent on the mildest mutation.59 The most common CFTR mutations have been organized according to the effect on clinical phenotype (severe, mild-variable, borderline, benign) and the effect on CFTR protein structure and function (classes 1 to 5).50,53 (Examples are given in Table 57-1.) Class 1 to 3 mutations result in no functional protein and therefore are associated with a severe phenotype when combined with a second severe CFTR mutation. Class 4 and 5 CFTR mutations result in CFTR proteins with altered, but residual function, and are associated with mild-variable or borderline phenotypes. There is current debate as to whether additional classes are justified. If a class 4 or 5 mutation associates with a class 1 to 3 mutation, the phenotype is mild CF, with only a subset of organs affected.60 The resulting conditions are often called atypical CF.59 If a class 1 to 3 or some class 4 mutations are combined with wild-type CFTR or a benign polymorphism, the overall function of CFTR is reduced by up to 50%, but the phenotype is usually normal because more than 90% of overall CFTR function must be lost before clinical features of atypical CF are seen.59 Recurrent acute pancreatitis requires, at minimum, a partially functioning pancreas and is therefore seen in some cases of CF with pancreatic sufficiency and atypical CF.
Table 57-1 Classification of CFTR Mutations, Resulting Defect, and Associated Degree of Pancreatic Dysfunction

Many of the features of CF cannot be explained by variations in CFTR sequence. Instead, these features are caused by specific environmental factors or modifier genes.50,61 Environmental factors, such as bacterial colonization of the respiratory system, tobacco smoke,62 and poor nutritional status contribute to the severity of lung disease.63 The risk of liver disease appears to be independent of CFTR genotype; it is associated with meconium ileus.64 Careful consideration must be given to patient with either classic CF symptoms, or atypical presentations resulting from less common combinations of genetic and environmental factors.
In 1998 two groups65,66 demonstrated that CFTR mutations were also very common in idiopathic and alcoholic chronic pancreatitis, which indicates that some of the more than 1600 known CFTR gene sequence variants cause milder disease (atypical CF59), pancreas-specific injury or that CFTR gene mutations may be part of a more complex trait.1 In some cases, recurrent acute pancreatitis and chronic pancreatitis appeared to be associated with heterozygous CFTR genotypes. Because heterozygous CFTR mutations and polymorphisms are common in the general European and American population, and because the parents of CF children (obligate CFTR mutation carriers without CF) do not have an increased incidence of acute or chronic pancreatitis compared with the normal population,67 it is likely that a second factor that specifically targets the pancreas is required.1 In early onset idiopathic pancreatitis this second factor may be a SPINK1 mutation (discussed later), a potent environmental factor,68 or other complex genetic combinations.69–73
SHWACHMAN-BODIAN-DIAMOND GENE MUTATIONS
The Shwachman-Bodian-Diamond syndrome gene (SBDS) is a gene of unknown function that is mutated in most cases of Shwachman-Diamond syndrome (SDS).74,75 This gene has five exons and encodes a predicted protein of 250 amino acids. The genetic defect in most cases of SDS is caused by gene conversion between the normal SBDS gene with a nonfunctional pseudogene designated SBDSP.74 The SBDSP pseudogene DNA code is 97% identical to SBDS gene code. The differences between the SBDS gene and SBDSP pseudogene are critical nucleotide deletions and nucleotide changes that render the SBDS gene product nonfunctional. Fourteen distinct mutations were initially identified in these kindreds, with the most common being the conversion mutations 183-184TA→CT and 258+2T→C. Interestingly, most patients had compound heterozygous mutations, and no patient was homozygous for the common 183-184TA→CT mutation. Although the function of the gene is unknown, there is significant homology with genes in other species that regulate or facilitate mRNA utilization or metabolism.74 Recent studies suggest that the gene encodes a protein that interacts with ribosomal RNA and helps regulate the maturation of the 60s ribosomal subunit and may also stabilize the mitotic spindle.76–78 The genetic defect results in an acinar cell–specific defect with markedly reduced zymogen synthesis and pancreatic insufficiency rather than susceptibility to pancreatitis. The other clinical features of the SDS are discussed later.
MODIFIER GENE
PANCREATIC SECRETORY TRYPSIN INHIBITOR GENE MUTATIONS
Pancreatic secretory trypsin inhibitor (PSTI, UniGene name: serine protease inhibitor, Kazal type 1; SPINK1) is a 56 amino acid peptide that specifically inhibits trypsin by physically blocking its active site (see Fig. 57-2). SPINK1 is synthesized by pancreatic acinar cells along with trypsinogen and it co-localizes with trypsinogen in the zymogen granules. In the mechanistic models of pancreatic acinar cell protection, SPINK1 acts as the first line of defense against prematurely activated trypsinogen in the acinar cell.9,32,79 SPINK1 is an acute phase reactant and concentrations in serum rise markedly with inflammation.80,81 Under normal conditions there is a great excess of potential trypsin compared with SPINK1 so that the inhibitory capacity of SPINK1 is limited, but with inflammation the expression of SPINK1 is increased dramatically.82
A number of SPINK1 mutations have been identified, but a high-risk haplotype (five polymorphisms that are inherited together) defined by SPINK1 N34S is by far the most common, being present in 1% to 4% of most populations throughout the world.79,83,84 Several other variants of the SPINK1 gene also have been described.25 Pancreatitis associated with SPINK1 gene mutations is associated with early onset recurrent acute and chronic pancreatitis in children,79 familial pancreatitis,83 and TP,85 and is often a feature of polygenic pancreatitis–associated genotype.
Because SPINK1 is a specific trypsin inhibitor, and because expression of SPINK1 is normally very low,82 it follows that SPINK1 cannot be a significant inhibitor of prematurely activated trypsin until the acute inflammatory process has become well established. Thus, the effect of loss-of-function SPINK1 polymorphisms would only become apparent late in the course of acute pancreatitis, or in the case of recurrent premature trypsinogen activation, as is seen with PRSS1 or CFTR mutations. This hypothesis was tested using multiple meta-analyses on data that were classified by proximal etiology.86 This analysis suggested that the strongest effect of SPINK1 N34S was in pancreatitis etiologies that were linked with recurrent trypsin activation, but the effect was low in other etiologies (e.g., alcohol-associated pancreatitis). If this is mechanistically true, then up-regulation of mutant SPINK1 will fail to prevent trypsin-associated recurrent pancreatic injury. Furthermore, suppression of recurrent proinflammatory responses with macrophage-derived anti-inflammatory cytokines (e.g., TGF-β1) will drive the pancreatic stellate cells to produce fibrosis.7,14,87 The implication is that the discovery of a SPINK1 mutation in an unaffected individual is of minimal importance, whereas the effect of a SPINK1 mutation in driving fibrosis in someone with PRSS1 or CFTR mutations is very strong. A summary of the mechanisms for trypsin activation and inactivation is illustrated in Figure 57-3.
PANCREATIC DISEASE IN CHILDREN
Whereas pancreatic disease in children was once considered uncommon, evidence suggests that the incidence is increasing. Although diagnostic modalities and physician awareness continue to improve, this does not appear to account for the increase.88 Acute pancreatitis occurs in all age groups, including infants.89 The most common causes of acute pancreatitis in adults are excessive alcohol use and gallstones. These risk factors are less often seen in children, although biliary pancreatitis is now recognized in this age group. The majority of cases of recurrent acute and chronic pancreatitis in children have a structural or genetic basis. The genetic factors predisposing to acute pancreatitis appear to be similar to those associated with chronic pancreatitis and are discussed in detail in the following sections.
ETIOLOGY OF ACUTE PANCREATITIS
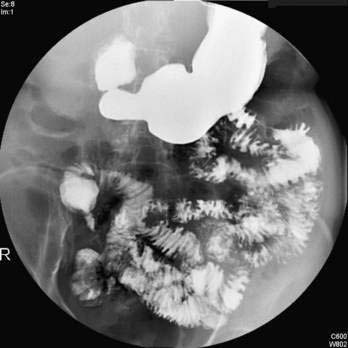
The primary causes of acquired pancreatitis in children are listed in Table 57-2. In a recent review of 1276 children with acute pancreatitis compiled from five studies, the most common causes were idiopathic (22.2%), association with systemic disease (20.8%), trauma (18.6%), structural (e.g., pancreas divisum) (10.6%), and medications (10.2%); gallstones, post–endoscopic retrograde cholangiopancreatography (ERCP), familial, hypercalcemia, hyperlipidemia, diabetic ketoacidosis, and “other” causes made up the remaining etiologies.88 Growing evidence suggests that some of these cases occur in children with high-risk genetic alterations, especially pancreatic-specific combinations of SPINK1 and CFTR mutations. Genetic testing, discussed later in this chapter, is usually performed after recurrent episodes and when other common causes have been excluded.
Table 57-2 Reported Causes of Acquired Pancreatitis in Children
AIDS, acquired immunodeficiency syndrome; ERCP, endoscopic retrograde cholangiopancreatography; HIV, human immunodeficiency virus.
* Class 1 drug: pancreatitis occurs with rechallenge(see Chapter 58).
† Probable cause of pancreatitis.
Trauma
Trauma is a frequent cause of acute pancreatitis despite the fact that the pancreas is well protected from minor injury by its retroperitoneal location. The trauma is usually blunt, associated with injuries to other abdominal viscera and becomes evident soon after the injury,90 although injury may apparently precede the manifestation or recognition of pancreatitis by several weeks. In such an instance, a precise relationship is unclear. Perhaps of more importance is that the possibility of injury to the pancreas is often not considered in a severely injured or battered child.91
Structural Abnormalities
Structural abnormalities are being recognized earlier as imaging techniques such as magnetic resonance imaging (MRI) and magnetic resonance cholangiopancreatography (MRCP) improve. Pancreas divisum is the most common anatomic aberration, although a wide variety of other structural abnormalities of the bile and pancreatic duct also have been observed (see Chapter 55).88
The widespread availability of MRCP has drastically reduced the use of diagnostic ERCP. Post-ERCP pancreatitis has been a significant cause of pancreatitis in several series,92,93 and this etiology is seen wherever ERCP is performed in children. ERCP remains invaluable for therapeutic intervention (see Chapter 61).
Biliary Tract Disease
Gallstone pancreatitis is less common in children than in adults and is probably a reflection of the relative infrequency of cholelithiasis before puberty. However, because almost 10% (as high as 30% in a single study) of children with pancreatitis in some series had cystic duct stones or common bile duct disease, this diagnosis must be considered, regardless of age.94 Little is known about the natural history of this disease in children.
Medications
Medications remain a frequent cause of acute pancreatitis in children, although the disease underlying the prescription must also be considered in the differential diagnosis.95–97 Recent studies identified valproate as the most frequent drug associated with pancreatitis in children, followed by L-asparaginase, prednisone, or multiple medications.92,93,98 The development of persistent abdominal pain in a child receiving any medication should suggest the possibility of drug-induced pancreatitis. This is confirmed only by documentation of pancreatic disease, improvement on drug withdrawal, and return of disease when the drug is reintroduced.
