Chapter 82 Hepatic tumors in childhood
Overview
An appreciation of hepatic segmental anatomy (see Chapter 1A, Chapter 1B ) has led to major advances in hepatic surgery, especially for tumors. In addition, the irresistible but still mysterious stimulus to hepatic regeneration has allowed larger and more extensive resections. In small infants, 85% of the liver can be removed safely, greatly increasing the scope for cure. Advances have also been made in understanding tumor biology and clinical behavior. This chapter addresses benign and malignant tumors of the liver and biliary tract encountered in infancy, childhood, and adolescence.
History
Between 310 and 280 bce, Herophilus and Erasistratus first presented a description of hepatic anatomy. In the late 1880s, hepatic resection was attempted, but advances in anesthesia and antisepsis would be required before a successful outcome could be realized. Wendel used avascular anatomic planes in the liver to perform a hepatic resection in 1910 (McClusky et al, 1997), and progress in hepatic surgery has been based on an appreciation of hepatic segmental anatomy as described by Couinaud (Bismuth, 1982; Couinaud, 1986, 1992; see also the Introduction to this text). The distribution of the portal and hepatic veins delimits each hepatic segment, which has a unique portal vein and hepatic artery branch and bile duct. Knowledge of this anatomy allows control of the vascular structures before division of the hepatic parenchyma, making major hepatic resections feasible (see Chapter 1A, Chapter 1B ).
Bloodless hepatic dissection is crucial in infants and small children, who may have a total blood volume of less than 1 L. In the pediatric literature, Martin and Woodman (1969) reported that hepatoblastomas could be treated by hepatic lobectomy, and modern hepatic resection is soundly based on principles of segmental hepatic anatomy.
A second important historic finding was the sensitivity of some tumors, especially hepatoblastoma, to systemic chemotherapy (Fegiz et al, 1977). Chemotherapy caused significant reductions in tumor volume, and previously unresectable hepatoblastomas became resectable (Filler et al, 1991; Reynolds, 1995). Presently, the standard of practice is to administer neoadjuvant systemic chemotherapy to patients with hepatoblastoma, unless the tumor is clearly resectable at diagnosis.
In addition, appreciation of the biology of hepatic epithelial malignancies has increased, especially the differences between hepatoblastoma and hepatocellular carcinoma (HCC). These differences include the relatively good prognosis of hepatoblastoma compared with HCC in childhood, the importance of complete surgical resection of the primary hepatic tumor, and the association of hepatoblastoma with certain clinical syndromes (Exelby et al, 1975; Koishi et al, 1996; Schneid et al, 1997; Simms et al, 1995; Tsai et al, 1996; Vaughan et al, 1995). Finally, the first application of hepatic transplantation to a childhood liver tumor was reported by Heimann and colleagues in 1987, and a series of pediatric liver tumor patients treated by hepatic transplantation was reported by Tagge and colleagues in 1992. There is continued interest in using this modality for unresectable hepatic malignancies in childhood and adolescence (Pichlmayr et al, 1995; Pinna et al, 1997; Superina & Bilik, 1996).
Malignant Tumors
Primary malignant liver tumors constitute approximately 1.1% of childhood malignancies in Western nations. The overall incidence of primary liver cancer, as published by the Surveillance Epidemiology and End Results (SEER) program, is 5 cases per 1 million children in the 0 to 4 year age group and 1 case per 1 million in the 5 to 9, 10 to 14, and 14 to 19 year age groups (National Cancer Institute, 1995). Liver cancers constitute 0.5% to 2% of all pediatric solid tumors and about 5% of abdominal tumors in childhood (Weinberg & Finegold, 1983). The distribution of the most common malignant hepatic tumors is depicted in Figure 82.1. Hepatoblastoma is the most common, and its treatment is a success story in pediatric oncology.
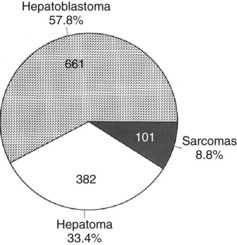
FIGURE 82.1 The frequency distribution of malignant hepatic tumors in childhood as compiled from reported large series.
(From Exelby PR, et al, 1975: Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey 1974. J Pediatr Surg 10:329-337; and Weinberg AG, Finegold MJ, 1983: Primary hepatic tumors of childhood. Hum Pathol 14:512-537.)
Hepatoblastoma
Incidence
Hepatoblastomas are the most common primary hepatic tumors of childhood, constituting 43% to 64% of all hepatic neoplasms in one large series (Mann et al, 1990; Stocker, 1994; Weinberg & Finegold, 1983). Hepatoblastoma accounts for 91% of primary hepatic tumors in children younger than 5 years (Darbari et al, 2003) but comprises less than 1% of hepatic malignancies when adult age groups are included (Kaczynski et al, 1996). The Liver Cancer Study Group (LSCG) of Japan (1987) identified 30 hepatoblastomas (0.6%) in a cohort of 4,658 patients of all ages diagnosed over a 2-year period.
Hepatoblastoma affects 1 child younger than 15 years old in 1 million per year (Finegold, 1994), and approximately 50 to 70 new cases per year are reported in the United States, with a male/female ratio of 1.7 : 1 (Lampkin et al, 1985). Although hepatoblastoma has been reported sporadically in adults (Bortolasi et al, 1996; Harada et al, 1995; Inoue et al, 1995; Kacker et al, 1995; Parada et al, 1997), the median age at diagnosis is about 18 months, and most cases occur before the age of 3 years (Exelby et al, 1975). Hepatoblastoma is the most prevalent malignant neoplasm of the fetus and neonate and results in death within 2 years if not treated (Dehner, 1978; DeMaioribus 1990; Isaacs, 1985, 2007; Patterson et al, 1985). Finally, the incidence of hepatoblastoma may be increasing (Blair & Birch, 1994). The incidence of hepatoblastoma was 0.6 per 1 million in the years 1973 through 1977, and it increased to 1.2 per 1 million from 1993 through 1997 (Darbari et al, 2003).
Hepatoblastoma may occur in siblings (Fraumeni et al, 1969; Ito et al, 1987; Napoli & Campbell, 1977; Surendran et al, 1989). It is most strongly associated with familial polyposis (Giardiello et al, 1996; Iwama & Mishima, 1994), Gardner syndrome (Hartley et al, 1990), and Beckwith-Wiedemann syndrome (Koishi et al, 1996; Tsai et al, 1996). In familial polyposis, incidence of hepatoblastoma seems to be increased in first-degree relatives of the patients with polyposis. Beckwith-Wiedemann syndrome is associated with Wilms tumor, rhabdomyosarcoma, adrenocortical carcinoma, and hepatoblastoma with a possible association between hepatoblastoma and trisomies 2, 8, 18, and 20 (Bove et al, 1996; Tomlinson, 2005).
Hepatoblastoma is also associated with low birth weight (Ikeda et al, 1997; Reynolds et al, 2004). It is unknown whether the causative agent is developmental abnormalities associated with prematurity or interventions, such as early total parenteral nutrition. These tumors also are reported in patients with congenital anomalies, such as cleft palate, and cardiovascular and renal anomalies, including multicystic kidney and absence of the right adrenal gland (Rao et al, 1989). There are also at least two reports of hepatoblastoma occurring in patients with biliary atresia (Taat et al, 2004). No evidence associates hepatoblastoma with hepatitis B or C infection or any other chronic viral hepatitis. These patients usually do not have cirrhosis or inborn errors of metabolism.
Pathology
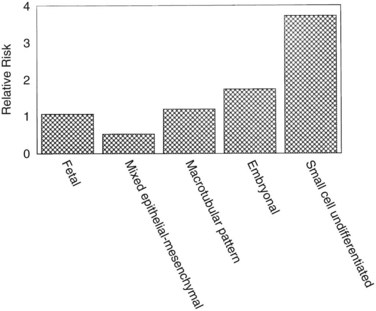
Hepatoblastomas are large tumors that can contain fibrous bands, producing a spoked-wheel appearance (Jha et al, 2009). The five histologic subtypes observed in hepatoblastoma are 1) fetal, 2) embryonal, 3) mixed epithelial, 4) mesenchymal/macrotubular, and 5) anaplastic or small-cell undifferentiated. These subtypes are differentiated based on the findings with light microscopy, but all tumor cells appear smaller than nonneoplastic hepatocytes. Extramedullary hematopoiesis is notably present and may be related to constitutive cytokine production by the tumor cells (von Schweinitz et al, 1995a). The fetal type grows in trabeculae and resembles fetal hepatic cells, whereas embryonic hepatoblastoma cells grow in incohesive sheets and resemble embryonic cells. Some hepatoblastomas contain mesenchymal tissue along with the epithelial component. Calcification also may appear in these tumors, and one patient was reported with osteosarcomatous elements in the hepatoblastoma and associated pulmonary metastases (Alcantar, 1985). The anaplastic or small-cell type consists of small, round blue cells reminiscent of neuroblastoma. This subtype is rare but particularly virulent, with a strong metastatic potential (Dehner & Manivel, 1988). The importance of subtyping in hepatoblastoma is due to the association between prognostic risk and subtype, illustrated in Figure 82.2 (Gonzalez-Crussi et al, 1982; Lack et al, 1982). Some studies have indicated that the fetal histologic subtype has a better prognosis; in contrast, patients with the rare anaplastic variant usually do poorly (Dehner & Manivel, 1988).
Basic Biology
Few cellular models of hepatoblastoma exist, and immortalized cell lines have been difficult to establish. One cell line, isolated from a human hepatoblastoma, clearly expresses the MYC and Hras1 oncogenes and epidermal growth factor receptor (EGFR) (Manchester et al, 1995). Antibodies that blocked the EGFR inhibited cell growth, but the significance of oncogene expression remains to be determined. A new cell line was established from primary hepatoblastoma tumor tissue and contains an identical genotype to tumor cells, with morphologic, molecular, and immunohistochemical confirmation.
In immunodeficient mice, hepatoblastoma is tumorigenic (Chen, 2009). Elevated hepatocyte growth factor levels have been demonstrated in the serum of 10 (45%) of 22 patients with hepatoblastoma (von Schweinitz et al, 1997a). Addition of hepatocyte growth factor causes proliferation in hepatoblastoma-derived cell lines.
Small epithelial cells, characteristic of hepatic stem cells, have been observed in human hepatoblastomas of various subtypes (Ruck & Xiao, 2002). Additionally, various genetic abnormalities have been reported in hepatoblastoma. Chromosome 8q amplification is associated with a worsened prognosis and has been correlated with overexpression of the transcription factor pleomorphic adenoma gene 1 (PLAG1) (Zatkova et al, 2004). Telomerase and its regulatory protein expression levels have been correlated with poor outcome in human hepatoblastoma (Hiyama et al, 2004), and tamoxifen may inhibit hepatoblastoma cells by reducing telomerase levels (Brandt et al, 2005).
