[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 78 Hepatitis B and D
HEPATITIS B
An estimated 400 million persons are carriers of hepatitis B virus (HBV) in the world today; of these, 75% reside in Asia and the Western Pacific. Effective vaccines against HBV have been available since the early 1980s, but perinatal and early life exposures continue to be major sources of infection in high-prevalence areas. High-risk behaviors such as promiscuous heterosexual contact and injection drug use account for many new cases in young adults. Fulminant acute hepatitis B accounts for several hundred deaths per year in the United States, and chronic HBV infection accounts for one million deaths worldwide each year from complications of end-stage liver disease, including hepatocellular carcinoma (HCC). Hepatitis B is the chief cause of cirrhosis and HCC in the world today, and nationwide vaccination has been shown to diminish greatly the number of new cases of infection and HCC in Taiwanese children.1 Universal hepatitis B vaccination is likely to have the greatest impact on liver disease–related mortality in future generations.
EPIDEMIOLOGY
Geographic Distribution and Sources of Exposure
The prevalence of hepatitis B varies markedly around the world. In highly endemic regions, such as Southeast Asia (excluding Japan), China, and much of Africa, 8% or more of the population are chronic HBV carriers, and the lifetime risk of infection ranges from 60% to 80%.2 In these areas, perinatal transmission and horizontal spread among children are the major sources of infection. Approximately 60% of the world’s population reside in these areas where HBV is highly endemic.3 Regions of intermediate risk include parts of southern and eastern Europe, the Middle East, Japan, the Indian subcontinent, much of the former Soviet Union, and northern Africa. In intermediate-risk areas, the lifetime risk of infection is between 20% and 60%. Persons of all age groups are infected, but as in high-risk areas, most infections occur during infancy or early childhood. Regions of low prevalence are North America, western Europe, certain parts of South America, and Australia. In these areas, the lifetime risk of HBV infection is less than 20%, and transmission is primarily horizontal (i.e., between young adults). Sexual transmission is the main mode of transmission in Europe and North America, and injection drug use is a major contributor to new cases as well.4
Transmission of infection from an HBV carrier mother to her neonate accounts for the majority of new infections in the world today. Sixty to ninety percent of hepatitis B surface antigen (HBsAg)-positive mothers who are hepatitis B e antigen (HBeAg)-positive transmit the disease to their offspring, whereas mothers who are positive for antibody to HBeAg (anti-HBe) transmit the disease less frequently (15% to 20%) (see later discussion of diagnosis). Other less common sources of infection are household contact with an HBV carrier, hemodialysis, exposure to infected health care workers, tattooing, body piercing, artificial insemination, and receipt of blood products or organs. Since routine screening of the blood supply was implemented in the early 1970s, transfusion-associated hepatitis B has become rare in the United States. Hepatitis B can be transmitted by blood that tests negative for HBsAg but positive for antibody to hepatitis B core antigen (anti-HBc) because of low levels of circulating HBV DNA in such blood.5 HBsAg-negative blood that is positive for anti-HBc is excluded from the donor pool in the United States and many countries around the world. In 0% to 30% of persons who are seropositive for anti-HBc alone, HBV DNA is detectable in serum by polymerase chain reaction (PCR) testing.6
HBV is transmitted efficiently by percutaneous and mucous membrane exposure to infectious body fluids. The virus is 100 times as infectious as human immunodeficiency virus (HIV) and 10 times as infectious as hepatitis C virus (HCV). HBeAg seropositivity indicates a higher risk of transmission from mother to child, after needlestick exposure, and in the setting of household contact. HBV DNA has been detected by sensitive techniques such as PCR testing in most body fluids, except for stool that has not been contaminated with blood. Although HBV replicates primarily in hepatocytes, the presence of replicative intermediates and virally encoded proteins in other sites, such as the adrenal gland, testis, colon, nerve ganglia, and skin, suggests that a vast extrahepatic reservoir for infectious virus exists.7 Small amounts of HBV DNA have been demonstrated in peripheral mononuclear cells and liver tissue years after apparent resolution of chronic infection.8,9 Extrahepatic localization of low levels of replicating virus explains the relatively high rate of transmission of infection from anti-HBc–positive organ donors.10
Rates of Infection in the United States
The incidence of hepatitis B has been declining in the United States over the past decade because of universal vaccination of newborns, adult vaccination programs for high risk persons, changes in sexual lifestyle, refinements in blood screening procedures, and the availability of virus-inactivated blood components.11 Most striking has been the decrease among children and health care workers, groups with the highest rates of vaccination. Data from the Centers for Disease Control and Prevention (CDC) indicate that more than 95% of pregnant women in the United States are tested for HBsAg, and infant vaccine coverage levels are now equivalent to those of other vaccines in the childhood schedule. Nonetheless, an estimated 78,000 new HBV infections occurred in 2001, with the highest incidence rates among sexually active young adults (20 to 29 years old) and higher rates occurring among black and Hispanic persons than in white persons.12 Since 1995, approximately 40% of cases of acute hepatitis B reported to the CDC were caused by intimate contact among heterosexuals, 15% to 20% were related to intravenous drug use, and 12% occurred in men who have sex with men. No identifiable source of exposure was demonstrated in approximately 15% of cases. Nearly one third of prison inmates have been infected with hepatitis B, and 2% are chronically infected.
According to the third National Health and Nutrition Examination Survey (1988 to 1994), one or more serologic markers of HBV infection were demonstrated in 4.9% of the U.S. population, and the prevalence of chronic infection was 0.2%.13 Traditional estimates based on the results of blood donation screening in the late 1970s also indicated a prevalence rate for chronic infection of 0.2% to 0.4% in the United States. Although traditional estimates are that the number of HBV carriers in the United States is between 1.25 and 1.5 million, this figure is likely to be a serious underestimate because of changing immigration patterns and underrepresentation of certain minority groups in field surveys. For example, 15 million Asians live in the United States, and even a conservative estimate that the prevalence in this group is 5% would raise the overall number of HBV carriers in the United States by more than 750,000.
CLINICAL OUTCOMES
Definitions
In common usage, the term HBV carrier has often been used to refer to persons persistently infected with HBV who have normal serum aminotransferase levels. (They are sometimes inappropriately referred to as “healthy” HBV carriers.) Because the nomenclature is potentially confusing, the proposal has been made that the carrier state be categorized as inactive or active. Inactive carriers are patients who have evidence of HBV replication on a PCR-based assay only (but not with a less sensitive non–PCR-based assay) and normal or only mildly elevated serum aminotransferase values (see later).14 Long-term follow-up of inactive carriers suggests that the majority of these patients do not have progressive liver disease and do not experience complications. Some of these patients, however, ultimately have one or more episodes of reactivated hepatitis in which the levels of viremia and serum aminotransferase activity increase. Also, some patients with the inactive carrier state may demonstrate HCC. Active carriers, on the other hand, have evidence of HBV replication on non–PCR-based assays for HBV DNA, intermittently or persistently elevated serum aminotransferase levels, and evidence of chronic hepatitis in a liver biopsy specimen.
Clinical Sequelae of Acute Hepatitis B Virus Infection
The age at which a person becomes infected with HBV is a principal determinant of the clinical outcome. HBV infection in adults with an intact immune system is likely to cause clinically apparent acute hepatitis B; only 1% to 5% of these persons become chronically infected.4 By contrast, as many as 95% of infected neonates become chronic HBV carriers because of immunologic tolerance to the virus.
In adults, fulminant liver failure caused by acute hepatitis B occurs in less than 1% of cases, but this group still accounts for 5% of all cases of acute liver failure and approximately 400 deaths annually in the United States.15 Rapid viral elimination may result in clearance of HBsAg from serum by the time of initial presentation. In these cases, the accurate diagnosis of fulminant hepatitis B may require testing with immunoglobulin (Ig) M antibody to hepatitis B core antigen (HBcAg) (IgM anti-HBc) (see later discussion of serologic markers of infection).16 The rate of spontaneous survival in acute liver failure caused by hepatitis B is only approximately 20%. Liver transplantation has resulted in survival rates of 50% to 60%. Recurrent disease in the allograft is now uncommon because of administration of hepatitis B immune globulin (HBIG) and potent orally administered antiviral agents (see later and Chapters 93 and 95).
Clinical Sequelae of Chronic Hepatitis B Virus Infection
The presence of active viral replication and long-standing necroinflammatory liver disease caused by HBV strongly influences the rate of progression to cirrhosis. The major determinant of survival is the severity of the liver disease when the patient first comes to medical attention.17 Cirrhosis is associated with decreased survival and an increased frequency of HCC. Five- and 20-year survival rates of 55% and 25%, respectively, have been reported in patients with cirrhosis at presentation, whereas rates of 97% and 63%, respectively, have been reported for those with mild (noncirrhotic) disease.18 Survival rates differ most dramatically between patients with compensated and decompensated cirrhosis. In one study, an 84% five-year survival rate was reported for patients with compensated HBV-related cirrhosis, compared with 14% for patients with cirrhosis complicated by ascites, jaundice, encephalopathy, or a history of variceal bleeding.19 Multivariate analyses in several large cohort studies have identified age, ascites, hyperbilirubinemia, and other features of advanced liver disease as correlating independently with survival in patients with HBV-related cirrhosis. Interferon-induced clearance of HBeAg (see later) has been associated with prolongation of survival without complications or the need for liver transplantation.20
Clearance of HBsAg in patients with HBV-related cirrhosis has been associated with an excellent prognosis, including improvement in liver histology and function, a decreased chance of viral reactivation, and prolonged survival.17 Even HBsAg clearance, however, is not an absolute safeguard against the future development of HCC in persons who already have cirrhosis.21
MOLECULAR BIOLOGY
HBV is a small DNA virus that belongs to the Hepadnaviridae family. Other members of this virus family are human HBV-like agents that infect the woodchuck, ground and tree squirrels, woolly monkey, crane, heron, Ross goose, and duck. HBV is a small (3.2-kilobase [kb]) virus with a DNA genome that has a relaxed, circular, partially double-stranded configuration (Fig. 78-1). The genome is composed of four open reading frames (ORFs) and has a compact design in which several genes overlap and use the same DNA to encode different viral proteins. The four viral genes are the core, surface, X, and polymerase genes. The core gene encodes the core nucleocapsid protein, which is important in viral packaging and production of HBeAg. The surface gene encodes the pre-S1, pre-S2, and S proteins (comprising the large [L], middle [M], and small [S] surface proteins). The X gene encodes the X protein, which has transactivating properties and may be important in hepatic carcinogenesis. The polymerase gene has a large ORF (approximately 800 amino acids) and overlaps the entire length of the surface ORF. It encodes a large protein with functions that are critical for packaging and DNA replication (including priming, RNA- and DNA-dependent DNA polymerase, and RNase H activities).
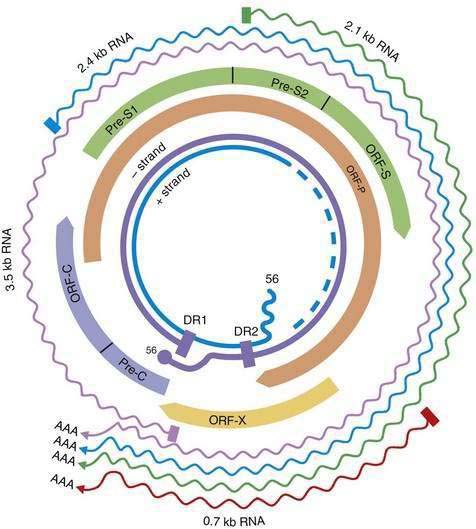
Although HBV is a DNA virus, replication occurs through an RNA intermediate and requires an active viral reverse transcriptase/polymerase enzyme. The mutation rate is higher for HBV than for other DNA viruses (an estimated 1010 to 1011 point mutations per day).22 Complete HBV genomic sequencing has identified a large number of mutations within the HBV genome, many of which are silent or do not alter the amino acid sequence of encoded proteins. Because of genomic overlap, however, some of the silent mutations in one ORF (for example, the polymerase gene) may result in an amino acid substitution in an overlapping ORF (surface gene), although with currently uncertain clinical implications.
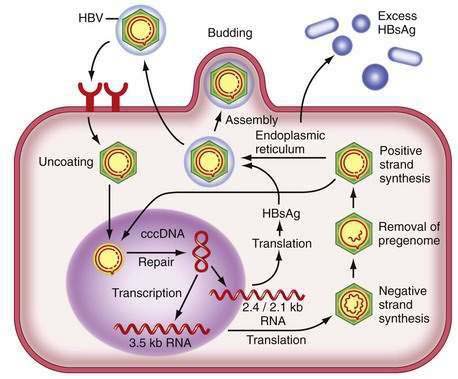
Figure 78-2 illustrates the life cycle of HBV. The initial phase of hepadnaviral infection involves the attachment of mature virions to host cell membranes. The human receptor for HBV remains unknown. Entry of the virus results from fusion of the viral and host membranes as the nucleocapsid is released into the cytoplasm. Mechanisms of intracellular transport of viral genome into the nucleus are poorly understood, but the first step in genomic replication involves conversion of the relaxed circular form of HBV DNA into a double-stranded, covalently closed circular form (cccDNA). The cccDNA, which serves as the template for viral transcription, is the major form of viral DNA in the nucleus of infected hepatocytes. Subgenomic (0.7 to 2.4 kb) and pregenomic (3.5 kb) RNA molecules are transcribed from this template. The L protein is translated from the 2.4 kb RNA, the M and S proteins from the 2.1 kb RNA, and the X protein from the 0.7 kb transcript. The pregenomic RNA serves as the template for reverse transcription as well as the messenger RNA (mRNA) for translation of the core and polymerase proteins; the precore RNA codes for the precore gene product.
Hepatitis B Virus Genotypes
A genetic classification based on comparisons of complete genomes has demonstrated eight genotypes of HBV, designated A through H (Table 78-1).23 Several methods have been used for HBV genotyping, including a commercially available line probe assay. Genotypic differences are based on an intergroup divergence of 8% or more in the complete nucleotide sequence. Genotype A is the predominant genotype in northern Europe and the United States. Genotypes B and C are confined to populations in eastern Asia and the Far East, but changes in immigration patterns have resulted in an influx of Asian HBV carriers with these genotypes into the United States.24 Genotype D is found worldwide but is especially prevalent in the Mediterranean area, Middle East, and south Asia. Genotype E is indigenous to western sub-Saharan areas, and genotype F prevails in Central America. Cases of genotype G have been reported in the United States and France. Genotype H has been described in Mexico.
Table 78-1 Hepatitis B Genotypes (A-H) and Their Possible Clinical Associations
| Geographic Distributions |
HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus.
Clinical associations appear to exist with the various genotypes (see Table 78-1). Currently, the strongest clinical associations appear to be that (1) HBeAg seroconversion occurs earlier in patients with genotype B than in those with genotype C and (2) response to therapy with interferon varies with genotype (see later).25 The viral genotype also has implications for the frequency of precore and core mutations (see later) and may have an effect on the frequency of HCC.
Mutations of the Hepatitis B Virus Genome
Hepatitis B Surface Antigen Mutants
Mutations in the surface gene can result in changes in the antibody-binding domain. Accordingly, both virus neutralization by polyclonal antibody to HBsAg and testing for HBsAg by methods that depend on antibody binding can be affected. Large-scale vaccination programs in regions endemic for HBV have revealed a 2% to 3% frequency of vaccine escape mutants that result from alterations in the “a” determinant of the HBsAg protein, which is the major neutralizable epitope. The typical mutation results in the substitution of glycine for arginine at amino acid position 145; this substitution prevents binding of neutralizing antibodies (i.e., antibody to HBsAg [anti-HBs]). The clinical significance of these mutants for neonatal vaccination programs is highly controversial because the frequency of these variants among HBV-infected mothers whose infants respond to vaccination has been found to be similar to that of mothers whose infants do not respond. The “a” determinant mutants also are proposed to have clinical relevance after liver transplantation for hepatitis B. As many as 50% of patients in whom recurrent HBV infection develops despite the use of HBIG have been shown to have these escape mutants, and the rate at which the mutations are detected appears to correlate with the length of time over which HBIG is repeatedly administered.26
Mutations in the Precore, Basal Core Promoter, and Core Genes
Mutations in the precore and basal core promoter regions of the HBV genome can influence the production of HBeAg. A precore mutation results in a stop codon at nucleotide 1896 that abolishes the synthesis of HBeAg,27 whereas mutations in the basal core promoter at nucleotides 1762 and 1764 decrease HBeAg synthesis by approximately 70% while maintaining pregenomic RNA levels.28 Both types of mutations have been observed in cases of severe or fulminant hepatitis, which has been attributed to the loss of the immune-tolerizing effects of HBeAg antigen (see later). The presence of core promoter mutations has been linked to a significantly increased risk of HCC.29 Precore and basal core promoter mutants have been described in the same patients and are particularly common in Asian and European patients with chronic hepatitis B.30 A large serosurvey of HBV carriers residing in the United States has found that precore and core promoter mutations are common (frequencies of 27% and 44%, respectively), depending on the ethnicity and places of birth of the patients. Both mutant forms of HBV were observed to occur far more commonly in HBeAg-negative patients (precore mutation in 38% of HBeAg-negative versus 9% of HBeAg-positive patients; core promoter mutation in 51% versus 36%).31 In addition to these mutations, upstream mutations in the core gene can influence immunologic responses to HBV. Core gene mutations have been shown to block recognition of HBV by cytotoxic T lymphocytes (CTLs), a key mode of viral clearance. Therefore, the mutations contribute to HBV immune escape and possibly influence the response to interferon.32 Core gene mutations within the immunodominant epitopes of the HBV nucleocapsid also can affect CD4+ T-cell reactivity.33
Hepatitis B Virus DNA Polymerase Mutants
The polymerase gene encodes a DNA polymerase enzyme needed for encapsidation of viral RNA into core particles, conversion of the pregenomic viral RNA into a negative strand of viral DNA (reverse transcription), and conversion of this first HBV DNA strand into a second DNA strand of positive polarity. In general, the HBV reverse transcriptase function of the polymerase gene is highly conserved because major mutations that impair the efficiency of viral replication lead to selection pressure against such variant forms. As indicated earlier, HBV has a high rate of replication (1011 virions per day) and low replication fidelity, meaning that it has a propensity to mispair nucleotide bases when it reverse transcribes viral RNA to DNA. HBV DNA polymerase also lacks any proofreading activity, so it cannot repair its mistakes. Therefore, when a nucleotide base is misplaced, it remains in the growing viral DNA strand as a base mutation, and the new HBV DNA molecule has a different sequence from the original (wild-type) genome. The overall error rate of HBV DNA polymerase is estimated to be 1 per 10,000 nucleotides copied, which translates to the potential for 10 million base-pair errors per day in an infected person. All possible single-base mutations can be produced in a 24-hour period, although many such mutations will yield nonviable viruses.34
Mutations in the sequence of HBV polymerase can lead to drug resistance to nucleoside analogs used to treat HBV infection because some HBV polymerase mutants have decreased susceptibility to these drugs and are selected during treatment. The mutations in the sequence of HBV DNA polymerase that confer drug resistance result in amino acid substitutions in the reverse transcriptase domain of the enzyme. The changes in the structure of the enzyme, in turn, are thought to sterically inhibit binding of the drugs to their active sites. The amino acids in the reverse transcriptase are numbered from 1 to 344, and an amino acid identity is given by the single letter amino acid code. By convention, substitutions in reverse transcriptase are designated by the wild-type amino acid, followed by the number of the amino acid, followed by the substituted amino acid. For example, for the nucleoside analog lamivudine (see later), two types of mutations occur at nucleotide position 204 of domain C (the catalytic site of the polymerase) that result in substitution of the amino acid methionine (M) for either isoleucine (I) or valine (V). These mutations are designated M204I and M204V, respectively, and are referred to collectively as YMDD mutants; the letters stand for the amino acids (Y = tyrosine, M = methionine, D = aspartate) in the C domain. The M204V mutation tends to occur in conjunction with a mutation in domain B that results in substitution of leucine (L) with methionine (L180M). The M204I mutation or the combined M204V-L180M mutations result in marked resistance to the effect of lamivudine (>10,000-fold reduction in susceptibility). After prolonged exposure to adefovir, drug resistant mutations in domains B and D are selected (A181V/T and N236T). Other nucleotide substitutions have been described that are instrumental for telbivudine and entecavir resistance (Fig. 78-3).

The inherent mutability of HBV indicates that single and even double polymerase mutants preexist as minor “quasispecies” even before treatment of HBV infection is begun. Because of the limitations in the sensitivities of current genotype assays, these mutants would not be detectable until they are selected and expanded under the pressure of drug treatment. Resistance to lamivudine is found in approximately 20% of patients after one year of treatment but in nearly 70% after five years (see Fig. 78-3).35 Rates of resistance to adefovir, a nucleotide analog, are 0% at one year and 29% after five years.36 The efficacy of both of these drugs against HBV is impaired by a single nucleotide substitution. The more mutations necessary for drug resistance (indicating a higher genetic barrier to resistance), the slower the emergence of and lower overall rate of resistance. For example, resistance to entecavir, another nucleoside analog, occurs in less than 1% of patients at five years because the preexistence of lamivudine-resistant mutations and one or more additional mutations in the viral polymerase gene are required for resistance.37 Persistent infection with drug-resistant HBV ultimately is associated with progression of disease and blunting of hepatic histologic improvement with antiviral therapy.38 Severe flares of hepatitis have also been reported after the emergence of drug-resistant mutants,39 and acquisition of these mutants may lead to rapidly progressive liver disease after liver transplantation (Table 78-2).40 Horizontal transmission of these mutants, which can complicate drug therapy in secondarily infected persons, is also possible.
Table 78-2 Hepatitis Flares in Patients with Chronic Hepatitis B
| CAUSE OF FLARES | COMMENT |
|---|---|
| Spontaneous | Factors that precipitate antecedent viral replication are unclear |
| Immunosuppressive therapy | Flares are often observed during withdrawal; requires preemptive antiviral therapy |
| Antiviral therapy for HBV | |
| Interferon | Flares are often observed during the second to third month; may herald virologic response |
| Lamivudine | |
| During treatment | Flares are no more common than with placebo |
| YMDD mutant | Can have severe consequences in patients with advanced liver disease |
| On withdrawal* | Flares are caused by rapid re-emergence of wild-type HBV; can have severe consequences in patients with advanced liver disease |
| HIV treatment | Flares can occur with HAART or with immune reconstitution; in addition, HBV increases the risk of antiretroviral drug hepatotoxicity |
| Genotypic variation | |
| Precore and core promoter mutants | Fluctuations in serum ALT levels are common with precore mutants |
| Superinfection with other hepatitis viruses | May be associated with suppression of HBV replication |
ALT, alanine aminotransferase; HIV, human immunodeficiency virus; HAART, highly active antiretroviral therapy; HBV, hepatitis B virus; YMDD, tyrosine-methionine-aspartate-aspartate.
PATHOGENESIS
HBV is generally not a cytopathic virus, and the severity of HBV-associated liver disease is considered to be related to the intensity of the host immunologic response to the virus. Whereas both humoral and cellular immune responses are needed for effective clearance of the virus, the cellular immune response appears to be the arm principally involved in the pathogenesis of disease. The immunologic response to HBV encompasses both an innate, or nonspecific, response (for example, natural killer cells and interferons) and an adaptive immune response, including antibodies to viral antigens, human leukocyte antigen (HLA) class II–restricted CD4+ T cells, and HLA class I–restricted CD8+ CTLs.41 Induction of the antigen-specific T-cell response is thought to occur in lymphoid organs, where the host T cells encounter viral peptide antigens (or epitopes) that are presented by antigen-presenting cells such as dendritic cells, B cells, and macrophages. This process results in the maturation and expansion of T cells that are specific for these viral epitopes and is followed by their migration to the liver, where they perform their effector function.