Infection
Infections, particularly with viruses, are a frequently associated with childhood pancreatitis; a partial list of putative agents appears in Table 57-2. Enteroviruses, particularly coxsackievirus and echovirus, have been documented by stool isolation and concomitant serum titer rise in up to 8% of adults with idiopathic acute pancreatitis. Only about half of virus isolations are associated with an antibody rise.99–101 Pancreatitis has been reported in children with Epstein-Barr virus infections, often appearing after an initial clinical improvement.102,103 Interstitial pancreatitis has been described in the congenital rubella syndrome.104 Pancreatitis in children is often attributed to mumps virus on the basis of abdominal pain and an elevated serum amylase value, with parotitis or waxing mumps antibody titers or both.105 Confirmation by serum isoamylase or lipase determinations and by abdominal ultrasonography is lacking, however, and the frequency of this entity may be overestimated. Bacterial pancreatitis has been described in one patient,106 although in that patient there may have been other causes for development of pancreatitis, including antecedent hypotension. Mycoplasma pneumoniae infection, followed one to two weeks later by clinically apparent pancreatitis, has been seen in an estimated 8% of patients with this infection. Complement-fixation titers and serum immunoglobulin M values were elevated, and other causes of pancreatitis were absent.107 Typhoid fever often manifests with abdominal pain; pancreatitis has been suggested as one possible cause.108 Although uncommon in the United States, ascariasis is among the most frequent causes of pancreatitis in children in regions such as South Africa and India. Worms can be found within the pancreatic duct and can be vomited as the initial diagnostic clue. Malaria also has been reported to cause pancreatitis.109 Pancreatitis is 35 to 800 times more common in patients with acquired immunodeficiency syndrome (AIDS).110 This extremely high risk for acute pancreatitis is attributed to several factors (see Chapter 33). A number of medications that are frequently prescribed to human immunodeficiency virus (HIV)–infected patients are associated with pancreatitis (see Table 57-2), possibly due to direct toxicity to pancreatic acinar cells. In addition, immunodeficiency itself predisposes patients to pancreatic infection.
Acute Pancreatitis in Systemic Diseases
Acute pancreatitis is often seen in patients with severe systemic illnesses.88,93 Hemolytic uremic syndrome (HUS) was the most common cause of acute pancreatitis of all systemic diseases in two major studies.92,93 The mechanism is unknown and likely multifactorial, and uremia itself is a risk factor for pancreatic injury.111–113 Systemic lupus erythematosus has been reported in association with pancreatitis.90 Two cases of clinically significant pancreatitis have been documented in association with Kawasaki’s disease.114 Histologic changes have been known to occur in the pancreas during Reye’s syndrome115 but it is unclear if these changes were specific to the disease. Usually this complication has been signaled by hypotension and rapid clinical deterioration during the treatment of advanced illness. Acute pancreatitis after organ transplantation is also common (see Chapter 34).93 Acute pancreatitis should be considered in the intensive care unit when the child is not responding to other therapies or appears to have an unexplained acute inflammatory process. The evaluation of a child with acute pancreatitis should include measurement of serum calcium and triglyceride levels, and these causes must be addressed to prevent recurrence. Hypercalcemia during parenteral nutrition leading to pancreatitis was first described in a child; similar reports have followed.116 Other causes of pancreatitis were not apparent in these patients, although it has been suggested that the calcium content of the solutions infused may not have been the only factor involved in the development of hypercalcemia and pancreatic disease.116 Pancreatitis is occasionally observed in a number of other metabolic disorders, such as diabetic ketoacidosis92,93,117 and various inborn errors of metabolism.118
Acquired Metabolic Derangements
Multiple metabolic derangements are associated with the development of pancreatic disease in children. Perhaps the most common of these is protein-calorie malnutrition. In severely malnourished children, pancreatic enzyme secretion is often compromised, whereas volume and bicarbonate secretion are preserved. Recovery of pancreatic function is said to occur more promptly after kwashiorkor than after marasmus, but in either case the pancreatic disease may contribute to malabsorption during convalescence. Vigorous early refeeding of malnourished children has been associated with the development of clinically significant pancreatitis. Malnutrition was considered a major contributing factor to TP, but this has now been questioned because TP is observed primarily in well-nourished patients.3,119
CLINICAL FEATURES
The diagnosis of pancreatitis is based on the syndrome of sudden onset of typical abdominal pain plus elevation of serum amylase or lipase to at least three times the upper limit of normal levels.120 The diagnosis of acute pancreatitis can be difficult because there is no readily available confirmatory test. Although there have been multiple attempts to determine the sensitivity and specificity of elevations in both enzymes in adults, the studies all suffer from the absence of a method to separately and absolutely document pancreatitis. It is clear that both enzymes can be normal when there is radiographic and clinical evidence of pancreatitis. Also, both enzymes can be elevated by other conditions unrelated to pancreatitis. The level of elevation is also not diagnostic, although the higher the level above the upper reference limit the more likely there is to be pancreatic inflammation. Levels just above the upper reference limits may still be secondary to pancreatitis, especially in patients presenting several days after the onset of symptoms.
The pain is usually supraumbilical, worsens with eating, and may be accompanied by nausea, vomiting, and occasionally jaundice. A transient fever is often present. In infants and toddlers, vomiting, fever, irritability, and abdominal distention can be presenting symptoms.89 Laboratory diagnosis centers on elevated serum amylase and lipase values. Normal amylase values increase with age, which is explained perhaps by the delayed appearance of pancreatic isoamylase, which is usually not present before the age of 3 months and often not detected until the age of 11 months; even then it is not present at adult levels until the age of 10 years. Salivary isoamylase appears and matures much sooner. The serum amylase concentration may be normal, however, despite other evidence of pancreatitis. The evaluation and treatment of acute pancreatitis are covered in Chapter 58.
RECURRENT ACUTE PANCREATITIS
Recurrent acute pancreatitis is seen in about 10% of children after a first episode of acute pancreatitis.93,121 The most common diagnoses in patients with recurrent acute pancreatitis are structural abnormalities, idiopathic pancreatitis, or familial pancreatitis.93,121 A careful evaluation aimed at identifying or ruling out reversible causes should be undertaken to prevent further attacks and to reduce the risk for developing chronic pancreatitis and its complications.
CLINICAL ASPECTS OF GENETIC DISORDERS AFFECTING THE PANCREAS
Several genes that are critical to pancreatic function manifest genetic variations and polymorphisms at variable frequencies among different populations and may lead to major pancreatic disorders (Table 57-3). The major clinical syndromes include CF caused by mutations in the CFTR gene, hereditary pancreatitis caused by mutations in the cationic trypsinogen (PRSS1) gene, and familial pancreatitis, usually caused by homozygous SPINK1 mutations or polygenic CFTR–SPINK1 genotypes. The last two disorders can also appear as sporadic pancreatitis in children. Disorders with a strong genetic basis typically have a younger age of onset than disorders requiring significant environmental exposure, such as alcoholic chronic pancreatitis. SDS is an uncommon pancreatic insufficiency syndrome without pancreatitis. Rare syndromes, including Johanson-Blizzard syndrome, Pearson’s marrow-pancreas syndrome, and other disorders are also recognized as having a genetic basis and are summarized in the subsequent section.
Table 57-3 Hereditary and Congenital Disorders of the Exocrine Pancreas
| DISORDER | DEFECTIVE GENE OR PROTEIN |
|---|---|
| Exocrine Pancreatic Insufficiency | |
| Pancreas agenesis | PDX1 or PTF1A (recessive) |
| Cystic fibrosis | CFTRsev/CFTRsev (recessive) |
| Shwachman-Diamond syndrome | SBDS (recessive) |
| Johanson-Blizzard syndrome | UBR1 (recessive) |
| Pearson’s marrow-pancreas syndrome | Mitochondrial DNA |
| Isolated enzyme deficiency | See text |
| Pancreatitis | |
| Hereditary | PRSS1 (autosomal dominant) |
| Unknown | |
| Familial | SPINK1/SPINK1 (autosomal recessive) |
| CFTRsev or CFTRbl/SPINK1 (complex) | |
| Atypical cystic fibrosis | CFTRsev/CFTRm-v (recessive) |
| Tropical pancreatitis | SPINK1/unknown (complex) |
| Sporadic (risk factors) | CTRC |
| CASR | |
| Metabolic | |
| Hyperlipidemias | Liproprotein lipase |
| Apolipoprotein C-II | |
| Hyperparathyroidism | |
CFTR, cystic fibrosis transmembrane conductance regulator; CFTRsev, severe CFTR mutations (classes 1 to 3); CFTRm-v, mild or variable CFTR mutations (class 4 or 5); CFTRbl, borderline CFTR mutations (class 4 or 5); CTRC, chymotrypsin-C; CASR, calcium-sensing receptor; UBR1, ubiquitin protein ligase E3 component n-recognin1; PDX1, pancrease and duodenal homeobox 1; PTF1A, pancreas specific transcription factor, 1a. SBDS, Shwachman-Bodian-Diamond syndrome gene; SPINK1, serine protease inhibitor, Kazal type 1.
CYSTIC FIBROSIS
CF (OMIN 2197000122) is the most common lethal genetic defect of white populations and is seen in about 1 in 2500 to 1 in 3200 live births. The incidence of CF is about 1 in 15,000 African American and 1 in 31,000 Asian American newborns.123 Expected survival for typical CF children born up to the early 1900s was only a matter of months. Fortunately, with improved pulmonary care and nutrition, the prognosis has dramatically improved, with median survival extending beyond 36.5 years of age in 2006 and many patients living into their 50s. Although CF affects many organs, the primary focus of this chapter is on manifestations of CFTR gene mutations on the pancreas, with brief discussions of liver and intestinal problems that are also seen by the gastroenterologist.
Clinical Features
CF is diagnosed within the first year of life in more than 70% of patients and in more than 85% by age 5. However, 8% of cases remain undiagnosed until after the age of 10 years.123 A small percentage remain undiagnosed until early adulthood. Median survival is more than 40 years with more than 95% living past age 15.60 Newborn screening for CF is done routinely in 46 of 50 states and sporadically in the others.89 The early clinical features are those of maldigestion or other pancreatic and intestinal manifestations of CFTR mutations, whereas the latter course is dominated by pulmonary complications. The presenting features during infancy include meconium ileus, malabsorption with frequent foul stools, failure to thrive, or rectal prolapse.52 Pulmonary function is normal in patients with CF at birth but accounts for much of the morbidity and almost all of the mortality associated with CF beyond the neonatal period. The severity of lung disease depends on known and unknown factors, including chronic infection with Pseudomonas aeruginosa and nutritional status, and probably the effect of unidentified modifier genes because severity of lung disease differs among patients with identical CFTR genotypes. The phenotype-genotype relationship between some childhood disorders and CFTR mutations is often striking, with severe CFTR mutations detected in more than 85% of all children presenting with pancreatic insufficiency and in the majority of infants presenting with meconium ileus.53,124 In older patients, presenting symptoms of CF may include pulmonary disease, nasal polyps, congenital bilateral absence of the vas deferens (CBAVD) with male infertility, liver disease, recurrent acute pancreatitis, or chronic pancreatitis,59,125 although the prevalence of CFTR mutations in patients who have these common disorders is low. Clinical features of CF are listed in Table 57-4 and frequencies of the various gastrointestinal (GI) manifestations of CF are listed in Table 57-5. The variations in CF presentation and clinical features reflect different combinations of CFTR gene mutations, modifier genes, and environmental factors. In cases where only a fraction of the organ systems that CF typically targets are affected, the condition is referred to as atypical cystic fibrosis (aCF). In these cases a careful family history may also provide important clues to the diagnosis of CF.