Loss of heterozygosity on chromosome 11p15.5, the region associated with Beckwith-Wiedemann syndrome, and on chromosome 1p36 has been described in hepatoblastoma (Albrecht et al, 1994; Kraus et al, 1996). Investigations into both of these regions suggest that each may contain a tumor suppressor gene, but this has not been proven. Trisomy 20 and trisomy of all or part of chromosome 2 also have been reported (Swarts et al, 1996). In addition, an abnormality of chromosome 2q may provide a common genetic link between hepatoblastoma and rhabdomyosarcoma (Rodriguez et al, 1991). Finally, frequent genetic losses found using comparative genomic hybridization included regions 13q21-q22 (28%) and 9p22-pter (22%), and the most frequent genetic gains were on chromosomes 2q23-q23 (33%) and 1q24-q25 (28%; Gray et al, 2000). Recently, differentially expressed microRNA has been shown to be deregulated in hepatoblastoma (Magrelli et al, 2009), and mutations of β-catenin have also been recognized (Curia et al, 2008).
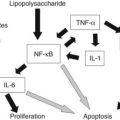
Perhaps the most exciting insight is the association between hepatoblastoma and familial adenomatous polyposis syndrome (Bala et al, 1997; Cetta et al, 1997). In one study of 13 hepatoblastomas obtained from nonfamilial adenomatous polyposis patients, 69% had mutations in the adenomatous polyposis coli (APC) gene (Oda et al, 1996). In one case of siblings with hepatoblastoma, a shared APC gene mutation was identified (Thomas et al, 2003). In addition, the well-known thrombocytosis associated with untreated hepatoblastoma is fascinating, as is the presence of extramedullary hematopoiesis in these tumors. It was shown that hepatoblastoma cells secrete interleukin (IL)-1β, which causes secretion of IL-6 from surrounding fibroblasts and endothelial cells (von Schweinitz et al, 1993). Other factors, such as erythropoietin and stem cell factor, have been localized to the cytoplasm of hepatoblastoma cells. Thrombopoietin has been identified in hepatoblastoma tissue and serum from a patient, but its correlation with the thrombocytosis associated with this neoplasm is unclear (Komura et al, 1998).
Clinical Findings
The most common presenting sign of hepatoblastoma is an asymptomatic abdominal mass. The child is often in good health, and the tumor usually is discovered incidentally, when an attentive parent, grandparent, or clinician discovers the mass on a routine examination or while bathing the child (Fabre et al, 2004). Patients with the anaplastic variant of hepatoblastoma, who often have distant metastases at diagnosis, are more frequently symptomatic. Accompanying symptoms such as pain, irritability, minor gastrointestinal disturbances, fever, and pallor occur in smaller numbers of patients. Significant weight loss is unusual, although patients may fail to thrive. In most series of hepatoblastomas and HCCs, a few patients present acutely with tumor rupture and intraperitoneal hemorrhage (Brown et al, 1993). Rarely, hepatoblastoma manifests with sexual precocity secondary to a β-human chorionic gonadotropin (hCG)-secreting tumor (Muraji et al, 1985), and one patient with a hepatoblastoma was reported presenting with a biliary fistula (Daniel & Kifle, 1989). Finally, hepatoblastoma may present as a cardiac tumor (Wang et al, 2003).
Measurement of serum α-fetoprotein (AFP) is well established as an initial tumor marker in the diagnosis of hepatoblastoma and a means of monitoring the therapeutic response (Van Tornout et al, 1993). The normal level in most laboratories is less than 20 ng/mL, whereas the AFP level at diagnosis in hepatoblastoma patients can range from normal to significantly elevated (7.7 × 106 ng/mL); it is estimated that the AFP is elevated in 84% to 91% of patients with hepatoblastoma (Lack et al, 1982). One study reported a mean AFP level in hepatoblastoma of 3 million ng/mL, whereas the mean in pediatric patients with HCC was about 200,000 ng/mL (Ortega et al, 1991). In infants younger than 1 year, the AFP is normally elevated and is highest at birth (Fig. 82.3).
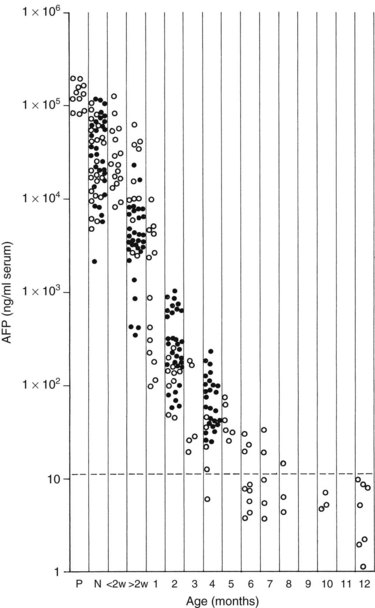
FIGURE 82.3 Graph shows the time decay of α-fetoprotein (AFP) levels in normal infants during the first year of life.
(Data from Wu JT, et al, 1981: Serum alpha fetoprotein [AFP] levels in normal infants. Pediatr Res 15:50-52.)
Some authors suggest that subfractionation more reliably indicates whether the increased AFP is secondary to a hepatoblastoma or HCC, an endodermal sinus tumor, or benign liver disease (Tsuchida et al, 1997). The half-life of AFP is about 6 days, and in one study, 24 (77%) of 31 patients had levels decline by at least 1 log before second-look surgery (Walhof et al, 1988). Of these patients, 16 (50%) of 32 eventually had AFP levels decline to normal at the end of therapy and had no clinical or radiographic evidence of hepatoblastoma at this point. Finally, 15 (94%) of 16 patients who attained a complete response also showed a decline in AFP levels of 2 logs or more before second-look surgery (Van Tornout et al, 1997). A large, early decline in AFP levels was an independent predictor of survival in multivariate analysis; it has also been stated that a low initial AFP level is associated with worse survival (von Schweinitz et al, 1995b), but this has not been confirmed in multivariate analysis (von Schweinitz et al, 1994a). Anaplastic hepatoblastomas may also be associated with lower AFP levels (Tsunoda et al, 1996).
When interpreting the AFP level, it is important to realize that normal levels are very high at birth and decrease over the first 6 months of life. Premature newborns may have AFP levels in the range of 100,000 ng/mL. Term newborns also can have relatively high levels (104 to 105 ng/mL). By 2 months of age, most infants have levels ranging from 100 to 1000 ng/mL, and by 6 months, levels should be less than 100 ng/mL. Usually, levels decrease to normal (<20 ng/mL) after 6 to 7 months, but levels may remain elevated for 1 year after birth (Ohama et al, 1997). AFP may also be elevated in the setting of liver damage, liver regeneration, or in the presence of another tumor.
Imaging
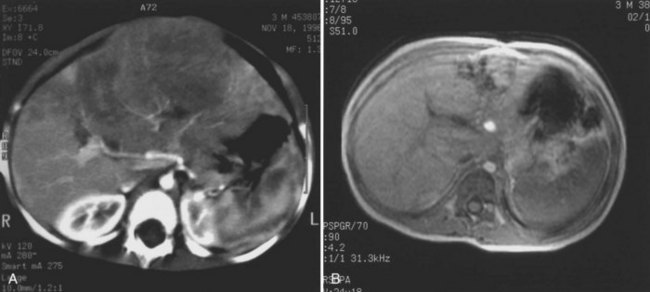
The first imaging study is usually an abdominal ultrasound (US) (see Chapter 13). If duplex technique is used, tumor vascularity can be gauged, and the hepatic veins can be assessed (Bates et al, 1990). The ultrasonographer also should search for anomalies of the genitourinary system and rule out tumor thrombus in either the vena cava or the hepatic veins. Computed tomography (CT) (see Chapter 16) is useful to identify pulmonary metastases, identify diffuse hepatic involvement, and determine resectability. Oral and intravenous contrast material is used (Fig. 82.4A). CT scans can be performed quickly and can be completed in less than 2 minutes in helical scanners; this greatly shortens the required period of sedation for infants or small children, and it has the added advantage of being a quick and reliable screening method for pulmonary metastases. Hepatoblastoma will appear as a well-demarcated tumor, without a capsule. CT angiography (CT portography) uses CT with fine cuts and an increased amount of intravenous contrast material to image hepatic tumors and the hepatic venous anatomy. CT portography may provide as much information as magnetic resonance imaging (MRI) (see Chapter 17), which is useful for evaluating hepatic lesions and their relationship to vascular structures. MRI can show the hepatic veins, vena cava, and bile ducts (Ohnuma et al, 1991). MRI of a hepatoblastoma patient after neoadjuvant chemotherapy is shown in Figure 82.4B. Positron emission tomography (PET) has been used to identify hepatoblastoma recurrence and to search for sites of metastatic disease, but it may not be reliable for lesions smaller than 6 to 10 mm (Wong et al, 2004).
Staging
In the United States, the commonly used staging system is that from the Children’s Oncology Group (COG), based on operative findings (Table 82.1). A tumor-node-metastasis (TNM) classification has also been used (Table 82.2; Brower et al, 1998). The PRE-Treatment EXTent (PRETEXT) system of disease staging has been used extensively by the International Society of Pediatric Oncology liver group (SIOPEL) (Fig. 82.5; Aronson et al, 2005). It relies on radiographic findings prior to any therapy, including surgery, and does not take into account the independent surgeon’s judgment at the time of surgery regarding resectability. This staging system is based on Couinaud’s system of segmentation of the liver and is thought to predict the degree of tumor infiltration, the extent of surgical resection, and the complexities involved in the resection (Couinaud, 1992; Otte, 2010). This system classifies the tumor into one of four categories, depending on which sections of the liver do not include tumor (Table 82.3; Roebuck et al, 2007). Additional criteria added in 2005 (Table 82.4) further classify these tumors (Otte, 2010; Roebuck, 2007).
Table 82.1 Children’s Oncology Group Staging for Hepatoblastoma
| Stage I | Complete Resection |
|---|---|
| Favorable histology | Purely fetal histology with a low mitotic index |
| Other histology | All other stage I tumors |
| Stage II | Gross total resection with microscopic residuals or total resection with preoperative or intraoperative rupture |
| Stage III | Unresectable tumors as determined by the attending surgeon, partially resected tumors with macroscopic residual involvement or any tumor with lymph node involvement |
| Stage IV | Measurable metastatic disease to lungs or other organs |
From Finegold MJ, et al, 2008: Liver tumors: pediatric population. Liver Transpl 14(11):1545-1556.
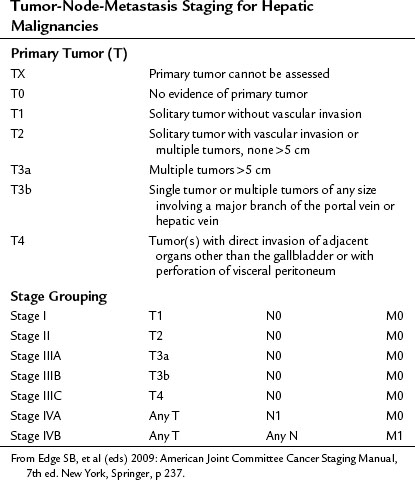
FIGURE 82.5 The PRE-Treatment EXTent (PRETEXT) staging system used by the International Society of Pediatric Oncology.
Table 82.3 PRETEXT Staging System
| PRETEXT Number | Definition |
|---|---|
| I | One section is involved, and three adjoining sections are free |
| II | One or two sections are involved, but two adjoining sections are free |
| III | Two or three sections are involved, and no two adjoining sections are free |
| IV | All four sections are involved |
PRETEXT, PRE-Treatment EXTent
From Roebuck DJ, et al, 2005: PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol 37:123-132.
Table 82.4 PRETEXT Staging: Additional Criteria
| C: Caudate lobe involvement |
IVC, inferior vena cava; HVs, hepatic veins
Reprinted with permission from Otte JB, 2010: Progress in the surgical treatment of malignant liver tumors in children.