During acute HBV infection, most HBV DNA molecules are cleared rapidly from the liver via noncytopathic mechanisms mediated by cytokines that are released initially by cells of the innate immune system42 and later by liver-infiltrating HBV-specific CD8+ cells. Cell-mediated immune responses are efficient in self-limited infection because the responses are vigorous, multispecific, and oriented toward type 1 helper T (Th1) cells. Persons with chronic HBV infection, by contrast, exhibit infrequent, narrowly focused, and weak HBV-specific T-cell responses.43 In chronic hepatitis B, the majority of mononuclear cells in liver infiltrates of patients with chronic hepatitis B at any given time are non–antigen-specific.44
CD8+ CTLs are thought to contribute to the disease process in the liver and result in apoptosis of infected hepatocytes. To be recognized by the CD8+ CTLs, targeted hepatocytes must present viral epitopes as short peptides that have been endogenously processed and fit within the peptide-binding groove of the class I major histocompatibility complex (MHC) molecules.45 The binding of the CTL T-cell receptor (TCR) to the peptide-MHC complex on the hepatocyte surface can then result in the direct killing of the infected cell and release of potent antiviral cytokines by the activated CTL. Recognition by MHC class II–restricted CD4+ helper T cells requires the appropriate presentation of viral peptides in the context of class II MHC molecules. The CD4+ cells produce antiviral cytokines and provide help in neutralizing antibody production. Antibody neutralization limits intrahepatic spread of virus during primary infection and serves an important role in preventing reinfection.
NATURAL HISTORY
Four phases of HBV infection have been described: immune tolerance, immune clearance, the inactive carrier state, and reactivation (Fig. 78-4).
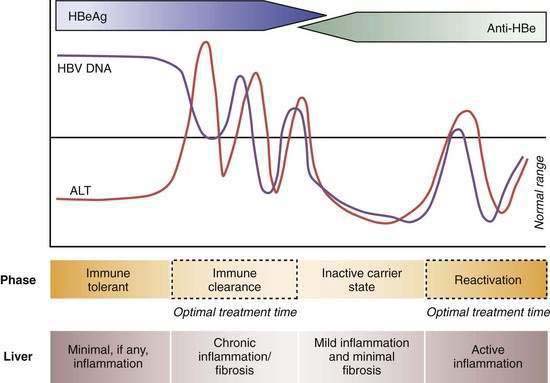
Patients who acquire the infection in the perinatal period often have high serum levels of HBV DNA without biochemical evidence of active hepatitis and are considered to be immune tolerant to HBV. When followed longitudinally, many of these patients ultimately exhibit elevated serum aminotransferase levels in association with histologic evidence of chronic hepatitis. The trigger mechanisms for this apparent change in tolerance are poorly understood but likely reflect changes in the immune reactivity of the host. Experiments in transgenic mice suggest that HBeAg induces a state of immunologic tolerance to HBV in neonates.46 Perinatal transmission of HBeAg has been considered to be a potential mechanism for the immune-tolerant state. As persons enter the immune clearance phase, HBV DNA concentrations diminish, serum alanine aminotransferase (ALT) levels rise, and hepatic histologic activity, reflecting immune-mediated lysis of infected hepatocytes, increases. The duration of this second phase varies, often lasting many years.
The third phase (inactive HBV carrier state) occurs after seroconversion from HBeAg to anti-HBe and is usually preceded by a marked reduction in serum HBV DNA to levels that are detectable only by PCR methodology, followed by normalization of serum ALT levels and resolution of liver necroinflammation. This phase may last a lifetime, but a proportion of patients ultimately undergo spontaneous or immunosuppression-mediated reactivation of HBV replication with reappearance of high levels of HBV DNA in serum, with or without HBeAg seroreversion and a rise in serum ALT levels. For unclear reasons, precore or core promoter mutants that prevent or down-regulate HBeAg production may be selected during or after HBeAg seroconversion (see earlier).47
A key event in the natural history of HBeAg-positive chronic hepatitis is seroconversion of HBeAg to anti-HBe, which is associated with marked reduction in HBV replication and biochemical and histologic remission in the majority of patients. Regression of liver fibrosis occurs gradually months to years after HBeAg seroconversion.48 Most studies have found that the mean annual rate of spontaneous HBeAg seroconversion ranges from 8% to 15% in HBV-infected children or adults with serum ALT elevations.
Longitudinal studies of untreated patients with predominantly HBeAg-positive chronic hepatitis B have shown that the frequency of development of cirrhosis ranges from 2 to 5 per 100 person-years and the five-year cumulative frequency of progression to cirrhosis from 8% to 20%.49 The rate of cirrhosis has been suggested to be higher in HBeAg-negative patients than in HBeAg-positive patients. Risk factors for the development of cirrhosis have been identified; of these, older age, the stage of fibrosis at presentation, and ongoing HBV replication with persistent or intermittent detection of HBV DNA by a non–PCR-based assay are perhaps the most important clinically. Combined infection with hepatitis D virus (HDV [see later]), HCV, or HIV and concomitant alcohol abuse have also been linked to a higher rate of development of cirrhosis.
When cirrhosis develops, two major complications may occur: hepatic decompensation and HCC. In a large European cohort with HBV-related compensated cirrhosis, the five-year cumulative frequency of hepatic decompensation was 16%, and the incidence per 100 person-years was 3.3.50 Similar rates have been reported in Asians. The cumulative five-year frequency of HCC can be as high as 14%.50 Factors associated with an increased risk of HCC include male gender, age more than 45 years, having a first-degree relative with HCC, the presence of cirrhosis, HBeAg positivity, and reversion from anti-HBe to HBeAg positivity.51 HCC can still develop in HBsAg-positive persons with none of the identified risk factors, but less frequently. In addition, HCC has been described in persons who lose HBsAg. Recommendations about ultrasonography and alpha fetoprotein screening for HCC are controversial, but in general, screening is recommended in all patients with cirrhosis and in male HBV carriers older than age 40 years in whom the likely route of transmission has been perinatal or early childhood exposure; some authorities recommend screening after age 30 years in highly viremic patients when perinatal acquisition is suspected (see Chapter 94).
ALT as a Surrogate Marker for Disease Activity
The serum ALT level has been used conventionally as a measure of disease activity in patients with chronic hepatitis B. Use of the standard reference range (0 to 40 U/L), however, can be misleading for evaluating HBV-related disease activity. A serum ALT level within the normal range is an imperfect surrogate marker for the absence of disease activity because determination of standard reference ranges has not traditionally taken into account increased body mass index (BMI), diabetes mellitus, and other features, such as alcohol intake, that tend to inflate values in a “normal” reference population (see Chapter 73). An insurance record-based study in Korea that included more than 140,000 persons who were followed for eight years demonstrated that all-cause liver-related mortality is increased when the serum ALT level exceeds 20 U/L in women and 30 U/L in men.52 This finding is particularly relevant to the management of Asians with hepatitis B and viremia. Because these patients tend to have a small body mass, a normal serum ALT value according to the standard laboratory reference range may be misleading. Such patients often have been excluded from treatment and assumed to be immune tolerant (that is, without liver disease). Studies in Asia and the United States have shown that as many as 20% to 30% of HBV carriers with persistently normal serum ALT levels and serum HBV DNA levels >104 copies/mL have stage 2 or greater (of 4) inflammation and stage 2 or greater (of 4) fibrosis on a liver biopsy specimen.53 Moreover, Asian HBV carriers with high-normal serum ALT levels (>0.5 × upper limit of normal [ULN]) have been shown to have more fibrosis on liver biopsy specimens than do those with low-normal serum ALT levels (<0.5 × ULN) and often demonstrate higher serum ALT elevations on prolonged follow-up.53–55 These data are consistent with the hypothesis that viremic HBV carriers who acquired HBV infection early in life and who have high-normal serum ALT levels may represent a subgroup of patients who have already entered the immune clearance phase and are not in the immune-tolerant phase. This occurrence should be suspected in particular in persons older than age 35 to 40 because immune tolerance often subsides after two to three decades of infection.54 Liver biopsy can be a useful tool to distinguish persons in the immune clearance phase despite normal or near-normal ALT levels but with active liver disease from those in the immune tolerant phase and absence of active liver disease (see Fig. 78-4).
HBV DNA Level and Long-Term Complications
Population-based Asian cohort studies have established that the serum HBV DNA level is the single best predictor of future progression to cirrhosis and HCC in HBV-infected persons.56,57 In a prospective cohort study, more than 3600 HBV carriers from Taiwan, of whom 60% were male, 70% were older than age 40, 85% were HBeAg negative, and 95% had normal serum ALT levels, were followed for a mean of 11 years. The calculated relative risks for cirrhosis and HCC were shown to correlate with the level of HBV DNA on entry into the study when compared with a reference population of HBV carriers in whom HBV DNA was undetectable in serum by a PCR assay.57 Even serum HBV DNA levels as low as 10,000 copies/mL (equivalent to 2000 IU/mL) were associated with a higher relative risk of cirrhosis and HCC. The relative risk of HCC was highest in persons with a serum HBV DNA level of more than 1 million copies/mL and intermediate in those in whom follow-up serum samples indicated spontaneous reduction of the serum HBV DNA level from greater than 100,000 copies/mL to less than 10,000 copies/mL. These data can be interpreted to mean that both the duration and level of viremia are important risk factors for the development of HCC. The data also suggest that suppression of serum HBV DNA levels, whether spontaneously or as a result of antiviral therapy, lowers the risk of HCC. The serum HBV DNA level remained a significant predictor of cirrhosis and HCC even after adjustment for patient age, gender, serum ALT level, and HBeAg status. Other studies in Chinese HBV carriers have demonstrated that persons in whom liver decompensation and HCC develop often have modest serum ALT elevations that are frequently below the recommended threshold (2 × ULN) for antiviral treatment in current practice guidelines (see later).58
On the basis of these data, some authorities have suggested that all persons who acquire HBV infection early in life and who are age 45 to 50 years or older and demonstrate serum HBV DNA levels of 100,000 or more copies/mL should receive long-term therapy with a nucleoside analog to prevent cirrhosis and HCC.59,60 In a landmark study,61 more than 600 Asian patients with advanced fibrosis and a serum HBV DNA level greater than 100,000 copies/mL were randomized in a ratio of 2 : 1 to active treatment with lamivudine or placebo. Treatment was planned for 5 years, but the study was discontinued after a mean duration of only 32 months because disease progression and HCC occurred significantly more frequently in the group of patients randomized to placebo.61 Therapeutic benefit was independent of the serum ALT level at baseline. These data strongly suggest that continuing suppression of serum HBV DNA by antiviral therapy can alter the long-term outcome of active chronic hepatitis B.
CLINICAL FEATURES
Acute Hepatitis B
The incubation period of acute hepatitis B varies from a few weeks to 6 months (average, 60 to 90 days), depending on the amount of replicating virus in the inoculum. The disease may be more severe in patients coinfected with other hepatitis viruses and in those with established underlying liver disease.62 Abstention from alcohol is usually recommended, but the chance of an uneventful recovery does not appear to be affected by the consumption of moderate amounts of alcohol (20 to 30 g daily) during the convalescent phase.63 Acute infections are heralded by a serum sickness–like prodrome of fever, arthralgia or arthritis, and rash, which is most commonly maculopapular or urticarial, in 10% to 20% of patients. This prodrome results from circulating HBsAg–anti-HBs complexes that activate complement and are deposited in the synovium and walls of cutaneous blood vessels.64 These features generally abate before the manifestations of liver disease and peak serum aminotransferase elevations are observed. Jaundice develops in only about 30% of patients.
Clinical symptoms and jaundice generally disappear after one to three months, but some patients have prolonged fatigue even after serum ALT levels return to normal. In general, elevated serum ALT levels and serum HBsAg titers decline and disappear together, and in approximately 80% of cases, HBsAg disappears by 12 weeks after the onset of illness.65 In 5% to 10% of cases, HBsAg is cleared early and is no longer detectable by the time the patient first presents to a health care provider. Persistence of HBsAg after six months implies development of a carrier state, with only a small likelihood of recovery during the next 6 to 12 months. Delayed clearance of HBsAg has been reported to be preceded by a decline in HBsAg titers.
After clinical recovery from acute hepatitis B and HBsAg seroconversion, HBV DNA often remains detectable in serum as determined by a PCR assay (see later discussion of diagnosis). After resolution of acute hepatitis, the numbers of HBV-specific CD4+ and CD8+ cells in blood and liver decrease rapidly. Nonetheless, T-cell responsiveness remains high on re-encounter with HBV antigens, indicating that traces of virus can maintain the CTL response indefinitely after clinical recovery, thereby exerting control over the virus and preventing reactivated infection.41,66
Fulminant hepatitis occurs in less than 1% of cases (see Chapter 93). Fulminant hepatitis B generally occurs within four weeks of the onset of symptoms and is associated with encephalopathy, multiorgan failure, and a high mortality rate (>80%) if not treated by liver transplantation. Patients older than age 40 years appear to be more susceptible than younger persons to “late-onset liver failure,” in which encephalopathy, renal dysfunction, and other extrahepatic complications of severe liver insufficiency become manifest over the course of several months. The pathogenic mechanisms of fulminant hepatitis are poorly understood but are presumed to involve massive immune-mediated lysis of infected hepatocytes. This proposed mechanism may explain why many patients with fulminant hepatitis B have no evidence of HBV replication in serum at presentation.
Chronic Hepatitis B
Physical findings may be normal, or hepatosplenomegaly may be found. In decompensated cirrhosis, spider angiomata, jaundice, ascites, and peripheral edema are common. Liver biochemical test results are usually completely normal during the inactive HBV carrier state. In contrast with patients in the immune-tolerant phase of HBV infection, most patients in the immune-clearance phase of chronic HBV infection have mild to moderate elevations in serum AST and ALT levels. During exacerbations of disease, serum ALT levels may be as high as 1000 U/L or more, and the clinical and laboratory picture is indistinguishable from that of acute hepatitis B, including the presence in serum of IgM anti-HBc. Progression to cirrhosis should be suspected whenever hypersplenism, hypoalbuminemia (in the absence of nephropathy), or prolongation of the prothrombin time is found. The serum AST level is typically higher than the serum ALT level in patients with advanced cirrhosis (see Chapter 73).
Extrahepatic Manifestations
Extrahepatic syndromes seen in association with acute or chronic hepatitis B are important to recognize because they may occur without clinically apparent liver disease and can be mistaken for independent disease processes in other organ systems. The pathogenesis of these extrahepatic disorders has not been fully elucidated but likely involves an aberrant immunologic response to extrahepatic viral proteins.67 Many of the extrahepatic manifestations (e.g., arthritis, dermatitis, glomerulonephritis, polyarteritis nodosa, cryoglobulinemia, papular acrodermatitis, and polymyalgia rheumatica) are observed in association with circulating immune complexes that activate serum complement. Antiviral therapy may be indicated for persistent symptoms.
Glomerulonephritis
Several types of glomerular lesions have been described in patients with chronic HBV infection; membranous glomerulonephritis and membranoproliferative glomerulonephritis are the most common.68 Renal biopsy specimens have demonstrated immune complex deposition and cytoplasmic inclusions in the glomerular basement membrane. The immune complexes activate complement and production of cytokines with a subsequent inflammatory response. Nephrotic syndrome is the most common presentation of HBV-associated glomerulonephritis. In affected children, renal failure at presentation is almost always mild, and a history of clinical liver disease is uncommon. Nevertheless, liver biopsy specimens almost always demonstrate varying degrees of chronic hepatitis. The diagnosis of HBV-associated glomerulonephropathy is usually established by serologic evidence of HBV antigens or antibodies, the presence of immune-complex glomerulonephritis in a renal biopsy specimen, and the demonstration of glomerular deposits of one or more HBV antigens, such as HBsAg, HBcAg, or HBeAg, by immunohistochemistry. Most patients have detectable HBeAg in serum and, in addition, demonstrate low serum C3 and occasionally low C4 levels. The renal disease typically resolves in months to several years, especially in children. Often, resolution occurs in conjunction with HBeAg seroconversion. Rarely, however, renal failure may ensue. The natural history of HBV-related glomerulonephritis in adults has not been well defined, but several reports suggest that glomerular disease is often slowly and relentlessly progressive.69 Successful treatment has been accomplished with interferon alpha and has been linked to long-term control of HBV replication.70 Therapy with nucleoside analogs has resulted in improved renal function and diminished proteinuria.
Cryoglobulinemia
Type II cryoglobulins consist of a polyclonal IgG and monoclonal IgM, whereas type III cryoglobulins contain polyclonal IgG and rheumatoid factor. Type II and type III cryoglobulinemia have been associated with hepatitis B, but the association is uncommon. In a large patient cohort, the frequency of cryoglobulinemia was significantly higher in patients with chronic HCV infection (54%) than in patients with chronic HBV infection (15%) (see Chapter 79). Cryoglobulinemia may be associated with systemic vasculitis (purpura, arthralgias, peripheral neuropathy, and glomerulonephritis) but is often paucisymptomatic or asymptomatic. Interferon has been used successfully to treat symptomatic cryoglobulinemia in association with chronic hepatitis B. Experience with nucleoside analog therapy has not been reported.
Histopathologic Features
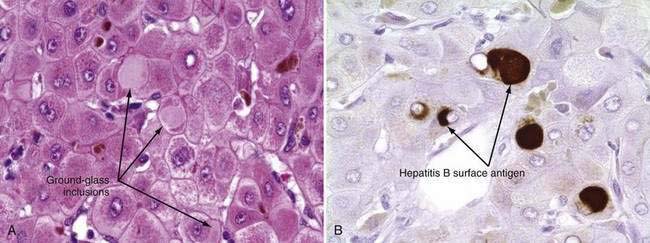
The only histologic feature noted on routine light microscopy that is specific for chronic hepatitis B is the presence of ground-glass hepatocytes (Fig. 78-5). This morphologic finding results from accumulation of HBsAg particles (20 to 30 nm in diameter) in the dilated endoplasmic reticulum. Because of high levels of cysteine in HBsAg, ground-glass cells have a high affinity for certain dyes, such as orcein, Victoria blue, and aldehyde fuchsin. Ground-glass hepatocytes also may be seen in HBV carriers, in whom they may be detected in up to 5% of cells. When present in abundance, ground-glass hepatocytes often indicate active viral replication.71 Immunofluorescence and electron microscopic studies have shown HBcAg inside the hepatocyte nuclei of affected cells. During periods of intense hepatitis activity, cytoplasmic core antigen staining is generally observed. After successful treatment of HBV infection with a nucleoside analog, the cytoplasmic core antigen staining often disappears, but nuclear core antigen staining may remain, indicating persistence of the HBV cccDNA template.
Acute Flares in Chronic Hepatitis B
Chronic hepatitis B is often punctuated by sudden flares of disease activity that are reflected by a rise in serum aminotransferase levels. Although a uniform definition is lacking, a flare has frequently been described as a rise in serum ALT levels to at least two times the baseline value. Spontaneous flares are an important part of the natural history of hepatitis B because when they occur repeatedly, they lead to histologic progression. Acute flares in chronic hepatitis B occur in association with a number of circumstances and clinical situations (see Table 78-2). Most flares result from a change in the balance between immunologic responses to HBV and the level of viral proliferation. Acute flares in chronic hepatitis B that are not explainable by infection with other hepatotropic viruses often occur as a secondary response to increased levels of replicating wild-type or mutant HBV or as a result of therapeutic intervention with immunologic modifiers such as interferon, glucocorticoids, and cancer chemotherapy. In some instances, the event that initiates an acute exacerbation of chronic hepatitis B may not be readily identifiable, and the flare is considered spontaneous.
Spontaneous Flares
Spontaneous exacerbations of chronic hepatitis B often result from reactivated infection, and an increase in serum HBV DNA levels often precedes an increase in serum aminotransferase levels. Histologic evidence of acute lobular hepatitis superimposed on the changes of chronic viral hepatitis is frequently observed during these flares.72 IgM anti-HBc, a marker that is often diagnostic of acute viral hepatitis, may also appear in serum at this time.73
The reasons for reactivated infection are unknown but likely relate to subtle changes in the immunologic control of viral replication. Reactivation seems to occur more commonly in persons who are infected with HIV.74 In persons who acquire HBV infection early in life, flares become more common during adulthood, presumably because of a breakdown in immune tolerance to HBV.75
Flares also can occur in patients who are in the replicative phase of infection (i.e., already positive for HBV DNA and HBeAg in serum). In these instances, HBV replication intensifies, serum HBV DNA levels rise, and liver biochemical deterioration occurs, often without the subsequent loss of HBeAg. Multiple episodes of reactivation and remission have been shown to accelerate the progression of chronic hepatitis B and are particularly likely to occur in patients infected with the precore mutant form of chronic hepatitis B (see earlier).47
Immunosuppressive Therapy-Induced Flares
Reactivation of HBV replication is a well-recognized complication in patients with chronic HBV infection who receive cytotoxic or immunosuppressive therapy.76 Suppression of the normal immunologic responses to HBV leads to enhanced viral replication and is thought to result in widespread infection of hepatocytes by HBV. On discontinuation of immunosuppressive medications, such as cancer chemotherapy, antirejection drugs, and glucocorticoid therapy, immune competence is restored and infected hepatocytes are rapidly destroyed. The more potent the immunosuppression, the higher the level of viral replication and, thus, the greater the potential for serious clinical consequences of sudden withdrawal of the therapy and restoration of immunologic competence. Postmortem studies of liver tissue from patients with severe liver injury have documented sparse staining of viral antigens, suggesting that the patients were in an active state of immune clearance.77
The vast majority of patients who experience immunosuppressive therapy–induced flares have been positive for HBsAg in serum before treatment, but some studies have described the reappearance of HBsAg in patients who were initially positive for anti-HBs, anti-HBc, or both.78 Reactivated hepatitis in patients who are negative for HBsAg and positive for either anti-HBc or anti-HBs is explainable by the possible latency of HBV in liver and mononuclear cells and the large extrahepatic reservoir of HBV. Chemotherapy given to patients with cancer who are HBV carriers is associated with an increased risk of liver-related morbidity and mortality.79
Reactivated hepatitis B also occurs in patients who are given immunosuppressive medications to prevent organ transplant rejection. The frequency of reactivated hepatitis appears to be particularly high in patients who undergo bone marrow transplantation because of extensive immunologic conditioning before transplantation and treatment of graft-versus-host disease.80 Rarely, fibrosing cholestatic hepatitis, a rapidly progressive form of liver injury associated with inordinately high levels of HBsAg and HBcAg in liver tissue, may develop in such patients.81
Acute flares of hepatitis B resulting from cancer chemotherapy and other immunosuppressive drugs are often detected after substantial increases in serum aminotransferase levels have been noted. Initiation of antiviral treatment after detection of such biochemical abnormalities has little effect on reducing liver injury because much of the immunologic response to HBV and viral elimination has already occurred. Instead, the key to management lies in anticipating the occurrence of a flare, initiating antiviral treatment preemptively (e.g., 4 to 6 weeks before the start of chemotherapy), and continuing the treatment for 6 to 12 months after completion of chemotherapy.82
Antiviral Therapy–Induced Flares
Interferon
Interferon-induced flares of chronic hepatitis B occur in approximately one third of treated patients and result from the immunostimulatory properties of the drug. Flares generally occur during the second or third month of treatment with conventional preparations of interferon. Flares also occur in patients treated with pegylated interferon and have been reported to occur more frequently in patients infected with HBV genotype A than with other genotypes. This finding may explain the higher rate of sustained virologic response and HBsAg clearance in patients infected with HBV genotype A.83 Serum ALT flares have been shown to be a predictor of sustained virologic response, particularly in patients with high levels of viremia.84 Flares tend to be particularly common in patients who have decompensated liver disease, with rates as high as 50% reported in one series.85 Such flares are frequently associated with clinical deterioration in the patient.
Nucleoside Analogs
Serum ALT flares occur in approximately 20% to 25% of patients after withdrawal of nucleoside analogs such as lamivudine and adefovir. These flares probably are caused by rapid resurgence of wild-type HBV, and although generally well tolerated, they have been associated with serious clinical exacerbations in patients with advanced liver disease.86 Reinstitution of therapy is often associated with a decline in serum HBV DNA and aminotransferase levels. Flares have been seen to follow the emergence of YMDD mutant HBV during therapy with lamivudine (see earlier).87 Initial reports emphasized the temporal occurrence of these flares at the time of or shortly after detection of lamivudine resistance. Further follow-up of patients with lamivudine-resistant HBV mutants, however, has indicated that the frequency of moderate or severe serum ALT flares (defined as >5 and >10 × UNL, respectively) increases with time after detection of lamivudine resistance. In one long-term study, the cumulative frequencies of such ALT flares were as follows: 24% at less than one year, 29% at one to two years, 30% at two to three years, 37% at three to four years, and 61% at more than four years after detection of lamivudine resistance.88
Glucocorticoid Withdrawal
Serum ALT levels increase, often with an inverse decline in HBsAg concentration and HBV DNA level, after withdrawal of glucocorticoids in patients with chronic HBV infection.89 In clinical trials of HBV therapy, a short course of glucocorticoid therapy given before conventional antiviral therapy was reported to enhance virologic response rates.90,91 The immune rebound following withdrawal of glucocorticoid therapy after a four- to eight-week course may result from increased activation of lymphocytes that promote Th1 cytokine responses at a time when viral antigen expression is increased. Serious hepatic decompensation has been reported in patients with advanced disease, however, and this therapeutic approach is no longer used.