Table 57-4 Clinical Manifestations of Cystic Fibrosis
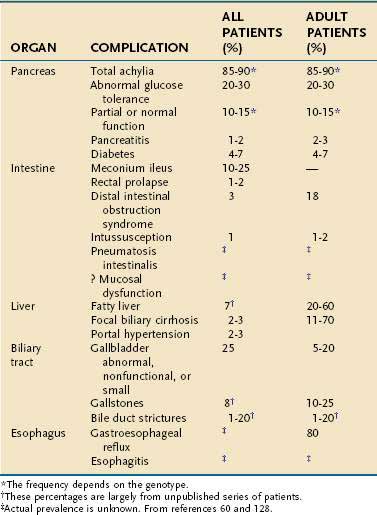
Clinical confirmation of a CF diagnosis rests on the demonstration of elevated sweat chloride concentrations126 (see Table 57-4) or demonstration of an abnormal nasal bioelectric response in specific testing protocols,127 reflecting abnormal CFTR function. When performed appropriately, these tests are reliable. However, false-positive as well as false-negative results may be observed in newborns, in patients with malnutrition, in the presence of some medications, or if inadequate sweat is obtained (see Table 57-5).128 Thus, most experts insist on using the standardized methods performed at CF centers, who use these testing methods frequently. The consensus of a Cystic Fibrosis Foundation panel suggested a diagnosis of CF could be made by the presence of one or more characteristic clinical features, a history of CF in a sibling, or a positive newborn screening test result with confirmation by laboratory evidence of CFTR dysfunction. Furthermore, they suggest that either sweat chloride or nasal bioelectrical responses should be abnormal on two separate days before the diagnosis is confirmed by one of these methods.123 Genetic testing is also commercially available to confirm the clinical diagnosis (two severe mutations must be identified), but these results cannot always be interpreted apart from the clinical context and functional testing—especially in cases with atypical symptoms. Mutational screening of the entire CFTR gene should be considered in atypical cases.
Pancreatic Pathology
Of patients with CF, 85% to 90% present with evidence of exocrine pancreatic dysfunction. Although pancreatic dysfunction in an infant with CF may initially appear minimal, it usually progresses to pancreatic exocrine failure. When severely affected, the pancreas is shrunken, cystic, fibrotic, and fatty.128 Histologically, hyperplasia and eventual necrosis of ductular and centroacinar cells, together with inspissated secretions, lead to blockage of pancreatic ductules and subsequently encroach on acini, causing flattening and atrophy of the epithelium (see Fig. 57-5). Cystic spaces are filled with calcium-rich eosinophilic concretions. A mild inflammatory reaction may be present around obstructed acini, and progressive fibrosis gradually separates and replaces the pancreatic lobules. The islets of Langerhans are spared in most cases until late in the process and are concentrated in the shrinking pancreas.128 Calcification, although rare, may be apparent on radiographs. Ultrasonography, MRI, and computed tomographic (CT) scanning can document the progression of pancreatic disease in CF. Radiographically, the pancreas can appear normal, as incomplete or complete lipomatosis, as a cystic pancreas, as a macrocystic pancreas, or as an atrophic pancreas.129,130 The greatest sensitivity is provided either by MRI or CT scanning, but even with these methods the correlation of abnormalities with the degree of exocrine dysfunction is poor.129
Exocrine Pancreas Dysfunction
Pancreatic enzymes play a critical but partial role in the digestion and absorption of nutrients. Patients with CF are usually pancreatic insufficient (PI), a problem that is compounded by intestinal pathology, high-caloric demands, and poor appetite. Fat and protein maldigestion with fecal losses are the primary, pancreatic manifestations of CF, although there may be considerable variation in severity from one patient to another. Steatorrhea and azotorrhea are generally greater with pancreatic insufficiency than with mucosal malabsorption. Exocrine pancreatic insufficiency may be recognized only when the secretion of lipase and trypsin falls to less than 10% of normal.131 Most patients with CF exhibit this pattern of pancreatic insufficiency. Recurrent acute pancreatitis may complicate the course of CF in patients who do not experience complete loss of pancreatic function in infancy. Among patients with typical CF, the incidence of pancreatitis over a 30-year follow-up is less than 2%.60 Pancreatitis tends to be more problematic in older patients, with the reported incidence among patients older than 30 years being about 2.4%.132
Endocrine Pancreas Dysfunction
Glucose intolerance has been reported in 30% to 75% of patients with CF, and clinically significant diabetes mellitus occurs in up to 10% of young patients.133,134 The previously reported estimates of 1% to 2% incidence135 may have reflected younger patients and poor survival among patients with CF before recent advances in treatment.134 CF-related diabetes mellitus (CFRD) develops with increasing age. At 20 years of age, 30% of CF patients will require insulin and 40% require insulin by age 30.136 The development of CFRD differs in etiology and presentation from typical type 1 or type 2 diabetes mellitus and may reflect destruction of the islets of Langerhans137 similar to what is observed in other forms of chronic pancreatitis. However, the severity of the endocrine deficiency lags behind the exocrine deficiency because the islets are relatively spared until later in the course of pancreatic destruction (see Fig. 57-5). CFRD is associated with deterioration in both respiratory and nutritional status, the development of late microvascular complications, and increased mortality.134 No well-designed studies have addressed this significant disease complication. However, most experts recognize the need for a multidisciplinary team approach, utilization of a high-energy diet (>100% of the recommended daily intake), and appropriate adjustment of insulin doses.134 Overnight enteral feedings may be necessary to maintain adequate nutrition.
Treatment
Pancreatic Enzyme Supplements
Treatment of maldigestion from pancreatic exocrine failure in CF rests on the delivery of active digestive enzymes to the proximal small intestine with meals. Numerous pancreatic preparations are available commercially, but enzyme activities vary considerably from one product to another, and reduced activity of lipase remains a problem for some patients.138 Enteric-coated minimicrospheres are now the preferred form of replacement because they protect the digestive enzymes from destruction by gastric acid (pH < 4) and are effective in treating steatorrhea.139 The size of the microspheres must be considered. If the majority of the spheres are too large (>1 mm), emptying of the spheres/enzymes can be delayed until after food is well into the small intestine.140 The use of histamine-2 (H2) receptor blockers or proton pump inhibitors along with uncoated or enteric-coated pancreatic enzyme supplements also should be considered in patients with CF, especially because the pancreatic and duodenal bicarbonate transport systems are disrupted.141,142 However, even with optimized treatment, fat absorption may not return completely to normal. In large part, the inability to normalize fat absorption may reflect decreased uptake of fatty acids by the abnormal intestinal mucosa.143 In contrast with other forms of pancreatic insufficiency, bicarbonate secretion within the duodenum and biliary tree is also impaired in CF, resulting in a significantly lower than normal duodenal pH.144–146 Thus, without acid suppression, the uncoated enzymes are susceptible to inactivation by gastric acid, and enteric-coated products may not release their contents.147 The use of antacids containing calcium carbonate or magnesium hydroxide should be avoided because they may interfere with the pancreatic enzyme supplements.
Initial therapy for pancreatic exocrine insufficiency in CF includes pancreatic enzyme replacement at doses ranging from 500 to 2000 units of lipase activity per kilogram of body weight per meal, given just before a meal and with snacks.148 The amount is usually advanced to 1000 to 2500 units of lipase activity per kilogram, with final dosage depending on the age, the degree of pancreatic insufficiency, the amount of fat ingested, and the commercial preparation chosen. Adequacy of treatment is typically determined on clinical grounds. Frequent, bulky, and fatty stools; excessive bloating and flatus; and excessive appetite or inadequate growth velocity are signs of inadequate treatment.
Pancreatic enzyme replacement is not without potential complications. Perioral and perianal irritation are common in infants, although less common with the microsphere preparations. Because of the high purine content of pancreatic extracts, hyperuricosuria may develop in some patients taking large doses of enzyme preparations.149 Powdered preparations of pancreatic extracts have caused immediate hypersensitivity reactions in parents of patients with CF.150,151
Colonic strictures and fibrosing colonopathy have been reported with very high-dose administration of pancreatic enzymes and have led to a withdrawal of all the high-dose formulations of enzymes.152,153 Fibrosing colopathy was first recognized in 1994152 and nearly disappeared by 1996.154 It usually develops as an ascending colon stricture causing intestinal obstruction and appears pathologically as postischemic ulceration with mucosal and submucosal fibrosis.152 Nearly all patients were younger than the age of 12, had prior GI surgery, had prior distal intestinal obstruction syndrome, and used H2-receptor antagonists, glucocorticoids, and recombinant human deoxyribonuclease.153 However, the most striking risk was the use of high doses of lipase-containing enzyme supplements. Compared with daily doses of pancreatic enzyme supplements containing up to 2400 units of lipase per kilogram per day, the relative risk of fibrosing colonopathy was 10.9 with a daily dose of 2401 to 5000 units of lipase per kilogram per day and 199.5 for patients taking more than 5000 units per kilogram per day.153 However, because the cases and controls were taken from the same centers where a single brand of enzyme supplement was generally used, it was never conclusively determined whether the problem was primarily related to the lipase content or the acid-resistant coating of the many capsules that were ingested.
Vitamin Supplements
Vitamin deficiencies may develop as a consequence of fat maldigestion and malabsorption, and therefore patients with CF are at risk. Nearly half of all newly diagnosed CF patients have a deficiency of vitamins A, D, or E.155,156 Vitamin A deficiency in CF rarely manifests with clinical abnormalities.128 Vitamin D levels are dependent on sunlight exposure and intake, and the bone demineralization seen in older CF patients may be more of a reflection of general malnutrition128 and the effects of glucocorticoid treatment. Chronic vitamin E deficiency is associated with hemolytic anemia (predominantly in infants) and neuroaxonal dystrophy with prominent neuromuscular symptoms, although these clinical symptoms are rare.128 Vitamin K deficiency and the consequent coagulopathy may occur at any age. Its manifestation may vary from mildly increased bruisability or purpura to catastrophic intracranial hemorrhage in the neonatal period. CF patients who have hepatic involvement are particularly prone to coagulation abnormalities from vitamin K deficiency.157 All CF patients should receive a multivitamin preparation daily; many patients require vitamin A, E, K and D supplements.158 However, frequent and serial monitoring of the serum concentrations of fat-soluble vitamins is important in children with CF because deficiencies, especially those of vitamin E, may occur during therapy.156
Intestinal Manifestations
There are myriad recognized gastrointestinal manifestations of CF (see Table 57-5). Although pancreatic failure (just reviewed) and meconium ileus (discussed following) initially dominate the clinical picture, these additional manifestations cause significant morbidity for many patients.
Pathology
The mucosal glands of the small intestine of patients with CF may contain variable quantities of inspissated secretions within the lumen but rarely have increased numbers of goblet cells. Brunner’s glands may show dilation, flattening of epithelial lining cells, and stringy secretions within their lumens. Severe alterations in the intestinal glands of the small bowel are found in meconium ileus.159 However, even in patients without meconium ileus, these findings are common and appear unrelated to the severity of GI symptoms or changes in other organs. The small intestinal mucosa in older CF patients often shows widely dilated crypts packed with mucus; the mucus frequently appears laminated or may extrude from a gaping crypt. Bulging goblet cells seem to crowd out the intervening columnar epithelium. Variable cellular infiltration may be present in the lamina propria. Mucus in CF is more abundant, stains more intensely, and contains more weak acid groups and protein. Mucus has increased fucosylation and sulfation and decreased sialylation.160–162
Characteristic changes of CF occur in the appendix. Increased numbers of goblet cells distended with mucus line dilated crypts. Eosinophilic casts of these crypts are extruded into the lumen of the appendix. The diagnosis of CF may be suspected on the basis of the histologic appearance of the appendix.163 Although chronic changes in the appendix are a common finding at autopsy, the incidence of acute appendicitis is apparently not increased in CF, inasmuch as only about 1.5% of patients in three large series were found to have appendicitis.164–166 The diagnosis of appendicitis in CF is often delayed and confused with distal intestinal obstruction syndrome, which results in a higher frequency of appendiceal perforation found at the time of diagnosis. The use of chronic antibiotics may also mask typical appendiceal signs.167,168 A smaller subset of patients present with chronic, intermittent pain and tenderness in the right lower quadrant, which results from appendiceal distention by inspissated mucus (but there are no findings of appendicitis on histologic examination). These symptoms are relieved by appendectomy.164 Appendicitis must be considered in all CF patients who have right lower quadrant abdominal pain.