PRETEXT was compared with pathologic findings in 110 patients and was correct in 51%, overstaged in 37%, and understaged in 12%. The authors compared this system with the Children’s Cancer Group/Pediatric Oncology Group (CCG/POG) and TNM schemes and claimed a better correlation with risk status. This study analyzed data from patients who had neoadjuvant chemotherapy, whereas a recent study from the COG analyzed data from patients with a hepatoblastoma at diagnosis and reported that both the COG stage and PRETEXT were useful prognostic indicators. The PRETEXT system has been described as showing improved predictive value for survival compared with other staging classifications (Aronson et al, 2005). Moreover, this system can be valuable for recognizing patients who are surgical candidates (PRETEXT stages I and II) and those who may benefit from lower dose chemotherapy (Meyers et al, 2009). It is recommended that all liver tumor patients in future COG studies undergo PRETEXT staging.
Treatment
Multiple studies support the effectiveness of systemic chemotherapy combined with complete surgical resection of the primary hepatic tumor (Gauthier et al, 1986; von Schweinitz et al, 1994a, 1994b, 1995b). Survival depends on removal of the primary liver tumor, when imaging suggests that complete resection is feasible.
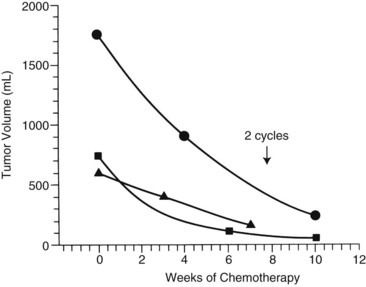
The first clinical decision is whether to initiate neoadjuvant chemotherapy or proceed with resection. Often, resection is not feasible if tumors are large and involve both hepatic lobes. Preoperative (neoadjuvant) chemotherapy results in tumor shrinkage and makes subsequent resection easier (Reynolds, 1995). In one study, the rate of shrinkage was high after initiation of chemotherapy, but it declined after two cycles had been administered (Fig. 82.6; Medary et al, 1996). Exquisite clinical judgment and good communication between members of the multidisciplinary team are crucial, because about 60% of hepatoblastomas are resectable at diagnosis.
Complete resection of the primary tumor is necessary for survival and may require extended hepatic lobectomies or complex biliary reconstructions. For hepatoblastoma, reports have suggested that gross total resection of the primary lesion may be adequate for cure in chemoresponsive tumors (Dicken et al, 2004; Schnater et al, 2002).
For resected tumors (stage I) with fetal histology, further therapy is not recommended. All other stage I tumors without pure fetal histology, in addition to stage II patients, should receive four cycles of cisplatin, 5-fluorouracil (5-FU), and vincristine (C5V). Patients with stage III and IV disease should receive four cycles of chemotherapy, followed by resection or transplantation, followed by two more cycles of chemotherapy. C5V is administered, followed by doxorubicin if there is minimal response to C5V. Recent reports have suggested the use of doxorubicin from the start in this subgroup (Finegold et al, 2008; Malogolowkin et al, 2008). The combination of cisplatin plus doxorubicin was compared with cisplatin plus 5-FU plus C5V in a combined CCG/POG (intergroup) study (Ortega et al, 2000). The efficacy was thought to be similar, but more complications resulted with the regimen containing doxorubicin, accounting for equivalent event-free survivals; however, a more detailed review of the analysis suggested that the doxorubicin-containing arm had an improved disease-specific survival. This finding implied that with better management of toxicity, patient outcome might be better with a doxorubicin-containing regimen. In addition, a clinical trial that used a combination of cisplatin and carboplatin had to be terminated because of poor tumor response, suggesting that platinum-based monotherapy, as proposed by SIOPEL, was not as effective as combination chemotherapy. Clinical trials by the COG and SIOPEL are planned to evaluate the use of doxorubicin, irinotecan, and other agents, especially in high-risk patients.
In patients with unresectable primary tumors, the use of liver transplantation is increasing. A recent analysis stated an approximate 80% long-term disease-free survival in those receiving transplantation in large, solitary, or multifocal tumors invading all four sectors of the liver (Otte et al, 2005). The United Network for Organ Sharing (UNOS) database consists of over 200 patients, with a median age of 2.9 years, who underwent orthotopic liver transplantation (OLT) for hepatoblastoma between 1987 and 2006. Approximately half of the patients had a recurrence. Overall survival was 80%, 69%, and 66% at 1, 5, and 10 years, respectively (Austin et al, 2006). In a recent multicenter review, 147 patients with hepatoblastoma were analyzed after liver transplantation. In almost three quarters of these patients, the original surgery was OLT; the remaining patients either had residual disease after prior resection or had recurrent tumor. The first group of patients had an improved outcome, with 82% overall disease-free survival, compared with 30% in the second group. Smaller, single-center reports have reinforced findings that liver transplantation for hepatoblastoma has the best outcome when done as the primary procedure rather than as a salvage (D’Alessandro et al, 2007; Otte et al, 2004; Pham et al, 2007; Reyes et al, 2000).
Transplantation, however, does require the use of immunosuppressive treatment, which comes along with its own set of side effects. Moreover, there is in increased chance of thrombosis of the hepatic artery after transplantation in children (Jain et al, 2006). The main causes of mortality after transplantation, accounting for 54% of this population, are metastases and recurrence (Austin et al, 2006). The COG is continuing to investigate the role of liver transplantation in hepatoblastoma. A global database has been instituted to aid in this endeavor.
In one study, the 1-year survival for patients presenting with metastases was no different from that in patients with localized tumors (Van Tornout et al, 1997). In another study by SIOPEL, the 5-year overall and event-free survival for children with hepatoblastoma who presented with pulmonary metastases were 57% and 28%, respectively (Perilongo et al, 2000). This study suggested that 25% to 30% of patients with synchronous pulmonary metastases are curable. It is still necessary to resect the primary liver tumor, and pulmonary metastasectomy should be done only if the primary site is controlled (Schnater et al, 2002). Many pulmonary metastases resolve with chemotherapy, but thoracotomy and resection are sometimes required for larger or persistent metastatic lesions (Passmore et al, 1995).
No current prospective studies are underway for pulmonary metastasectomy, but one recent study describes the advantage of pulmonary metastasectomy for diagnosed lesions that remain after neoadjuvant therapy (Meyers, 2007). Some radiation oncologists have treated pulmonary metastases with external-beam radiotherapy in an approach similar to that used for Wilms tumors, but with 18 to 20 Gy administered (Habrand et al, 1992); however, this may be associated with significant pulmonary toxicity and has not resulted in cure. There is no reported cure in patients with spread to sites outside of the lung or local lymph nodes.
Outcome
Following a gross total resection, the 5-year event-free survival is 83%, but this drops to 41% in patients with tumor remaining after surgery (Ortega et al, 2000). Some patients with microscopic residual tumor are curable with continued chemotherapy and may benefit from external-beam radiotherapy to the primary hepatic site. Resection of many hepatoblastomas may be easier after chemotherapy, and complete resection of the primary hepatic tumor is necessary for survival. In multivariate analysis, factors that have been independent predictors of worse prognosis include a high TNM stage, unresectable tumor, bilobar involvement and multifocality, AFP less than 100 ng/mL or greater than 105 ng/mL, distant metastases, embryonal versus fetal histology, and vascular invasion (von Schweinitz et al, 1997b). The COG has reported a 3-year event-free survival of 90%, 50%, and 20% for stage I to II, III, and IV, respectively (Malogolowkin et al, 2008).
Future Directions
The uses of various novel treatments are currently being investigated. First, transcatheter selective arterial chemoembolization, which involves the direct injection of chemotherapeutic agents to the tumor, hypothetically decreases systemic toxicity. The average decrease in tumor size was 84% in one study (Clouse et al, 1994), and unresectable hepatoblastomas may become resectable with this intervention (Berthold et al, 1986). Doxorubicin, cisplatin, and fluorodeoxyuridine have been promising, as these agents have a high hepatic extraction; case reports exist with striking results. For example, Yokomori and colleagues (1991) describe the total regression of a tumor in a 4-month-old infant with fetal hepatoblastoma treated for 1.5 years with 5-FU, vincristine, doxorubicin, and cisplatin. No recurrence was seen after 6 years of follow-up. Risks involved with this technique include infection, thrombosis, or shifting of the catheter. Furthermore, it is challenging to carry out in children, and prospective studies are needed. Other new approaches include treatment with anti-AFP antibodies, IL-2, and viral transfection vectors to attack malignant hepatic cells (Geiger, 1996; Huber & Richards, 1996; Ji & St, 1997; Ramani et al, 1997).
Hepatocellular Carcinoma (Hepatoma)
Epidemiology
HCC (see Chapter 80) accounts for 23% of pediatric liver tumors but is rare in infancy (Finegold, 1994). Approximately 1.4 cases per 1 million children exist in the United States (Ni et al, 1997). The LCSG (1987) reported no cases in children age 4 years or younger in a series of 2286 patients with histologically reviewed tumors. Historic series without pathologic review may report a higher rate of infantile HCC owing to misdiagnosis of some early hepatoblastomas (Exelby et al, 1975). We have had personal experience of infants with well-documented HCC, and it affects about 0.5 children younger than age 15 years per 1 million annually. SEER data indicate that HCC accounts for 87% of primary hepatic malignancies in the 15- to 19-year age group (Darbari et al, 2003). The incidence is bimodal, with an early peak that is lower than that of hepatoblastoma. Most of these early cases occur before 5 years of age. A second peak occurs between 13 and 15 years of age. The median age at presentation of fibrolamellar carcinoma, a variant of HCC, is 20 years, and it is rarely observed before age 10 years. HCC has a male predominance (1.3 to 3.2 : 1), and in areas endemic for hepatitis B, the male/female ratio may be reversed, at 0.2 : 1. Approximately 35 to 40 new HCCs are diagnosed per year in the pediatric age group in the United States. Incidence reported for the years 1973 through 1977 versus 1993 through 1997 showed a decrease from 0.45 to 0.29 cases per 1 million (Darbari et al, 2003).
Hepatitis B and C infection correlates with the incidence of HCC. In Asia, 85% of HCC patients, both adult and pediatric, are hepatitis B surface antigen positive, whereas this is found in only 10% to 25% of patients in the United States. The relative risk for the development of HCC is 250 : 1 for patients with chronic active hepatitis compared with patients without hepatitis surface antigen positivity (Brower et al, 1998). Hepatitis C antibodies are found in 20% of patients with HCC. In one report, an infant with a history of neonatal hepatitis developed HCC (Moore et al, 1997a). Other conditions associated with the development of HCC include cirrhosis, α1-antitrypsin deficiency, tyrosinemia, aflatoxin ingestion, hemochromatosis, hepatic venous obstruction, androgen and estrogen exposure, Alagille syndrome (arteriohepatic dysplasia), and Thorotrast administration (Wegmann et al, 1996). One case of childhood HCC developing in a patient with neurofibromatosis has also been reported (Ettinger & Freeman, 1979).