Antiretroviral Therapy
Serum ALT flares occur in patients coinfected with HIV and HBV who receive highly active antiretroviral therapy (HAART).92 A number of potential causes have been identified. Lamivudine resistance and withdrawal may be associated with ALT flares. HBV infection raises the risk of toxicity from antiretroviral therapy, usually within six months after the initiation of treatment. Immune reconstitution resulting from HAART may also be associated with ALT flares. Affected patients may also be particularly susceptible to flares because of infection with other hepatitis viruses.
Flares Associated with Genotypic Variation
Chronic infection with precore mutant HBV (referred to as HBeAg-negative chronic hepatitis B) often is associated with multiple flares of liver cell necrosis interspersed with periods of normal serum ALT and low serum HBV DNA levels.47 Approximately 45% of patients have episodic serum ALT flares with normal levels between episodes, and 20% have flares superimposed on persistent ALT elevations.93 These flares have been attributed to rises in the concentration of precore mutants in the liver and changes in the ratio of concentrations of precore to wild-type HBV.
Mutations at the basal core promoter region of the HBV genome are associated with greater HBeAg synthesis, histologic evidence of liver inflammation, and increased viral replication.94 Multiple exacerbations of hepatitis resulting from reactivated HBV infection have been described in patients with basal core promoter mutations, either alone or in association with precore mutations. HBeAg-negative patients who have both precore and core promoter mutants may be particularly predisposed to episodes of severe reactivation after cancer chemotherapy.95
Flares Caused by Infection with Other Viruses
Patients with chronic HBV infection may exhibit severe flares in serum aminotransferase levels and even frank liver failure when superinfected with other hepatotropic viruses, such as hepatitis A virus (HAV), HCV, and HDV. Increased mortality has been reported when HDV superinfection is superimposed on chronic hepatitis B, and chronic HDV infection is often associated with multiple fluctuations in serum aminotransferase levels (see later discussion of HDV).96 Acute hepatitis C superimposed on chronic hepatitis B has been reported to be as severe as HDV superinfection and has been associated with a high rate of liver failure (34%) and death (10%).97 A cumulative frequency of cirrhosis and HCC that is higher than that attributable to chronic HDV infection or chronic HBV infection alone has been demonstrated. Acute hepatitis C often leads to chronic HCV infection, and the subsequent course also may be characterized by frequent fluctuations in serum aminotransferase levels.
DIAGNOSIS
The disappearance of HBsAg is followed several weeks later by the appearance of anti-HBs. In most patients, anti-HBs persists for life and provides long-term immunity. In some patients, anti-HBs may not become detectable after disappearance of HBsAg, but these patients do not appear to be susceptible to recurrent infection.98 Anti-HBs may not be detectable during a window period of several weeks to months after the disappearance of HBsAg. During this period, the diagnosis of acute HBV infection is made by the detection of IgM anti-HBc in serum.99
Coexistence of HBsAg and anti-HBs in serum has been reported in approximately 25% of HBsAg-positive persons and occurs more commonly in persons with chronic hepatitis B than in those with acute hepatitis B.100 In most instances, the anti-HBs is present in a low level, non-neutralizing, and heterotypic—that is, directed against a subtype of HBsAg different from the subtype present in the infected patient. The mechanisms behind this finding are not clear but relate to antibody formed against minor variants of the HBsAg protein. The presence of these heterotypic antibodies is not associated with specific risk factors or changes in clinical course and may occur in patients with or without active liver disease and viral replication.
In areas where HBV is not endemic, isolated anti-HBc in serum has been detected in 1% to 4% of the general population. Isolated reactivity for anti-HBc may occur in the following situations: (1) during the window period of acute hepatitis B, when anti-HBc is predominantly of the IgM class; (2) many years after recovery from acute hepatitis B, when anti-HBs has fallen to undetectable levels; (3) as a false-positive serologic test result; (4) after many years of chronic HBV infection, when the HBsAg titer has fallen below the level of detection; (5) in HBV-infected persons who are coinfected with HCV; and (6) rarely, as a result of varying sensitivity of HBsAg assays.101 Evidence for coinfection with HCV has been demonstrated in as many as 60% of persons in whom anti-HBc is the only marker of HBV.102
Results of PCR testing of sera have shown that 0% to 30% of patients with isolated anti-HBc have HBV DNA in serum. Usually, the HBV DNA is detectable at a low level and not by standard hybridization assays, which are less sensitive than PCR assays.103 The presence of low-level viremia in these HBsAg-negative subjects has clinical implications with regard to potential infectivity. For example, in the past, anti-HBc testing of blood donors prevented some cases of post-transfusion hepatitis B.104 Also, the risk of transmission of HBV infection from a liver donor with isolated anti-HBc has been found to be as high as 50% to 70% in some series; lower rates of transmission have been observed in other forms of solid organ transplantation.105
Low-level viral replication also has implications with regard to the possibility of underlying liver disease. HBV DNA in serum and liver tissue has been confirmed by PCR methodology in some HBsAg-negative patients with cirrhosis and HCC and in some patients with fulminant non-A, non-B, non-C hepatitis as defined by conventional serologic testing.106
HBeAg is a soluble viral protein that is found in serum early during acute HBV infection. HBeAg reactivity usually disappears at or soon after the peak in serum aminotransferase levels, and persistence of HBeAg three or more months after the onset of illness indicates a high likelihood of transition to chronic HBV infection. The finding of HBeAg in the serum of an HBV carrier indicates greater infectivity, a high level of viral replication, and the need for antiviral therapy. With a commercially available PCR assay, nearly 90% of patients with HBeAg-positive chronic hepatitis B were found to have serum HBV DNA levels persistently above 105 copies/mL, with a mean value of 8.37 log10 (>108) copies/mL.107 By contrast, anti-HBe–positive patients had much lower serum HBV DNA levels; higher values were found in those with persistently or intermittently elevated serum ALT levels (mean of 5.1 log10 copies/mL) than in those with persistently normal ALT levels (3.10 log10 copies/mL).
Most HBeAg-positive patients have active liver disease; the exceptions are HBeAg-positive children and young adults with perinatally acquired HBV infection, who usually have normal serum ALT levels and minimal inflammation of the liver.14 In general, seroconversion from HBeAg to anti-HBe is associated with a reduction in serum HBV DNA levels of 3 log10 copies/mL or greater and remission of liver disease. Some patients, however, continue to have active liver disease and detectable HBV DNA in serum because of low levels of wild-type virus or the selection of precore or core promoter mutations that impair HBeAg secretion.
The measurement of serum HBV DNA is commonly used to evaluate a patient’s candidacy for antiviral therapy and to monitor response during treatment. Patients with a high serum HBV DNA level at baseline less commonly respond to therapy with conventional interferon than patients with a low level.108 With the use of solution hybridization testing, a baseline HBV DNA level of 200 pg/mL (roughly equivalent to 56 million copies/mL on a PCR assay) or greater has been found to be associated with a low rate of response to standard interferon. By contrast, baseline serum HBV DNA levels have not been shown to correlate with response to nucleoside analog therapy because of the more potent inhibition of viral replication by these agents. Monitoring of HBV DNA levels at key intervals during therapy allows one to predict the likelihood of HBeAg clearance. Several studies have found that the level of serum HBV DNA at 12 weeks of nucleoside analog treatment may help predict the likelihood of HBeAg seroconversion.109,110 Other studies have suggested that measuring the HBV DNA level at baseline or during treatment can be used to evaluate the likelihood of relapse after treatment is discontinued and development of resistance to lamivudine.111,112 Reappearance of HBV DNA in serum during treatment suggests that drug resistance has occurred,113 and high pretreatment levels of serum HBV DNA have been shown to correlate with a higher rate of recurrent HBV infection in liver transplant recipients who are treated with lamivudine.114
Qualitative PCR is an even more sensitive method of detecting HBV DNA than quantitative PCR. Use of qualitative PCR has altered traditional concepts about the clearance of HBV DNA from serum in acute and chronic HBV infection. Small amounts of HBV DNA can be detected in serum and peripheral mononuclear cells years after recovery from acute hepatitis B.66 Even after disappearance of HBsAg and apparent loss of HBV DNA from serum in patients with chronic hepatitis B, small amounts of HBV DNA persist in liver tissue and peripheral mononuclear cells years later.9 Detection of HBV DNA in serum by a qualitative PCR assay before liver transplantation may identify patients who are at increased risk of apparent de novo hepatitis after transplantation and may pinpoint HBV as the cause of liver disease in HBsAg-negative patients.115,116 Finally, detection of minute amounts of HBV DNA may be particularly important in diagnosing patients with fulminant hepatitis B—who frequently have cleared HBsAg by the time they seek medical attention.117
TREATMENT
Virologic Endpoints and Definitions of Response
The primary goal of treatment for chronic hepatitis B is durable suppression of serum HBV DNA to levels below those associated with liver disease. This goal can be accomplished with either interferon alpha or nucleoside analogs. The level at which serum HBV DNA is suppressed adequately is generally considered to be less than 105 copies/mL (<20,000 IU/mL) for patients with HBeAg-positive chronic hepatitis B and often lower for those with HBeAg-negative hepatitis.14 Definitions of response vary, but the most important clinically is a lasting or durable suppression of serum HBV DNA when the patient is no longer receiving treatment (Table 78-3). HBeAg seroconversion (loss of HBeAg and appearance of anti-HBe in serum) is an additional endpoint that can be used to determine the appropriate length of treatment with a nucleoside analog.
Table 78-3 Treatment of Chronic Hepatitis B: Definitions of Response to Antiviral Therapy
| Virologic response | Decrease in serum HBV DNA level to <105 copies/mL or <20,000 IU/mL in HBeAg-positive cases and <104 copies/mL or <2000 IU/mL in HBeAg-negative cases |
| Loss of HBeAg with or without seroconversion to anti-HBe* | |
| Biochemical response | Normalization of serum ALT levels |
| On-treatment response | |
| Initial response | Suppression of HBV DNA levels to <104-5 copies/mL with or without loss of HBeAg, in addition to normalization of serum ALT levels |
| Maintained response | Requiring continuation of therapy |
| Off-treatment response | |
| Sustained response | Virologic and biochemical response observed for 6-12 months after treatment is discontinued |
| Durable response | Indefinite virologic and biochemical response after treatment is discontinued |
ALT, alanine aminotransferase; HBeAg, hepatitis B e antigen; HBV, hepatitis B virus.
* Pertains to HBeAg-positive patients only.
Although most experts agree that achieving HBsAg seroconversion is more desirable than HBV suppression, the former occurs so infrequently with current antiviral therapies that it is considered an impractical endpoint. Even so, a systematic review of clinical trials with interferon alpha indicated that early HBsAg seroconversion occurs significantly more frequently in treated than in nontreated patients. In a meta-analysis of 16 randomized, controlled trials, loss of HBsAg from serum occurred six times as frequently in interferon-treated patients as in nontreated patients.118 Long-term follow-up (mean, 6.2 years; range, 1 to 11 years) of HBeAg-positive patients treated with standard interferon alpha demonstrated that 71% of sustained responders became HBsAg negative.119 By contrast, a one-year course of lamivudine or adefovir does not result in a higher rate of HBsAg seroconversion than does placebo, and the frequency of HBsAg seroconversion with prolonged therapy has yet to be evaluated extensively. The observation that early HBsAg seroconversion occurs more often with interferon than with nucleoside analogs emphasizes that the mechanisms of action of the two treatments differ fundamentally and provides a rationale for the use of combination therapy using both types of drugs (see sections on the individual agents and combination therapy).
Factors Involved in the Choice of Agents
In deciding on the appropriate type of therapy for patients with chronic hepatitis B, the physician should consider the serum ALT level, serum HBV DNA level, and liver histology at baseline as well as the expense of treatment, potential for and ability of the patient to withstand adverse effects, age and other comorbid conditions of the patient, and realistic expectations about the need for monitoring. Interferon and nucleoside analogs each have advantages and disadvantages, and no one therapy is suitable for all patients (Table 78-4). One major advantage of therapy with interferon is that it tends to be time limited, in that durable responses do not require maintenance therapy. By contrast, prolonged treatment with a nucleoside analog is often necessary to maintain viral suppression.
Table 78-4 Advantages and Disadvantages of Currently Available Antiviral Agents
| AGENT(S) | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Peginterferon | Finite duration of treatment | Given by injection |
| Durable off-treatment response | Frequent side effects | |
| Disappearance of HBsAg (5%-8%) | Expensive | |
| Low response rate patients with a high level of viremia | ||
| Nucleos(t)ide analogs | Negligible side effects | Oral delivery |
| Potent inhibition of virus replication | Drug resistance | |
| Less expensive than interferon | Long or indefinite treatment duration | |
| Low rate of HBsAg disappearance | ||
| Moderately expensive when given long term* |
HBsAg, hepatitis B surface antigen.
* Average retail price is approximately $200-$700 (USD) per month, depending on drug.
Guidelines for the Management of Hepatitis B
Consensus guidelines for the treatment of hepatitis B have been published by the American Association for the Study of Liver Diseases (AASLD), Asian-Pacific Association for the Study of the Liver, and European Association for the Study of the Liver.120–122 In general, the three sets of guidelines are quite similar. As innovations in medical therapy are made, the guidelines are updated, and differences among the guidelines represent, in part, differences in the availability of the various therapeutic agents around the world as well as unavoidable delays in publication that prevent the incorporation of new data. Because the practice guidelines of the liver societies are evidence based, recommendations have not been made about combination therapy with more than one nucleoside analog or interferon plus a nucleoside analog. In fact, combination therapy has not been shown in the available clinical trials to have additional therapeutic benefit (see later). The three guidelines have changed as new information has become available.
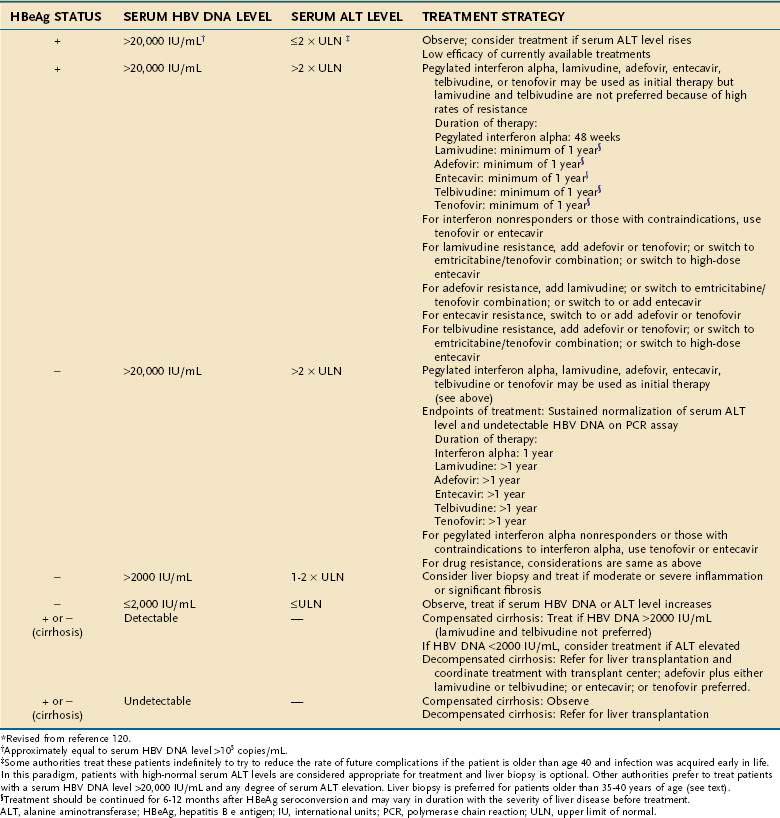
The recommendations made in the three sets of published guidelines have many similarities. In general, the published guidelines recommend treatment of persons who have both biochemical evidence of liver injury and serum HBV DNA levels in excess of 20,000 IU/mL, or roughly 100,000 copies/mL.123 Nucleoside analog therapy is recommended specifically in patients with decompensated cirrhosis. Emphasis also is given to the treatment of patients with serum ALT levels that are at least double the upper limit of normal (Table 78-5), although some experts disagree with setting arbitrary serum ALT and HBV DNA thresholds.59,123 This recommendation is based on the observation that rates of sustained virologic response in patients with minimal pretreatment serum ALT elevations are low with either interferon or nucleoside analogs.108,124 All the guidelines indicate that treatment decisions ideally should be made in the context of liver histologic findings and that treatment should be directed preferentially to patients with histologically moderate to severe hepatitis, although currently treatment decisions are not based on specific grading or staging of liver histology.
Antiviral Agents
Interferons
Interferon alpha
Interferon alpha was licensed for the treatment of chronic hepatitis B in 1992. Interferon is effective after a relatively short course of treatment (four months to one year) and, unlike the nucleoside analogs, has not been associated with drug resistance. Also, unlike nucleoside analogs, interferon has direct immunomodulatory properties. Interferon enhances HLA class I antigen expression on the surface of infected hepatocytes and augments CD8+ CTL activity. These effects could be important mechanistically in reducing the amount of the HBV cccDNA (the genomic template for viral transcription) and may thereby explain the loss of HBsAg that occurs in approximately 5% to 8% of interferon-treated patients. The major disadvantages of interferon relate to its poorer acceptance because of side effects (see Chapter 79) and lower level of HBV DNA suppression.
Flares of serum ALT have been described during therapy with interferon alpha, and although these flares are potentially important in achieving a virologic response, they occur unpredictably and are inconsistently associated with antiviral efficacy. The magnitude of an ALT flare has been shown to predict the likelihood of a sustained virologic response in patients with high levels of viremia, suggesting that vigorous cell-mediated immune responses often are required to overcome high levels of viral replication.84
Pegylated Interferon Alpha
Pegylated interferon has been found to be more potent than conventional or standard interferon and is currently licensed in more than 75 countries.125 Doses of 1.0 µg per kg of body weight of peginterferon alfa-2b and 180 µg of peginterferon alfa-2a given once weekly have been studied in clinical trials.126–128 No data are available for judging whether the increased effectiveness of pegylated interferon is primarily a function of a more pronounced effect on viral replication or of greater immunomodulatory action.
Viral genotype appears to affect the response to interferon. In a report from Taiwan, patients with genotype-B, HBeAg-positive chronic hepatitis B were found to have a response to conventional interferon more frequently than patients with genotype-C chronic hepatitis B.129 A relationship between virologic response and genotype was reaffirmed in a large multicenter study of peginterferon alfa-2b. In this study, HBeAg-positive patients infected with HBV genotype A demonstrated HBeAg loss more frequently than those with genotypes B, C, and D (47% vs. 44%, 28%, and 25%, respectively).126 A subsequent follow-up study of this cohort after a mean interval of three years demonstrated that patients infected with HBV genotypes A or B had the highest rates of durable virologic response (96% and 86%, respectively) and the highest rate of HBsAg clearance (58% and 14%, respectively).130 By contrast, rates for patients infected with HBV genotype D were 76% and 6%, respectively. These results confirm and extend those of earlier studies in HBeAg-positive patients that suggested that patients infected with HBV genotype A respond more frequently than those infected with genotype D. The relationship between sustained virologic response and HBV genotype is less clear in patients with HBeAg-negative hepatitis B, but a systematic analysis of more than 500 patients treated with either conventional or pegylated interferon alpha found that patients infected with HBV genotype C had the highest rates of sustained virologic and biochemical response and those infected with HBV genotype D had the lowest rates (50% and 21%, respectively).131 The effect that genotype exerts on the response to interferon could be particularly relevant to the treatment of North American patients with chronic HBV infection, in light of the influx of Asian HBV carriers infected with HBV genotypes B and C beginning in the last decades of the 20th century.
Nucleoside and Nucleotide Analogs
Nucleoside and nucleotide analogs replace natural nucleosides during the synthesis of the first or second strand (or both) of HBV DNA. They thus serve as competitive inhibitors of the viral reverse transcriptase and DNA polymerase. Because nucleos(t)ide analogs partially suppress viral replication, they have to be given for more than one year in most cases to achieve maximal efficacy. Unfortunately, drug resistance occurs with prolonged monotherapy. Figure 78-3 illustrates the common HBV nucleotide substitutions associated with drug resistance and the potential for cross resistance. Nucleos(t)ide analogs have several other limitations as well. With these agents, demonstrating the clearance of HBV cccDNA has been difficult, and in contrast to treatment with interferon, HBsAg clearance rarely occurs after one year of treatment with nucleoside analogs. These problems may result, in part, from the fact that these agents, unlike interferon, do not have a direct, enhancing effect on the immunologic response to HBV.78 Also, as indicated earlier, postwithdrawal serum ALT flares have been seen in approximately 25% of cases after discontinuation of nucleoside analog therapy.
Lamivudine
The approval of the nucleoside analog lamivudine in 1998 was a major breakthrough in the treatment of hepatitis B. The drug has been shown to be a relatively potent inhibitor of viral replication, convenient to administer, and free of severe adverse effects. Clinical trials demonstrated that a one-year course of lamivudine resulted in suppression of viral replication and improvement in histologic findings in the liver.132,133 In one study, HBeAg loss and HBeAg seroconversion occurred in 32% and 17% of patients, respectively.133 A two-year course of lamivudine proved to be more effective, resulting in an increase in the rate of HBeAg seroconversion from 17% at one year to 27% at two years.134 Prolongation of treatment beyond one year, however, has been associated with incremental changes in viral resistance (38% at two years), and the longer treatment is continued, the more frequently resistance is seen (65% at year five).88 Resistance is even more commonly encountered (90% at four years) in patients coinfected with HIV because of the early use of lamivudine in HAART regimens.92 Lamivudine resistance for more than two years has been associated with a blunted histologic response, and patients in whom lamivudine resistance has developed experience more hepatitis flares.38,88 For these reasons, the drug is no longer recommended as first-line therapy. Fortunately, a number of alternative nucleos(t)ide analogs have considerably lower resistance profiles.
Adefovir Dipivoxil
Adefovir dipivoxil is the acyclic phosphonate nucleotide analog of adenosine monophosphate. The drug was approved in 2002 for the treatment of HBeAg-positive and HBeAg-negative chronic hepatitis B on the basis of the findings of randomized, controlled trials in the United States, Europe, and Asia.135,136 In these pivotal studies, treatment with adefovir for 48 weeks resulted in median serum HBV DNA reductions of 3.52 log10 and 3.91 log10 copies/mL in HBeAg-positive and HBeAg-negative patients, respectively. The rates of HBeAg loss and HBeAg seroconversion were slightly lower than those achieved with lamivudine for 52 weeks. A rise in the frequency of HBeAg seroconversion and nondetectability of HBV DNA by PCR methodology has been observed during the second year of adefovir treatment.137 The level of HBV DNA suppression has been the same irrespective of viral genotype.138
Although the extent of viral suppression is only 0.5 to 1.0 log value less with adefovir than with lamivudine, the two drugs differ greatly in their resistance profiles. Point mutations (A181V, N236T) in the B and D domains, respectively, of the HBV polymerase gene that affect HBV susceptibility to adefovir occur in only 3% of patients after two years of treatment but increase thereafter, with rates of 6% to 8% at three years, 15% to 18% at four years, and 20% to 29% at five years.139–143 HBV isolates with the N236T mutation have remained susceptible to lamivudine and appear to be sensitive to entecavir and telbivudine in vitro (see later).144
Adefovir has been shown to be clinically and virologically effective in patients with lamivudine-resistant HBV, whether they have clinically stable disease, decompensated cirrhosis, or recurrent hepatitis B after liver transplantation.145,146 Adefovir-resistant mutants, on the other hand, remain susceptible to lamivudine, and adefovir resistance has been shown to occur more frequently when lamivudine is discontinued in lamivudine-resistant patients than when lamivudine is continued.147,148 Therefore, most experts choose to continue lamivudine when starting adefovir therapy in patients who have become resistant to lamivudine. The sooner resistance to lamivudine is appreciated, the more rapid and complete the virologic response to the addition of adefovir.149 Adefovir has the disadvantage of potential nephrotoxicity, and dose reductions may be necessary in patients with or at risk of compromised renal function.146 Adefovir is still used frequently, but incomplete viral suppression occurs in 30% of patients, particularly those with high viral levels at the initiation of treatment. Therefore, adefovir is being replaced as first-line therapy with more potent drugs such as entecavir and tenofovir (see later).