Radiographic Features
Characteristic radiographic features of the intestine are frequently observed in CF. In approximately 80% of patients, thickened duodenal folds, nodular filling defects, mucosal smudging, and areas of dilation and redundancy are seen.169 The findings are not age related, and duodenal biopsies do not adequately explain the radiographic appearance. Similar changes occur in the more distal small bowel, including thickening and distortion of jejunal folds and variable dilation of intestinal loops from the jejunum to the rectum.170 Pneumatosis coli, a benign condition secondary to chronic pulmonary disease and fecal impaction, may be seen.
Functional Abnormalities
Small bowel mucosal dysfunction in CF has been suggested by studies that demonstrate absorption defects that are apparently unexplained by exocrine pancreatic insufficiency or that persist after adequate pancreatic replacement therapy. Decreased activity of certain cytoplasmic peptide hydrolases in intestinal mucosa and reduced uptake of phenylalanine, isoleucine, and glycine have been found in CF patients in comparison with control subjects.171
Basal and stimulated duodenal bicarbonate secretion is largely dependent on functional CFTR, and patients with CF suffer several consequences of diminished duodenal bicarbonate secretion. The importance of CFTR in bicarbonate secretion was first demonstrated in CFTR-deficient knockout mice.172–175 The same abnormalities in duodenal bicarbonate secretion are also present in CF patients, which partially explains the lower postprandial pH (one or two units) in the proximal duodenum of CF patients compared with normal subjects.144,145 Therefore, CFTR-dependent duodenocyte bicarbonate secretion, and, likely, other mechanisms of alkaline secretion, are defective in CF and contribute to the inability to maintain normal proximal duodenal pH.146
Unlike the small bowel and the respiratory system, the CFTR defect in the colon cannot be compensated by any other chloride channel.176 Therefore, the defect in colonic function closely relates to the CFTR genotype.
Lactase deficiency in patients with CF is not related to the disease entity per se but merely reflects a normal ethnic- and age-related phenomenon. Young children with CF often have elevated lactase values in comparison with age-matched controls. This finding may be a consequence of pancreatic insufficiency with slower turnover of microvillus membrane hydrolases.177 Xylose absorption is normal in CF patients.178
Meconium Ileus
Meconium ileus is the presenting symptom in 10% to 20% of infants with CF and appears to be related, in part, to genotype.179,180 Meconium ileus rarely occurs in infants without CF but has been reported in infants with stenosis of the pancreatic duct or partial pancreatic aplasia, with Hirschsprung’s disease, and in infants with otherwise normal GI tracts as a familial occurrence or as an isolated incident.
Pathology
Uncomplicated meconium ileus characteristically demonstrates a narrow distal ileum with beaded appearance caused by waxy, gray pellets of inspissated meconium, beyond which the colon is unused.181 Proximally, the ileal wall is hypertrophied; it then becomes greatly distended with extremely sticky, dark green to black meconium. As many as half of the cases of meconium ileus are complicated by volvulus, atresia, or meconium peritonitis from extravasation of meconium into the peritoneal cavity after intestinal perforation; it may manifest clinically merely as intra-abdominal calcifications, a meconium pseudocyst, generalized adhesive meconium peritonitis, or meconium ascites. Fetal volvulus and vascular compromise may cause atresia. Identification of the role of the meconium ileus modifier genes should clarify the pathophysiology of these various presentations.182
Radiologic Features
Characteristic radiologic findings reveal unevenly distended loops of bowel with absent or scarce air-fluid levels.183 Small bubbles of gas trapped in the sticky meconium may be scattered throughout the distal small bowel. Barium enema demonstrates a microcolon and may outline the obstructing meconium mass in the distal ileum (Fig. 57-6). Abdominal calcification reflects meconium peritonitis, and a meconium pseudocyst may displace loops of bowel.
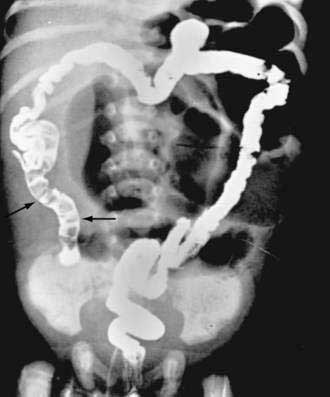
Clinical Features
Meconium ileus classically manifests with signs of intestinal obstruction within 48 hours of birth in an infant who is otherwise well; complicated meconium ileus manifests earlier, and infants appear much sicker. Hydramnios is a common prenatal finding. A family history of CF is helpful in establishing the diagnosis. The increased frequency of meconium ileus in some families with histories of CF is strongly associated with a yet-to-be-identified modifier gene on chromosome 19.184 Dilated, firm, rubbery loops of bowel may be visible and palpable through the abdominal wall, particularly in the right lower quadrant.
Sweat tests should be performed in all infants with meconium ileus, with jejunal or ileal atresia, or with volvulus (Tables 57-6 and 57-7). Results are likely to be positive in 30% of patients with meconium peritonitis and in 15% to 20% of those with atresia of the small intestine.183 Although occasional infants with meconium plug syndrome have CF, meconium plug syndrome and meconium ileus must be carefully differentiated.
Table 57-6 Indications for the Sweat Test (Quantitative Pilocarpine Iontophoresis)
Table 57-7 Conditions Associated with Elevated Sweat Electrolyte Concentration
Treatment
Meconium ileus was considered invariably fatal until 1948, when the first patients were successfully treated by surgery. More recent reports indicate a very low operative mortality, and long-term survival approaches 90% for uncomplicated meconium ileus.180 Various irrigating solutions have been used during the operation and postoperatively to dissolve and dislodge the abnormal meconium. N-acetylcysteine (Mucomyst), which reduces the viscosity of mucoprotein solutions by cleaving disulfide bonds in the mucoprotein molecule, and polysorbate 80 (Tween 80), a mild industrial detergent and preservative, are now generally recognized as safe and effective. Nonoperative relief of obstruction with diatrizoate (Gastrografin or Hypaque) enemas is also possible and has virtually eliminated prolonged hospitalization and early respiratory complications for most infants with uncomplicated meconium ileus.165,180 However, water-soluble hypertonic enemas may cause dangerous fluid and electrolyte shifts, especially in small, sick infants and can cause colonic perforation. Complicated meconium ileus requires surgical therapy. A diagnostic barium enema should precede therapeutic Gastrografin enemas.180,183,185 Infants with CF and meconium ileus who survive beyond six months of age have the same prognosis as any patient with CF and do not tend to have more severe disease.
Distal Intestinal Obstruction Syndrome
Pathogenesis
Mechanisms other than inspissated intestinal sections and pancreatic achylia are probably operative in the pathogenesis of the distal intestinal obstruction syndrome and include undigested food residues; possible disturbances of motility; dilation of the bowel, leading to fecal stasis; and dehydration. Intussusception and, less frequently, volvulus may complicate the distal intestinal obstruction syndrome. The incidence of distal intestinal obstruction syndrome is estimated to be as high as 10% among CF patients, although more recent data indicate a prevalence of 3% or less.165,186–189 Distal intestinal obstruction syndrome may in fact be the presenting disease symptom.
Clinical Features
A spectrum of clinical conditions results from partial or complete obstruction of the bowel by abnormal intestinal contents, including (1) abdominal pain caused by constipation or fecal impaction, (2) palpable cecal masses that may eventually pass spontaneously, and (3) complete obstruction of the bowel by firm putty-like fecal material in the terminal ileum or right colon or both.186,189,190
Abdominal pain, typically recurrent and cramping in nature, is the most common symptom of the distal intestinal obstruction syndrome. This type of pain may be the only symptom, and it may persist for years before distinct obstructive symptoms occur. Insufficient doses or cessation of pancreatic enzyme replacement, recent or concomitant respiratory infection, and dietary changes have been incriminated as precipitating factors.191 Patients with inadequately controlled steatorrhea may be at higher risk for development of this problem.189 Frequently, however, symptoms occur without warning in patients receiving presumably adequate medical management. The distal intestinal obstruction syndrome should be suspected in any CF patient who has abdominal pain, a palpable mass in the right lower abdominal quadrant, or bowel obstruction. When no acute symptoms are present, the soft, indentable, nontender nature of the palpable fecal mass on examination of the abdomen may be a diagnostic aid. The plain radiograph of the abdomen characteristically shows the proximal colon and distal small bowel packed with bubbly appearing fecal material. The fecal bolus can be identified on barium enema but may have to be differentiated from a cecal neoplasm or appendiceal abscess. It is also important to consider the diagnosis of appendicitis in these patients.
Treatment
Once a surgical issue, uncomplicated distal intestinal obstruction syndrome now usually responds to medical management. A stepwise approach with therapeutic trials of more than one modality should be attempted in each patient before a consideration of surgery.181 Vigorous medical therapy includes regular oral doses of pancreatic enzymes and stool softeners, oral or rectal administration of 10% N-acetylcysteine, and Gastrografin enemas. Maintenance treatment with oral doses of N-acetylcysteine, increased doses of pancreatic enzymes, and lactulose has been used successfully to prevent recurrent episodes of the syndrome. Treatment of this disorder with balanced intestinal lavage solutions has also proven beneficial.187,192
Intussusception (see also Chapter 119)
Intussusception, most often ileocolic, is a complication of the distal intestinal obstruction syndrome reported in approximately 1% of patients with CF.165,193 Presumably a tenacious fecal bolus adherent to the intestinal mucosa acts as the lead point of the intussusception. Most of the patients present acutely with intermittent, severe, cramping abdominal pain, although some experience pain for several months before the diagnosis is recognized. Only 25% of the patients note blood in their stools. Efforts should be made to reduce intussusceptions by using radiologic techniques. Intussusception has been reported as the presenting symptom of CF, and CF is a major cause of intussusception after infancy.
Rectal Prolapse (see also Chapter 124)
In the past, rectal prolapse in the setting of CF was quite common, with a frequency of about 20%. The Cystic Fibrosis Registry now reports this complication occurs in 1% to 2% of patients.194 CF accounts for about 11% of’ all cases of rectal prolapse.195 Onset of rectal prolapse is usually in the first few years of life, is often the presenting symptom of CF, and many times is recurrent. Patients in whom CF is diagnosed early in life are much less likely to experience rectal prolapse than those diagnosed later in life except when stools are voluminous. Additional factors thought to be responsible for the high rate of rectal prolapse in CF patients include frequent bowel movements, varying degrees of malnutrition, and increased intra-abdominal pressure secondary to coughing. Medical management is almost always successful, and adequate replacement of pancreatic enzymes typically results in rapid improvement. However, up to 10% of patients may require surgical correction.