A universal vaccination program against hepatitis B has reduced the incidence of HCC in Taiwan. The average annual incidence of HCC in children 6 to 14 years old declined from 0.70 per 100,000 children in the years 1981 through 1986 to 0.36 in the years 1990 through 1994 (P < .01) coincident with widespread administration of the hepatitis B vaccine (Chang et al, 1997). The mortality rate also decreased during this period. Antiviral therapy with lamivudine reduced the risk of development of HCC in patients with chronic hepatitis B infection and cirrhosis or fibrosis (Liaw et al, 2004). In contradistinction to HCC in adults, however, conditions associated with cirrhosis occur in only 30% to 40% of children with HCC; the remaining 60% to 70% of tumors are present without any cirrhosis (De Potter et al, 1987; Fattovich et al, 2004; Ismail et al, 2009; Llovet & Beaugrand, 2003; Marsh et al, 2004; Reynolds, 2001).
In one comparative study of pediatric HCC and hepatoblastoma, numerous discriminating features were reported (Chan et al, 2002). The mean age at presentation was 18 months for hepatoblastoma versus 10 years for HCC. The initial resectability of HCC was 45% and did not improve with chemotherapy, whereas 91% of hepatoblastomas could be completely resected before or after chemotherapy. Tumor rupture occurred in 36% of hepatoblastomas versus 9% of HCCs. Most importantly, the survival of patients with HCC was much worse.
Pathology (See Chapter 78)
HCCs are highly invasive and often multicentric at diagnosis with frequent hemorrhage and necrosis. Nuclear pleomorphism, nucleolar prominence, and the absence of extramedullary hematopoiesis are observed, and the cells are larger than normal hepatocytes. Low-grade HCCs may look similar to normal hepatocytes, especially if a limited amount of tissue is sampled. Invasiveness, and vascular invasion in particular, is a hallmark of these tumors. Extrahepatic dissemination to portal lymph nodes, lungs, and bones is frequent at diagnosis and strongly affects survival. HCCs naturally progress from capsular invasion to extracapsular extension, then vascular invasion, and finally to intrahepatic metastases (Toyosaka et al, 1996). A strong correlation has been found between intrahepatic metastases and portal vein thrombosis; this suggests that efferent tumor vessels anastomose to the portal, rather than the hepatic, veins, allowing intrahepatic spread and explaining the multicentricity that is a hallmark of HCC. Fibrolamellar HCC is a histologic variant seen in older children and young adults. It was once thought that this variant was associated with a better prognosis (Greenberg & Filler, 1989); however, studies indicate that when stage is controlled for, the survival is similar between standard HCC and its fibrolamellar variant.
Biology and Molecular Biology
Most investigations into the basic biology of HCC involve the study of hepatitis B and its relationship to carcinogenesis (Scaglioni et al, 1996). In one in vivo model in which rats developed HCC after prolonged feeding with glyceryl trinitrate, KRAS point mutations were identified in 8 of 18 animals that developed tumors (Tamano et al, 1996), and no TP53 mutations were seen. Cytogenetic data indicate that chromosomal abnormalities are complex, but no consistent patterns have been established (Terris et al, 1997).
Clinical Findings
Children and adolescents with HCC are often seen initially with palpable abdominal masses (40%), but many are symptomatic at diagnosis (Ni et al, 1997). Pain is frequent (38%) and may occur in the absence of an obvious mass. Constitutional disturbances such as anorexia, malaise, nausea and vomiting, and significant weight loss occur with greater frequency. Jaundice is an uncommon feature of either disease, but AFP is elevated in approximately 85% of patients, with most levels greater than 1000 ng/mL (Brower et al, 1998). Although elevated, these levels are usually less than those measured in hepatoblastoma patients; 10% may come to medical attention with tumor rupture and a hemoperitoneum (Brower et al, 1998).
Treatment
In over a quarter century, no significant progress has been made in treatment of the pediatric population with HCC (Ismail et al, 2009). This tumor remains extremely resistant to current chemotherapy agents, and long-term survival is impossible without complete resection. Because of a high incidence of multifocality within the liver, extrahepatic extension to regional lymph nodes, vascular invasion, and distant metastases, complete resection is often impossible. Infiltration with thrombosis of portal and hepatic venous branches is common, and even the vena cava may be involved. Furthermore, the cirrhosis found in a number of HCC livers may preclude an extensive resection. Most analyses report a 10% to 20% resectability rate among children with HCC. In contrast, patients with a fibrolamellar histologic pattern have a higher resection rate and essentially do not have cirrhotic livers. Even with complete resection, however, the prognosis remains poor secondary to the high rate of recurrence.
Historically, the same chemotherapy protocols used for hepatoblastoma were also applied to HCCs in childhood; however, HCC is rather unresponsive to chemotherapy overall, although cisplatin in particular has shown activity against it (Bower et al, 1996). The utility of external-beam radiation therapy is unclear; it can aid with temporary control of gross disease, but it has not been shown to reduce the risk of relapse in patients with residual disease after resection. Because of the poor survival of patients with HCC, present thinking is to apply new and innovative approaches to these cancers.
Unresectable HCCs can be palliated with embolization with or without added chemotherapeutic agents or radioisotopes (Maini et al, 1996; see Chapter 83). Percutaneous intralesional injection of ethanol also has been of palliative benefit when lesions are small (Ryu et al, 1997; see Chapter 85A). Radiofrequency ablation (RFA) of these tumors, percutaneously or at laparoscopy/laparotomy, has been associated with tumor resolution and prolonged survival (Inamori et al, 2004; Koda et al, 2004; Raut et al, 2005; Santambrogio et al, 2003; see Chapter 85C). Lin and colleagues (2004) reported that radiofrequency ablation was superior to ethanol injection in HCCs 4 cm or less in diameter. Moreover, preliminary research using metronomic chemotherapy and adjuvant antiangiogenic treatments is currently underway (Gille et al, 2005; Meng et al, 2007; Meyers, 2007; Pang & Poon, 2006).
As standard therapies have proven unsuccessful, liver transplantation has been used more widely. The Milan criteria used for adults have been extrapolated to children. They stipulate a single tumor no larger than 5 cm in diameter, or up to three tumors each 3 cm or less in diameter, an absence of macroscopic portal vein invasions, and absence of extrahepatic disease; however, no current data support the efficacy of using the Milan criteria in this population. The UNOS database analyzed 41 patients with HCC who received OLT from 1987 through 2006. The overall survival was 86%, 63%, and 58% for 1, 5, and 10 years, respectively. Analogous to hepatoblastoma, mortality was mainly secondary to recurrence, which occurred even more often than it did in hepatoblastoma (86% vs. 54%) (Austin et al, 2006).
Outcome
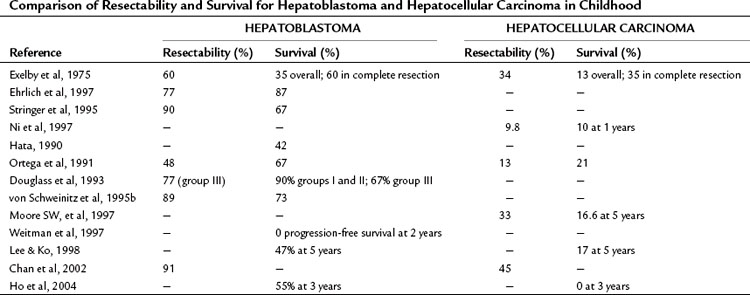
The overall survival from HCC in childhood approaches zero, and it remains a therapeutic problem (Table 82.5). Occasionally, resection of localized lesions results in long-term survival. Results from the LCSG (1987) are consistent with these findings, showing a 1-year survival rate of 40% for resectable disease and 10% for unresectable lesions. The trend is to separate HCC from hepatoblastoma in clinical studies because of its greatly differing biologic behavior.
Future Directions
Gene therapy with viral vectors that attack dividing cells is being investigated. Hepatocytes rarely divide unless stimulated by liver resection. Viruses such as herpes attack dividing cells and can be molecularly manipulated to contain cytotoxic genes, and modified herpesvirus can be transfected efficiently into hepatoma cells (Carew et al, 1998; Tung et al, 1996). One group has used an adenovirus vector to deliver murine endostatin to tumors in nude mice injected with HCC cells with a resultant reduction in tumor growth (Li et al, 2004).
Rhabdomyosarcoma of the Extrahepatic Bile Ducts
Incidence
Although embryonal rhabdomyosarcoma of the extrahepatic bile ducts is extremely rare, it is the most frequently seen malignancy in the biliary tree of children (Martinez et al, 1982). Ten cases were reported in intergroup rhabdomyosarcoma studies I and II, constituting 0.8% of confirmed tumors in those studies (Ruymann et al, 1985). Fewer than 50 cases have been reported since 1975 (Zampieri et al, 2006).
Pathology
Most tumors are of the embryonal histopathologic subtype. They often show botryoid characteristics similar to other rhabdomyosarcomas that arise in a hollow viscus. Immunohistochemistry and nuclear staining are informative for diagnosis of embryonal rhabdomyosarcoma (Ali et al, 2009; Morotti et al, 2006). About 40% of patients develop distant metastases, but mortality is most often due to the effects of local invasion (Lack et al, 1981). Rhabdomyosarcoma of the liver and the ampulla of Vater, but not involving the bile ducts, also has been reported but is very rare (Horowitz et al, 1987; Perera et al, 2009).
Presentation
Patients range in age from 1 to 9 years at diagnosis, and this tumor is seen more frequently in males. The typical presentation includes intermittent jaundice and may include loss of appetite and episodes of cholangitis (Charcot’s triad) (Perisic et al, 1991). Hepatomegaly and a palpable abdominal mass are usual (Lack et al, 1981; Nagaraj et al, 1977). The diagnosis may be confused with hepatitis with resultant delay in specific therapy, but rhabodmyosarcoma of the bile ducts can also be mistaken for a choledochal cyst (Sanz et al, 1997).
Imaging
Ultrasonography (US) shows a hilar or intrahepatic mass that may be confused with a choledochal cyst (Friedburg et al, 1984; Geoffray et al, 1987). Although CT or MRI may provide information about extension and metastases (Ng et al, 1997), they do not establish the diagnosis. Imaging may be more accurate for extrahepatic ductal involvement (Roebuck et al, 1998).
Treatment
Surgical exploration and biopsy are necessary to establish the diagnosis in most cases. Often a hilar lymph node provides diagnostic material, but carefully sampling the primary tumor may be necessary. A core needle biopsy or aspiration cytology may be adequate for diagnosis, and entry into the bile ducts should be avoided if possible. Intraoperative US (see Chapter 21) is helpful in identifying the course of biliary structures. Initial complete resection before chemotherapy may be difficult or impossible because of the extensive nature of these tumors. Also, microscopic submucosal extension is common, and resection margins are often microscopically positive despite the normal appearance of the intrahepatic bile ducts.
It is probably better to first establish the diagnosis with a biopsy and then begin systemic chemotherapy, which reduces the tumor size and allows a cleaner resection at second-look surgery. During the initial biopsy, hilar and left gastric lymph node sampling is performed to determine whether these nodal echelons require added radiotherapy. In the series reported from the intergroup rhabdomyosarcoma study, six of the 10 patients underwent initial resection, but all had microscopic or minimal gross disease left behind (Ruymann et al, 1985). The only four survivors were in this group; however, none of the four patients survived in whom resection was not attempted. In another report of three consecutive cases from a single institution, all patients presented with jaundice, cachexia, and abdominal mass (Martinez et al, 1982). The tumor arose in the common hepatic duct in two children and in the left hepatic duct in the third. The technical difficulty of prechemotherapy resection is illustrated by the fact that intrahepatic extension was noted in all, making complete resection impossible. Two of these patients are alive and disease-free at 9 months and 14 years, whereas the third died as a result of progressive disease at 33 months. All received radiotherapy in addition to multiagent chemotherapy. Jaundice was not a late problem.