Entecavir
Entecavir is a deoxyguanine nucleoside analog that inhibits HBV replication selectively. The drug blocks HBV replication by inhibiting the priming of HBV DNA polymerase and the synthesis of the first and second strands of HBV DNA. Entecavir was approved by the U.S. Food and Drug Administration (FDA) in 2005 on the basis of registration trials that demonstrated no resistance after one year of treatment and improved virologic efficacy when compared with lamivudine.150–151 Entecavir is effective against both wild-type and lamivudine-resistant HBV. In phase 3 clinical trials, an entecavir dose of 0.5 mg was used to treat HBeAg-positive and HBeAg-negative patients who were previously untreated with a nucleoside analog, whereas 1 mg was used in patients who were resistant to lamivudine. Entecavir is more potent than either lamivudine or adefovir and results in nondetectable HBV DNA in 67% and 90% of previously untreated (“treatment-naïve”) HBeAg-positive and HBeAg-negative patients, respectively, after one year. In HBeAg-positive patients treated for an additional year, the cumulative rate of HBV DNA negativity by a PCR assay was 80% for entecavir-treated patients compared with 37% for lamivudine-treated patients.152 Entecavir resistance is rare (approximately 1% at five years) in treatment-naïve patients but is common in patients with prior lamivudine resistance. Virologic “rebound” as a result of resistance to entecavir has been demonstrated to occur in 1% of lamivudine-refractory patients after one year of treatment and in an additional 9% after two years, with further increases as treatment is continued.153
Telbivudine
Telbivudine is an l-nucleoside analog of thymidine that has been shown to be more potent than lamivudine in randomized, controlled trials in patients with HBeAg-positive and HBeAg-negative hepatitis B.154,155 In the registration trial, 1367 patients with HBeAg-positive or HBeAg-negative hepatitis B were randomized to receive telbivudine 600 mg or lamivudine 100 mg, each once daily, for 104 weeks.155 Virologic and biochemical responses associated with telbivudine were superior to those with lamivudine. Several randomized studies also have reported rapid and marked reductions in serum HBV DNA levels in patients who had been switched from adefovir to telbivudine.
Unfortunately, after one and two years of treatment, genotypic resistance was found in 5% and 25% of HBeAg-positive patients, respectively, and 2% and 11% of HBeAg-negative patients, respectively.155,156 The highest rates of virologic response and, conversely, the lowest rates of resistance, were found in patients who had less than 3 log10 copies of HBV DNA at week 24 of treatment, with the best responses found in those with a negative HBV DNA result by a PCR assay. Resistance to telbivudine is conferred by the M204I mutation either alone or in conjunction with the L180M mutation. These mutations also confer resistance to lamivudine, and for this reason switching to telbivudine is not preferred in cases of lamivudine resistance. Despite the greater antiviral potency and lower resistance profile of telbivudine compared with lamivudine, telbivudine has a higher rate of resistance than entecavir and tenofovir (see later) and has fallen out of favor as a treatment for HBV infection. In addition, a multicenter study of telbivudine in combination with pegylated interferon was stopped prematurely because of the development of myopathy and elevations in serum creatinine kinase levels in a higher percentage of patients than reported with other nucleoside analogs.
Tenofovir Disoproxil Fumarate
Tenofovir, an acyclic nucleotide inhibitor of HBV polymerase and HIV reverse transcriptase, is similar chemically to adefovir dipivoxil but is produced in a 300-mg rather than a 10-mg formulation. Tenofovir was originally licensed for the treatment of HIV infection and approved in 2008 for the treatment of HBV infection. Its antiviral activity against HBV has been reported to be significantly greater than that of the 10-mg dose of adefovir in lamivudine-resistant and treatment-naïve patients.157,158 Resistance to tenofovir has not occurred after two years of treatment in either HBeAg-positive or HBeAg-negative patients.158 Preliminary studies have suggested that adefovir-resistant HBV may be less sensitive than wild-type HBV to tenofovir.159 Whether unique nucleotide substitutions in the HBV polymerase will ultimately be found that confer resistance to tenofovir in either previously untreated or adefovir-resistant cases is unknown.
Prolonged tenofovir therapy has been associated with bone loss and decreased bone density in HIV-infected patients.160 As with adefovir, renal tubular damage and Fanconi syndrome have been associated with prolonged use of tenofovir in a small number of cases and may be a particular risk in the elderly or persons with existing mild renal disease.161 Serum creatinine levels should be monitored regularly during prolonged use of tenofovir, and if the serum creatinine level rises, the dose may need to be modified or the drug discontinued to reduce the risk of further nephrotoxicity. Concomitant use of ritonavir or didanosine in HIV-infected patients has been found to be associated with a greater risk of Fanconi syndrome.162
Other Nucleoside Analogs
Emtricitabine
Emtricitabine is a fluorinated cytosine analog that inhibits HBV DNA polymerase and HIV reverse transcriptase. This drug is currently licensed in the United States and other countries for the treatment of HIV type 1 (HIV-1) infection. Preliminary results from a placebo-controlled, phase 3 study in previously untreated patients with chronic HBV infection have demonstrated that emtricitabine, 200 mg daily for 48 weeks, reduces serum HBV DNA levels by a median of 3 log10 copies/mL and improves liver histology significantly.163 Emtricitabine treatment of patients coinfected with HBV and HIV has led to levels of suppression of HBV DNA similar to those that occur in treated patients infected with HBV alone.163 The drug is structurally related to lamivudine, however, and has similar mutational sites and rates of resistance. In HBV monoinfected patients, the frequencies of YMDD mutations in persons who receive emtricitabine, 200 mg daily, have been shown to be between 9% and 13% at week 48 and 19% at week 96.164 On the basis of these findings, this agent is unlikely to play an important role in the management of HBV monoinfection.
Clevudine
Clevudine, a pyrimidine analog, is a potent inhibitor of HBV replication both in vitro and in vivo. This drug has been studied in woodchucks and in humans and is licensed for treatment of hepatitis B in Korea on the basis of 24-week clinical trials. In one study, therapy with clevudine given daily for 12 weeks resulted in a reduction in serum HBV DNA levels of greater than 4 log10 copies/mL.165 After therapy was discontinued, rebound to pretreatment HBV DNA levels occurred slowly in this and other studies.166 In one study, a 3-log10 reduction in HBV DNA levels was still evident 6 months after discontinuation of clevudine after a 24-week course of treatment.167 This pattern is different from that reported with other nucleoside analogs and might be explainable by a suppressive effect of clevudine on HBV cccDNA, the genomic template that has been shown to be relatively resistant to treatment with other oral agents. To date, little is known about the safety and efficacy of long-term treatment with clevudine. In 2009, clinical trials were halted because of the occurrence of myopathy in patients treated with clevudine for more than 24 weeks.
Viral Resistance to Nucleoside and Nucleotide Analogs
Registration trials for nucleos(t)ide analogs have incorporated serial genotypic testing for drug-resistant HBV mutants. These studies have shown quite clearly that genotypic resistance to a nucleos(t)ide analog often occurs weeks to months before a virologic breakthrough (defined as greater than a 1-log10 [10-fold] increase in serum HBV DNA levels above the previous nadir) (Fig. 78-6). Ultimately, if the patient is continued on the drug, virologic rebound (defined as an increase in serum HBV DNA levels to greater than 100,000 copies or 20,000 IU/mL) will occur and will be followed by elevation of the serum ALT level (see Fig. 78-6). Rescue therapy can modify this sequence of events if a second agent that lacks cross resistance to the original drug is used alone to replace the first drug or added to the first drug. many experts prefer to add another nucleos(t)ide analog that does not share the same resistance pattern rather than switch to an alternative monotherapy in this clinical situation.
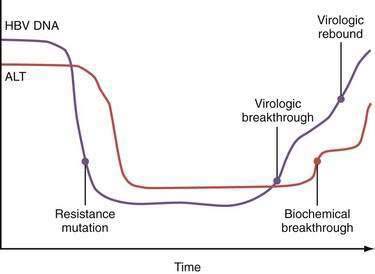
A patient treated with a nucleos(t)ide analog should be tested at periodic intervals to assess for virologic breakthrough.168 Such testing enables assessment of suppression of viremia and permits detection of viral breakthrough as early during the course of treatment as possible. To do this, the serum HBV DNA level should be monitored at three-month intervals. Patients who do not have at least a 1-log10 decline in the serum HBV DNA level after three months of treatment are considered to have primary treatment failure and should be given an alternative agent. The next key interval is at 24 weeks of treatment. In the GLOBE trial, the relative efficacies of lamivudine and telbivudine were compared, and persons who remained positive in serum for HBV DNA at 24 weeks of treatment with either drug were more likely to have a failed response and eventually developed resistance; the risk of a negative outcome to treatment was proportional to the level of detectable HBV DNA in serum at the 24-week point.155 In clinical practice, patients often have a serum HBV DNA level of greater than 10,000 copies/mL at week 24 if their baseline level of viremia is high, even with very potent drugs. If such a patient is taking a nucleos(t)ide analog with a high genetic barrier to resistance (e.g., entecavir or tenofovir), continuing the drug is reasonable, with the expectation of an eventual response. By contrast, if such a patient is taking a nucleos(t)ide analog that is associated with either a high rate of resistance (e.g., lamivudine) or limited antiviral potency (e.g., adefovir), switching to an alternative drug is probably best.169 Unless the choice of a particular nucleos(t)ide analog is restricted or the baseline serum HBV DNA level is relatively low (less than 106 copies/mL), first-line therapy with a highly potent nucleos(t)ide analog such as entecavir or tenofovir is preferred because the rapidity of HBV DNA suppression associated with these agents makes it far less likely that drug-resistant mutants will emerge.
Whenever a virologic breakthrough occurs, the patient should be questioned about adherence to therapy. If poor adherence is not a factor, genotypic testing for resistance should be done, as described earlier. If drug resistance is confirmed, the treating clinician may prescribe an alternative nucleos(t)ide analog that lacks cross resistance to the first drug or add the new drug while continuing the first. Clinical experience has indicated, however, that adding a second drug may be preferred to switching to another single agent because sequential monotherapy can result in multidrug-resistant HBV.170 When multidrug resistance occurs, combination therapy is not likely to be effective.
Combination Nucleoside or Nucleotide Analog Treatment
Somewhat surprisingly, the results of early clinical trials of combination therapy in previously untreated patients with HBeAg-positive chronic hepatitis B have shown that the combination of two nucleoside analogs (telbivudine and lamivudine) or the combination of a nucleoside analog and a nucleotide analog (lamivudine and adefovir) does not lead to greater viral inhibition during the first year of treatment than that seen with monotherapy.154,171 The reasons for the lack of apparent additive effect in these studies remain unexplained. Possibly, nucleoside analogs such as telbivudine and lamivudine compete sterically for binding to the HBV DNA polymerase or compete for phosphorylation enzymes (kinases) required for drug activation. Another possible explanation is that a measurable increase in viral suppression may be difficult to demonstrate whenever a drug with substantially less antiviral activity is added to a more potent drug (for example, when lamivudine is added to telbivudine or when adefovir is added to lamivudine). A study in which the combination of lamivudine and adefovir was compared with lamivudine alone, each given for two years, clearly demonstrated a lower rate of lamivudine resistance in the combination therapy arm (15% vs. 43%, respectively).171 On the basis of these and similar observations and the clinical experience with combination reverse transcriptase inhibitors in HIV infection, some authorities have recommended combination therapy as a first-line approach to prevent drug resistance. With the newer nucleos(t)ide agents, however, resistance occurs so infrequently that combination therapy might be better reserved for patients with clinical and laboratory features that have been associated with drug resistance, such as a high level of viremia and high body mass index.168
Combination Interferon and Nucleoside Analog Treatment
From a conceptual standpoint, treatment with the combination of interferon and a nucleoside analog might prove to be more effective than either drug alone because these agents have different mechanisms of action and might also permit a shorter course of nucleoside analog therapy, thereby reducing the risk of viral resistance. Three large multicenter studies evaluated these effects in patients given a combination of pegylated interferon and lamivudine. In one study, HBeAg-positive patients received peginterferon alfa-2b with either lamivudine or placebo for one year.126 At the end of treatment, 44% of the patients who received combination therapy had lost HBeAg, whereas only 29% who received peginterferon alone had done so; however, response rates in the two groups were no longer significantly different 26 weeks after the end of treatment (35% and 36%, respectively). It is possible that the low dose of peginterferon used in this study (100 µg weekly for eight months followed by 50 µg weekly until the end of treatment) may have contributed to the relatively high relapse rates after remission. In the second study,127 HBeAg-positive patients were treated with peginterferon alfa-2a, 180 µg weekly for 48 weeks, or combined therapy with lamivudine or with lamivudine monotherapy. Although the greatest degree of HBV DNA suppression was observed in the group that received combination therapy, the proportion of patients who underwent HBeAg seroconversion did not differ significantly between the two interferon-containing arms six months after completion of therapy. In the third study,128 patients with HBeAg-negative chronic hepatitis B were treated with peginterferon alfa-2a in an identical design strategy to that of the HBeAg-positive trial. Again, viral suppression was greater in the combination therapy arm, but this advantage did not translate into a higher rate of sustained virologic response. An interesting finding from the three-arm studies that evaluated peginterferon alfa-2a was that the rate of lamivudine resistance was significantly lower with combination therapy than with lamivudine monotherapy. Taken together, these three studies provide proof of concept that pegylated interferon alpha and lamivudine have additive antiviral effects during treatment. Trials of combinations of pegylated interferon and entecavir or tenofovir given for more than one year are in progress.
Antiviral Therapy in Special Populations
Pregnant Women
Nucleos(t)ide analog therapy may be considered during pregnancy for two reasons: to protect the health of the mother and to prevent breakthrough HBV infection in HBV-vaccinated newborns. Several studies have shown that treatment of mothers who have high serum levels of HBV DNA during the last 4 to 12 weeks of pregnancy decreases the rate of HBV infection in newborns vaccinated at birth against HBV.172,173 These studies have been small and generally have had problematic study designs or incomplete follow-up. Furthermore, breakthrough HBV infections develop in only 5% of newborns when the newborns are given a three-dose regimen of HBV vaccine and single dose of HBIG (see later). Therefore, antiviral treatment of the mother to prevent newborn HBV infection remains highly controversial and cannot be recommended at this time.
Persons with Acute Hepatitis
Because of the high rate (>95%) of complete immunologic recovery from acute hepatitis B, definitive recommendations about the treatment of acute hepatitis B cannot be made. Some experts recommend nucleos(t)ide analog therapy when HBeAg remains detectable in serum for more than 10 to 12 weeks because of the high likelihood of evolution to the chronic HBV carrier state without treatment. A National Institutes of Health–sponsored clinical workshop on hepatitis B proposed that persons with acute viral hepatitis complicated by an increase in INR above 1.5 and deep jaundice persisting for more than four weeks should receive antiviral therapy.174 Antiviral treatment of patients with fulminant hepatitis B is recommended in the AASLD guidelines because of the safety of nucleos(t)ide analog therapy and the need for liver transplantation in many of these patients.120
Persons with Cirrhosis
Nucleos(t)ide analog therapy has been shown to be safe in patients with cirrhosis and has made a major difference in the care of patients with advanced liver disease. Interferon is contraindicated in patients with even mildly decompensated cirrhosis because immune-mediated flares of serum ALT levels may occur and may be associated with further clinical deterioration. Also, serious infections have been reported in treated patients.175 Practice guidelines of the AASLD suggest that nucleos(t)ide analog therapy is preferred in all cases of HBV-related cirrhosis because of a greater chance of dose-limiting side effects and clinical worsening in the event of an ALT flare with interferon. Nevertheless, some studies have shown that patients with stable, well compensated cirrhosis can be treated safely and actually may have a higher rate of virologic response when compared with patients without cirrhosis.176,177 Treatment of patients with HBV cirrhosis needs to be individualized, and all of those treated with interferon should be carefully monitored.
Persons with Human Immunodeficiency Virus–Hepatitis B Virus Coinfection
With improved control of HIV disease with HAART, liver disease has emerged as one of the leading causes of death in patients with HIV.178 Antiviral therapy for hepatitis B should be considered for all HIV-HBV–coinfected patients with evidence of liver disease, irrespective of the CD4 count. In coinfected patients not requiring HAART, therapy for HBV should be based on agents with no HIV activity such as adefovir or pegylated interferon.179 Entecavir treatment is associated with a decline in HIV RNA levels; thus, it also is not recommended for use in patients who are not receiving concomitant HIV treatment.180 In patients with CD4 counts less than 350/mm3, the use of agents with dual anti-HIV and anti-HBV activity should be considered. Combination therapy with either emtricitabine and tenofovir or lamivudine and tenofovir should ideally be used to avoid or delay the development of antiviral resistance.
Persons with Hepatitis B Virus–Hepatitis C Virus Coinfection
When compared with monoinfected patients, HBV-HCV–coinfected patients tend to have more severe liver injury and a higher probability of cirrhosis.181 Limited data are available, however, to define the best approach to treatment in this group of patients. In most instances, one virus, often HCV, is dominant through the process of viral interference. The typical patient is positive for HCV RNA but negative for HBV DNA in serum. Close monitoring has been recommended before treatment is initiated, however, because some patients exhibit alternating viremia. In a prospective clinical trial of 19 patients with combined HBV-HCV infection, all were positive for HCV RNA, and only 5 were positive for HBV DNA prior to treatment. A high rate of sustained virologic response (74%) was observed after a 48-week course of pegylated interferon alpha and ribavirin (see Chapter 79), and two of the five HBV DNA-positive patients also had a virologic response. Unfortunately, HBV DNA became detectable again in four patients who were initially negative, suggesting that a risk exists that HBV may reactivate with eradication of HCV.182 The optimal therapy for patients who are positive in serum for both HBV DNA and HCV RNA is also unclear. In such instances, the author has had success in treating both infections simultaneously with a combination of a nucleos(t)ide analog, pegylated interferon alpha, and ribavirin.
Unresolved Issues
Major advances in antiviral therapy have occurred since 2000, but many unresolved issues remain. Although clearance of HBsAg from serum occurs in some patients after a relatively short course of interferon therapy, interferon is used infrequently because of its adverse effects. Nevertheless, data based on serial monitoring of HBsAg and HBeAg concentrations during therapy with pegylated interferon strongly suggest that failure to achieve a decline in serum concentrations of these viral parameters at key treatment intervals (12 and 24 weeks) has a high negative predictive value for a sustained response and could provide stopping rules similar to those used for the treatment of HCV infection (see Chapter 79). Clinical trials also have suggested that serial monitoring of the HBsAg concentration allows determination of the appropriateness of extending the duration of interferon therapy in patients with HBeAg-negative hepatitis, a condition in which HBsAg clearance is the most reliable endpoint. Multinational trials to evaluate the clinical utility of these markers to monitor virologic response during therapy with interferon are in progress. Commercial assays for HBsAg or HBeAg concentrations are not yet available in the United States but are widely available in Europe and elsewhere in the world and are likely to become available in the United States.
PREVENTION
Hepatitis B Vaccine
Currently marketed HBV vaccines make use of DNA recombinant technology by introducing the gene for HBsAg (S gene) into the genome of the yeast Saccharomyces cerevisiae. The two vaccines available in the United States are Recombivax HB (Merck, licensed in 1986) and Engerix-B (GlaxoSmithKline, licensed by SmithKline Beecham in 1989). Aluminum hydroxide is used as an adjuvant in both vaccines. Because thimerosal, a preservative used in the vaccines, contains mercury, thimerosal-free vaccines have become available, especially for use in infants. The HBV vaccine is administered intramuscularly in the deltoid muscle of adults and the anterolateral thigh of infants and neonates. The vaccines induce HBsAg-specific helper T cells and T cell–dependent B cells to produce neutralizing antibody against the “a” epitope (amino acid sequence 124 to 148) of HBsAg as early as two weeks after the first injection.183 HBV vaccines are highly efficacious in preventing HBV infection.184 Because the vaccines contain HBsAg only, anti-HBs is the sole antibody produced. Consequently, a vaccinee who tests positive for anti-HBc after vaccination should be considered to have had a subclinical HBV infection.
The vaccines typically achieve an anti-HBs titer greater than 100 mIU/mL. Antibody titers greater than 100 mIU/mL confer 100% protection against HBV infection, and a lower antibody titer (up to 10 mIU/mL) is seroprotective in most instances. Peak antibody titers and persistence of antibody levels vary among different persons. The titers drop steadily over the first two years after vaccination, sometimes to levels less than 10 mIU/mL. Two studies in different populations have demonstrated that anti-HBs titers decrease to nonprotective levels in at least 25% to 50% of recipients over a period of 5 to 10 years.185
Although protective anti-HBs response rates after HBV vaccination typically exceed 90%, a number of factors can impede an adequate antibody response. Smoking, obesity, injection into the buttock, chronic liver disease, presence of HLA-DR3, DR7, and DQ2 alleles, absence of the HLA-A2 allele, and extremes of age may be associated with reduced immunogenicity. Such “hyporesponders” may benefit from a higher dose of vaccine. Response rates also are lower in immunocompromised patients, such as transplant recipients, patients receiving chemotherapy, and those with end-stage liver disease. Only 50% to 60% of patients on hemodialysis respond adequately to vaccination. Therefore, patients with chronic kidney disease should be vaccinated early in the course of their disease, before renal disease progresses, to ensure optimal response to vaccination.186
Five percent to 8% of HBV vaccine recipients do not achieve detectable anti-HBs levels (“nonresponders”). Studies conducted mostly in animals indicate that intradermal injection of the vaccine may produce a stronger humoral and cellular immune response than conventional intramuscular administration.187,188 Intradermal injection, by recruiting “professional” dendritic cells, stimulates primary MHC class I- and class II–restricted T cell responses. In one study, intradermal vaccination resulted in protective anti-HBs responses in nonresponders to intramuscular administration.188 Repeated dosing with intradermal vaccination (5 µg every two weeks to provide an anti-HBs titer of 1000 mIU/mL or greater or a total of 52 doses) has resulted in a protective antibody response rate of nearly 100% in patients undergoing long-term hemodialysis. At present, intradermal HBV vaccination has not been recommended officially—in part because of concerns about standardization of the technique for intradermal delivery.
Because HBV vaccination results in strong immunologic memory capable of preventing infection even in patients with low or undetectable antibody titers, no role exists for a booster vaccine dose in immunocompetent adults and children. Current recommendations include booster doses only for patients undergoing hemodialysis, in whom anti-HBs titers should be tested annually and a booster dose given if the titer is lower than 10 mIU/mL.189
Targeted High-Risk Groups
Table 78-6 lists the high-risk groups for whom HBV vaccination is recommended. Targeted vaccination has not achieved its objective in certain high-risk groups, such as injection drug users, but has achieved great success among health care workers and newborns. The CDC has extended its original recommendations for routine HBsAg screening to include persons born in countries in which the prevalence of hepatitis B exceeds 2%.190 This new recommendation will facilitate identification of susceptible persons who are in need of vaccination and those in need of antiviral therapy.
Table 78-6 High-Risk Groups for Whom Hepatitis B Virus (HBV) Vaccination Should Be Considered
Vaccination Schedule
The doses of currently available HBV vaccines and recommendations for the schedules of administration are shown in Table 78-7. The typical vaccination schedule is zero, one, and six months. The first two doses have no effect on the final anti-HBs titer. The third dose acts as a booster to achieve a high anti-HBs titer. In immunocompromised patients and patients undergoing hemodialysis, four vaccine doses are recommended, with the fourth dose given to ensure the highest possible anti-HBs titer. If vaccination is interrupted, the second dose should be administered as soon as possible after the first.190 If the third dose is not given on schedule, it should be given at least two months after the second dose.
Table 78-7 Recommended Dosing for the Currently Available Hepatitis B Vaccines*
| RECOMBIVAX HB (10 µg/mL) | ENGERIX-B (20 µg/mL) | |
|---|---|---|
| Infants† and children age <11 yr | 2.5 µg | 10 µg |
| Children age 11-19 yr | 5 µg | 20 µg |
| Adults (≥20 yrs) | 10 µg | 20 µg |
| Hemodialysis patients | 40 µg (1.0 mL)‡ | 40 µg (2.0 mL)§ |
| Immunocompromised patients | 40 µg (1.0 mL)‡ | 40 µg (2.0 mL)§ |
* The standard schedule is 0, 1, and 6 months.