Gastroesophageal Reflux
Up to 20% of patients with CF complain of heartburn or regurgitation.196 In adults, the incidence of gastroesophageal reflux symptoms may be as high as 80%. Although the overall incidence of esophagitis is not known, esophagitis has been documented in up to 50% of patients who have significant respiratory problems. Barrett’s esophagus has also been observed in numerous CF patients.197 It is important to recognize and treat gastroesophageal reflux in these patients, but it can be difficult because many of the complaints can be attributed to CF alone and are consequently ignored. Approaches to treatment should be the same as in any other patient population (see Chapter 43).
Cancer Risk
Until the early 1990s, the idea that CF could be associated with the subsequent development of cancer was controversial. One study including 712 patients found no increased risk,198 whereas a second of 412 individuals suggested an increased risk of pancreatic and small intestinal tumors.199 In a more recent, prospective study of 38,000 CF patients, these discrepancies appear to be resolved. The investigators documented an increase in tumors of the digestive tract, but did not observe an increase in the risk of cancer in relation to the general population for all types of cancer.200 Cancer tended to occur in the third decade of life and involved the esophagus, small and large intestines, stomach, liver, biliary tract, pancreas, and rectum. Their pathogenesis is uncertain, but an increased risk of pancreatic cancer has been seen in patients with chronic pancreatic inflammation from other causes including alcohol,201 hereditary pancreatitis,202 and TP.203 Indeed, pancreatic cancer arising in the context of chronic inflammation is being increasingly recognized.204 This heightened cancer risk should be kept in mind as the survival of persons with CF continues to increase. Adolescents and adults with unexplained complaints, especially relating to the abdominal organs, should be evaluated for occult malignancy.
Liver Disease
The frequency of hepatic abnormalities in CF has decreased since the 1950s, with newer surveys noting a prevalence of about 15%. According to older literature, hepatic involvement in CF varied from 20% to 50% of cases studied, although only about 5% of CF patients developed cirrhosis and approximately 2% progressed to clinically apparent liver disease requiring treatment.132 More recent literature suggests that most patients with mild liver abnormalities do not progress and the high frequency of abnormal liver injury test results noted in infancy spontaneously resolves. Nevertheless, approximately 10% of patients develop some degree of cirrhosis, usually prior to or during puberty.205 Although no genotypic association between liver involvement and CF has yet been confirmed, a familial tendency to develop cirrhosis has been observed in some patients,206 and there is now strong evidence that liver disease is associated with a modifier gene. In addition, some risk factors may predispose patients to the development of biliary and liver problems. Such factors include neonatal liver disease, pancreatic insufficiency, and possibly human leukocyte antigen (HLA) class206,207 and meconium ileus.64 Malnutrition may also predispose patients to fatty liver and specific nutrient deficits (protein, fat-soluble vitamins, minerals, essential fatty acids, carnitine). However, in a recent longitudinal four-year study of 124 children with CF, despite the finding that 92% showed some evidence of liver abnormality (6% based on clinical exam, 42% based on elevated serum aminotransferase activity, and 35% based on an abnormal hepatic ultrasonography), liver abnormalities did not correlate with a decline in nutritional status.208 A study from Sweden, however, suggests that essential fatty acid deficiency is more common in CF patients with marked hepatic steatosis.205 Altered drug metabolism in CF209 is characterized mainly by increased hepatic clearance of drugs.210
Newer studies identify the frequency of liver involvement in CF as follows: palpable liver (11%), elevated levels of liver enzymes (2.4%), low serum albumin levels (7.4%), cirrhosis with portal hypertension (2.5%), fatty liver (7%), neonatal liver disease (6%), and palpable spleen (2.2%).194 A second study found cirrhosis in 28% of adults with CF, two thirds of whom had associated portal hypertension.211 The prevalence of liver abnormalities in patients with pancreatic sufficiency is markedly lower.
Pathology
Hepatic changes may be present at any age and may be progressive. Excessive biliary mucus associated with mild periportal inflammation and early fibrosis is common in infants less than 1 year of age. Focal biliary fibrosis, characterized by inspissated granular eosinophilic material in ductules, bile duct proliferation, chronic inflammatory infiltrates, and variable fibrosis, is uncommon in infants but is present in more than 20% of surviving children and adolescents (Fig. 57-7). In time, focal lesions coalesce in some patients and progress to multilobular biliary cirrhosis.212 Bile stasis within lobules is conspicuously rare beyond the neonatal period, even in advanced liver disease caused by CF. Cholestasis is not uncommon in neonates and young infants; it may be prolonged and associated with excessive biliary mucus and mild periportal changes. Approximately half the reported cases were associated with meconium ileus.213
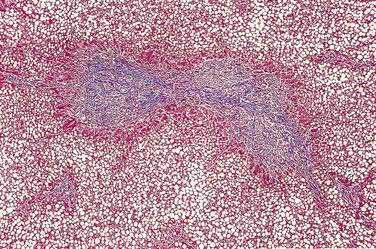
Fatty liver, often independent of nutritional status, remains one of the most common hepatic abnormalities encountered in CF.212 Unexplained hemosiderin deposits in hepatocytes, as well as Kupffer cells, may be prominent in infants and persist beyond four to six months of age.
Radiologic Features
Ultrasonography is the best method for identifying abnormalities of the liver in CF, and may well reveal valuable information regarding the liver parenchyma.214–216 Cirrhosis produces increased coarse echogenicity in many cases and, in some patients, an irregular liver margin. Fatty infiltration is associated with an increase in fine echoes within the liver, with marked attenuation of the ultrasound beam in comparison to normal. A dilated portal vein is indicative of portal hypertension.214,216 Enlarged hepatic veins may be seen as a consequence of congestive heart failure or poor venous outflow from the liver due to constriction of the inferior vena cava by an enlarged liver at or above the entrances of the hepatic veins. CT can also be valuable in assessing the liver parenchyma and is most useful before liver transplantation.
Hepatobiliary scintigraphy with scanning agents derived from iminodiacetic acid (IDA) is the best functional test available for imaging bile flow and can provide valuable information about hepatocyte function, liver size, and the presence of gallbladder filling. Qualitative examination has incorporated the use of deconvolution analysis to measure the hepatic extraction fraction and the hepatic half-clearance time. The hepatic extraction fraction provides a quantitative measure of hepatocyte uptake of tracer to reflect hepatocyte function. The hepatic half-clearance time provides a quantitative measure of clearance of tracer from hepatocytes and bile flow through the ducts.217,218
Functional Abnormalities
Tests of hepatic function in CF may be normal even in cases of overt cirrhosis.219 Serum enzymes reflecting hepatocellular injury may be moderately elevated and fluctuate over the course of the illness. Up to 20% of CF patients with pancreatic insufficiency have elevated serum alanine aminotransferase (ALT) values and 40% to 50% of patients have intermittently increased serum aminotranferases. An elevated serum alkaline phosphatase activity is the next most common chemical abnormality indicative of hepatic involvement, and the high serum activities frequently noted in normal infants and children (mainly resulting from the bone isoenzyme) may conceal increased levels of the hepatic isoenzyme.220 Fasting bile acid levels are elevated in many CF patients, and this may be among the more sensitive measures of liver function in this disease.
Bile acid metabolism is disturbed in patients with CF and exocrine pancreatic insufficiency.219,221,222 Fecal bile salt losses are high, often approaching those of patients with ileal resection (see Chapter 64). Pancreatic enzyme replacement improves fat digestion and absorption, thereby reducing fecal bile acid excretion and steatorrhea. The fractional turnover rate of the bile acid pool is increased and the total bile acid pool size diminished in the absence of pancreatic enzymes,222 whereas the biliary lipid composition and saturation index approach those of patients with cholelithiasis.221 Treatment with pancreatic supplements returns abnormal biliary lipid values toward normal.
Clinical Features
Evidence for liver disease in patients with CF is often subtle; a variety of symptoms can be the presenting complaint. Although there is a broad spectrum of liver disease in CF patients, there are three predominant forms: (1) neonatal cholestasis, manifesting with or without meconium ileus or intestinal atresia223; (2) fatty liver syndrome; and (3) cirrhosis, manifesting either as portal hypertension or as liver failure. Notably, asymptomatic increases in serum liver enzymes or abnormal ultrasonographic findings may be the only clinical manifestations. More commonly, hepatomegaly or splenomegaly is the initial indication of hepatic disease. Esophageal varices or ascites are additional manifestations of hepatic involvement in CF, which may precede evidence of functional impairment (hepatocellular failure) by many years. Examination of the liver and spleen by percussion and palpation should be performed at each clinic visit, and the size and character of these organs recorded.
In patients with suspected liver involvement, the degree of liver dysfunction and injury should be assessed at least annually by tests of synthetic capacity and reflections of liver cell damage. Synthetic capacity of the liver is most readily measured through serum protein analysis (at least total protein and albumin) and prothrombin time (INR). Liver cell damage is reflected by increases in serum bile acids, bilirubin (direct and total), aspartate aminotransferase (AST) and ALT, and alkaline phosphatase. Elevated gamma glutamyl transpeptidase (GGT) may reflect liver damage when other enzyme levels are normal.220 Normal liver biochemistry tests at regular intervals have been proposed as a good negative predictor for liver disease.205
As with all patients with CF, nutritional status must be assessed regularly. Guidelines are available from the Cystic Fibrosis Foundation.224 Plasma carnitine levels also should be measured.225 Nutritional rehabilitation should be accomplished in all patients with liver and biliary disease to eliminate avoidable complications of malnutrition.226
Treatment
The treatment of symptomatic liver disease in CF is a challenge and usually requires a team approach. Treatment of cholestasis is probably best accomplished with ursodeoxycholic acid (20 mg/kg/day), although controlled clinical trials have not yet confirmed that ursodeoxycholic acid can prevent the progression of liver disease. This agent also may improve liver function in patients with elevated serum aminotransferase levels who lack cholestasis. The benefits of ursodeoxycholic acid have been shown to be dose dependent, and, indeed, scintigraphically evident improvement in hepatobiliary excretory function has been observed with higher doses of this bile acid.218,227,228
Nutritional rehabilitation is required in patients in whom disease activity has produced malnutrition. Preventive nutritional management in patients with early liver involvement is indicated. Attention should be paid to the provision of adequate quantities of fat-soluble vitamins. Indeed, essential fatty acid deficiency has been suggested to contribute to liver damage in CF.229
Portosytemic shunts have been placed effectively in CF patients with portal hypertension. The same indications should be applied in CF as in any other disorder when deciding on shunt surgery. The distal splenorenal shunt is the procedure of choice. Prophylactic shunting for varices that have never bled is not recommended. If severe lung disease is not a contraindication for surgery and the clinical status of the patient is acceptable, end-stage liver disease in CF is an indication for liver transplantation.230
Gallbladder and Biliary System
The gallbladder and biliary tract are abnormal in approximately 25% of patients with CF, independent of age, clinical course, or hepatic pathology.214,231–233 “Microgallbladders” are found in 23% and stones or sludge in 8% of patients. Data from the Cystic Fibrosis Registry suggests that only about 2% of individuals with CF eventually require gallbladder surgery.194
Radiologic Features
The plain file of the abdomen may demonstrate radiopaque gallstones. Ultrasound examination shows gallbladder size, content (sludge, gallstones, or bile), and wall thickness. It is excellent for determining dilation of the biliary tract and may help detect cholecystitis.214 Scintigraphy is valuable in delineating functional, biliary system abnormalities in patients with liver disease, although test results are usually normal in CF patients without liver disease. ERCP reveals bile duct abnormalities in some patients with liver disease, but the prevalence of lesions such as bile duct stenosis (approximately 1% to 10%) is probably lower than originally reported.232 It is said that these lesions are not apparent in patients with normal liver function. There are no studies reporting the use of MRCP in patients with CF although it seems reasonable that MRCP should replace diagnostic ERCP in this patient population as it has for others. MRCP should be considered in patients with one or more of the following: unexplained abdominal pain, evidence of biliary tract disease, an abnormal ultrasonogram showing intrahepatic bile duct abnormalities, and a cholangitis-like illness. MRCP may also reveal irregular filling defects throughout the biliary tree with cystic dilations of the intrahepatic bile ducts and intrahepatic cholelithiasis. Irregularities of the smaller proximal ducts have also been noted by ERCP, presumably caused by focal biliary cirrhosis and these abnormalities may be detected by MRCP as well. CT scanning is of additional value in the assessment of biliary abnormalities and may be useful prior to MRCP. Therapeutic ERCP should be reserved for patients with gallstone disease or biliary strictures amenable to stenting.