The need for aggressive surgical resection of biliary rhabdomyosarcomas was challenged in a report by Spunt and colleagues (2000). They reported 25 patients with this diagnosis from intergroup rhabdomyosarcoma studies I through IV and noted that although only 29% of these patients underwent a gross total resection before or after multiagent chemotherapy, the overall 5-year survival rate was 78%. They concluded that surgery was important for diagnosis and staging but that it was not necessary for long-term survival with present chemotherapeutic agents. They also noted that the use of biliary drains in patients undergoing aggressive surgery was associated with a high rate of serious infections.
Embryonal Sarcoma
Incidence
Embryonal sarcoma, also known as malignant mesenchymoma or undifferentiated sarcoma in the older literature, is a rare primary hepatic neoplasm that occurs in older children. In one study, two cases were reported out of 1102 (0.2%) primary liver tumors analyzed (Flemming et al, 1995). The age at diagnosis ranges from 5 to 16 years (Newman et al, 1989; Vetter et al, 1989). Embryonal sarcoma constitutes 14% of malignant liver tumors occurring in children 6 to 10 years of age (Yedibela et al, 2000).
Pathology
Microscopically these tumors appear as a pleomorphic or undifferentiated sarcoma, and areas of the liver may be entrapped. Primitive mesenchymal cells with occasional small cysts and ducts lined with benign-appearing epithelium are sometimes present at the periphery (Gallivan et al, 1983). Some reports have suggested that embryonal sarcoma of the liver may represent malignant degeneration of a mesenchymal hamartoma (Begueret et al, 2001; O’Sullivan et al, 2001).
Clinical Presentation
A right upper quadrant abdominal mass and pain are significant presenting symptoms, and fever also may be prominent. Spontaneous rupture has been reported (Yedibela et al, 2000).
Imaging
Embryonal sarcoma often appears hypodense (dark) on CT and has a bright peripheral fibrous pseudocapsule, as shown in Figure 82.7. They can be very bulky tumors and sometimes are confused with cystic liver disease (Orozco et al, 1991; Tozzi et al, 1992). These tumors may have the appearance of a solitary liver cyst in childhood (Chowdhary et al, 2004) or may mimic a hydatid cyst (Aggarwal et al, 2001).
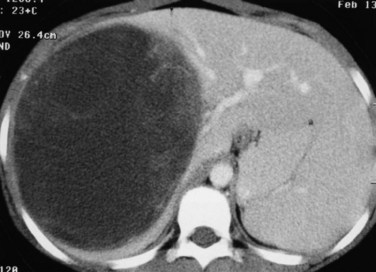
Treatment and Outcome
Embryonal sarcomas of the liver are treated with resection and adjuvant chemotherapy (Kadomatsu et al, 1992). Cisplatin, doxorubicin, cyclophosphamide, dacarbazine, actinomycin, and other agents have been used in combination with radiotherapy (Newman et al, 1989; Vetter et al, 1989). Complete surgical resection should be attempted and usually requires a major hepatectomy for tumor clearance. Although past reports have cited a poor prognosis, we have seen long-term survival with a combination of modern multiagent chemotherapy and complete resection.
Leiomyosarcoma
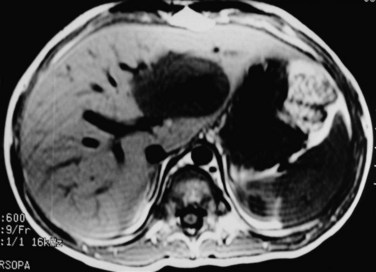
Patients with human immunodeficiency virus (HIV) infection experience an increasing incidence of smooth muscle tumors (Norton et al, 1997; Ross et al, 1992). Leiomyosarcoma of the liver has been reported in more than 20 patients; the youngest was 9 years old. Adjuvant therapy is ineffective; these tumors must be resected for control. They tend to be low grade or have an indeterminate malignant potential. Supportive and antiretroviral therapy is given through the perioperative period. A case of HIV-associated leiomyosarcoma is shown in Figure 82.8.
Primary Hepatic Rhabdoid Tumor
Incidence
Rhabdoid tumors are very rare, highly malignant, sarcoma-like neoplasms that usually involve the kidney or central nervous system (Vujanic et al, 1996). They can be primary in extrarenal sites, including extremity, paraspinal, and cervical soft tissues and the liver (Honda et al, 1996; Jimenez-Heffernan et al, 1998; Kelly et al, 1998). In a recent literature review of 19 cases of primary hepatic rhabdoid tumor, the median patient age was 16.7 months, and 89% of patients were younger than 2 years. The overall mortality rate was 89%, and the median time to death was 15.3 weeks (Yuri et al, 2004).
Pathology
Histopathologic examination shows a high-grade round-cell neoplasm with abundant cytoplasm and containing cells with cytoplasmic filamentous inclusions (White et al, 1999). The cells are frequently positive for vimentin and cytokeratins, and deletions at chromosome 22q11 are often seen (White et al, 1999).
Clinical Presentation
The diagnosis may be suspected if widespread metastases, including central nervous system dissemination, are found with a hepatic mass. Cases with spontaneous rupture have been reported (Clairotte et al, 2006; Kelly et al, 1998; Ravindra, 2002; Yuri et al, 2004).
Angiosarcoma
Some authors describe angiosarcoma as the malignant form of hemangioendothelioma in children (Falk et al, 1981; Noronha & Gonzalez-Crussi, 1984). Arsenic exposure has been associated with malignant progression from hemangioendothelioma to hemangiosarcoma (Falk et al, 1981); however, no clear association has been reported between angiosarcoma and arsenic, Thorotrast, or vinyl chloride (Dimashkieh et al, 2004). Angiosarcoma is typically evidenced by a swiftly growing liver. Histologically, hypercellular whorls of spindled sarcoma cells are observed interspersed with bile ducts, blood vessels, and collagen. Intracellular periodic acid-Schiff–positive globules are present in most cases, and focal factor VIII staining may be seen.
Complete resection is required for cure of angiosarcoma, but recurrence rates are high, and the tumor is often not resectable; it is typically present in both lobes of the liver, and consideration should be given to adjuvant chemotherapy. Early metastasis, most commonly to the lung, is seen frequently (Awan et al, 1996; Falk et al, 1981; Kaufmann & Stout, 1961; Selby et al, 1992; Weinberg & Finegold, 1983). A recent review stated an average survival of 16 months (Dimashkieh et al, 2004). In one report of 10 cases of hepatic angiosarcoma in childhood, six patients were female, and four were male (Selby et al, 1992). The median age at diagnosis was 3.7 years (range, 1.5 to 7 years), with a mean follow-up of 10 months, and six of seven patients with follow-up died from disease.
Malignant Germ Cell Tumors
Primary malignant germ cell tumors of the liver are very rare and may present as teratomas, choriocarcinomas, or yolk sac tumors (Theegarten et al, 1998). In childhood, these may respond well to neoadjuvant chemotherapy followed by resection. Usually, a regimen containing cisplatin, ifosfamide, and bleomycin is employed.
Primary Hepatic Non-Hodgkin Lymphoma
Primary non-Hodgkin lymphoma of the liver occurs in childhood and may comprise 5% of primary hepatic malignancies in this age group (Gururangan et al, 1992). Burkitt lymphoma and other types of small-cell, noncleaved lymphoma have been reported (Huang et al, 1997; Mills, 1988). One or many lesions may be involved (Mantadakis et al, 2008). The primary treatment of lymphoma in childhood is chemotherapy, which usually results in complete resolution of the hepatic mass. Resection is rarely, if ever, indicated.
Hepatic Metastases
We have had experience with 11 patients (age range, 4.3 to 21 years) who underwent hepatic metastasectomy. The overall survival in this group was 20%, but local hepatic control was greater than 85%. Advantages of hepatic metastasectomy include amelioration of liver function abnormalities, improvement of biliary obstruction, and possibly prolonged survival (Su et al, 2007).
Neuroblastoma
Hepatic metastases from neuroblastoma may be encountered in stage IV neuroblastoma and in newborns and infants with stage IV-S disease. In the latter situation, the liver involvement is a hallmark of the disease (Komuro et al, 1998). In stage IV-S, hepatic tumors generally resolve, although they may increase alarmingly in size before this happens. The increase in size usually causes pulmonary and vena caval impairment and may require relief by placement of an abdominal silo similar to that used for congenital abdominal wall defects. For stage IV disease, it may be advantageous for patients to undergo hepatic metastasectomy, which may aid in diminution of treatment or prolonged survival (Su et al, 2007). One patient with neuroblastoma and hepatic metastasectomy had disease-free survival with a follow-up time of 59 months (Su et al, 2007).
Wilms Tumor
Wilms tumor metastasizes to the liver in about 12% of patients (Cohen & Siddiqui, 1982), and this usually is associated with unfavorable histology (Breslow et al, 1986; Thomas et al, 1991). Resection of localized residual disease may be beneficial in well-selected cases. In one report, 15 cases of metastatic Wilms tumor with hepatic metastasectomy were analyzed and showed a 2-year survival of 62% and a 5-year survival of 44% (Foster, 1978). In one report, four patients with Wilms tumors metastatic to the liver underwent hepatic resection with a 2-year survival of 80%, and two patients survived 14 and 17 years, respectively (Morrow et al, 1982). This experience is also noted in other reports of Wilms tumor (Hagiwara et al, 1982). Experience with three patients, however, did not show survival greater than 9 months (Su et al, 2007).
Osteogenic Sarcoma
Calcified hepatic metastases sometimes may be observed in patients with osteogenic sarcoma (Shapiro et al, 1988). Although pulmonary metastasectomy for this disease is well described (La Quaglia, 1993; Saltzman et al, 1993; Torre et al, 2004), the role for hepatic metastasectomy has not been clarified. We reported a case of a child with osteogenic sarcoma with isolated liver metastases resected by a formal right lobectomy. This patient remained without disease for 2 years afterward and then developed recurrence in a limb, which required amputation. Hepatic metastases returned 46 months after this, and the patient died thereafter (Su et al, 2007).
Desmoplastic Small Round-Cell Tumor
Hepatic involvement with desmoplastic small round-cell tumor is frequent and is usually associated with a fatal outcome (Kushner et al, 1996; Mazuryk et al, 1998; Ordonez, 1998). The pattern of metastases is diffuse, leaving no spared segments that would allow resection, and complete resection is associated with a significant survival advantage in this disease (Schwarz et al, 1998). Future strategies include chemoembolization and arterial infusion therapy. Hepatic radiation to a level of 3000 cGy is ineffective.
Rhabdomyosarcoma
The liver follows regional lung, lymph nodes, and bone as a site of rhabdomyosarcoma metastases (Shimada et al, 1987). Resection of liver metastases is usually not feasible because of diffuse hepatic involvement and the presence of metastases in other sites.
Colon Cancer
Colon cancer can occur in childhood (La Quaglia et al, 1992). About 50% of these patients have signet ring tumors. The pattern of spread is over peritoneal surfaces, rather than through the portal system, and hepatic metastases usually occur late despite massive peritoneal disease.