† Infants born to hepatitis B surface antigen-negative mothers.
‡ Special formulation (40 µg/mL).
§ Two 1.0-mL doses administered at one site in four-dose schedule (0, 1, 2, 6 months).
The HBV vaccine is currently administered to all children and infants as a part of the universal immunization program. Combination HBV vaccines with diphtheria-pertussis-tetanus (DPT) and Haemophilus influenzae type B (Hib) (DTPw-HB/Hib), the vaccines in current use for immunization of infants, do not reduce the immunogenicity of any of the components of HBV infection.191 Adolescents who have not been vaccinated in infancy or childhood should also be vaccinated.
Postexposure and Perinatal Prophylaxis
Table 78-8 summarizes recommendations for prevention of perinatal transmission of HBV. Table 78-9 lists recommendations for prophylaxis after exposure to a known HBsAg-positive source. Postexposure vaccination should be considered for any percutaneous, ocular, or mucous membrane exposure. The type of immunoprophylaxis is determined by the HBsAg status of the source and the vaccination-response status of the exposed person.

Table 78-9 Postexposure Prophylaxis of Hepatitis B if the Source Is HBsAg Positive
| VACCINATION STATUS OF EXPOSED PERSON | IMMUNE PROPHYLAXIS |
|---|---|
| Unvaccinated | HBIG (0.06 mL/kg) and initiate hepatitis B vaccine series |
| Previously vaccinated: | |
| Known responder* | No treatment |
| Known nonresponder | HBIG × 2 doses (one month apart) |
| OR | |
| HBIG × 1 dose and initiate revaccination | |
| Antibody response unknown |
Anti-HBs, antibody to hepatitis B surface antigen; HBIG, hepatitis B immune globulin; HBsAg, hepatitis B surface antigen.
Bivalent Vaccine
A combined HAV and HBV vaccine has been licensed commercially (TWINRIX, GlaxoSmithKline, Research Triangle Park, NC) and has been shown to be highly immunogenic and protective against both infections (see Chapter 77). This vaccine offers ease of administration for persons at increased risk of both HAV and HBV infection (e.g., world travelers or men who have sex with men) and in patients with underlying chronic liver disease.192
HEPATITIS D
Hepatitis D (delta) virus (HDV) was discovered by Rizzetto and associates in 1977, as a unique nuclear antigen in the hepatocytes of patients infected with HBV.193 The antigen was identified subsequently as a novel pathogen and was linked to severe chronic hepatitis B and fulminant HBV infection.
EPIDEMIOLOGY
HDV is distributed globally with wide variations in prevalence. At least 5% of HBV carriers worldwide are estimated to be infected with HDV, and therefore, the overall burden of HDV infection is between 15 and 20 million cases. The highest prevalence is seen in South America and the Mediterranean basin. The prevalence is low in Northern Europe and North America, where HDV infection is confined to injection drug users. The rate of infection among HBsAg-positive blood donors in the United States has been found to be 3.8%.194 The incidence of transfusion-associated HDV infection has been declining steadily because of HBV vaccination and screening of donor sera for HBsAg. In fact, many epidemiologists believe that the epidemic of HDV, which started in the 1970s, is coming to an end. For example, epidemiologic data from Italy show that the prevalence of HDV infection in HBsAg carriers by 2000 was 8.3%, compared with 25% in the early 1970s.195–197 HDV infection remains an important problem among injection drug users, however.198
Among the three genotypes of HDV (I to III), genotype I is the most prevalent and is the most common genotype in Mediterranean countries, Africa, Europe, and North America.199,200 Different subtypes within this genotype may exist in certain parts of Africa. Genotype II is reported mostly in Japan and Taiwan and is associated with milder liver disease than that seen with genotype I.201 Genotype III has been isolated from epidemics in South America.202 Different genotypes of HDV may interact variably with different HBV genotypes. Whether the interaction between HDV and HBV genotypes specifically increases the severity of HDV infection is unclear, although infection with HDV genotype III and HBV genotype F is reported to cause severe hepatitis. The mode of HDV transmission is linked closely to that of HBV transmission, with the parenteral route being the most efficient. Sexual transmission of HDV has been reported, and familial clustering of cases has been seen in endemic areas.
VIROLOGY
HDV is a unique agent that bears no similarity to other transmissible agents that infect animals. In fact, the 1.7-kb single-strand HDV RNA genome shares several features with plant viroids, such as intramolecular base pairing and autocatalytic RNA segments.203 Unlike plant viroids, however, HDV RNA encodes a protein, hepatitis delta antigen (HDAg). The virion consists of the HDV genome complexed with approximately 70 copies of HDAg in an envelope protein composed of lipids and HBsAg. The protein envelope that is contributed by HBV protects the HDV RNA-HDAg complex. The protein envelope is not required for replication of HDV and is the only helper function provided by HBV. Once HDV with its HBV envelope protein enters the host, the HDV RNA-HDAg complex migrates to the nucleus. Viral replication then proceeds in the nucleus according to a double-rolling model, aided possibly by host DNA-dependent RNA polymerase.204
During translation, two forms of HDAg (encoded by the same regions of RNA) are produced, a short form (HDAg-S) and a long form (HDAg-L). HDAg-L has 19 to 20 more amino acids than HDAg-S. The additional amino acids in HDAg-L are incorporated by a process of RNA editing, another unique aspect of the HDV genome.205 Interestingly, HDAg-S and HDAg-L have opposite effects on viral replication; HDAg-S acts as a facilitator, and HDAg-L as an inhibitor.206 The extent of RNA editing determines the amount of HDAg-L formed and, consequently, influences the rate of replication. In states of high replication, only HDAg-S is produced. Ultimately, the intracellular ratio of HDAg-S to HDAg-L determines the rate of replication, assembly, and transport from infected hepatocytes.
PATHOGENESIS
The pathogenic mechanisms of HDV hepatitis remain poorly understood. Because HBV is not known to be directly cytotoxic, the severity of combined infection with HBV and HDV may be attributed either to a direct cytotoxic effect of HDV or an enhanced immune response against the two viruses. Direct cytotoxicity of HDV has been questioned on the basis of studies in transgenic mice. Mice expressing either HDAg-L or HDAg-S show no evidence of hepatocyte injury. The lack of a direct cytotoxic effect also is supported by the observation that liver transplant recipients who express HDAg in their allografts do not manifest evidence of cellular damage.207 Instead, the pathogenic mechanism of HDV-induced liver damage is most likely related to the immunologic response to HDV. The occurrence of classic necroinflammatory changes in the liver and several autoantibodies, such as antibodies to liver-kidney microsome (anti-LKM), thymocytes, and nuclear lamin C, also suggest a role for immune-mediated liver injury. One fact is certain: The ability of HDV to cause hepatic necrosis is determined by expression of HBV, as illustrated after liver transplantation, when HDV infection becomes pathogenic only if HBV infection also recurs.207
DIAGNOSIS
The most useful markers of HDV infection include HDAg, antibody to HDAg (anti-HDV), HDV RNA, and immunohistochemical staining of HDAg in liver tissue. Detection of HDV RNA by reverse transcriptase PCR amplification (RT-PCR), with a detection limit of 50 to 100 copies/mL, is the most reliable diagnostic technique, with nearly 100% sensitivity. HDV RNA is the earliest marker to appear during the course of HDV infection and may be seen in the absence of other markers.208 Higher levels of HDV RNA in serum may be associated with more severe disease. The level of HDV RNA in serum is a reliable marker for monitoring the efficacy of treatment and documenting viral eradication. HDV RNA also can be detected in liver cells by hybridization techniques, which are generally less sensitive than RT-PCR.
The most readily available marker of HDV infection has been anti-HDV. Anti-HDV does not confer protection against HDV. Either IgM anti-HDV or total anti-HDV, which is composed of both IgM and IgG anti-HDV, can be detected. IgM anti-HDV appears in serum at the time of acute infection, and IgG anti-HDV develops later in the course. IgM anti-HDV often persists as the disease progresses to chronicity and is detectable in high titers in patients with chronic HDV infection. It is frequently regarded as a marker of serious liver damage.209 As the infection evolves from the acute to the chronic phase, the type of IgM antibody also changes from a monomeric (S) form to a multimeric (19S) form. IgG anti-HDV persists for a long time in immunocompetent persons and may indicate chronic or previous HDV infection. Some patients with IgG anti-HDV may not have active infection and test negative for HDV RNA.210
NATURAL HISTORY
Early epidemiologic studies suggested that HDV infection aggravates the severity of HBV infection in coprimary infection, but subsequent reports have disputed this claim.211 In a European multicenter study on prognostic factors in 366 patients with chronic hepatitis B and compensated cirrhosis, HDV infection did not influence the prognosis.212 In a long-term follow-up study of 302 patients with chronic hepatitis B (76 with HDV infection), HDV infection was not an independent predictor of mortality.213 Therefore, HDV infection appears to have a varying influence on the course of hepatitis B and is not necessarily associated with severe hepatitis. The severity of HDV infection may vary with the frequency of HDV in a population, with the level of HBV viremia, and with interactions between specific HBV and HDV genotypes.
Coprimary infection is seen most often in injection drug users (Fig. 78-7). Because HBV infection resolves in a majority of patients, HDV also disappears in most patients, and the risk of chronicity after coprimary infection is less than 5%. Some data suggest, however, that coinfection with HDV enhances the risk of fulminant hepatitis B.214
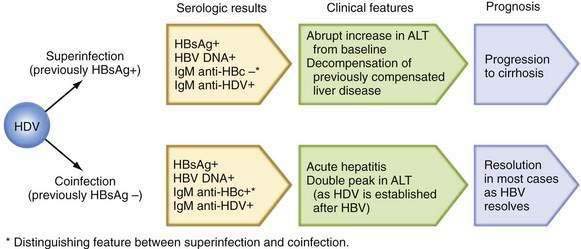
Superinfection of HDV in an HBV carrier can lead to severe hepatitis and acute decompensation of preexisting liver disease. Affected patients often express a high level of HDV viremia because high serum levels of HBsAg readily protect the replicating HDV genome. Superinfection may also coincide with a decline in serum HBV DNA levels because HDV replication inhibits HBV replication.215 In a study involving 185 patients with HDV superinfection, HDV RNA was detectable in 63 of 64 patients with acute HDV infection, but HBV DNA was detectable in only 40% of patients.215 Rarely, HDV superinfection may lead to disappearance of HBsAg and appearance of anti-HBs.215 In contrast to coinfection, chronic HDV infection develops frequently after HDV superinfection. HDV superinfection evolves to chronic HDV infection in 70% of patients and is characterized by persistent HDV viremia and detectable HDV RNA in serum. Although the clinical course of chronic HDV infection varies, persistent replication of HDV and HBV often leads to progressive hepatitis and cirrhosis within a few years. More rapid clinical progression leading to end-stage liver disease within two years may be seen in some injection drug users; HCC also may develop.216
TREATMENT
Despite developments in the treatment of HBV monoinfection, results of therapy for HDV-HBV infection have been disappointing. Hepatitis D is the least common cause of chronic viral hepatitis worldwide, but it is the most severe form. The only therapeutic option currently available is interferon alpha, the efficacy of which is related to the dose and duration of treatment. Nucleos(t)ide analogs, currently the mainstay of treatment for HBV infection, are not effective in HDV infection. The lack of efficacy of nucleos(t)ide analogs may be explained in part by the observation that nucleos(t)ide analogs seldom lead to disappearance of HBsAg, the only HBV protein that is required by HDV. The rate of sustained HDV clearance after a one-year course with high doses of conventional interferon alpha (9 million units three times a week) is low (20%).217 Better results have been reported with peginterferon alpha, both in interferon-naïve patients and previous nonresponders to conventional interferon-α. Doses of 1.5 ug/kg of peginterferon alfa-2b and 180 ug of peginterferon alfa-2a have been used successfully.217 Current recommendations from an Italian workshop include the use of pegylated interferon alpha for 48 to 72 weeks.218 Because the ultimate goal of treatment is the eradication of both HBV and HDV, some authorities have advocated that therapy be continued as long as possible in responders and preferably until the loss of HBsAg occurs.217 Nucleos(t)ide analog therapy used in conjunction with interferon does not improve response rates compared with interferon alone.219 Reliable predictors of a long-term response have not been identified, although patients with disease of short duration or without cirrhosis are more likely to respond, thereby underscoring the importance of early diagnosis and treatment.
Buster EH, Flink HJ, Cakaloglu Y, et al. Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon a-2b. Gastroenterology. 2008;135:459-67. (Ref 130.)
Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65-73. (Ref 57.)
Doo E, Liang TJ. Molecular anatomy and pathophysiologic implications of drug resistance in hepatitis B virus infection. Gastroenterology. 2001;120:1000-8. (Ref 34.)
Farci P, Chessa L, Balistrieri C, et al. Treatment of chronic hepatitis D. J Viral Hepatitis. 2007;14(Suppl 1):58-63. (Ref 217.)
Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J Hepatol. 2008;48:335-52. (Ref 17.)
Hoofnagle JH, Doo E, Liang TJ, et al. Management of hepatitis B: Summary of a clinical research workshop. Hepatology. 2007;45:1056-75. (Ref 174.)
Iloeje UH, Yang HI, Su J, et al. Predicting liver cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology. 2006;130:678-86. (Ref 56.)
Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 Update. Clin Gastroenterol Hepatol. 2008;6:1315-41. (Ref 123.)
Keeffe EB, Zeuzem S, Koff RS, et al. Report of an international workshop: Roadmap for management of patients receiving oral therapy for chronic hepatitis B. Clin Gastroenterol Hepatol. 2007;5:890-7. (Ref 169.)
Lai CL, Yuen MF. The natural history and treatment of chronic hepatitis B: A critical evaluation of standard treatment criteria and end points. Ann Intern Med. 2007;147:58-61. (Ref 60.)
Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682-95. (Ref 127.)
Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-31. (Ref 61.)
Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714-22. (Ref 35.)
Lok AS, McMahon BJ. AASLD Practice Guidelines. Chronic hepatitis B. Hepatology. 2007;45:507-39. (Ref 120.)
Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351:1206-17. (Ref 128.)
Perrillo RP. Acute flares in chronic hepatitis B. The natural and unnatural history of an immunologically mediated liver disease. Gastroenterology. 2001;120:1009-22. (Ref 78.)
Zoulim F, Perrillo R. Hepatitis B: Reflections on the current approach to antiviral therapy. J Hepatol. 2008;48:S2-19. (Ref 168.)
1. Chang MH, Chen CJ, Lai MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855-9.
2. Gust ID. Epidemiology of hepatitis B infection in the Western Pacific and South East Asia. Gut. 1996;38(Suppl 2):S18-23.
3. Shepard CW, Simard EP, Finelli L, et al. Hepatitis B virus infection: Epidemiology and vaccination. Epidemiol Rev. 2006;28:112-25.
4. Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733-45.
5. Dickson RC, Everhart JE, Lake JR, et al. Transmission of hepatitis B by transplantation of livers from donors positive for antibody to hepatitis B core antigen. The National Institute of Diabetes and Digestive and Kidney Diseases Liver Transplantation Database. Gastroenterology. 1997;113:1668-74.
6. Grob P, Jilg W, Bornhak H, et al. Serological pattern “anti-HBc alone”: Report of a workshop. J Med Virol. 2000;62:450-5.
7. Mason A, Wick M, White H, et al. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology. 1993;18:781-9.
8. Mason A, Yoffe B, Noonan C, et al. Hepatitis B virus DNA in peripheral-blood mononuclear cells in chronic hepatitis B after HBsAg clearance. Hepatology. 1992;16:36-41.
9. Kuhns M, McNamara A, Mason A, et al. Serum and liver hepatitis B virus DNA in chronic hepatitis B after sustained loss of surface antigen. Gastroenterology. 1992;103:1649-56.
10. Fong TL, Bunnapradist S, Jordan SC, et al. Impact of hepatitis B core antibody status on outcomes of cadaveric renal transplantation: Analysis of United Network of Organ Sharing database between 1994 and 1999. Transplantation. 2002;73:85-9.
11. Centers for Disease Control and Prevention. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. Morb Mortal Wkly Rep. 2005;54(RRI6):1-23.
12. Alter MJ. Epidemiology and prevention of hepatitis B. Semin Liver Dis. 2003;23:39.
13. McQuillan GM, Coleman PJ, Kruszon-Moran D, et al. Prevalence of hepatitis B virus infection in the United States: The National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health. 1999;89:14-18.
14. Lok AS, Heathcote EJ, Hoofnagle JH. Management of hepatitis B: 2000-summary of workshop. Gastroenterology. 2001;120:1828-53.
15. Lee WM. Acute liver failure. N Engl J Med. 1993;329:1862-72.
16. Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transpl Surg. 1999;5:29-34.
17. Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J Hepatol. 2008;48:335-52.
18. Weissberg JI, Andres LL, Smith CI, et al. Survival in chronic hepatitis B: An analysis of 379 patients. Ann Intern Med. 1984;101:613-16.
19. de Jongh FE, Janssen HL, de Man RA, et al. Survival and prognostic indicators in hepatitis B surface antigen-positive cirrhosis of the liver. Gastroenterology. 1992;103:1630-4.
20. Niederau C, Heintgen T, Lange S, et al. Long-term follow-up of HBeAg-positive patients treated with interferon alfa for chronic hepatitis B. N Engl J Med. 1996;334:1422-7.
21. Yuen MF, Wong DKH, Sablon E, et al. HBsAg seroclearance in chronic hepatitis B in the Chinese: Virological, histological, and clinical aspects. Hepatology. 2004;39:1694-701.
22. Nowak MA, Bonhoeffer S, Hill AM, et al. Viral dynamics in hepatitis B virus infection. Proc Natl Acad Sci U S A. 1996;93:4398-402.
23. Bartholomeusz A, Schaefer S. Hepatitis B virus genotypes: Comparison of genotyping methods. Rev Med Virol. 2004;14:3-16.
24. Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: Results of a nationwide study. Gastroenterology. 2003;125:444-51.
25. Chu CJ, Lok AS. Clinical significance of hepatitis B virus genotypes. Hepatology. 2002;35:1274-6.
26. Ghany MG, Ayola B, Villamil FG, et al. Hepatitis B virus S mutants in liver transplant recipients who were reinfected despite hepatitis B immune globulin prophylaxis. Hepatology. 1998;27:213-22.
27. Locarnini S, McMillan J, Bartholomeusz A. The hepatitis B virus and common mutants. Semin Liver Dis. 2003;23:5-20.
28. Li J, Buckwold VE, Hon MW, Ou JH. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. J Virol. 1999;73:1239-44.
29. Guo X, Jin Y, Qian G, et al. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J Hepatol. 2008;49:718-25.
30. Lindh M, Andersson AS, Gusdal A. Genotypes, nt 1858 variants, and geographic origin of hepatitis B virus: Large scale analysis using a new genotyping method. J Infect Dis. 1997;175:1285-93.
31. Chu CJ, Keeffe EB, Han SH, et al. Prevalence of HBV precore/core promoter variants in the United States. Hepatology. 2003;38:619-28.
32. Bertoletti A, Sette A, Chisari FV, et al. Natural variants of cytotoxic epitopes are T-cell receptor antagonist for antiviral cytotoxic T cells. Nature. 1994;369:407-10.
33. Torre F, Cramp M, Owsianka A, et al. Direct evidence that naturally occurring mutations within hepatitis B core epitope alter CD-4 T-cell reactivity. J Med Virol. 2004;72:370-6.
34. Doo E, Liang TJ. Molecular anatomy and pathophysiologic implications of drug resistance in hepatitis B virus infection. Gastroenterology. 2001;120:1000-8.
35. Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714-22.
36. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to five years. Gastroenterology. 2006;131:1743-51.
37. Colonno RJ, Rose R, Baldick CJ, et al. Entecavir resistance is rare in nucleoside naïve patients with hepatitis B. Hepatology. 2006;44:1656-65.
38. Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105-17.
39. Liaw YF. Impact of YMDD mutations during lamivudine therapy in patients with chronic hepatitis B. Antivir Chem Chemother. 2001;12(Suppl 1):67-71.
40. Mutimer D, Pillay D, Shields P, et al. Outcome of lamivudine resistant hepatitis B virus infection in the liver transplant recipient. Gut. 2000;46:107-13.
41. Rehermann B. Immune responses in hepatitis B virus infection. Semin Liver Dis. 2003;23:21-38.
42. Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65-91.
43. Ferrari C, Missale G, Boni C, Urbani S. Immunopathogenesis of hepatitis. Br J Hepatol. 2003;39(Suppl 1):S36-42.
44. Curry MP, Koziel M. The dynamics of the immune response in acute hepatitis B: New lessons using new techniques. Hepatology. 2000;32:1177-9.
45. Bertoletti A, Ferrari C, Fiaccadori F, et al. HLA class I-restricted human cytotoxic T cells recognize endogenously synthesized hepatitis B virus nucleocapsid antigen. Proc Natl Acad Sci U S A. 1991;88:10445-9.
46. Milich DR, Chen MK, Hughers JL, et al. The secreted hepatitis B precore antigen can modulate the immune response to the nucleocapsid: A mechanism for persistence. J Immunol. 1998;160:2013-21.
47. Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen-negative chronic hepatitis B. Hepatology. 2001;34:617-24.
48. Perrillo RP, Brunt EM. Hepatic histologic and immunohistochemical changes in chronic hepatitis B after prolonged clearance of hepatitis B surface antigen. Ann Intern Med. 1991;115:113-15.
49. Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis. 2003;23:47-58.
50. Fattovich G, Pantalena M, Zagni I, et al. Effect of hepatitis B and C virus infection on the natural history of compensated cirrhosis: A cohort study of 297 patients. Am J Gastroenterol. 2002;97:2886-95.
51. McMahon BJ. The natural history of chronic hepatitis B virus infection. Semin Liver Dis. 2004;24(Suppl 1):17-21.
52. Kim HC, Nam CM, Jee SH, et al. Normal serum aminotransferase concentration and risk of mortality from liver diseases: Prospective-cohort study. BMJ. 2004;323:983.
53. Lai M, Hyatt BJ, Nasser I, et al. The clinical signficance of persistently normal ALT in chronic hepatitis B infection. J Hepatol. 2007;47:760-7.
54. Lin CL, Liao LY, Liu CJ, et al. Hepatitis B viral factors in HBeAg-negative carriers with persistently normal serum alanine aminotransferase levels. Hepatology. 2007;45:1193-8.
55. Hui CK, Leung N, Yuen ST, et al. Natural history and disease progression in Chinese chronic hepatitis B patients in immune-tolerant phase. Hepatology. 2007;46:395-401.
56. Iloeje UH, Yang HI, Su J, et al. Predicting liver cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology. 2006;130:678-86.
57. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65-73.
58. Yuen MF, Yuan HJ, Wong DKH, et al. Prognsotic determinants for chronic hepatitis B in Asians: Therapeutic implications. Gut. 2005;54:1610-14.
59. Perrillo R. Indications for hepatitis B treatment should be relaxed. In: Arroyo V, Sanchez-Fueyo A, Fernandez-Gomez J, et al, editors. Advances in the Therapy of Liver Diseases. Barcelona: Ars Medica; 2007:211.
60. Lai CL, Yuen MF. The natural history and treatment of chronic hepatitis B: A critical evaluation of standard treatment criteria and end points. Ann Intern Med. 2007;147:58-61.
61. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-31.
62. Koff RS. Risks associated with hepatitis A and B in patients with hepatitis C. J Clin Gastroenterol. 2001;33:20-6.
63. Tozun N, Forbes A, Anderson MG, et al. Safety of alcohol after viral hepatitis. Lancet. 1991;337:1079-80.
64. Gocke DJ. Extrahepatic manifestations of viral hepatitis. Am J Med Sci. 1975;270:49-52.
65. Krugman S, Overby LR, Mushahwar IK, et al. Viral hepatitis type B: Studies on natural history and prevention-reexamined. N Engl J Med. 1979;300:101-6.
66. Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med. 1996;2:1104-8.
67. Pyrsopoulos NT, Reddy KR. Extrahepatic manifestations of chronic viral hepatitis. Curr Gastroenterol Rep. 2001;3:71-8.
68. Johnson RJ, Couser WG. Hepatitis B infection and renal disease: Clinical, immunopathogenetic and therapeutic considerations. Kidney Int. 1990;37:663-76.
69. Lai KN, Li PK, Lui SF, et al. Membranous nephropathy related to hepatitis B virus in adults. N Engl J Med. 1991;324:1457-63.