Genital Abnormalities in Male Patients
Evaluation of patients not clinically suspected of having CF, but who have congenital absence of the vas deferens have a high frequency of CFTR mutations.234,235 Up to 70% of men with the sole finding of congenital absence of the vas deferens have a detectable mutation in at least one allele of CFTR. Other work has suggested that alterations in transcription also may be associated with this defect, inasmuch as a mutation, 5T, which reduced functional messenger ribonucleic acid (mRNA) transcripts of wild-type CFTR, is found in high frequency in men with congenital absence of the vas deferens.234,235 This group of men, without other manifestations of CF, likely represents a very mild form of the disease. On the other hand, the rate of CFTR mutations in patients with primary testicular failure is not elevated, and CFTR gene mutation screening is not warranted for this condition.236
Nutritional Management of Patients with Cystic Fibrosis
In the routine clinical setting, the nutritional management of patients with CF is based on an assessment of nutritional requirements (see Chapter 4), considering age, height, weight and anthropometrics, and severity of lung disease, as well as anorexia, pancreatic insufficiency, and mucosal dysfunction.158,226,237 Ideally, a normal age-appropriate diet should be encouraged, with adequate pancreatic enzyme replacement therapy provided (with gastric acid suppression, if indicated) to achieve as normal a fat balance as possible.238 However, children with end-stage lung disease may require in excess of 150% of the recommended dietary allowances (RDA) for age for calories and protein to promote normal growth. High-calorie, high-fat, and liberal salt diets are also encouraged by many CF centers. Several studies have suggested that improved nutritional therapy improves or at least slows progression of the pulmonary disease.226 All centers now place an emphasis on nutritional intervention before severe malnutrition is evident.
In 2005, the Cystic Fibrosis Foundation revised the nutrition classification guidelines to eliminate the use of percentage of ideal body weight (% IBW) to define nutritional failure based on recommendations from consensus committees.239–241 The guidelines were reviewed and updated in 2008.242 For children younger than 2 years of age, weight-for-length percentile should be maintained at or above the 50%. Up to 20 years, a body mass index (BMI) percentile equal to or greater than the 50th percentile was recommended. It was recommended that adult women should have a BMI greater than or equal to 22 and that adult men should maintain a BMI greater than or equal to 23.
Patients whose growth does not meet these guidelines or who are unintentionally losing weight should undergo a careful nutritional assessment. Malnutrition in CF can result from a variety of factors that increase nutrient loss, reduce energy intake, and increase energy expenditure.237 Increased losses are primarily related to underlying pancreatic insufficiency but are also influenced by conditions such as poorly controlled diabetes mellitus, vomiting and/or regurgitation, excess intestinal mucus, and inadequate bile salt secretion. Energy intake can be affected both by disease complications and by psychosomatic issues.237 Severe respiratory symptoms can be accompanied by anorexia, nausea, and vomiting. GI symptoms or complications such as abdominal pain, gastroesophageal reflux with chest pain, anorexia, and vomiting can lead to reduced caloric intake. In some patients, clinical depression, physical fatigue, a disordered sense of smell (food is unappetizing), and altered body image can lead to reduced food intake. Increased energy expenditure also frequently accompanies the severe respiratory disease of CF and is likely related to variables including chronic infections, fever, increased respiratory effort, and bronchodilator medications.237
The optimal dietary intake for a CF patient is greater than the RDA of healthy children and adults. Ideally, a normal diet for age should be encouraged, except that fat intake should represent 35% to 40% of ingested calories, balanced with adequate pancreatic replacement therapy.238,239 However, a daily calorie and protein intake between 110% to 200% of the standard RDA for age is necessary to promote normal growth, and children with end-stage lung disease may require in excess of 150% of the RDA.239,243,12 Nutritional intervention begins with addition of high-calorie foods to the usual diet, and use of nutritional supplements.239 When these methods fail, enteral feedings should be started via nasogastric, gastrostomy, or jejunostomy tubes. The presence and severity of gastroesophageal reflux disease (GERD) symptoms may influence the decision on the preferred route. Standard formulas are usually well tolerated. Nocturnal infusion is encouraged to promote normal eating patterns during the day. Initially, 30% to 50% of the estimated caloric needs should be provided overnight. Very low-fat, elemental formulas may be used without enzyme supplements for patients with an enteral feeding tube, and should be given by continuous infusion.239 Pancreatic enzyme supplements taken orally in the usual premeal dose are recommended before all nocturnal enteral feedings if nonelemental formulas are used. Patients receiving enteral feedings should be monitored for carbohydrate intolerance on at least two separate nights by measuring blood glucose levels two to three hours into the feeding and at the end of the feeding. Insulin may be required to prevent hyperglycemia, with adjustment of the insulin dosage during acute illness, glucocorticoid therapy, or other changes in health status.
A great deal has been written regarding defined-formula diets as supplements or replacement for food in patients with CF. Although there is no evidence that these defined-formula diets are superior to a balanced diet in providing appropriate protein, energy, and essential nutrients, liquid formulas are easy to administer by tube and may provide added nutrients when infused at night. Nutritional status should be followed carefully, and therapy instituted early. Some adolescents learn to pass soft Silastic feeding tubes nightly in order to administer nasogastric feedings. Gastrostomy feedings may be preferred by some families and patients, especially in younger children, for chronic administration of enteral supplements. Currently, gastrostomy or jejunostomy feedings are instituted at the first sign of nutritional failure.226,244,245 Finally, in some cases, parenteral nutrition may be necessary, but it should be reserved for acute support with a return to some form of enteral nutrition as soon as possible.
Prognosis
More than 50% of patients with CF survive to 36 years of age.194,246 Most significant morbidity and mortality are related to chronic obstructive pulmonary disease.194 The relative influence of nutritional support, pancreatic enzyme replacement, and aggressive treatment of pulmonary disease in improving the quality and duration of life remain under study. Patients with intact pancreatic function have better pulmonary status than those with pancreatic insufficiency,179 which suggests that there is a heterogeneous form of the disease (consistent with evolving genetic information) and/or that survival is longer with better nutrition and treatment.
As survival improves, the problems facing these patients will likely differ and will be begin to spill over into the domain of caretakers predominantly focused on the issues of adults. These medical problems will include such entities as pancreatitis, continued difficulties with adequate nutrition, cirrhosis with portal hypertension, diabetes with its long-term complications, osteopenia, and reproductive issues, as well as all of the more common problems seen in childhood.247,248 In a report of the Cystic Fibrosis Registry, gallbladder disease (0.9% of patients), peptic ulcer disease (0.7%), pancreatitis (0.8%), and cirrhosis with portal hypertension (1.2%) were more common in adults than in children. Pulmonary disease is more severe in adults than in children and malnutrition continues to be a problem in about 35% of adults with CF.194 Increasingly, these patients will require evaluation for potential malignancies of the digestive tract, evaluation for liver disease, or for other complications that will necessitate the specialized attention of a gastroenterologist. It is possible that in the near future, the prognosis of these patients will be changed dramatically by new therapies aimed at recovering specific functions through either drug treatment or gene therapy.
HEREDITARY PANCREATITIS
Hereditary pancreatitis is a syndrome of recurrent acute pancreatitis often leading to chronic pancreatitis that develops in an individual from a family in which the pancreatitis phenotype appears to be inherited through a disease-causing gene mutation expressed in an autosomal dominant pattern.9,249 The most common cause is a mutation in the cationic trypsinogen gene (PRSS1) that appears to cause a gain-of-function through altering the regulatory domains usually controlled by calcium. (The details the PRSS1 gene mutations were presented earlier [see Fig. 57-2].) The majority of documented kindreds with PRSS1 mutations are from the United States and Europe, with a few families in Japan and South America but none identified in southern Asia. Most but not all kindreds with autosomal dominant appearing inheritance pattern of pancreatitis have PRSS1 mutations. In two large representative studies11,250 19% and 35% of pancreatitis-affected patients in hereditary pancreatitis families had no identifiable PRSSI mutations, suggesting that other genes or factors may be responsible for the high risk of pancreatitis in these families.
Clinical Features
The phenotypic features of hereditary pancreatitis caused by PRSS1 mutations are confined to the pancreas because the pancreas is the primary site of trypsinogen expression. Several studies suggest that there are small differences in the clinical features of hereditary pancreatitis depending on the genotype, with PRSS1 R122H being slightly more severe than N29I and patients in whom no gene mutation can be identified.10,11,251 Although about 20 disease-associated PRSS1 mutations have been identified, only the PRSS1 R122H and N29I are seen frequently enough to determine modifier-independent characteristics.
Acute Pancreatitis
The primary clinical phenotypic feature is recurrent acute pancreatitis. The severity of attacks is variable, with severe cases resulting in all of the complications commonly seen in other forms of acute pancreatitis. The patients develop typical epigastric abdominal pain, nausea, and vomiting, with elevated serum amylase and lipase levels. Some families appear to have more severe attacks, with nearly 90% reporting more than five hospitalizations10; an unusually high incidence of major complications such as splenic vein thrombosis was seen in another family.252 However, in other families the phenotype may be mild with attacks of pain characterized as “of nuisance value only.”253 In Europe, patients reported that the vast majority of attacks lasted less than seven days and on the average had 2 (R122H) or 1.4 (N29I) attacks per year. The hospital admission rate was significantly greater with the R122H mutation (0.33 per year) than with the N29I mutations (0.19 per year).11 An uncommon finding is prolonged, persistent, or smoldering acute pancreatitis in which the patient may remain hospitalized for weeks or months. This problem has not been systematically studied and there are no clear treatment recommendations.
Hereditary pancreatitis affects both sexes equally.11 The age of symptom onset is much younger than in most other causes of acute pancreatitis. The first mutation-specific studies reported an age of onset before 5 years in 58% of subjects with R122H mutations,251 but in only 27% of subjects with N29I,10 suggesting that the N29I mutation was slightly milder with fewer hospitalizations. However, patients with N29I mutations had more surgery than R122H patients, although their average age was older. A multicenter European study (EUROPAC) of 418 subjects from 112 families found that the median age of onset for subjects was 10 years with PRSS1 R122H, 14 years for N29I, and 14.5 years for patients with no identified mutations.11 The penetrance of the phenotype in gene mutation carriers is incomplete. Disease penetrance has been consistently reported at approximately 80%.251,253–255 However, there is no maximal age of disease onset, so that the cumulative risk of disease symptoms may be higher, with 96% penetrance by age 50 years.11 The apparent incomplete penetrance and variable expression appear to be determined by genetic and environmental factors,254 with earlier age of onset and more severe clinical course seen in patients with multiple mutations (e.g., PRSS1 plus SPINK1).256,257
The treatment of an acute attack of pancreatitis is currently identical to the treatment of nonhereditary pancreatitis (see Chapter 58). The best approach is to prevention recurrent attacks as much as possible. Multiple small meals, avoidance of fatty meals, and use of antioxidants and vitamins has been used as one approach.2,258 A small open-label trial using antioxidants and vitamins appeared to reduce the number of days of pain attacks in one hereditary pancreatitis family.259 More definitive studies will be needed with antioxidants and other treatments before clear recommendations become available.