Malignant Peripheral Nerve Cell Tumor
Malignant peripheral nerve cell tumors are likely to metastasize to the liver in childhood and adolescence. The pattern of metastases is usually miliary, and the tumors do not respond well to chemotherapy (Probst-Cousin et al, 1997).
Adrenocortical Carcinoma
Adrenocortical carcinoma also may metastasize to the liver, although concomitant pulmonary and retroperitoneal metastases are usually also present (Arico et al, 1992). Treatment is usually with platinum-based chemotherapy, and surgery is rarely done.
Rhabdoid Tumor
Rhabdoid tumor most commonly originates from the kidney and can metastasize to liver (White et al, 1999). It also may be primary to the liver, but this is rare. The pattern for hepatic metastases is usually diffuse and is not amenable to surgical resection. Chemotherapy protocols suitable for sarcoma are usually used (Vujanic et al, 1996).
Hepatic Evaluation and Resection
Surgical Anatomy
The schema of hepatic anatomy most useful for the surgeon is based on the work of Couinaud; this is described in other chapters (see Chapter 1A, Chapter 1B ). The principles of hepatic resection in small children and infants are the same as those for adults (see Chapters 90A through 90F).
Hepatic Regeneration
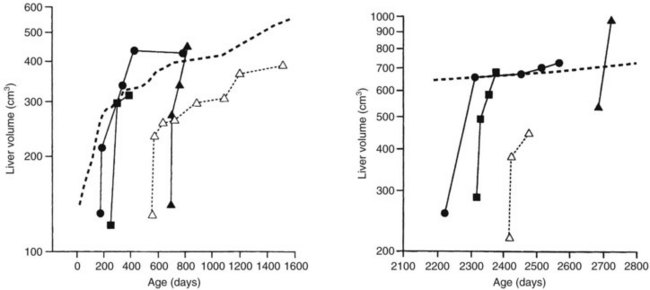
The liver in a child is able to regenerate quickly, even after massive resection (see Chapter 5) and administration of systemic chemotherapy (Shamberger et al, 1996; Wheatley et al, 1996). In most patients, recovery to the normal volume for age is rapid (Fig. 82.9).
Benign Hepatic Tumors
Benign hepatic tumors (see Chapter 79A) accounted for less than 35% of 1250 pediatric liver tumors in one study (Finegold, 1994). Benign tumors of the liver that occur in childhood include hemangiomata or vascular malformations, hepatocytic adenomas, focal nodular hyperplasia, mesenchymal hamartomas, and various types of cysts and cystic disease. The distribution of common benign hepatic tumors of childhood is illustrated in Figure 82.10.
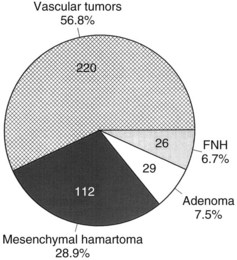
FIGURE 82.10 The frequency distribution of benign hepatic tumors in childhood (Exelby et al, 1975; Weinberg & Finegold, 1983). This graph is based on surgical or pathologic series of patients. With modern imaging techniques, the frequency of asymptomatic vascular tumors discovered incidentally is undoubtedly much higher (Flemming et al, 1995). FNH, focal nodular hyperplasia.
Hemangiomata and Vascular Malformations
Hemangiomata are lesions characterized by endothelial-lined vascular spaces that vary in size and extent (Ehren et al, 1983; Ishak, 1976). They are sometimes classified as hamartomas and are the most common skin lesions observed in childhood. Hemangioendotheliomas are highly proliferative cellular lesions of variable malignant potential. In contrast, venous malformations and cavernous hemangiomata are distinguished by a lack of cellularity and large vascular spaces. Arteriovenous malformations are the rarest pathologic subtype and are distinguished by abnormal anastomoses between arteries and veins. Venous malformations, cavernous hemangiomata, and arteriovenous malformations may be associated with significant shunting that results in congestive heart failure.
Incidence
The overall incidence of endothelial-lined vascular tumors of the liver in childhood is probably unknown, because many are asymptomatic. Vascular lesions taken together represent 13% to 18% of symptomatic hepatic tumors in childhood (Exelby et al, 1975; Finegold, 1994). Hepatic hemangiomata are more common in girls by 2 : 1 (Mulliken, 1988).
Presentation and Diagnosis
An abdominal mass is probably the most frequent sign of a vascular tumor of the liver. Multiple hemispheric, cutaneous hemangiomata may be present and warn the physician of possible visceral lesions. A systolic bruit sometimes can be heard over the enlarged liver. Infants with large, actively perfused vascular lesions may come to medical attention with congestive heart failure. Rarely, jaundice, disseminated intravascular coagulation (DIC), or hemorrhagic shock from intraperitoneal rupture may be present, and rupture can be precipitated by percutaneous needle biopsy (Hobbs, 1990).
Imaging studies are usually all that is required to confirm the diagnosis. Tumor extent and tissue characterization are assessed using MRI with standard spin echo T1-weighted and T2-weighted imaging (Powers et al, 1994). Intravenous administration of gadopentetate dimeglumine (gadolinium) can produce greater resolution. Vascular lesions can be seen as intensely white on T2-weighted images, and this study is often accurate enough to be definitive. Contrast-enhanced CT scan will demonstrate a focus with decreased density, with contrast enhancement from the edges toward the middle of the tumor, which will then be homogeneously enhanced (Meyers et al, 2007). Arteriography is rarely indicated given the detail and accuracy of modern imaging techniques, and open biopsy can result in massive hemorrhage.
Treatment
Asymptomatic lesions do not require treatment, and many hepatic hemangiomata regress after the first year of life. Patients with congestive heart failure are admitted to the intensive care unit and treated with digitalis and furosemide. If the heart failure remains refractory to these maneuvers, hepatic artery embolization or direct surgical ligation may be necessary (de Lorimier et al, 1967; Herlin et al, 1988). Hepatic arterial embolization may be successful in controlling symptoms, but rapid development of collateral vessels can make subsequent resection or embolization difficult. Transfusion of blood and platelets may be required for DIC. Interferon-alfa therapy for symptomatic angiomatosis and hemangioendothelioma is being used with success in individual patients (Le Luyer et al, 2000).
Patients who are seen in hemorrhagic shock with rupture of a vascular tumor usually require hepatic resection; however, initial hepatic arterial embolization may control bleeding temporarily and may allow stabilization and safer surgery (Takvorian et al, 1988). Hepatic resection may also be required in some patients in association with congestive heart failure. Blood loss in these complicated procedures may be reduced using hemodilution techniques (Kitahara et al, 1995; Schaller et al, 1984).
Outcome
The overall prognosis for benign hepatic vascular lesions is good. Most patients do not require operative intervention, and most cellular lesions start to regress after the first year of life. Angiosarcomatous degeneration of benign hemangioendotheliomas has been reported in five patients from one institution (Weinberg & Finegold, 1983). Three of the five were treated with radiotherapy to the benign hemangioendotheliomas before the development of angiosarcoma. This and other reports from the literature have correlated radiotherapy of benign vascular tumors with subsequent malignant degeneration, usually to angiosarcoma (Costello & Seywright, 1990).
Hemangioendothelioma
The incidence of hemangioendothelioma is 1%, and it is the most prevalent hepatic vascular tumor in pediatric patients younger than 6 months (Sari et al, 2006). In one report of 16 infants and children with hemangioendothelioma, 15 were seen initially with hepatomegaly, 7 with congestive heart failure, and 4 with associated cutaneous lesions (Holcomb et al, 1988). Kasabach-Merrit syndrome, a platelet-trapping coagulopathy, also has been observed (von Schweinitz et al, 1995c). These lesions may appear very cellular, but they do not metastasize. If a primary lesion produces symptoms, resection is indicated for relief.
Hemangioblastoma
Hemangioblastoma of the liver usually is associated with von Hippel-Lindau disease (Rojiani et al, 1991). In infancy and childhood, these lesions appear very cellular, but distant metastases are uncommon. Complete resection should be performed and is usually curative. Hemangioblastomas of the central nervous system and retina have been treated with interferon alfa-2a, but without striking resolution, although two retinal lesions showed shrinkage (Niemela et al, 2001).
Mesenchymal Hamartoma
Mesenchymal hamartomas are usually solitary hepatic masses occurring in infants. They are usually multicystic, and the cysts are lined with flattened biliary epithelium or endothelium. Actively growing mesenchyme is abundant, associated with distended lymphatics. It is postulated that mesenchymal hamartomas arise in areas of focal intrahepatic biliary atresia; this results in distal bile duct obstruction and hepatocellular necrosis (Cooper et al, 1989). Others have hypothesized that these lesions arise in conjunction with anomalies of vascular development. This explains the occurrence of small hemangiomata observed in close proximity to mesenchymal hamartomas (Srouji et al, 1978). Cytogenetic analysis has only rarely been performed on these tumors, but a consistent 19q13.4 breakpoint has been identified (Mascarello & Krous, 1992).
Epidemiology
Mesenchymal hamartomas account for 6% of primary liver tumors in childhood, and a male predominance is reported. In one study, four of 134 patients with space-occupying liver lesions had mesenchymal hamartomas (Yen et al, 2003). Two thirds of these tumors are diagnosed in infants younger than 1 year, although presentation in the teenage years has been described. In one study of 18 such tumors, the mean age at diagnosis was 16 months (DeMaioribus et al, 1990).
Presentation and Diagnosis
Most mesenchymal hamartomas are seen as an enlarging abdominal mass or hepatomegaly and are usually not otherwise symptomatic (Srouji et al, 1978), but they can grow to great size and may cause respiratory distress or evidence of caval obstruction. Mesenchymal hamartomas are typically found within the right lobe of the liver, and US, CT, and MRI are the most useful diagnostic studies, although an open biopsy is often necessary to make the diagnosis. Figure 82.11 shows an MRI of a huge mesenchymal hamartoma. Giant hepatic cysts in fetuses also have been diagnosed on ultrasound (Tsao et al, 2002).
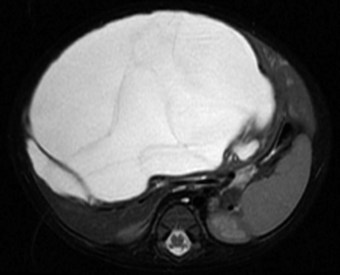
Treatment
Anatomic resection (hepatic lobectomy) is effective treatment, especially for large lesions. Because of the mesenchymal component, these lesions have a definite capsule that facilitates enucleation of large, central mesenchymal hamartomas not amenable to lobectomy. Enucleation was done in the case illustrated in Figure 82.11. Occasionally, it is necessary to marsupialize a large cystic lesion, but only if it is not feasible to remove this lesion completely. This approach may not be successful (Meinders et al, 1998). One report using sequential CT scans documented initial expansion followed by involution of a mesenchymal hamartoma (Barnhart et al, 1997). Because many reports exist describing spontaneous regression, the role of surgery is being contested in the asymptomatic patient (Meyers, 2007; Leary et al, 1989). It has been postulated that in utero decompression of giant hepatic cysts would improve the outcome in these patients (Tsao et al, 2002).