70. Conjeevaram HS, Hoofnagle JH, Austin HA, et al. Long-term outcome of hepatitis B virus-related glomerulonephritis after therapy with interferon alfa. Gastroenterology. 1995;109:540-6.
71. Shikata T, Uzawa T, Yoshiwara N, et al. Staining methods of Australia antigen in paraffin section detection of cytoplasmic inclusion bodies. Jpn J Exp Med. 1974;44:25-36.
72. Perrillo RP, Campbell CR, Sanders GE, et al. Spontaneous clearance and reactivation of chronic hepatitis B virus infection among male homosexuals with chronic type B hepatitis. Ann Intern Med. 1984;100:43-6.
73. Mels GC, Bellati G, Leandro G, et al. Fluctuations in viremia, aminotransferases and IgM antibody to hepatitis B core antigen in chronic hepatitis B patients with disease exacerbations. Liver. 1994;14:175-81.
74. Di Martino V, Thevenot T, Colin JF, et al. Influence of HIV infection on the response to interferon therapy and the long-term outcome of chronic hepatitis B. Gastroenterology. 2002;123:1812-22.
75. Liaw YF, Tsai SL. Pathogenesis and clinical significance of spontaneous exacerbations and remissions in chronic hepatitis B virus infection. Viral Hepatitis. 1997;3:143.
76. Xunrong L, Yan AW, Liang R, et al. Hepatitis B virus (HBV) reactivation after cytotoxic or immunosuppressive therapy: Pathogenesis and management. Rev Med Virol. 2001;11:287-99.
77. Lau JY, Lai CL, Lin HJ, et al. Fatal reactivation of chronic hepatitis B virus infection following withdrawal of chemotherapy in lymphoma patients. Q J Med. 1989;73:911-17.
78. Perrillo RP. Acute flares in chronic hepatitis B. The natural and unnatural history of an immunologically mediated liver disease. Gastroenterology. 2001;120:1009-22.
79. Liang R, Lau GK, Kwong YL. Chemotherapy and bone marrow transplantation for cancer patients who are also chronic hepatitis B carriers: A review of the problem. J Clin Oncol. 1999;17:394-8.
80. Lau GK, Liang R, Chiu EK, et al. Hepatic events after bone marrow transplantation in patients with hepatitis B infection: A case controlled study. Bone Marrow Transplant. 1997;19:795-9.
81. Davies SE, Portmann BC, O’Grady JG, et al. Hepatic histological findings after transplantation for chronic hepatitis B virus infection, including a unique pattern of fibrosing cholestatic hepatitis. Hepatology. 1991;13:150-7.
82. Lau GK, Yiu HH, Fong DY, et al. Early is superior to deferred preemptive lamivudine therapy for hepatitis B patients undergoing chemotherapy. Gastroenterology. 2003;125:1742-9.
83. Flink HJ, Sprengers D, Hansen BE, et al. Flares in chronic hepatitis B patients induced by the host or the virus? Relation to treatment response during Peg-interferon alpha-2b therapy. Gut. 2005;54:1604-9.
84. Nair S, Perrillo R. Serum alanine aminotransferase flares during interferon treatment of chronic hepatitis B: Is sustained clearance of HBV DNA dependent on levels of pretreatment viremia? Hepatology. 2001;34:1021-6.
85. Hoofnagle JH, Di Bisceglie AM, Waggoner JG, et al. Interferon alfa for patients with clinically apparent cirrhosis due to chronic hepatitis B. Gastroenterology. 1993;104:1116-21.
86. Honkoop P, de Man RA, Niesters HG, et al. Acute exacerbation of chronic hepatitis B virus infection after withdrawal of lamivudine therapy. Hepatology. 2000;32:635-9.
87. Liaw YF, Chien RN, Yeh CT, et al. Acute exacerbation and hepatitis B virus clearance after emergence of YMDD motif mutation during lamivudine therapy. Hepatology. 1999;30:567-72.
88. Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125:1714-22.
89. Perrillo RP, Regenstein FG, Peters MG, et al. Prednisone withdrawal followed by recombinant alpha interferon in the treatment of chronic type B hepatitis: A randomized, controlled trial. Ann Intern Med. 1988;109:95-100.
90. Krogsgaard K, Marcellin P, Trepo C, et al. Prednisolone withdrawal therapy enhances the effect of human lymphoblastoid interferon in chronic hepatitis B. INTREPED Trial Group. J Hepatol. 1996;25:803-13.
91. Liaw YF, Tsai SL, Chien RN, et al. Prednisolone priming enhances Th1 response and efficacy of subsequent lamivudine therapy in patients with chronic hepatitis B. Hepatology. 2000;32:604-9.
92. Thio CL. Hepatitis B in the human immunodeficiency virus-infected patient: Epidemiology, natural history, and treatment. Semin Liver Dis. 2003;23:125-36.
93. Bonino F, Brunetto MR. Chronic hepatitis B e antigen (HBeAg) negative, anti-HBe positive hepatitis B: An overview. J Hepatol. 2003;39(Suppl 1):S160-3.
94. Lindh M, Gustavson C, Mardberg K, et al. Mutation of nucleoside 1,762 in the core promoter region during hepatitis B e seroconversion and its relation to liver damage in hepatitis B e antigen carriers. J Med Virol. 1998;55:185-90.
95. Steinberg JL, Yeo W, Zhong S, et al. Hepatitis B virus reactivation in patients undergoing cytotoxic chemotherapy for solid tumours: Precore/core mutations may play an important role. J Med Virol. 2000;60:249-55.
96. Hadziyannis SJ. Hepatitis delta: An overview. In: Rizzetto M, Purcell RH, Gerin JL, Verme G, editors. Viral Hepatitis and Liver Disease: Proceedings of IX Triennial International Symposium on Viral Hepatitis and Liver Disease. Turin: Edizioni Minerva Medica; 1997:283.
97. Liaw YF, Tsai SL, Chang JJ, et al. Displacement of hepatitis B virus by hepatitis C virus as the cause of continuing chronic hepatitis. Gastroenterology. 1994;106:1048-53.
98. Hoofnagle JH, Schafer DF. Serologic markers of hepatitis B virus infection. Semin Liver Dis. 1986;6:1-10.
99. Perrillo RP, Chau KH, Overby LR, et al. Anti-hepatitis B core immunoglobulin M in the serologic evaluation of hepatitis B virus infection and simultaneous infection with type B, delta agent, and non-A, non-B viruses. Gastroenterology. 1983;85:163-7.
100. Shiels MT, Taswell HF, Czaja AJ, et al. Frequency and significance of concurrent hepatitis B surface antigen and antibody in acute and chronic hepatitis B. Gastroenterology. 1987;93:675-80.
101. Grob P, Jilg W, Bornhak H, et al. Serological pattern “anti-HBc”: Report on a workshop. J Med Virol. 2000;62:450-5.
102. Weber B, Melchior W, Gehrke R, et al. Hepatitis B virus markers in anti-HBc only positive individuals. J Med Virol. 2001;64:312-19.
103. Jilg W, Sieger E, Zachoval R, et al. Individuals with antibodies against hepatitis B core antigen as the only serological marker for hepatitis B infection: High percentage of carriers of hepatitis B and C virus. J Hepatol. 1995;23:14-20.
104. Mosley JW, Stevens CE, Aach RD, et al. Donor screening for antibody to hepatitis B core antigen and hepatitis B virus infection in transfusion recipients. Transfusion. 1995;35:5-12.
105. Dodson SF, Issa S, Araya V, et al. Infectivity of hepatic allografts with antibodies to hepatitis B virus. Transplantation. 1997;64:1582-4.
106. Paterlini P, Gerken G, Nakajima E, et al. Polymerase chain reaction to detect hepatitis B virus DNA and RNA sequences in primary liver cancers from patients negative for hepatitis B surface antigen. N Engl J Med. 1990;323:80-5.
107. Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology. 2002;36:1408-15.
108. Perrillo RP, Schiff ER, Davis GL, et al. A randomized, controlled trial of interferon alfa-2b alone and after prednisone withdrawal for the treatment of chronic hepatitis B. The Hepatitis Interventional Therapy Group. N Engl J Med. 1990;323:295-301.
109. Gauthier J, Bourne EJ, Lutz MW, et al. Quantitation of hepatitis B viremia and emergence of YMDD variants in patients with chronic hepatitis B treated with lamivudine. J Infect Dis. 1999;180:1757-62.
110. Werle B, Cinquin K, Marcellin P, et al. Evolution of hepatitis B viral load and viral genome sequence during adefovir dipivoxil therapy. J Viral Hepat. 2004;11:74-83.
111. van Nunen AB, Hansen BE, Suh DJ, et al. Durability of HBeAg seroconversion following antiviral therapy for chronic hepatitis B: Relation to type of therapy and pretreatment serum hepatitis B virus DNA and alanine aminotransferase. Gut. 2003;52:420-4.
112. Lai CL, Dienstag J, Schiff E, et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis. 2003;36:687-96.
113. Hunt CM, McGill JM, Allen MI, et al. Clinical relevance of hepatitis B viral mutations. Hepatology. 2000;31:1037-44.
114. Mutimer D, Pillay D, Dragon E, et al. High pre-treatment serum hepatitis B virus titre predicts failure of lamivudine prophylaxis and graft re-infection after liver transplantation. J Hepatol. 1999;30:715-21.
115. Roche B, Samuel D, Gigou M, et al. De novo and apparent de novo hepatitis B virus infection after liver transplantation. J Hepatol. 1997;26:517-26.
116. Brechot C, Degos F, Lugassy C, et al. Hepatitis B virus DNA in patients with chronic liver disease and negative tests for hepatitis B surface antigen. N Engl J Med. 1985;312:270-6.
117. Wright TL, Mamish D, Combs C, et al. Hepatitis B virus and apparent fulminant non-A, non hepatitis. Lancet. 1992;339:952-5.
118. Wong DK, Cheung AM, O’Rourke K, et al. Effect of alpha interferon treatment in patients with hepatitis B e antigen-positive chronic hepatitis B: A meta-analysis. Ann Intern Med. 1993;119:312-23.
119. Lau DT, Everhart J, Keiner DE, et al. Long-term follow up of patients with chronic hepatitis B treated with interferon alfa. Gastroenterology. 1997;113:1660-7.
120. Lok AS, McMahon BJ. AASLD Practice Guidelines. Chronic hepatitis B. Hepatology. 2007;45:507-39.
121. Liaw YF, Leung N, Guan R, et al. Asian-Pacific consensus statement on the management of chronic hepatitis B: A 2005 update. Liver International. 2005;25:472-89.
122. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of chronic hepatitis B. J Hepatol. 2009;50:227-42.
123. Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 Update. Clin Gastroenterol Hepatol. 2008;6:1315-41.
124. Perrillo RP, Lai CL, Liaw YF, et al. Predictors of HBeAg loss after lamivudine treatment for chronic hepatitis B. Hepatology. 2002;36:186-94.
125. Cooksley WG. Treatment with interferons in patients with hepatitis B. Semin Liver Dis. 2004;24(Suppl 1):45-53.
126. Janssen HL, van Zonneveld M, Senturk H, et al. Pegylated interferon alfa-2b, alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomized trial. Lancet. 2005;365:123-9.
127. Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682-95.
128. Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351:1206-17.
129. Kao JH, Wu NH, Chen PJ, et al. Hepatitis B genotypes and the response to interferon therapy. J Hepatol. 2000;33:998-1002.
130. Buster EH, Flink HJ, Cakaloglu Y, et al. Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon a-2b. Gastroenterology. 2008;135:459-67.
131. Erhardt A, Ludwig AD, Brunetto M, et al. HBV genotypes are the strongest predictors of response to interferon-alfa treatment: Multivariate evaluation in 1229 hepatitis B patients. [abstract]. Hepatology, 48, 2008:700A
132. Lai CL, Chien RN, Leung NW, et al. A one-year trial of lamivudine for chronic hepatitis. Engl J Med. 1998;339:61-8.
133. Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 1999;341:1256-63.
134. Liaw YF, Leung NW, Chang TT, et al. Effects of extended lamivudine therapy in Asian patients with chronic hepatitis B. Gastroenterology. 2000;119:172-80.
135. Marcellin P, Chang TT, Lim SG, et al. Adefovir dipivoxil for the treatment of hepatitis antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808-16.
136. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med. 2003;348:800-7.
137. Marcellin P, Chang T, Lim S, et al. Adefovir dipivoxil (ADV) 10 mg for the treatment of patients with HBeAg+ chronic hepatitis B: Continued efficacy beyond 48 weeks [Abstract 840]. In: Programs and Abstracts from the 53rd Annual Meeting of the American Association for the Study of Liver, November 1-5, 2002, Boston.
138. Westland C, Delaney W4th, Yang H, et al. Hepatitis B virus genotypes and virologic response in 694 patients in phase III studies of adefovir dipivoxil. Gastroenterology. 2003;125:107-16.
139. Angus P, Vaughan R, Xiong S, et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292-7.
140. Hadziyannis S, Tassopoulos N, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B. N Engl J Med. 2005;352:2673-81.
141. Locarnini S, Qi X, Arterburn S, et al. Incidence and predictors of emergence of adefovir resistant HBV during 4 years of adefovir dipivoxil (ADV) therapy for patients with chronic hepatitis B [abstract]. J Hepatol. 2005;42:17.
142. Marcellin P, Chang TT, Lim SG, et al. Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2008;48:750-8.
143. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131:1743-51.
144. Yang H, Westland CE, Das K, et al. In vitro characterization and molecular modeling analysis of adefovir resistance mutation rtN236T in the HBV polymerase [Abstract 054]. In: Programs and Abstracts from the Hep Dart Meeting, December 14-8, 2003, Kauai, Hawaii.
145. Perrillo R, Hann HW, Mutimer D, et al. Adefovir dipivoxil added to ongoing lamivudine in chronic hepatitis B with YMDD mutant hepatitis B virus. Gastroenterology. 2004;126:81-90.
146. Schiff ER, Lai CL, Hadziyannis S, et al. Adefovir dipivoxil therapy for lamivudine-resistant hepatitis B in pre- and post-liver transplantation patients. Hepatology. 2003;38:1419-27.
147. Fung SK, Chae HB, Fontana RJ, et al. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J Hepatol. 2006;44:283-90.
148. Lampertico P, Vigano M, Manenti E, et al. Low resistance to adefovir combined with lamivudine: A 3 year study of 145 lamivudine-resistant hepatitis B patients. Gastroenterology. 2007;133:1445-51.
149. Lampertico P, Vigano M, Manenti E, et al. Adefovir rapidly suppresses hepatitis B in HBeAg-negative patients developing genotypic resistance to lamivudine. Hepatology. 2005;42:1414-19.
150. Chang TT, Gish RG, de Man R, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med. 2006;354:1001-10.
151. Lai CL, Shouval D, Lok AS, et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2006;354:1011-20.
152. Gish RG, Lok AS, Chang TT, et al. Entecavir therapy for up to 96 weeks in patients with HBeAg-positive chronic hepatitis B. Gastroenterology. 2007;133:1437-44.
153. Tenney DJ, Rose RE, Baldick CJ, et al. Two-year assessment of entecavir resistance in lamivudine-refractory hepatitis B virus patients reveals different clinical outcomes depending on the resistance substitutions present. Antimicrob Agents Chemother. 2007;51:902-11.
154. Lai CL, Leung N, Teo EK, et al. A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology. 2005;129:528-36.
155. Lai CL, Gane E, Liaw YF, et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357:2576-88.
156. Liaw YF, Gane E, Leung N, et al. 2-year GLOBE trial results: Telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology. 2009;136:486-95.
157. Heathcote EJ, Gane EJ, deMan RA. Two year tenofovir disoproxil fumarate (TDF) treatment and adefovir dipivoxil (ADV) switch data in HBeAg-positive patients with chronic hepatitis B (study 103), preliminary analysis. [abstract]. Hepatology, 48, 2008:376A
158. Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442-55.
159. Van Bommel F, Trojan J, Feucht HH, et al. Tenofovir shows limited efficacy in treatment of HBV infections resistant against adefovir. [abstract]. Hepatology, 46, 2007:664A
160. Parsonage MJ, Wilkins EG, Snowden N, et al. The development of hypophosphatemic osteomalacia with myopathy in two patients with HIV infection receiving tenofovir therapy. HIV Med. 2005;6:341-6.
161. Winston J, Deray G, Hawkins T, et al. Kidney disease in patients with HIV infection and AIDS. Clin Infect Dis. 2008;47:1449-57.
162. Gupta SK. Tenofovir-associated Fanconi syndrome: Review of the FDA adverse event reporting system. AIDS Patient Care STDS. 2008;22:99-103.
163. Raffi F, Snow A, Borroto-Esoda K, et al. Anti-HBV activity of emtricitabine (FTC) in patients co-infected with HIV and hepatitis B virus [Abstract 215]. In: Programs and Abstracts of the 2nd IAS Conference on Pathogenesis and Treatment, July 13-16, 2003, Paris.
164. Gish R, Leung N, Wang C, et al. Antiviral activity, safety, and incidence of resistance in chronically infected hepatitis B patients (CHB) given once daily emtricitabine for 2 years [abstract]. Hepatology. 2003;36:372A.
165. Lee HS, Chung YH, Lee KS, et al. A 12-week randomized placebo controlled double blind trial of clevudine in patients with chronic hepatitis B with post-treatment follow up for 24 weeks [Abstract 052]. In: Programs and Abstracts from the Hep Dart Meeting, December 14-18, 2003, Kauai, Hawaii.
166. Marcellin P, Mommeja-Marin H, Sacks SL, et al. A phase II dose-escalating trial of clevudine in patients with chronic hepatitis B. Hepatology. 2004;40:140-8.
167. Yoo BC, Kim JH, Kim TH, et al. Clevudine is highly efficacious in hepatitis B e antigen-negative chronic hepatitis B with durable off-therapy viral suppression. Hepatology. 2007;46:1041-8.
168. Zoulim F, Perrillo R, Hepatitis B. Reflections on the current approach to antiviral therapy. J Hepatol. 2008;48:S2-19.
169. Keeffe EB, Zeuzem S, Koff RS, et al. Report of an international workshop: Roadmap for management of patients receiving oral therapy for chronic hepatitis B. Clin Gastroenterol Hepatol. 2007;5:890-7.
170. Yim HJ, Hussain M, Liu Y, et al. Evolution of multi-drug resistant hepatitis B virus during sequential therapy. Hepatology. 2006;44:703-12.
171. Sung JJ, Lai JY, Zeuzem S, et al. Lamivudine compared with lamivudine and adefovir dipivoxil for the treatment of HBeAg-positive chronic hepatitis B. J Hepatol. 2008;48:728-35.
172. Su GG, Pan KH, Zhao KF, et al. Efficacy and safety of lamivudine treatment for chronic hepatitis B in pregnancy. World J Gastroenterol. 2004;10:910-12.
173. van Zonneveld M, van Nunen AB, Niesters HG, et al. Lamivudine treatment during pregnancy to prevent perinatal transmission of hepatitis B virus infection. J Viral Hepat. 2003;10:294-7.
174. Hoofnagle JH, Doo E, Liang TJ, et al. Management of hepatitis B: Summary of a clinical research workshop. Hepatology. 2007;45:1056-75.
175. Perrillo R, Tamburro C, Regenstein F, et al. Low-dose, titratable interferon alfa in decompensated liver disease caused by chronic infection with hepatitis B virus. Gastroenterology. 1995;109:908-16.
176. Buster EH, Hansen BE, Buti M, et al. Peginterferon alfa-2b is safe and effective in HBeAg-positive chronic hepatitis B patients with advanced fibrosis. Hepatology. 2007;46:388-94.
177. Chu CM, Liaw YF. Hepatitis B virus-related cirrhosis: Natural history and treatment. Semin Liv Dis. 2006;26:142-52.
178. Bica I, McGovern B, Dhar R, et al. Increasing mortality due to end-stage liver disease in patients with HIV infection. Clin Infect Dis. 2001;32:492-7.
179. Thio CL, Locarnini S. Treatment of HIV/HBV coinfection: Clinical and virologic issues. AIDS Rev. 2007;9:40-53.
180. McMahon MA, Jilek BL, Brennan TP, et al. The HBV drug entecavir-effects on HIV-1 replication and resistance. N Engl J Med. 2007;356:2614-21.
181. Chu CJ, Lee SD. Hepatitis B virus/hepatitis C virus coinfection: Epidemiology, clinical features, viral interactions and treatment. J Gastroenterol Hepatol. 2008;23:512-20.
182. Potthoff A, Wedemeyer H, Boecher WO, et al. The HEP-NET B/C coinfection trial: A prospective multicenter study to investigate the efficacy of pegylated interferon alfa-2b and ribavirin in patients with HBV/HCV coinfection. J Hepatol. 2008;49:688-94.
183. Bocher WO, Herzog-Hauff S, Herr W, et al. Regulation of the neutralizing anti-hepatitis B surface (HBs) antibody response in vitro in HBs vaccine recipients and patients with acute chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 1996;105:52-8.
184. Szmuness W, Stevens CE, Harley EJ, et al. Hepatitis B vaccine: Demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N Engl J Med. 1980;303:833-41.
185. Hadler SC, Francis DP, Maynard JE, et al. Long-term immunogenicity and efficacy of hepatitis B vaccine in homosexual men. N Engl J Med. 1986;315:209-14.
186. Seaworth B, Drucker J, Starling J, et al. Hepatitis B vaccines in patients with chronic renal failure before dialysis. J Infect Dis. 1988;157:332-7.
187. Wilson CC, Olson WC, Tuting T, et al. HIV-1 specific CTL responses primed in vitro by blood derived dendritic cells and Th1-biasing cytokines. J Immunol. 1999;162:3070-8.
188. Rahman F, Dahmen A, Herzog-Hauff S, et al. Cellular and humoral immune responses induced by intradermal or intramuscular vaccination with the major hepatitis B surface antigen. Hepatology. 2000;31:521-7.
189. Hepatitis B virus: A comprehensive strategy for eliminating transmission in the United States through universal childhood vaccination. Recommendations of the Immunization Practices Advisory Committee (ACIP). MMWR Recomm Rep. 1991;40(RR-13):1-25.
190. Recommendations for identification and public health management of persons with chronic hepatitis B virus infection. MMWR Recomm Rep. 2008;57(RR-08):1-20.
191. Lopez P, Rubiano L, del Pilar Rubio M, et al. Immunogenicity and reactogenicity of DTPw-HB/Hib vaccine administered to Colombian infants after a birth dose of hepatitis B vaccine. Expert Rev Vaccines. 2002;1:277-83.
192. Murdoch DL, Goa K, Figgitt DP. Combined hepatitis A and B vaccines: A review of their immunogenicity and tolerability. Drugs. 2003;63:2625-49.
193. Rizzetto M, Canese MG, Arico S, et al. Immunofluorescence detection of a new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18:997-1003.
194. Nath N, Mushawar IK, Fang CT, et al. Antibodies to delta antigen in asymptomatic hepatitis B surface antigen-reactive blood donors in the United States and their association with other markers of hepatitis B virus. Am J Epidemiol. 1985;122:218-25.
195. Gaeta GB, Stroffolini T, Chiaramonte M, et al. Chronic hepatitis D: A vanishing disease? An Italian multicenter study. Hepatology. 2000;32:824-7.
196. Sagnelli E, Stroffolini T, Ascione A, et al. Decrease in HDV endemicity in Italy. J Hepatol. 1997;26:20-4.
197. Navascues CA, Rodriguez M, Sotorrio NG, et al. Epidemiology of hepatitis D virus infection: Changes in the last 14 years. Am J Gastroenterol. 1995;90:1981-4.
198. Coppola RC, Manconi PE, Piro R, et al. HCV, HIV HBV, and HDV infection in drug addicts. Eur J Epidemiol. 1994;10:279-83.
199. Shakil AO, Hadziyannis S, Hoofnagle JH, et al. Geographic distribution and genetic variability of hepatitis delta virus genotype I. Virology. 1997;234:160-7.
200. Zhang YY, Tsega E, Hansson BG. Phylogenetic analysis of hepatitis D viruses indicating a new genotype I subgroup among African isolates. J Clin Microbiol. 1996;34:3023-30.
201. Lee CM, Changchien CS, Chung JC, et al. Characterization of a new genotype II hepatitis delta virus from Taiwan. J Med Virol. 1996;49:145-54.
202. Casey JL, Niro GA, Engle RE, et al. Hepatitis B virus (HBV)/hepatitis D virus (HDV) coinfection in outbreaks of acute hepatitis in the Peruvian Amazon Basin: The roles of HDV genotype III and HBV genotype F. J Infect Dis. 1996;174:920-6.