Chronic Pancreatitis
Chronic pancreatitis is a process that is initiated by attacks of acute pancreatitis and characterized by inflammatory destruction of the normal parenchyma and progressive fibrosis.1 Reasons that some individuals with recurrent acute pancreatitis progress to chronic pancreatitis and others do not remains unclear, although there is a clear relationship between the number of attacks and the degree of fibrosis. The onset of chronic pancreatitis is currently impossible to determine, but the time between the onset of symptoms (usually acute pancreatitis) and exocrine failure can be determined (Fig. 57-8), although the moment a patient transitions from nonfailure to failure is also poorly defined in patients and between studies. In the EUROPAC study, the cumulative risk of pancreatic exocrine failure was 2.0% at 10 years of age, 8.4% at 20 years, 33.6% at 40 years, and 60.2% at 70 years of age.11 Thus, the exocrine failure associated with end-stage chronic pancreatitis progressed at substantially different rates than the onset of symptoms, and in only in a subset of affected patients.
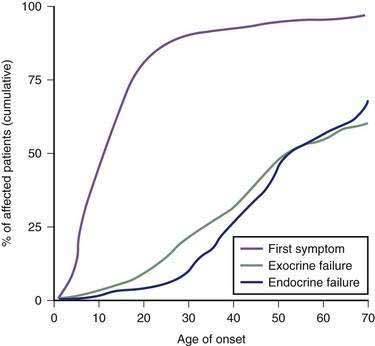
An unexpected observation in Europe was that surgery was much more common in female than male hereditary pancreatitis patients by age 50 years (24.3% vs. 10.5%), and more common with the N29I mutation (compared with R122H or no mutation) by age 50 years (34.7% vs. 12.6% and 13.2%)11 as previously suggested in the United States.10 Currently, the treatment of chronic pancreatitis and associated complications is identical to the treatment of chronic pancreatitis from other etiologies (see Chapter 59).
Diabetes Mellitus
In the EUROPAC study the cumulative risk of endocrine failure was 1.3% at age 10 years, 4.4% at 20 years, 8.5 % at 30 years, and 47.6% by age 50 years (see Fig. 57-6).11 The cumulative incidence of endocrine failure continue to increase after age 50 years, especially in subjects the N29I variant.11
Pancreatic Cancer
As discussed in Chapter 60, there are many reports of an increased incidence of pancreatic cancer in patients with hereditary pancreatitis.202,260 Pancreatic cancer appears to develop about 30 to 40 years after the onset of pancreatitis.202,261 The estimated accumulated risk of pancreatic cancer by age 70 in these families is about 40%,202,260,262,263 although it was slightly lower in the EUROPAC study.11
The reason for the high incidence of pancreatic cancer is unknown. The PRSS1 gene does not appear to play a role in sporadic pancreatic cancer,264 and our current knowledge of trypsin biology provides no rationale for how trypsin may act as an oncogene or other cancer-related factor. Rather, the recurrent pancreatic injury caused by unregulated trypsinogen activation, and subsequent inflammatory response appears to provide an environment that is oncogenic in nature.265
Treatment options for pancreatic cancer are very limited and the prognosis remains poor despite some recent advances. The most effective approach is prevention, with the recognition that the early onset pancreatitis in hereditary pancreatitis is one of the strongest known risk factors for the development of pancreatic cancer. The best target is tobacco smoking because it is a well known risk factor for pancreatic cancer—doubling the risk.266–268 Doubling (or halving) of risk becomes critically important when multiplied to an underlying risk of pancreatic cancer that is 50-fold the risk of the population. Indeed, in hereditary pancreatitis the age- and sex-adjusted odds ratio is doubled by tobacco smoking and the median age of diagnosis of pancreatic cancer is 20 years earlier in the smokers.202,261
Although hereditary pancreatitis families are clearly at high cancer risk, no effective screening methods have been established.260,269 The consensus guidelines of an expert panel suggested that if an individual in the pancreatic cancer risk age range was contemplating pancreatic surgery, a total pancreatectomy should be considered.270 This is based on the difficulty of early detection of pancreatic cancer in an anatomically distorted gland. Newer techniques for early detection and early diagnosis are being evaluated,271 but no controlled clinical trial results in hereditary pancreatitis have yet been reported.
Diagnosis
Prior to 1996, the diagnosis of hereditary pancreatitis was based purely on clinical criteria, including examination of the family pedigree.255 The discovery of the cationic trypsinogen gene mutation R122H opened the door to molecular diagnosis.1,9 Availability of a genetic test revealed that the clinical impression of mild cases was often wrong,251 and also that between 0% to 19% of patients presumed to have idiopathic chronic pancreatitis had hereditary pancreatitis–causing trypsinogen mutations.272–275 The phenotypic features were also clarified, in part because of the difference is proportion of the R122H and N29I mutations in different populations.11 Genetic testing for all of the trypsinogen mutations is now commercially available (Ambry Genetics, Irvine, Calif). Indications and limitations are discussed next.
Genetic Testing
Before clinicians order any test, they must determine the purpose of testing, have the experience to understand and interpret the test results, and anticipate how the results will guide patient management. These considerations are especially true for genetic testing because a genetic test result remains unchanged throughout the life of the patient, has implications for future descendants and other family members, and may affect social and reproductive choices, employment, and insurability.276,277 Thus, the clinician must fully comprehend the implications of testing, be prepared to provide pre- and post-test counseling to the patient (or refer the patient to a genetic counselor), and ensure that informed consent is obtained before testing.276,277
Reasons for cationic trypsinogen mutation testing also vary, but generally include verification of a clinical suspicion, helping a patient understand or validate his or her condition, and assisting individuals at assessing risk of pancreatitis and eventually pancreatic cancer.2,202 This information may also be useful in making life decisions to minimize risk of disease (e.g., reproduction, diet, smoking).276 Identification of an established pancreatitis-associated gene mutation can be valuable in expediting an expensive and prolonged evaluation of recurrent pancreatitis in children, and may preclude further evaluation of elusive causes of pancreatitis in adults.
Interpretation of Genetic Testing
The positive and negative predictive value of a genetic test in identifying specific mutations is almost perfect with properly applied modern techniques. Interpretation of test results and explanation of their meaning to the patient continue to be pivotal issues because the test result has implications for the patient as well as the patient’s extended family. The prognosis for these patients can be outlined in general terms from the clinical discussion earlier, noting that there is significant variability and the effectiveness of future treatments in preventing side effects is unknown. Finally, the mutation-positive individual has a 50% chance of passing on the mutation to each child. A positive test result in clinically unaffected person is interpreted as an increased risk of pancreatitis, with this risk possibly diminishing with age. A negative test result in a family with a known mutation in the PRSSI gene essentially eliminates the risk of this genetic form of pancreatitis. If a mutation has not been previously identified in the family, then a negative test result in an unaffected person is considered noninformative because one cannot distinguish whether the tested individual is free from genetic risk or whether he or she has inherited a different pancreatitis predisposing gene mutation.2 Up-to-date counseling information should always be available through the commercial genetic testing laboratory.
Genetic Testing of Children
The genetic testing of children raises unique issues. Unlike an adult patient, a child legally cannot provide informed consent. Thus, the decision for a child is essentially left to the parents or legal guardian. For children seven years and older, a parent or legal guardian may provide consent for genetic testing, although these older children should also provide assent, or agreement to the testing.278–280 The primary reason for testing children for cationic trypsinogen gene mutations is to assist in determining the cause of unexplained pancreatitis or to confirm suspected pancreatitis in a child at risk for hereditary pancreatitis, thereby limiting further investigations. The testing of purely asymptomatic children is strongly discouraged because currently there is no clear medical benefit in identifying carriers at a young age.276,281 Testing for the purpose of intervention with diet, medication, or surveillance for complications of a genetic disorder has been advocated.281 Because alcohol consumption, emotional stress, and fatty foods have been reported to precipitate pancreatitis attacks,253 and because smoking increases the risk of pancreatitis282–284 and pancreatic cancer,262,267,285 testing for the purpose of encouraging mutation-positive older children to avoid these excesses is advocated by some caregivers. However, the avoidance of fatty foods, alcohol, and tobacco represents excellent general health advice for all children and, therefore, provides no compelling reason for testing.276 In either case, the personal desires of older children to postpone testing or to proceed with testing to relieve their own anxieties and learn more about their own health must also be carefully considered.280 Ownership of test results in children must be addressed.2
FAMILIAL PANCREATITIS
Familial pancreatitis refers to pancreatitis from any cause that occurs in a family with an incidence that is greater than would be expected by chance alone, given the size of the family and incidence of pancreatitis within a defined population.286 Therefore, familial pancreatitis may or may not be caused by a genetic defect. Hereditary pancreatitis (discussed previously) is therefore a subset of familial pancreatitis in which the pancreatitis phenotype follows an autosomal dominant inheritance pattern. Familial pancreatitis is also seen in association with other inherited conditions, such as hypertryglyceridemia and hyperparathyroidism (see following). The etiologies of pure familial pancreatitis are becoming evident and include autosomal recessive conditions as well as complex genetic disorders with gene-gene or gene-environmental interactions.
Familial pancreatitis in which the initial symptoms occur at a young age (e.g., less than 20 years) is more likely associated with a strong genetic risk. The most common causes are autosomal recessive disorders associated with homozygous or compound heterozygous CFTR mutations (atypical CF) or SPINK1 mutations. The important distinction should be made through genetic testing because patients with atypical CF have other organ systems at risk.59 As noted, the diagnosis of any form of CF should be made with great caution and under rigorous protocol because of the implications for the patient and family in terms of the more intense and chronic medical interventions that will be required, as well as the significant social and financial considerations. That said, the appropriate diagnosis must be made.
TROPICAL PANCREATITIS
TP is a form of early onset idiopathic chronic pancreatitis with unique epidemiological and clinical features.3,119 TP is generally characterized by recurrent abdominal pain, pancreatic calculi, and diabetes mellitus, occurring mostly among poor children and young adults inhabiting many developing nations.119 However, there is no consensus on diagnostic criteria to distinguish TP (including TCP and FCPD) from other forms pancreatitis and perspectives on the epidemiology, clinical characteristics, and prognosis are heavily influenced by referral bias (e.g., those presenting to gastroenterologists for pain, to surgeons for severe pain, or to endocrinologists for diabetes mellitus). Furthermore, the age of onset and features of the “typical” patient have changed over the past 50 years, possibly due to improvements in sanitation and better nutrition.3,119,287 The current consensus is that the age of onset is older than in earlier reports, the nutritional status is usually normal at the onset of symptoms, and the distribution is well outside of tropical regions (e.g., northern India).