Focal Nodular Hyperplasia and Hepatocellular Adenoma
Focal nodular hyperplasia (FNH) and hepatocytic adenomas are benign hepatocellular proliferations that are more common in adults than in children (see Chapter 79A). Hepatic adenomas are expected with type I glycogen storage disease after the first decade of life and may be multiple (Saito et al, 1984). FNH has also been described in a patient with type I glycogen storage disease (Sakatoku et al, 1996). Hepatic adenomas also have grown in patients undergoing androgen therapy for hematologic disorders, after danazol use, and in woman on oral contraceptives (Fermand et al, 1990). Both tumors have been correlated with a high-estrogen environment (Sakatoku et al, 1996). One report in the literature is of FNH developing in children treated for other solid malignancies (Bouyn et al, 2003). These authors postulated that a vascular injury secondary to chemotherapy or radiation was the causal factor.
FNH is distinguished from adenoma by the presence of fibrous septa that contain bile ducts and an inflammatory infiltrate. In one report, five of 39 unresected hepatic adenomas developed HCC (Foster & Berman, 1994).
Epidemiology
Less than 2% of hepatic tumors in childhood are FNH or hepatocellular adenomas (Weinberg & Finegold, 1983). Both lesions have been described in infants and teenagers. Most patients are younger than 5 years at presentation and predominantly female (Nagorney, 1995). An association with contraceptive use has been found in adults, but no defined association between exogenous hormone administration in childhood and adolescence has been reported, although FNH has been reported in an infant antenatally exposed to corticosteroids (Prasad et al, 1995). In one report of 48 benign liver tumors in childhood, there were three cases (6%) of FNH and two cases of hepatocellular adenoma (4%) (Ehren et al, 1983).
Presentation and Diagnosis
Patients with hepatocellular adenomas usually come to medical attention with an asymptomatic abdominal mass; they are more likely to be symptomatic and may present with rupture and resultant hemorrhage, causing an acute abdomen. Both FNH and adenoma are well encapsulated on imaging studies. Because FNH is associated with fibrous septa, abdominal US and CT may show a distinctive central “scar.” Hepatic adenomas may show encapsulation on imaging studies, but histologic sections may be difficult to distinguish from HCC. In childhood, these tumors may attain great size, contributing to symptoms. In one report of six cases in childhood, the average tumor size was 7.5 cm (range, 2.5 to 10 cm) (Lack & Ornvold, 1986). The tumor was localized to the right lobe in four of the six and was bilobar in the remaining two. Superparamagnetic oxide-enhanced MRI may discriminate between hepatic adenoma and FNH (Beets-Tan et al, 1998).
Treatment
It is best to remove adenomas because of the difficulty of differentiating them from low-grade HCCs, uncertainty about future malignant degeneration, and the possibility of rupture and hemorrhage (Westaby et al, 1983). Resection also results in relief of preoperative symptoms (Hutton et al, 1993). An anatomic liver resection usually is required, and we have employed extended left hepatectomy successfully in children to treat this lesion (Glick et al, 2000). Laparoscopic resection of these lesions has also has been done (Marks et al, 1998; Samama et al, 1998), and percutaneous RFA has recently become an option for treatment (Rocourt et al, 2006). Embolization of unresectable lesions is another alternative, if the masses are large or symptomatic. Unresectable and asymptomatic cases of FNH are observed using serial abdominal US, MRI, and clinical examination. Spontaneous regression has been documented, and patients with symptoms are typically treated with either surgical resection or ablation (Meyers, 2007).
Outcome
Most patients do well after hepatic resection, and in general, anatomic resection provides the best result. In one study of six patients with FNH and two with an adenoma, six were long-term survivors (Lack & Ornvold, 1986). Three patients with FNH were alive 4 to 17 years after hepatic lobectomy, and two other patients were followed with observation alone and were alive and well 13 and 15 years later. The one death in this group was from leukemia with an incidental finding of FNH. One of the two patients with an adenoma survived for 10.5 years after lobectomy, whereas the second died as a result of postoperative hemorrhage.
Cysts and Cystic Disease (See Chapter 69A, Chapter 69B )
Multiple cases of solitary, congenital, nonparasitic liver cysts in childhood have been reported (Hernandez-Siverio et al, 1988; Pul & Pul, 1995). They are extremely rare but have been increasingly noted as incidental findings on US and CT performed for other reasons. Cysts are simple and not multilocular, they often have a bluish appearance at laparotomy, and the wall usually has three layers. The lining is generally cuboidal or columnar, although mucinous or squamous epithelium is reported. The middle layer comprises vascular elements, and the outer layer comprises compressed hepatocytes, collagen, muscle fibers, and bile ducts (Jones, 1994). The central contents are straw-colored and clear and are not under high pressure, as is the case with parasitic cysts. Most of these cysts are located in the right, anteroinferior hepatic lobe (segment V), and occasionally they are pedunculated.
Preoperative MRI or Doppler US is necessary to determine proximity to the portal and hepatic veins. Cysts that are neither adherent nor in proximity to these vascular structures are excised. Hepatic lobectomy may be necessary for symptomatic cysts that adhere to major veins, and marsupialization is sometimes a viable alternative. Injection of contrast media into the cyst can rule out a rare communication with the biliary tract. Cysts discovered incidentally at laparotomy should be left alone when less than 5 cm in diameter, but aspiration of cysts that are between 5 cm and 10 cm in diameter should be performed to confirm the diagnosis. Aspiration is followed by excision of the cyst wall. For large cysts—especially if there has been previous inflammation, or the cyst wall is near major venous or biliary branches—an anatomic liver resection is indicated (Iwatsuki et al, 1990). This resection avoids unnecessary bleeding and risk of postoperative biliary fistula.
Adult-type polycystic disease involving the liver has been reported with right lobar replacement and sparing of the left hepatic lobe (Marcellini et al, 1986); this was effectively treated by right hepatic lobectomy. Embryonal sarcoma of the liver sometimes can be confused with a solitary cyst (Chowdhary et al, 2004), and percutaneous needle aspiration and biopsy may be required to establish the diagnosis.
Aggarwal S, et al. Embryonal sarcoma of the liver mimicking a hydatid cyst in an adult. Trop Gastroenterol. 2001;22:141-142.
Albrecht S, et al. Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res. 1994;54:5041-5044.
Alcantar VC. Hepatoblastoma associated with osteosarcoma. Rev Mex Pediatr. 1985;52:389-392.
Ali S, Russo MA, Margraf L. Biliary rhabdomyosarcoma mimicking choledochal cyst. J Gastrointest Liver Dis. 2009;18:95-97.
Arico M, et al. Partial response after intensive chemotherapy for adrenal cortical carcinoma in a child. Med Pediatr Oncol. 1992;20:246-248.
Aronson DC, et al. Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol. 2005;23:1245-1252.
Austin MT, et al. Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg. 2006;41:182-186.
Awan S, et al. Angiosarcoma of the liver in children. J Pediatr Surg. 1996;31:1729-1732.
Bala S, et al. Childhood hepatocellular adenoma in familial adenomatous polyposis: mutations in adenomatous polyposis coli gene and p53. Gastroenterology. 1997;112:919-922.
Barnhart DC, et al. Conservative management of mesenchymal hamartoma of the liver. J Pediatr Surg. 1997;32:1495-1498.
Bates SM, et al. Hepatoblastoma: detection of tumor vascularity with duplex Doppler US. Radiology. 1990;176:505-507.
Beets-Tan RG, et al. Hepatic adenoma and focal nodular hyperplasia: MR findings with superparamagnetic iron oxide–enhanced MRI. Clin Imaging. 1998;22:211-215.
Begueret H, et al. Hepatic undifferentiated embryonal sarcoma: malignant evolution of mesenchymal hamartoma? Study of one case with immunohistochemical and flow cytometric emphasis. J Hepatol. 2001;34:178-179.
Berthold F, et al. Combination chemotherapy and chemoembolization in the treatment of primary inoperable hepatoblastoma. Klin Padiatr. 1986;198:257-261.
Bismuth H. Surgical anatomy and anatomical surgery of the liver. World J Surg. 1982;6:3-9.
Blair V, Birch JM. Patterns and temporal trends in the incidence of malignant disease in children: II. Solid tumours of childhood. Eur J Cancer. 1994;30A:1498-1511.
Bortolasi L, et al. Hepatoblastoma in adult age: a report of two cases. Hepatogastroenterology. 1996;43:1073-1078.
Bouyn CI, et al. Hepatic focal nodular hyperplasia in children previously treated for a solid tumor: incidence, risk factors, and outcome. Cancer. 2003;97:3107-3113.
Bove KE, et al. Hepatoblastoma in a child with trisomy 18: cytogenetics, liver anomalies, and literature review. Pediatr Pathol Lab Med. 1996;16:253-262.
Bower M, et al. Fibrolamellar hepatocellular carcinoma responsive to platinum-based combination chemotherapy. Clin Oncol (R Coll Radiol). 1996;8:331-333.
Brandt S, et al. The tamoxifen-induced suppression of telomerase activity in the human hepatoblastoma cell line hepG2: a result of post-translational regulation. J Cancer Res Clin Oncol. 2005;131:120-128.
Breslow NE, et al. Clinicopathologic features and prognosis for Wilms’ tumor patients with metastases at diagnosis. Cancer. 1986;58:2501-2511.
Brower ST, et al. Pancreatic cancer, hepatobiliary cancer, and neuroendocrine cancers of the GI tract. In: Pazdur, R, et al. Cancer Management: A Multidisciplinary Approach. Huntington, NY: Published Research & Representation; 1998:113-148.
Brown BF, et al. Hepatoblastoma: a rare pediatric neoplasm. Mil Med. 1993;158:51-55.
Carew JF, et al. Efficient gene transfer to human squamous cell carcinomas by the herpes simplex virus type 1 amplicon vector. Am J Surg. 1998;176:404-408.
Cetta F, et al. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut. 1997;41:417.
Chan KL, et al. Paediatric hepatoblastoma and hepatocellular carcinoma: retrospective study. Hong Kong Med J. 2002;8:13-17.
Chang MH, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855-1859.
Chen TT, et al. Establishment and characterization of a cancer cell line derived from an aggressive childhood liver tumor. Pediatr Blood Cancer. 2009;53:1040-1047.
Chowdhary SK, et al. Undifferentiated embryonal sarcoma in children: beware of the solitary liver cyst. J Pediatr Surg. 2004;39:E9-E12.
Clairotte A, et al. [Malignant rhabdoid tumor of the liver with spontaneous rupture: a case report]. Ann Pathol. 2006;26:122-125.
Clouse ME, et al. Hepatic arterial chemoembolization for metastatic neuroendocrine tumors. Digestion. 1994;55:92-97.
Cohen MD, Siddiqui A. Liver metastases in Wilms’ tumour. Clin Radiol. 1982;33:539-540.
Cooper K, et al. Mesenchymal hamartoma of the liver: a report of 5 cases. S Afr Med J. 1989;75:295-298.
Costello SA, Seywright M. Post-irradiation malignant transformation in benign haemangioma. Eur J Surg Oncol. 1990;16:517-519.
Couinaud C. Surgical anatomy of the liver: several new aspects. Chirurgie. 1986;112:337-342.
Couinaud C. The anatomy of the liver. Ann Ital Chir. 1992;63:693-697.
Curia MC, et al. Sporadic childhood hepatoblastomas show activation of beta-catenin, mismatch repair defects and p53 mutations. Mod Pathol. 2008;21:7-14.