203. Wang KS, Choo QL, Weiner AJ, et al. Structure, sequence and expression of hepatitis delta (delta) viral genome. Nature. 1986;323:508-14.
204. Taylor JM. The structure and replication of hepatitis delta virus. Annu Rev Microbiol. 1992;46:253-76.
205. Luo GX, Chao M, Hsieh SY, et al. A specific base transition occurs on replicating hepatitis delta virus RNA. J Virol. 1989;64:1021-7.
206. Chao M, Hsieh SY, Taylor J. Role of two forms of hepatitis delta virus antigen: Evidence for a mechanism of self-limiting genome replication. J Virol. 1990;64:5066-9.
207. Samuel D, Zignego AL, Reynes M, et al. Long-term clinical and virological outcome after liver transplantation for cirrhosis caused by chronic delta hepatitis. Hepatology. 1995;21:333-9.
208. Tang JR, Cova L, Lamelin JP, et al. Clinical relevance of the detection of hepatitis delta virus RNA in serum by RNA hybridization and polymerase chain reaction. J Hepatol. 1994;21:953-60.
209. Borghesio B, Rosina F, Smedile A, et al. Serum immunoglobulin M antibody to hepatitis D as a surrogate marker of hepatitis D in interferon-treated patients and in patients who underwent liver transplantation. Hepatology. 1998;27:873-6.
210. Huang YH, Wu JC, Sheng WY, et al. Diagnostic value of anti-hepatitis D virus (HDV) antibodies revisited: A study of total and IgM anti-HDV compared with detection of HDV-RNA by polymerase chain reaction. J Gastroenterol Hepatol. 1998;13:57-61.
211. Rizzetto M, Ponzetto A, Forzani I. Epidemiology of hepatitis delta virus: Overview. Prog Clin Biol Res. 1991;364:1-20.
212. Realdi G, Fattovich G, Hadziyannis S, et al. Survival and prognostic factors in 366 patients with compensated cirrhosis type B: A multicenter study. The investigators of the European Concerted Action on Viral Hepatitis (EUROHEP). J Hepatol. 1994;21:656-66.
213. DiMarco V, Lo Iacono O, Camma C, et al. The long-term course of chronic hepatitis B. Hepatology. 1999;30:257-64.
214. Tassopoulos NC, Koutelou MG, Macagno S, et al. Diagnostic significance of IgM antibody to hepatitis delta virus in fulminant hepatitis B. J Med Virol. 1990;30:174-7.
215. Wu JC, Chen TZ, Huang YS, et al. Natural history of hepatitis D viral superinfection: Significance of viremia detected by polymerase chain reaction. Gastroenterology. 1995;108:796-802.
216. Shakil AO, Hadziyannis S, Hoofnagle JH, et al. Geographic distribution and genetic variability of hepatitis delta virus genotype I. Virology. 1997;234:160-7.
217. Farci P, Chessa L, Balistrieri C, et al. Treatment of chronic hepatitis D. J Viral Hepatitis. 2007;14(Suppl 1):58-63.
218. Carosi G, Rizzetto M. Treatment of chronic hepatitis B: Recommendations from an Italian workshop. Dig Liver Dis. 2008;40:603-17.
219. Yurdaydin C, Bozkaya H, Onder FO, et al. Treatment of chronic delta hepatitis with lamivudine vs. lamivudine + interferon vs. interferon. J Viral Hepatitis. 2008;15:314-21.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 78 Hepatitis B and D
HEPATITIS B
An estimated 400 million persons are carriers of hepatitis B virus (HBV) in the world today; of these, 75% reside in Asia and the Western Pacific. Effective vaccines against HBV have been available since the early 1980s, but perinatal and early life exposures continue to be major sources of infection in high-prevalence areas. High-risk behaviors such as promiscuous heterosexual contact and injection drug use account for many new cases in young adults. Fulminant acute hepatitis B accounts for several hundred deaths per year in the United States, and chronic HBV infection accounts for one million deaths worldwide each year from complications of end-stage liver disease, including hepatocellular carcinoma (HCC). Hepatitis B is the chief cause of cirrhosis and HCC in the world today, and nationwide vaccination has been shown to diminish greatly the number of new cases of infection and HCC in Taiwanese children.1 Universal hepatitis B vaccination is likely to have the greatest impact on liver disease–related mortality in future generations.
EPIDEMIOLOGY
Geographic Distribution and Sources of Exposure
The prevalence of hepatitis B varies markedly around the world. In highly endemic regions, such as Southeast Asia (excluding Japan), China, and much of Africa, 8% or more of the population are chronic HBV carriers, and the lifetime risk of infection ranges from 60% to 80%.2 In these areas, perinatal transmission and horizontal spread among children are the major sources of infection. Approximately 60% of the world’s population reside in these areas where HBV is highly endemic.3 Regions of intermediate risk include parts of southern and eastern Europe, the Middle East, Japan, the Indian subcontinent, much of the former Soviet Union, and northern Africa. In intermediate-risk areas, the lifetime risk of infection is between 20% and 60%. Persons of all age groups are infected, but as in high-risk areas, most infections occur during infancy or early childhood. Regions of low prevalence are North America, western Europe, certain parts of South America, and Australia. In these areas, the lifetime risk of HBV infection is less than 20%, and transmission is primarily horizontal (i.e., between young adults). Sexual transmission is the main mode of transmission in Europe and North America, and injection drug use is a major contributor to new cases as well.4
Transmission of infection from an HBV carrier mother to her neonate accounts for the majority of new infections in the world today. Sixty to ninety percent of hepatitis B surface antigen (HBsAg)-positive mothers who are hepatitis B e antigen (HBeAg)-positive transmit the disease to their offspring, whereas mothers who are positive for antibody to HBeAg (anti-HBe) transmit the disease less frequently (15% to 20%) (see later discussion of diagnosis). Other less common sources of infection are household contact with an HBV carrier, hemodialysis, exposure to infected health care workers, tattooing, body piercing, artificial insemination, and receipt of blood products or organs. Since routine screening of the blood supply was implemented in the early 1970s, transfusion-associated hepatitis B has become rare in the United States. Hepatitis B can be transmitted by blood that tests negative for HBsAg but positive for antibody to hepatitis B core antigen (anti-HBc) because of low levels of circulating HBV DNA in such blood.5 HBsAg-negative blood that is positive for anti-HBc is excluded from the donor pool in the United States and many countries around the world. In 0% to 30% of persons who are seropositive for anti-HBc alone, HBV DNA is detectable in serum by polymerase chain reaction (PCR) testing.6
HBV is transmitted efficiently by percutaneous and mucous membrane exposure to infectious body fluids. The virus is 100 times as infectious as human immunodeficiency virus (HIV) and 10 times as infectious as hepatitis C virus (HCV). HBeAg seropositivity indicates a higher risk of transmission from mother to child, after needlestick exposure, and in the setting of household contact. HBV DNA has been detected by sensitive techniques such as PCR testing in most body fluids, except for stool that has not been contaminated with blood. Although HBV replicates primarily in hepatocytes, the presence of replicative intermediates and virally encoded proteins in other sites, such as the adrenal gland, testis, colon, nerve ganglia, and skin, suggests that a vast extrahepatic reservoir for infectious virus exists.7 Small amounts of HBV DNA have been demonstrated in peripheral mononuclear cells and liver tissue years after apparent resolution of chronic infection.8,9 Extrahepatic localization of low levels of replicating virus explains the relatively high rate of transmission of infection from anti-HBc–positive organ donors.10
Rates of Infection in the United States
The incidence of hepatitis B has been declining in the United States over the past decade because of universal vaccination of newborns, adult vaccination programs for high risk persons, changes in sexual lifestyle, refinements in blood screening procedures, and the availability of virus-inactivated blood components.11 Most striking has been the decrease among children and health care workers, groups with the highest rates of vaccination. Data from the Centers for Disease Control and Prevention (CDC) indicate that more than 95% of pregnant women in the United States are tested for HBsAg, and infant vaccine coverage levels are now equivalent to those of other vaccines in the childhood schedule. Nonetheless, an estimated 78,000 new HBV infections occurred in 2001, with the highest incidence rates among sexually active young adults (20 to 29 years old) and higher rates occurring among black and Hispanic persons than in white persons.12 Since 1995, approximately 40% of cases of acute hepatitis B reported to the CDC were caused by intimate contact among heterosexuals, 15% to 20% were related to intravenous drug use, and 12% occurred in men who have sex with men. No identifiable source of exposure was demonstrated in approximately 15% of cases. Nearly one third of prison inmates have been infected with hepatitis B, and 2% are chronically infected.
According to the third National Health and Nutrition Examination Survey (1988 to 1994), one or more serologic markers of HBV infection were demonstrated in 4.9% of the U.S. population, and the prevalence of chronic infection was 0.2%.13 Traditional estimates based on the results of blood donation screening in the late 1970s also indicated a prevalence rate for chronic infection of 0.2% to 0.4% in the United States. Although traditional estimates are that the number of HBV carriers in the United States is between 1.25 and 1.5 million, this figure is likely to be a serious underestimate because of changing immigration patterns and underrepresentation of certain minority groups in field surveys. For example, 15 million Asians live in the United States, and even a conservative estimate that the prevalence in this group is 5% would raise the overall number of HBV carriers in the United States by more than 750,000.
CLINICAL OUTCOMES
Definitions
In common usage, the term HBV carrier has often been used to refer to persons persistently infected with HBV who have normal serum aminotransferase levels. (They are sometimes inappropriately referred to as “healthy” HBV carriers.) Because the nomenclature is potentially confusing, the proposal has been made that the carrier state be categorized as inactive or active. Inactive carriers are patients who have evidence of HBV replication on a PCR-based assay only (but not with a less sensitive non–PCR-based assay) and normal or only mildly elevated serum aminotransferase values (see later).14 Long-term follow-up of inactive carriers suggests that the majority of these patients do not have progressive liver disease and do not experience complications. Some of these patients, however, ultimately have one or more episodes of reactivated hepatitis in which the levels of viremia and serum aminotransferase activity increase. Also, some patients with the inactive carrier state may demonstrate HCC. Active carriers, on the other hand, have evidence of HBV replication on non–PCR-based assays for HBV DNA, intermittently or persistently elevated serum aminotransferase levels, and evidence of chronic hepatitis in a liver biopsy specimen.
Clinical Sequelae of Acute Hepatitis B Virus Infection
The age at which a person becomes infected with HBV is a principal determinant of the clinical outcome. HBV infection in adults with an intact immune system is likely to cause clinically apparent acute hepatitis B; only 1% to 5% of these persons become chronically infected.4 By contrast, as many as 95% of infected neonates become chronic HBV carriers because of immunologic tolerance to the virus.
In adults, fulminant liver failure caused by acute hepatitis B occurs in less than 1% of cases, but this group still accounts for 5% of all cases of acute liver failure and approximately 400 deaths annually in the United States.15 Rapid viral elimination may result in clearance of HBsAg from serum by the time of initial presentation. In these cases, the accurate diagnosis of fulminant hepatitis B may require testing with immunoglobulin (Ig) M antibody to hepatitis B core antigen (HBcAg) (IgM anti-HBc) (see later discussion of serologic markers of infection).16 The rate of spontaneous survival in acute liver failure caused by hepatitis B is only approximately 20%. Liver transplantation has resulted in survival rates of 50% to 60%. Recurrent disease in the allograft is now uncommon because of administration of hepatitis B immune globulin (HBIG) and potent orally administered antiviral agents (see later and Chapters 93 and 95).
Clinical Sequelae of Chronic Hepatitis B Virus Infection
The presence of active viral replication and long-standing necroinflammatory liver disease caused by HBV strongly influences the rate of progression to cirrhosis. The major determinant of survival is the severity of the liver disease when the patient first comes to medical attention.17 Cirrhosis is associated with decreased survival and an increased frequency of HCC. Five- and 20-year survival rates of 55% and 25%, respectively, have been reported in patients with cirrhosis at presentation, whereas rates of 97% and 63%, respectively, have been reported for those with mild (noncirrhotic) disease.18 Survival rates differ most dramatically between patients with compensated and decompensated cirrhosis. In one study, an 84% five-year survival rate was reported for patients with compensated HBV-related cirrhosis, compared with 14% for patients with cirrhosis complicated by ascites, jaundice, encephalopathy, or a history of variceal bleeding.19 Multivariate analyses in several large cohort studies have identified age, ascites, hyperbilirubinemia, and other features of advanced liver disease as correlating independently with survival in patients with HBV-related cirrhosis. Interferon-induced clearance of HBeAg (see later) has been associated with prolongation of survival without complications or the need for liver transplantation.20
Clearance of HBsAg in patients with HBV-related cirrhosis has been associated with an excellent prognosis, including improvement in liver histology and function, a decreased chance of viral reactivation, and prolonged survival.17 Even HBsAg clearance, however, is not an absolute safeguard against the future development of HCC in persons who already have cirrhosis.21
MOLECULAR BIOLOGY
HBV is a small DNA virus that belongs to the Hepadnaviridae family. Other members of this virus family are human HBV-like agents that infect the woodchuck, ground and tree squirrels, woolly monkey, crane, heron, Ross goose, and duck. HBV is a small (3.2-kilobase [kb]) virus with a DNA genome that has a relaxed, circular, partially double-stranded configuration (Fig. 78-1). The genome is composed of four open reading frames (ORFs) and has a compact design in which several genes overlap and use the same DNA to encode different viral proteins. The four viral genes are the core, surface, X, and polymerase genes. The core gene encodes the core nucleocapsid protein, which is important in viral packaging and production of HBeAg. The surface gene encodes the pre-S1, pre-S2, and S proteins (comprising the large [L], middle [M], and small [S] surface proteins). The X gene encodes the X protein, which has transactivating properties and may be important in hepatic carcinogenesis. The polymerase gene has a large ORF (approximately 800 amino acids) and overlaps the entire length of the surface ORF. It encodes a large protein with functions that are critical for packaging and DNA replication (including priming, RNA- and DNA-dependent DNA polymerase, and RNase H activities).
Although HBV is a DNA virus, replication occurs through an RNA intermediate and requires an active viral reverse transcriptase/polymerase enzyme. The mutation rate is higher for HBV than for other DNA viruses (an estimated 1010 to 1011 point mutations per day).22 Complete HBV genomic sequencing has identified a large number of mutations within the HBV genome, many of which are silent or do not alter the amino acid sequence of encoded proteins. Because of genomic overlap, however, some of the silent mutations in one ORF (for example, the polymerase gene) may result in an amino acid substitution in an overlapping ORF (surface gene), although with currently uncertain clinical implications.
Figure 78-2 illustrates the life cycle of HBV. The initial phase of hepadnaviral infection involves the attachment of mature virions to host cell membranes. The human receptor for HBV remains unknown. Entry of the virus results from fusion of the viral and host membranes as the nucleocapsid is released into the cytoplasm. Mechanisms of intracellular transport of viral genome into the nucleus are poorly understood, but the first step in genomic replication involves conversion of the relaxed circular form of HBV DNA into a double-stranded, covalently closed circular form (cccDNA). The cccDNA, which serves as the template for viral transcription, is the major form of viral DNA in the nucleus of infected hepatocytes. Subgenomic (0.7 to 2.4 kb) and pregenomic (3.5 kb) RNA molecules are transcribed from this template. The L protein is translated from the 2.4 kb RNA, the M and S proteins from the 2.1 kb RNA, and the X protein from the 0.7 kb transcript. The pregenomic RNA serves as the template for reverse transcription as well as the messenger RNA (mRNA) for translation of the core and polymerase proteins; the precore RNA codes for the precore gene product.
Hepatitis B Virus Genotypes
A genetic classification based on comparisons of complete genomes has demonstrated eight genotypes of HBV, designated A through H (Table 78-1).23 Several methods have been used for HBV genotyping, including a commercially available line probe assay. Genotypic differences are based on an intergroup divergence of 8% or more in the complete nucleotide sequence. Genotype A is the predominant genotype in northern Europe and the United States. Genotypes B and C are confined to populations in eastern Asia and the Far East, but changes in immigration patterns have resulted in an influx of Asian HBV carriers with these genotypes into the United States.24 Genotype D is found worldwide but is especially prevalent in the Mediterranean area, Middle East, and south Asia. Genotype E is indigenous to western sub-Saharan areas, and genotype F prevails in Central America. Cases of genotype G have been reported in the United States and France. Genotype H has been described in Mexico.
Table 78-1 Hepatitis B Genotypes (A-H) and Their Possible Clinical Associations
| Geographic Distributions |
HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus.
Clinical associations appear to exist with the various genotypes (see Table 78-1). Currently, the strongest clinical associations appear to be that (1) HBeAg seroconversion occurs earlier in patients with genotype B than in those with genotype C and (2) response to therapy with interferon varies with genotype (see later).25 The viral genotype also has implications for the frequency of precore and core mutations (see later) and may have an effect on the frequency of HCC.
Mutations of the Hepatitis B Virus Genome
Hepatitis B Surface Antigen Mutants
Mutations in the surface gene can result in changes in the antibody-binding domain. Accordingly, both virus neutralization by polyclonal antibody to HBsAg and testing for HBsAg by methods that depend on antibody binding can be affected. Large-scale vaccination programs in regions endemic for HBV have revealed a 2% to 3% frequency of vaccine escape mutants that result from alterations in the “a” determinant of the HBsAg protein, which is the major neutralizable epitope. The typical mutation results in the substitution of glycine for arginine at amino acid position 145; this substitution prevents binding of neutralizing antibodies (i.e., antibody to HBsAg [anti-HBs]). The clinical significance of these mutants for neonatal vaccination programs is highly controversial because the frequency of these variants among HBV-infected mothers whose infants respond to vaccination has been found to be similar to that of mothers whose infants do not respond. The “a” determinant mutants also are proposed to have clinical relevance after liver transplantation for hepatitis B. As many as 50% of patients in whom recurrent HBV infection develops despite the use of HBIG have been shown to have these escape mutants, and the rate at which the mutations are detected appears to correlate with the length of time over which HBIG is repeatedly administered.26
Mutations in the Precore, Basal Core Promoter, and Core Genes
Mutations in the precore and basal core promoter regions of the HBV genome can influence the production of HBeAg. A precore mutation results in a stop codon at nucleotide 1896 that abolishes the synthesis of HBeAg,27 whereas mutations in the basal core promoter at nucleotides 1762 and 1764 decrease HBeAg synthesis by approximately 70% while maintaining pregenomic RNA levels.28 Both types of mutations have been observed in cases of severe or fulminant hepatitis, which has been attributed to the loss of the immune-tolerizing effects of HBeAg antigen (see later). The presence of core promoter mutations has been linked to a significantly increased risk of HCC.29 Precore and basal core promoter mutants have been described in the same patients and are particularly common in Asian and European patients with chronic hepatitis B.30 A large serosurvey of HBV carriers residing in the United States has found that precore and core promoter mutations are common (frequencies of 27% and 44%, respectively), depending on the ethnicity and places of birth of the patients. Both mutant forms of HBV were observed to occur far more commonly in HBeAg-negative patients (precore mutation in 38% of HBeAg-negative versus 9% of HBeAg-positive patients; core promoter mutation in 51% versus 36%).31 In addition to these mutations, upstream mutations in the core gene can influence immunologic responses to HBV. Core gene mutations have been shown to block recognition of HBV by cytotoxic T lymphocytes (CTLs), a key mode of viral clearance. Therefore, the mutations contribute to HBV immune escape and possibly influence the response to interferon.32 Core gene mutations within the immunodominant epitopes of the HBV nucleocapsid also can affect CD4+ T-cell reactivity.33
Hepatitis B Virus DNA Polymerase Mutants
The polymerase gene encodes a DNA polymerase enzyme needed for encapsidation of viral RNA into core particles, conversion of the pregenomic viral RNA into a negative strand of viral DNA (reverse transcription), and conversion of this first HBV DNA strand into a second DNA strand of positive polarity. In general, the HBV reverse transcriptase function of the polymerase gene is highly conserved because major mutations that impair the efficiency of viral replication lead to selection pressure against such variant forms. As indicated earlier, HBV has a high rate of replication (1011 virions per day) and low replication fidelity, meaning that it has a propensity to mispair nucleotide bases when it reverse transcribes viral RNA to DNA. HBV DNA polymerase also lacks any proofreading activity, so it cannot repair its mistakes. Therefore, when a nucleotide base is misplaced, it remains in the growing viral DNA strand as a base mutation, and the new HBV DNA molecule has a different sequence from the original (wild-type) genome. The overall error rate of HBV DNA polymerase is estimated to be 1 per 10,000 nucleotides copied, which translates to the potential for 10 million base-pair errors per day in an infected person. All possible single-base mutations can be produced in a 24-hour period, although many such mutations will yield nonviable viruses.34
Mutations in the sequence of HBV polymerase can lead to drug resistance to nucleoside analogs used to treat HBV infection because some HBV polymerase mutants have decreased susceptibility to these drugs and are selected during treatment. The mutations in the sequence of HBV DNA polymerase that confer drug resistance result in amino acid substitutions in the reverse transcriptase domain of the enzyme. The changes in the structure of the enzyme, in turn, are thought to sterically inhibit binding of the drugs to their active sites. The amino acids in the reverse transcriptase are numbered from 1 to 344, and an amino acid identity is given by the single letter amino acid code. By convention, substitutions in reverse transcriptase are designated by the wild-type amino acid, followed by the number of the amino acid, followed by the substituted amino acid. For example, for the nucleoside analog lamivudine (see later), two types of mutations occur at nucleotide position 204 of domain C (the catalytic site of the polymerase) that result in substitution of the amino acid methionine (M) for either isoleucine (I) or valine (V). These mutations are designated M204I and M204V, respectively, and are referred to collectively as YMDD mutants; the letters stand for the amino acids (Y = tyrosine, M = methionine, D = aspartate) in the C domain. The M204V mutation tends to occur in conjunction with a mutation in domain B that results in substitution of leucine (L) with methionine (L180M). The M204I mutation or the combined M204V-L180M mutations result in marked resistance to the effect of lamivudine (>10,000-fold reduction in susceptibility). After prolonged exposure to adefovir, drug resistant mutations in domains B and D are selected (A181V/T and N236T). Other nucleotide substitutions have been described that are instrumental for telbivudine and entecavir resistance (Fig. 78-3).
The inherent mutability of HBV indicates that single and even double polymerase mutants preexist as minor “quasispecies” even before treatment of HBV infection is begun. Because of the limitations in the sensitivities of current genotype assays, these mutants would not be detectable until they are selected and expanded under the pressure of drug treatment. Resistance to lamivudine is found in approximately 20% of patients after one year of treatment but in nearly 70% after five years (see Fig. 78-3).35 Rates of resistance to adefovir, a nucleotide analog, are 0% at one year and 29% after five years.36 The efficacy of both of these drugs against HBV is impaired by a single nucleotide substitution. The more mutations necessary for drug resistance (indicating a higher genetic barrier to resistance), the slower the emergence of and lower overall rate of resistance. For example, resistance to entecavir, another nucleoside analog, occurs in less than 1% of patients at five years because the preexistence of lamivudine-resistant mutations and one or more additional mutations in the viral polymerase gene are required for resistance.37 Persistent infection with drug-resistant HBV ultimately is associated with progression of disease and blunting of hepatic histologic improvement with antiviral therapy.38 Severe flares of hepatitis have also been reported after the emergence of drug-resistant mutants,39 and acquisition of these mutants may lead to rapidly progressive liver disease after liver transplantation (Table 78-2).40 Horizontal transmission of these mutants, which can complicate drug therapy in secondarily infected persons, is also possible.
Table 78-2 Hepatitis Flares in Patients with Chronic Hepatitis B
| CAUSE OF FLARES | COMMENT |
|---|---|
| Spontaneous | Factors that precipitate antecedent viral replication are unclear |
| Immunosuppressive therapy | Flares are often observed during withdrawal; requires preemptive antiviral therapy |
| Antiviral therapy for HBV | |
| Interferon | Flares are often observed during the second to third month; may herald virologic response |
| Lamivudine | |
| During treatment | Flares are no more common than with placebo |
| YMDD mutant | Can have severe consequences in patients with advanced liver disease |
| On withdrawal* | Flares are caused by rapid re-emergence of wild-type HBV; can have severe consequences in patients with advanced liver disease |
| HIV treatment | Flares can occur with HAART or with immune reconstitution; in addition, HBV increases the risk of antiretroviral drug hepatotoxicity |
| Genotypic variation | |
| Precore and core promoter mutants | Fluctuations in serum ALT levels are common with precore mutants |
| Superinfection with other hepatitis viruses | May be associated with suppression of HBV replication |
ALT, alanine aminotransferase; HIV, human immunodeficiency virus; HAART, highly active antiretroviral therapy; HBV, hepatitis B virus; YMDD, tyrosine-methionine-aspartate-aspartate.