Clinical Features
The most striking feature of TP is the strong propensity for diabetes mellitus to develop well before exocrine failure, marked calcifications in a grossly dilated main pancreatic duct, and in many patients, pancreatic atrophy. The severity of diabetes appears to correlate with the degree of calcification, suggesting that this represents a clearly different form of pancreatitis and that the pathophysiologic mechanisms are linked. The dilated main pancreatic duct and pancreatic atrophy may be due, in part, to proximal duct obstruction by stones.119 The variation seen in gross pathologic reports spans from complete fatty replacement of the pancreas to severe atrophy resulting in a thin membrane—like a bag filled with stones—to chronic pancreatitis that is indistinguishable from cases of alcoholic or hereditary chronic pancreatitis.
In tropical calcific pancreatitis (TCP), the presenting complaint is usually abdominal pain. Initially the abdominal pain episodes last for days and often are aggravated by small amounts of food so that patients refuse to eat. In the early stages, the bouts of pain are severe and are associated with vomiting.119 This pain is typical of recurrent acute pancreatitis. Some patients develop severe pain late in the course of the disease associated with an inflammatory mass in the head of the pancreas or other features. This characteristic type of pain is similar to B-type pain described by Ammann and colleagues288 in alcoholic chronic pancreatitis, and remains resistant to all but the most aggressive treatment including major surgery. Diabetes mellitus develops in about half of these patients by age 50 years, about 10 years after the initial onset of pain.119
In fibrocalculous pancreatic diabetes (FCPD), the patients are often first diagnosed after referral to an endocrinologist for diabetes. On investigation at this stage of the disease, the pancreas is often atrophic, with a grossly dilated main pancreatic duct that is filled with large, calcific stones, but the patient reports minimal pain. Although many patients with TCP appear to have diabetes from loss of pancreatic islets, patients with fibrocalculous pancreatic diabetes have preserved alpha-cell function.4 The destructive process is not associated with typical anti–beta-cell antibodies.289 Early pathology reports in severe cases of TP with marked pancreatic atrophy also noted marked islet cell hyperplasia (nesidioblastosis) composed primarily of beta cells, but it is unclear if the beta-cell mass remains insufficient (i.e., a few hyperplastic islets in the context of massive loss) or if there is a confounding deficiency or defect in insulin release. The diabetes is often brittle and difficult to manage, even though it is seldom associated with ketoacidosis.3 This latter problem may be related in part to erratic carbohydrate absorption that can be corrected with adequate oral enzyme replacement therapy.290
Etiology
Early attempts to determine the etiology of TP focused on protein and carbohydrate malnutrition and environmental factors such as eating cassava melon. Although there appears to be a weak epidemiologic association, these factors cannot be the primary cause because most individuals with TP are not malnourished and the effect of cassava is weak at best.291
There are some mechanistic similarities between TP in southern Asia and idiopathic chronic pancreatitis in Europe and North America. In both diseases, a significant fraction of subjects have SPINK1 mutations, and especially the N34S haplotype. Interestingly, this high-risk haplotype was only observed in a subset of children in Germany79 or families in North America,83 with the phenotype of the heterozygous and homozygous being identical.83 Because these mutations appear to lower the threshold for intrapancreatic trypsin activation, it appears that trypsin-related injury is a component of both of these disorders. The association between SPINK1 mutations and TP was first recognized in Bangladesh in 2001, including in fibrocalculous pancreatic diabetes and TCP,85,292 a finding confirmed in India in 2002,293 and extended by additional studies.294–296 Of note, a subset of patients with diabetes mellitus but without evidence of exocrine pancreatic disease also had SPINK1 mutations in Bangladesh, a finding that does not appear in diabetes populations tested in the United States.295 This finding suggests that the primary lesion is recurrent acute pancreatitis, and there is a spectrum of complications, include varying degrees of fibrosis, calcification, diabetes mellitus, and pain, that defines the varying phenotypic features.
Treatment
The fundamentals of treatment of TP are similar to other forms of chronic pancreatitis. In earlier stages of disease, pancreatic duct decompression and pancreatic enzyme supplements have been used for symptomatic relief,119,290 but adequate studies to prove their effectiveness are lacking. Oral pancreatic enzyme supplements remain useful for treating maldigestion and can improve glycemic control in patients with diabetes mellitus, presumably through providing a predictable pattern of nutrient absorption.290 The problem of unrelenting pain in TP is similar to other forms of chronic pancreatitis in which surgery is indicated, but not always fully effective in pain control.3,119
SHWACHMAN-DIAMOND SYNDROME
SDS (OMIM 260400122) is a rare autosomal recessive disorder associated with mutations in the SBDS gene (see earlier) and characterized by exocrine pancreatic insufficiency, hematologic abnormalities such as cyclic neutropenia, skeletal defects, short stature, and normal sweat electrolytes.52,297–301 Among these features exocrine pancreatic insufficiency and cyclic neutropenia are present in most patients.300 Myelodysplastic syndromes and acute leukemias develop in up to a third of patients302,303 and numerous other features have been reported (Table 57-8). Severe cases present in infancy with malabsorption, failure to thrive, or recurrent infections. Because of the variable expression of pancreatic, hematologic, and other features, the diagnosis in mild cases may be delayed.300,304 Several hundred families, most with a single affected member, have been identified.301 The discovery of the SBDS gene and genotype-phenotype studies have confirmed the variable phenotypic features that were previously described based on family studies. SDS remains the second most frequently recognized cause of pancreatic insufficiency in children after CF.52,128,298
| Pancreatic | Exocrine pancreatic hypoplasia | 91%-100% |
| Steatorrhea | 55%-88% | |
| Hematologic | Neutropenia | 88%-100% |
| Anemia | 42%-66% | |
| Thrombocytopenia | 24%-34% | |
| Pancytopenia | 44% | |
| Leukopenia | 52% | |
| Elevated fetal hemoglobin | 80% | |
| Myelodysplastic syndromes | 8%-33% | |
| Leukemia | 12% | |
| Skeletal | Metaphyseal dysostosis | 44% |
| Long-bone tubulation defects | * | |
| Short or flared ribs | * | |
| Thoracic dystrophy | 32% | |
| Others | <5% | |
| Growth | Short statue (normal growth velocity) | Common |
| Other | Psychomotor delay | Common |
| Mental retardation | 33% | |
| Renal tubular dysfunction | * | |
| Diabetes mellitus | <5% | |
| Dental abnormalities | * | |
| Ichthyosis | Reported | |
| Hepatomegaly | <5% | |
| Abnormal liver biochemical tests | Common | |
| Myocardial abnormalities | 50% (autopsy) |
Clinical Features
Pancreatic Insufficiency
The clinical features of SDS usually become evident in the first year of life.52,300,304,305 Severe pancreatic insufficiency, steatorrhea, and failure to thrive are frequent presenting symptoms.300,304 A normal sweat-chloride concentration or other normal measures of CFTR function distinguish SDS from CF.52 Serial assessments of exocrine pancreatic function reveal persistent deficits of enzyme secretion, but nearly half of patients showed moderate age-related improvements (more than four years of age) leading to pancreatic sufficiency,300,304 with some pancreatic digestive enzymes (e.g., trypsin) improving more than others (e.g., amylase). The pancreas itself may be small or even of normal size, but the acinar cells appear to have undergone fatty replacement.52 The extensive lipomatous changes result in characteristic changes during abdominal imaging by CT scan (Fig. 57-9), MRI, or ultrasound.306–308
Bone Marrow Dysfunction
Neutropenia-related infections are also an early problem and are severe in at least 85% of patients, occasionally leading to death.128,298 The neutropenia occurs in a cyclical fashion in two thirds of patients, and, when tested, the neutrophils appear to have impaired chemotaxis.302 However, one patient with severe neutropenia and recurrent infections was successfully treated with granulocyte colony-stimulating factor.309 Unidentified serum factors may also impair immune function.310 Common infections include otitis media, sinusitis, pneumonia, osteomyelitis, urinary tract infections, skin infections, and lymphadenitis.311 Thrombocytopenia and anemia are also frequently seen. Patients with hypoplasia of all three bone marrow cellular lines have the worst prognosis.300 Thus, the median age of survival for all patients with SDS is 35 years, but patients with pancytopenia have a median life expectancy of only 24 years. Pancytopenia appears with a mean age of onset of 6 years and occurs in 10% to 25% of patients.311 Up to a third of patients will develop myelodysplastic syndrome (MDS) and about 10% to 25% will develop acute myeloid leukemia (AML) or other leukemias.300,302,304 This is illustrated by a French study reporting that 8 of 71 persons with SDS developed MDS and/or leukemia over a 10-year period.312
Growth and Development
The birth weight of children with SDS is typically low (2.9 ± 0.5 kg, 25th percentile300), and by six months of age the mean heights and weights are characteristically below the 5th percentile. Thereafter, growth velocity appears normal.300 The short stature is independent of nutrition.52 Some clinically evident skeletal abnormalities may be present. For example, metaphyseal chondrodysplasia and dysostosis may be evident radiologically in 44% of patients, especially in the femoral head and proximal tibia.52,128 Thoracic dystrophy, short flared ribs, and other skeletal abnormalities also have been described.52,300,313 Most patients remain below the 3rd percentile for height and weight although some adults reach the 25th percentile for height.52,128 Although men and women are probably affected equally, men with mild disease and short stature are more likely to undergo thorough investigation than women, leading to a mild ascertainment bias.301,304
Molecular Pathology
The molecular defect in SSD is a novel gene of unclear function called the Shwachman-Bodian-Diamond gene (SBDS) (see earlier). The pancreatic lesion appears to result in developmental failure of the pancreatic acini in utero.52 Macroscopically the pancreas appears fatty and may be small or normal size. The main pancreatic ducts and islets are normal. Microscopically there is extensive fatty replacement of the pancreatic acinar tissue.52 Likewise, the hematologic disorder appears to affect cellular development and involves the progenitor cells and bone marrow stroma needed to support hematopoiesis.303 The defect appears to affect multiple specific lineages, although the hematologic deficit can be cured by bone marrow transplantation,310 or improved by granulocyte colony-stimulating factor.309 The long bone abnormalities in about 40% of patients with SDS appear to involve protein processing in the rough endoplasmic reticulum of cartilage chondrocytes, but the exact defect remains obscure.309,314 Although a number of specific cytogenetic abnormalities have been reported,302,315–317 none has been consistently observed. A recent molecular genetics study demonstrated that the majority of patients that were classified as having SDS according to rigorous clinical criteria had compound heterozygous mutations of the SBDS gene and the absence of full-length SBDS protein in leukocytes. However, a subgroup of patients had no SDBS gene mutations and had full-length SBDS protein in leukocytes, suggesting that SDS is heterogeneous.318
Treatment
The treatment of pancreatic exocrine deficiency is more straightforward with SDS than with CF because bicarbonate secretion in the pancreas and duodenum is spared. Optimal pancreatic enzyme replacement (500 to 2000 units lipase activity per kilogram before each meal, and half as much with snacks) should be initiated with an expectation of diminished steatorrhea and improved weight gain, but not necessarily enhanced growth.128 Fat-soluble vitamins, medium-chain triglycerides, and other high-calorie supplements may be needed, as discussed for CF.
During periods of granulocytopenia, febrile episodes should be evaluated and treated with antimicrobial drugs. Anecdotal information suggests that granulocyte colony-stimulating factor can be used in patients with severe neutropenia who have suppurative infections.309 In the case of those with recurrent respiratory infections, humoral immunologic defects should also be considered. Episodes of bleeding or severe anemia may necessitate transfusion. Hip disease should be monitored, with surgical intervention if progression occurs. The use of recombinant human growth hormone in this condition has not been systematically investigated, but anecdotal reports have shown efficacy in accelerating growth.