PATHOGENESIS
HBV is generally not a cytopathic virus, and the severity of HBV-associated liver disease is considered to be related to the intensity of the host immunologic response to the virus. Whereas both humoral and cellular immune responses are needed for effective clearance of the virus, the cellular immune response appears to be the arm principally involved in the pathogenesis of disease. The immunologic response to HBV encompasses both an innate, or nonspecific, response (for example, natural killer cells and interferons) and an adaptive immune response, including antibodies to viral antigens, human leukocyte antigen (HLA) class II–restricted CD4+ T cells, and HLA class I–restricted CD8+ CTLs.41 Induction of the antigen-specific T-cell response is thought to occur in lymphoid organs, where the host T cells encounter viral peptide antigens (or epitopes) that are presented by antigen-presenting cells such as dendritic cells, B cells, and macrophages. This process results in the maturation and expansion of T cells that are specific for these viral epitopes and is followed by their migration to the liver, where they perform their effector function.
During acute HBV infection, most HBV DNA molecules are cleared rapidly from the liver via noncytopathic mechanisms mediated by cytokines that are released initially by cells of the innate immune system42 and later by liver-infiltrating HBV-specific CD8+ cells. Cell-mediated immune responses are efficient in self-limited infection because the responses are vigorous, multispecific, and oriented toward type 1 helper T (Th1) cells. Persons with chronic HBV infection, by contrast, exhibit infrequent, narrowly focused, and weak HBV-specific T-cell responses.43 In chronic hepatitis B, the majority of mononuclear cells in liver infiltrates of patients with chronic hepatitis B at any given time are non–antigen-specific.44
CD8+ CTLs are thought to contribute to the disease process in the liver and result in apoptosis of infected hepatocytes. To be recognized by the CD8+ CTLs, targeted hepatocytes must present viral epitopes as short peptides that have been endogenously processed and fit within the peptide-binding groove of the class I major histocompatibility complex (MHC) molecules.45 The binding of the CTL T-cell receptor (TCR) to the peptide-MHC complex on the hepatocyte surface can then result in the direct killing of the infected cell and release of potent antiviral cytokines by the activated CTL. Recognition by MHC class II–restricted CD4+ helper T cells requires the appropriate presentation of viral peptides in the context of class II MHC molecules. The CD4+ cells produce antiviral cytokines and provide help in neutralizing antibody production. Antibody neutralization limits intrahepatic spread of virus during primary infection and serves an important role in preventing reinfection.
NATURAL HISTORY
Four phases of HBV infection have been described: immune tolerance, immune clearance, the inactive carrier state, and reactivation (Fig. 78-4).
Patients who acquire the infection in the perinatal period often have high serum levels of HBV DNA without biochemical evidence of active hepatitis and are considered to be immune tolerant to HBV. When followed longitudinally, many of these patients ultimately exhibit elevated serum aminotransferase levels in association with histologic evidence of chronic hepatitis. The trigger mechanisms for this apparent change in tolerance are poorly understood but likely reflect changes in the immune reactivity of the host. Experiments in transgenic mice suggest that HBeAg induces a state of immunologic tolerance to HBV in neonates.46 Perinatal transmission of HBeAg has been considered to be a potential mechanism for the immune-tolerant state. As persons enter the immune clearance phase, HBV DNA concentrations diminish, serum alanine aminotransferase (ALT) levels rise, and hepatic histologic activity, reflecting immune-mediated lysis of infected hepatocytes, increases. The duration of this second phase varies, often lasting many years.
The third phase (inactive HBV carrier state) occurs after seroconversion from HBeAg to anti-HBe and is usually preceded by a marked reduction in serum HBV DNA to levels that are detectable only by PCR methodology, followed by normalization of serum ALT levels and resolution of liver necroinflammation. This phase may last a lifetime, but a proportion of patients ultimately undergo spontaneous or immunosuppression-mediated reactivation of HBV replication with reappearance of high levels of HBV DNA in serum, with or without HBeAg seroreversion and a rise in serum ALT levels. For unclear reasons, precore or core promoter mutants that prevent or down-regulate HBeAg production may be selected during or after HBeAg seroconversion (see earlier).47
A key event in the natural history of HBeAg-positive chronic hepatitis is seroconversion of HBeAg to anti-HBe, which is associated with marked reduction in HBV replication and biochemical and histologic remission in the majority of patients. Regression of liver fibrosis occurs gradually months to years after HBeAg seroconversion.48 Most studies have found that the mean annual rate of spontaneous HBeAg seroconversion ranges from 8% to 15% in HBV-infected children or adults with serum ALT elevations.
Longitudinal studies of untreated patients with predominantly HBeAg-positive chronic hepatitis B have shown that the frequency of development of cirrhosis ranges from 2 to 5 per 100 person-years and the five-year cumulative frequency of progression to cirrhosis from 8% to 20%.49 The rate of cirrhosis has been suggested to be higher in HBeAg-negative patients than in HBeAg-positive patients. Risk factors for the development of cirrhosis have been identified; of these, older age, the stage of fibrosis at presentation, and ongoing HBV replication with persistent or intermittent detection of HBV DNA by a non–PCR-based assay are perhaps the most important clinically. Combined infection with hepatitis D virus (HDV [see later]), HCV, or HIV and concomitant alcohol abuse have also been linked to a higher rate of development of cirrhosis.
When cirrhosis develops, two major complications may occur: hepatic decompensation and HCC. In a large European cohort with HBV-related compensated cirrhosis, the five-year cumulative frequency of hepatic decompensation was 16%, and the incidence per 100 person-years was 3.3.50 Similar rates have been reported in Asians. The cumulative five-year frequency of HCC can be as high as 14%.50 Factors associated with an increased risk of HCC include male gender, age more than 45 years, having a first-degree relative with HCC, the presence of cirrhosis, HBeAg positivity, and reversion from anti-HBe to HBeAg positivity.51 HCC can still develop in HBsAg-positive persons with none of the identified risk factors, but less frequently. In addition, HCC has been described in persons who lose HBsAg. Recommendations about ultrasonography and alpha fetoprotein screening for HCC are controversial, but in general, screening is recommended in all patients with cirrhosis and in male HBV carriers older than age 40 years in whom the likely route of transmission has been perinatal or early childhood exposure; some authorities recommend screening after age 30 years in highly viremic patients when perinatal acquisition is suspected (see Chapter 94).
ALT as a Surrogate Marker for Disease Activity
The serum ALT level has been used conventionally as a measure of disease activity in patients with chronic hepatitis B. Use of the standard reference range (0 to 40 U/L), however, can be misleading for evaluating HBV-related disease activity. A serum ALT level within the normal range is an imperfect surrogate marker for the absence of disease activity because determination of standard reference ranges has not traditionally taken into account increased body mass index (BMI), diabetes mellitus, and other features, such as alcohol intake, that tend to inflate values in a “normal” reference population (see Chapter 73). An insurance record-based study in Korea that included more than 140,000 persons who were followed for eight years demonstrated that all-cause liver-related mortality is increased when the serum ALT level exceeds 20 U/L in women and 30 U/L in men.52 This finding is particularly relevant to the management of Asians with hepatitis B and viremia. Because these patients tend to have a small body mass, a normal serum ALT value according to the standard laboratory reference range may be misleading. Such patients often have been excluded from treatment and assumed to be immune tolerant (that is, without liver disease). Studies in Asia and the United States have shown that as many as 20% to 30% of HBV carriers with persistently normal serum ALT levels and serum HBV DNA levels >104 copies/mL have stage 2 or greater (of 4) inflammation and stage 2 or greater (of 4) fibrosis on a liver biopsy specimen.53 Moreover, Asian HBV carriers with high-normal serum ALT levels (>0.5 × upper limit of normal [ULN]) have been shown to have more fibrosis on liver biopsy specimens than do those with low-normal serum ALT levels (<0.5 × ULN) and often demonstrate higher serum ALT elevations on prolonged follow-up.53–55 These data are consistent with the hypothesis that viremic HBV carriers who acquired HBV infection early in life and who have high-normal serum ALT levels may represent a subgroup of patients who have already entered the immune clearance phase and are not in the immune-tolerant phase. This occurrence should be suspected in particular in persons older than age 35 to 40 because immune tolerance often subsides after two to three decades of infection.54 Liver biopsy can be a useful tool to distinguish persons in the immune clearance phase despite normal or near-normal ALT levels but with active liver disease from those in the immune tolerant phase and absence of active liver disease (see Fig. 78-4).
HBV DNA Level and Long-Term Complications
Population-based Asian cohort studies have established that the serum HBV DNA level is the single best predictor of future progression to cirrhosis and HCC in HBV-infected persons.56,57 In a prospective cohort study, more than 3600 HBV carriers from Taiwan, of whom 60% were male, 70% were older than age 40, 85% were HBeAg negative, and 95% had normal serum ALT levels, were followed for a mean of 11 years. The calculated relative risks for cirrhosis and HCC were shown to correlate with the level of HBV DNA on entry into the study when compared with a reference population of HBV carriers in whom HBV DNA was undetectable in serum by a PCR assay.57 Even serum HBV DNA levels as low as 10,000 copies/mL (equivalent to 2000 IU/mL) were associated with a higher relative risk of cirrhosis and HCC. The relative risk of HCC was highest in persons with a serum HBV DNA level of more than 1 million copies/mL and intermediate in those in whom follow-up serum samples indicated spontaneous reduction of the serum HBV DNA level from greater than 100,000 copies/mL to less than 10,000 copies/mL. These data can be interpreted to mean that both the duration and level of viremia are important risk factors for the development of HCC. The data also suggest that suppression of serum HBV DNA levels, whether spontaneously or as a result of antiviral therapy, lowers the risk of HCC. The serum HBV DNA level remained a significant predictor of cirrhosis and HCC even after adjustment for patient age, gender, serum ALT level, and HBeAg status. Other studies in Chinese HBV carriers have demonstrated that persons in whom liver decompensation and HCC develop often have modest serum ALT elevations that are frequently below the recommended threshold (2 × ULN) for antiviral treatment in current practice guidelines (see later).58
On the basis of these data, some authorities have suggested that all persons who acquire HBV infection early in life and who are age 45 to 50 years or older and demonstrate serum HBV DNA levels of 100,000 or more copies/mL should receive long-term therapy with a nucleoside analog to prevent cirrhosis and HCC.59,60 In a landmark study,61 more than 600 Asian patients with advanced fibrosis and a serum HBV DNA level greater than 100,000 copies/mL were randomized in a ratio of 2 : 1 to active treatment with lamivudine or placebo. Treatment was planned for 5 years, but the study was discontinued after a mean duration of only 32 months because disease progression and HCC occurred significantly more frequently in the group of patients randomized to placebo.61 Therapeutic benefit was independent of the serum ALT level at baseline. These data strongly suggest that continuing suppression of serum HBV DNA by antiviral therapy can alter the long-term outcome of active chronic hepatitis B.
CLINICAL FEATURES
Acute Hepatitis B
The incubation period of acute hepatitis B varies from a few weeks to 6 months (average, 60 to 90 days), depending on the amount of replicating virus in the inoculum. The disease may be more severe in patients coinfected with other hepatitis viruses and in those with established underlying liver disease.62 Abstention from alcohol is usually recommended, but the chance of an uneventful recovery does not appear to be affected by the consumption of moderate amounts of alcohol (20 to 30 g daily) during the convalescent phase.63 Acute infections are heralded by a serum sickness–like prodrome of fever, arthralgia or arthritis, and rash, which is most commonly maculopapular or urticarial, in 10% to 20% of patients. This prodrome results from circulating HBsAg–anti-HBs complexes that activate complement and are deposited in the synovium and walls of cutaneous blood vessels.64 These features generally abate before the manifestations of liver disease and peak serum aminotransferase elevations are observed. Jaundice develops in only about 30% of patients.
Clinical symptoms and jaundice generally disappear after one to three months, but some patients have prolonged fatigue even after serum ALT levels return to normal. In general, elevated serum ALT levels and serum HBsAg titers decline and disappear together, and in approximately 80% of cases, HBsAg disappears by 12 weeks after the onset of illness.65 In 5% to 10% of cases, HBsAg is cleared early and is no longer detectable by the time the patient first presents to a health care provider. Persistence of HBsAg after six months implies development of a carrier state, with only a small likelihood of recovery during the next 6 to 12 months. Delayed clearance of HBsAg has been reported to be preceded by a decline in HBsAg titers.
After clinical recovery from acute hepatitis B and HBsAg seroconversion, HBV DNA often remains detectable in serum as determined by a PCR assay (see later discussion of diagnosis). After resolution of acute hepatitis, the numbers of HBV-specific CD4+ and CD8+ cells in blood and liver decrease rapidly. Nonetheless, T-cell responsiveness remains high on re-encounter with HBV antigens, indicating that traces of virus can maintain the CTL response indefinitely after clinical recovery, thereby exerting control over the virus and preventing reactivated infection.41,66
Fulminant hepatitis occurs in less than 1% of cases (see Chapter 93). Fulminant hepatitis B generally occurs within four weeks of the onset of symptoms and is associated with encephalopathy, multiorgan failure, and a high mortality rate (>80%) if not treated by liver transplantation. Patients older than age 40 years appear to be more susceptible than younger persons to “late-onset liver failure,” in which encephalopathy, renal dysfunction, and other extrahepatic complications of severe liver insufficiency become manifest over the course of several months. The pathogenic mechanisms of fulminant hepatitis are poorly understood but are presumed to involve massive immune-mediated lysis of infected hepatocytes. This proposed mechanism may explain why many patients with fulminant hepatitis B have no evidence of HBV replication in serum at presentation.
Chronic Hepatitis B
Physical findings may be normal, or hepatosplenomegaly may be found. In decompensated cirrhosis, spider angiomata, jaundice, ascites, and peripheral edema are common. Liver biochemical test results are usually completely normal during the inactive HBV carrier state. In contrast with patients in the immune-tolerant phase of HBV infection, most patients in the immune-clearance phase of chronic HBV infection have mild to moderate elevations in serum AST and ALT levels. During exacerbations of disease, serum ALT levels may be as high as 1000 U/L or more, and the clinical and laboratory picture is indistinguishable from that of acute hepatitis B, including the presence in serum of IgM anti-HBc. Progression to cirrhosis should be suspected whenever hypersplenism, hypoalbuminemia (in the absence of nephropathy), or prolongation of the prothrombin time is found. The serum AST level is typically higher than the serum ALT level in patients with advanced cirrhosis (see Chapter 73).
Extrahepatic Manifestations
Extrahepatic syndromes seen in association with acute or chronic hepatitis B are important to recognize because they may occur without clinically apparent liver disease and can be mistaken for independent disease processes in other organ systems. The pathogenesis of these extrahepatic disorders has not been fully elucidated but likely involves an aberrant immunologic response to extrahepatic viral proteins.67 Many of the extrahepatic manifestations (e.g., arthritis, dermatitis, glomerulonephritis, polyarteritis nodosa, cryoglobulinemia, papular acrodermatitis, and polymyalgia rheumatica) are observed in association with circulating immune complexes that activate serum complement. Antiviral therapy may be indicated for persistent symptoms.
Glomerulonephritis
Several types of glomerular lesions have been described in patients with chronic HBV infection; membranous glomerulonephritis and membranoproliferative glomerulonephritis are the most common.68 Renal biopsy specimens have demonstrated immune complex deposition and cytoplasmic inclusions in the glomerular basement membrane. The immune complexes activate complement and production of cytokines with a subsequent inflammatory response. Nephrotic syndrome is the most common presentation of HBV-associated glomerulonephritis. In affected children, renal failure at presentation is almost always mild, and a history of clinical liver disease is uncommon. Nevertheless, liver biopsy specimens almost always demonstrate varying degrees of chronic hepatitis. The diagnosis of HBV-associated glomerulonephropathy is usually established by serologic evidence of HBV antigens or antibodies, the presence of immune-complex glomerulonephritis in a renal biopsy specimen, and the demonstration of glomerular deposits of one or more HBV antigens, such as HBsAg, HBcAg, or HBeAg, by immunohistochemistry. Most patients have detectable HBeAg in serum and, in addition, demonstrate low serum C3 and occasionally low C4 levels. The renal disease typically resolves in months to several years, especially in children. Often, resolution occurs in conjunction with HBeAg seroconversion. Rarely, however, renal failure may ensue. The natural history of HBV-related glomerulonephritis in adults has not been well defined, but several reports suggest that glomerular disease is often slowly and relentlessly progressive.69 Successful treatment has been accomplished with interferon alpha and has been linked to long-term control of HBV replication.70 Therapy with nucleoside analogs has resulted in improved renal function and diminished proteinuria.
Cryoglobulinemia
Type II cryoglobulins consist of a polyclonal IgG and monoclonal IgM, whereas type III cryoglobulins contain polyclonal IgG and rheumatoid factor. Type II and type III cryoglobulinemia have been associated with hepatitis B, but the association is uncommon. In a large patient cohort, the frequency of cryoglobulinemia was significantly higher in patients with chronic HCV infection (54%) than in patients with chronic HBV infection (15%) (see Chapter 79). Cryoglobulinemia may be associated with systemic vasculitis (purpura, arthralgias, peripheral neuropathy, and glomerulonephritis) but is often paucisymptomatic or asymptomatic. Interferon has been used successfully to treat symptomatic cryoglobulinemia in association with chronic hepatitis B. Experience with nucleoside analog therapy has not been reported.
Histopathologic Features
The only histologic feature noted on routine light microscopy that is specific for chronic hepatitis B is the presence of ground-glass hepatocytes (Fig. 78-5). This morphologic finding results from accumulation of HBsAg particles (20 to 30 nm in diameter) in the dilated endoplasmic reticulum. Because of high levels of cysteine in HBsAg, ground-glass cells have a high affinity for certain dyes, such as orcein, Victoria blue, and aldehyde fuchsin. Ground-glass hepatocytes also may be seen in HBV carriers, in whom they may be detected in up to 5% of cells. When present in abundance, ground-glass hepatocytes often indicate active viral replication.71 Immunofluorescence and electron microscopic studies have shown HBcAg inside the hepatocyte nuclei of affected cells. During periods of intense hepatitis activity, cytoplasmic core antigen staining is generally observed. After successful treatment of HBV infection with a nucleoside analog, the cytoplasmic core antigen staining often disappears, but nuclear core antigen staining may remain, indicating persistence of the HBV cccDNA template.
Acute Flares in Chronic Hepatitis B
Chronic hepatitis B is often punctuated by sudden flares of disease activity that are reflected by a rise in serum aminotransferase levels. Although a uniform definition is lacking, a flare has frequently been described as a rise in serum ALT levels to at least two times the baseline value. Spontaneous flares are an important part of the natural history of hepatitis B because when they occur repeatedly, they lead to histologic progression. Acute flares in chronic hepatitis B occur in association with a number of circumstances and clinical situations (see Table 78-2). Most flares result from a change in the balance between immunologic responses to HBV and the level of viral proliferation. Acute flares in chronic hepatitis B that are not explainable by infection with other hepatotropic viruses often occur as a secondary response to increased levels of replicating wild-type or mutant HBV or as a result of therapeutic intervention with immunologic modifiers such as interferon, glucocorticoids, and cancer chemotherapy. In some instances, the event that initiates an acute exacerbation of chronic hepatitis B may not be readily identifiable, and the flare is considered spontaneous.
Spontaneous Flares
Spontaneous exacerbations of chronic hepatitis B often result from reactivated infection, and an increase in serum HBV DNA levels often precedes an increase in serum aminotransferase levels. Histologic evidence of acute lobular hepatitis superimposed on the changes of chronic viral hepatitis is frequently observed during these flares.72 IgM anti-HBc, a marker that is often diagnostic of acute viral hepatitis, may also appear in serum at this time.73
The reasons for reactivated infection are unknown but likely relate to subtle changes in the immunologic control of viral replication. Reactivation seems to occur more commonly in persons who are infected with HIV.74 In persons who acquire HBV infection early in life, flares become more common during adulthood, presumably because of a breakdown in immune tolerance to HBV.75
Flares also can occur in patients who are in the replicative phase of infection (i.e., already positive for HBV DNA and HBeAg in serum). In these instances, HBV replication intensifies, serum HBV DNA levels rise, and liver biochemical deterioration occurs, often without the subsequent loss of HBeAg. Multiple episodes of reactivation and remission have been shown to accelerate the progression of chronic hepatitis B and are particularly likely to occur in patients infected with the precore mutant form of chronic hepatitis B (see earlier).47
Immunosuppressive Therapy-Induced Flares
Reactivation of HBV replication is a well-recognized complication in patients with chronic HBV infection who receive cytotoxic or immunosuppressive therapy.76 Suppression of the normal immunologic responses to HBV leads to enhanced viral replication and is thought to result in widespread infection of hepatocytes by HBV. On discontinuation of immunosuppressive medications, such as cancer chemotherapy, antirejection drugs, and glucocorticoid therapy, immune competence is restored and infected hepatocytes are rapidly destroyed. The more potent the immunosuppression, the higher the level of viral replication and, thus, the greater the potential for serious clinical consequences of sudden withdrawal of the therapy and restoration of immunologic competence. Postmortem studies of liver tissue from patients with severe liver injury have documented sparse staining of viral antigens, suggesting that the patients were in an active state of immune clearance.77
The vast majority of patients who experience immunosuppressive therapy–induced flares have been positive for HBsAg in serum before treatment, but some studies have described the reappearance of HBsAg in patients who were initially positive for anti-HBs, anti-HBc, or both.78 Reactivated hepatitis in patients who are negative for HBsAg and positive for either anti-HBc or anti-HBs is explainable by the possible latency of HBV in liver and mononuclear cells and the large extrahepatic reservoir of HBV. Chemotherapy given to patients with cancer who are HBV carriers is associated with an increased risk of liver-related morbidity and mortality.79
Reactivated hepatitis B also occurs in patients who are given immunosuppressive medications to prevent organ transplant rejection. The frequency of reactivated hepatitis appears to be particularly high in patients who undergo bone marrow transplantation because of extensive immunologic conditioning before transplantation and treatment of graft-versus-host disease.80 Rarely, fibrosing cholestatic hepatitis, a rapidly progressive form of liver injury associated with inordinately high levels of HBsAg and HBcAg in liver tissue, may develop in such patients.81
Acute flares of hepatitis B resulting from cancer chemotherapy and other immunosuppressive drugs are often detected after substantial increases in serum aminotransferase levels have been noted. Initiation of antiviral treatment after detection of such biochemical abnormalities has little effect on reducing liver injury because much of the immunologic response to HBV and viral elimination has already occurred. Instead, the key to management lies in anticipating the occurrence of a flare, initiating antiviral treatment preemptively (e.g., 4 to 6 weeks before the start of chemotherapy), and continuing the treatment for 6 to 12 months after completion of chemotherapy.82
Antiviral Therapy–Induced Flares
Interferon
Interferon-induced flares of chronic hepatitis B occur in approximately one third of treated patients and result from the immunostimulatory properties of the drug. Flares generally occur during the second or third month of treatment with conventional preparations of interferon. Flares also occur in patients treated with pegylated interferon and have been reported to occur more frequently in patients infected with HBV genotype A than with other genotypes. This finding may explain the higher rate of sustained virologic response and HBsAg clearance in patients infected with HBV genotype A.83 Serum ALT flares have been shown to be a predictor of sustained virologic response, particularly in patients with high levels of viremia.84 Flares tend to be particularly common in patients who have decompensated liver disease, with rates as high as 50% reported in one series.85 Such flares are frequently associated with clinical deterioration in the patient.
Nucleoside Analogs
Serum ALT flares occur in approximately 20% to 25% of patients after withdrawal of nucleoside analogs such as lamivudine and adefovir. These flares probably are caused by rapid resurgence of wild-type HBV, and although generally well tolerated, they have been associated with serious clinical exacerbations in patients with advanced liver disease.86 Reinstitution of therapy is often associated with a decline in serum HBV DNA and aminotransferase levels. Flares have been seen to follow the emergence of YMDD mutant HBV during therapy with lamivudine (see earlier).87
[/not-level-membership-for-gastroenterology-and-hepatology-category]