D’Alessandro A, et al. Liver transplantation in pediatric patients: twenty years of experience at the University of Wisconsin. Pediatr Transplant. 2007;11:661-670.
Daniel E, Kifle A. An unusual presentation of hepatoblastoma. Ethiop Med J. 1989;27:231-234.
Darbari A, et al. Epidemiology of primary hepatic malignancies in U.S. children. Hepatology. 2003;38:560-566.
Dehner LP. Hepatic tumors in the pediatric age group: a distinctive clinicopathologic spectrum. Perspect Pediatr Pathol. 1978;4:217-268.
Dehner LP, Manivel JC. Hepatoblastoma: an analysis of the relationship between morphologic subtypes and prognosis. Am J Pediatr Hematol Oncol. 1988;10:301-307.
de Lorimier AA, et al. Hepatic-artery ligation for hepatic hemangiomatosis. N Engl J Med. 1967;277:333-337.
DeMaioribus CA, et al. Mesenchymal hamartoma of the liver: a 35-year review. Arch Surg. 1990;125:598-600.
De Potter CR, et al. Hepatitis B related childhood hepatocellular carcinoma: childhood hepatic malignancies. Cancer. 1987;60:414-418.
Dicken BJ, et al. Association between surgical margins and long-term outcome in advanced hepatoblastoma. J Pediatr Surg. 2004;39:721-725.
Dimashkieh HH, et al. Pediatric hepatic angiosarcoma: case report and review of the literature. Pediatr Dev Pathol. 2004;7:527-532.
Douglass EC, et al. Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol. 1993;11:96-99.
Ehren H, et al. Benign liver tumors in infancy and childhood: report of 48 cases. Am J Surg. 1983;145:325-329.
Ehrlich PF, et al. Improved long-term survival with preoperative chemotherapy for hepatoblastoma. J Pediatr Surg. 1997;32:999-1002.
Ettinger LJ, Freeman AI. Hepatoma in a child with neurofibromatosis. Am J Dis Child. 1979;133:528-531.
Exelby PR, et al. Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey 1974. J Pediatr Surg. 1975;10:329-337.
Fabre M, et al. Hepatic tumors in childhood: experience on 245 tumors and review of literature. Ann Pathol. 2004;24:536-555.
Falk H, et al. Review of four cases of childhood hepatic angiosarcoma-elevated environmental arsenic exposure in one case. Cancer. 1981;47:382-391.
Fattovich G, et al. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35-S50.
Fegiz G, et al. A case report of hepatoblastoma treated by chemotherapy and hepatic lobectomy. World J Surg. 1977;1:407-414.
Fermand JP, et al. Danazol-induced hepatocellular adenoma. Am J Med. 1990;88:529-530.
Filler RM, et al. Preoperative chemotherapy in hepatoblastoma. Surgery. 110, 1991. 591–556
Finegold MJ. Tumors of the liver. Semin Liver Dis. 1994;14:270-281.
Finegold MJ, et al. Liver tumors: pediatric population. Liver Transpl. 2008;14:1545-1556.
Flemming P, et al. Mesenchymal tumors of the liver: their frequency and histopathological diagnostic problems in surgical investigations. Verh Dtsch Ges Pathol. 1995;79:116-119.
Foster JH. Survival after liver resection for secondary tumors. Am J Surg. 1978;135:389-394.
Foster JH, Berman MM. The malignant transformation of liver cell adenomas. Arch Surg. 1994;129:712-717.
Fraumeni JFJr, et al. Hepatoblastoma in infant sisters. Cancer. 1969;24:1086-1090.
Friedburg H, et al. Sonographic and computed tomographic features of embryonal rhabdomyosarcoma of the biliary tract. Pediatr Radiol. 1984;14:436-438.
Gallivan MV, et al. Undifferentiated (“embryonal”) sarcoma of the liver: ultrastructure of a case presenting as a primary intracardiac tumor. Pediatr Pathol. 1983;1:291-300.
Gauthier F, et al. Hepatoblastoma and hepatocarcinoma in children: analysis of a series of 29 cases. J Pediatr Surg. 1986;21:424-429.
Geiger JD. Surgery for hepatoblastoma in children. Curr Opin Pediatr. 1996;8:276-282.
Geoffray A, et al. Ultrasonography and computed tomography for diagnosis and follow-up of biliary duct rhabdomyosarcomas in children. Pediatr Radiol. 1987;17:127-131.
Giardiello FM, et al. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut. 1996;39:867-869.
Gille J, Spieth K, Kaufmann R. Metronomic low-dose chemotherapy as antiangiogenic therapeutic strategy for cancer. J Dtsch Dermatol Ges. 2005;3:26-32.
Glick RD, et al. Extended left hepatectomy (left hepatic trisegmentectomy) in childhood. J Pediatr Surg. 2000;35:303-307.
Gonzalez-Crussi F, et al. Hepatoblastoma: attempt at characterization of histologic subtypes. Am J Surg Pathol. 1982;6:599-612.
Gray SG, et al. Comparative genomic hybridization reveals population-based genetic alterations in hepatoblastomas. Br J Cancer. 2000;83:1020-1025.
Greenberg M, Filler RM. Hepatic tumors. In: Pizzo, PA, Poplack, DG. Principles and Practice of Pediatric Oncology. Philadelphia: Lippincott; 1989:697-711.
Gururangan S, et al. Primary hepatic tumours in children: a 26-year review. J Surg Oncol. 1992;50:30-36.
Habrand JL, et al. Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys. 1992;23:525-531.
Hagiwara M, et al. Multimodal treatment of advanced adult Wilms’ tumor. J Urol. 1982;127:535-538.
Harada T, et al. Adult hepatoblastoma: case report and review of the literature. Aust N Z J Surg. 1995;65:686-688.
Hartley AL, et al. Epidemiological and familial aspects of hepatoblastoma. Med Pediatr Oncol. 1990;18:103-109.
Hata Y. The clinical features and prognosis of hepatoblastoma: follow-up studies done on pediatric tumors enrolled in the Japanese Pediatric Tumor Registry between 1971 and 1980. Part I. Committee of Malignant Tumors, Japanese Society of Pediatric Surgeons. Jpn J Surg. 1990;20:498-502.
Heimann A, et al. Hepatoblastoma presenting as isosexual precocity: the clinical importance of histologic and serologic parameters. J Clin Gastroenterol. 1987;9:105-110.
Herlin T, et al. Treatment of unresectable hepatoblastoma with cisplatin, vincristine and 5-fluorouracil. Eur J Pediatr. 1988;147:514-515.
Hernandez-Siverio N, et al. Solitary, non-parasitic hepatic cyst in childhood. An Esp Pediatr. 1988;28:269-270.
Hiyama E, et al. High expression of telomerase is an independent prognostic indicator of poor outcome in hepatoblastoma. Br J Cancer. 2004;91:972-979.
Ho SY, et al. Hepatic angiosarcoma presenting as hepatic rupture in a patient with long-term ingestion of arsenic. J Formos Med Assoc. 2004;103:374-379.
Hobbs KE. Hepatic hemangiomas. World J Surg. 1990;14:468-471.
Holcomb GW, et al. Experience with hepatic hemangioendothelioma in infancy and childhood. J Pediatr Surg. 1988;23:661-666.
Honda M, et al. Case report: intrahepatic cholangiocarcinoma with rhabdoid transformation. J Gastroenterol Hepatol. 1996;11:771-774.
Horowitz ME, et al. Hepatic undifferentiated (embryonal) sarcoma and rhabdomyosarcoma in children: results of therapy. Cancer. 1987;59:396-402.
Huang CB, et al. Primary Burkitt’s lymphoma of the liver: report of a case with long-term survival after surgical resection and combination chemotherapy. J Pediatr Hematol Oncol. 1997;19:135-138.
Huber BE, Richards CA. Regulated expression of artificial chimeric genes contained in retroviral vectors: implications for virus-directed enzyme prodrug therapy (VDEPT) and other gene therapy applications. J Drug Target. 1996;3:349-356.
Hutton KA, et al. Focal nodular hyperplasia of the liver in childhood. Eur J Pediatr Surg. 1993;3:370-372.
Ikeda H, et al. Association between hepatoblastoma and very low birth weight: a trend or a chance? J Pediatr. 1997;130:557-560.
Inamori H, et al. Laparoscopic radiofrequency ablation of hepatocellular carcinoma in the caudate lobe by using a new laparoscopic US probe with a forward-viewing convex-array transducer. Gastrointest Endosc. 2004;60:628-631.
Inoue S, et al. Successful resection of a large hepatoblastoma in a young adult: report of a case. Surg Today. 1995;25:974-977.
Isaacs HJr. Perinatal (congenital and neonatal) neoplasms: a report of 110 cases. Pediatr Pathol. 1985;3:165-216.
Isaacs HJr. Fetal and neonatal hepatic tumors. J Pediatr Surg. 2007;42:1797-1803.
Ishak KG. Primary hepatic tumors in childhood. Prog Liver Dis. 1976;5:636-667.
Ismail H, et al. Liver transplantation in children with hepatocellular carcinoma: do Milan criteria apply to pediatric patients? Pediatr Transplant. 2009;13:682-692.
Ito E, et al. Type Ia glycogen storage disease with hepatoblastoma in siblings [published erratum appears in Cancer 60:723]. Cancer. 1987;59:1776-1780.
Iwama T, Mishima Y. Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer. 1994;73:2065-2068.
Iwatsuki S, et al. Excisional therapy for benign hepatic lesions. Surg Gynecol Obstet. 1990;171:240-246.
Jain A, et al. Thrombotic and nonthrombotic hepatic artery complications in adults and children following primary liver transplantation with long-term follow-up in 1000 consecutive patients. Transpl Int. 2006;19:27-37.
Jha P, et al. Pediatric liver tumors: a pictorial review. Eur Radiol. 2009;19:209-219.
Ji W, Si CW. Inhibition of hepatitis B virus by retroviral vectors expressing antisense RNA. J Viral Hepat. 1997;4:167-173.
Jimenez-Heffernan JA, et al. Pathological case of the month: primary hepatic malignant tumor with rhabdoid features. Arch Pediatr Adolesc Med. 1998;152:509-510.
Jones RS. Surgical management of non-parasitic liver cysts. In: Blumgart, LH, editor. Surgery of the Liver and Biliary Tract. London: Churchill Livingstone; 1994:1211-1218.
Kacker LK, et al. Hepatoblastoma in an adult with biliary obstruction and associated portal venous thrombosis. HPB Surg. 1995;9:47-49.
Kaczynski J, et al. Incidence of primary liver cancer and aetiological aspects: a study of a defined population from a low-endemicity area. Br J Cancer. 1996;73:128-132.
Kadomatsu K, et al. Undifferentiated (embryonal) sarcoma of the liver: report of three cases. Surg Today. 1992;22:451-455.
Kaufmann SL, Stout AP. Malignant hemangioendothelioma in infants and children. Cancer. 1961;14:1186-1196.
Kelly DM, et al. Spontaneous rupture of a malignant rhabdoid tumour of the liver. Pediatr Surg Int. 1998;14:111-112.
Kitahara S, et al. Successful left trisegmentectomy for ruptured hepatoblastoma using intraoperative transarterial embolization. J Pediatr Surg. 1995;30:1709-1712.
Koda M, et al. Percutaneous sonographically guided radiofrequency ablation with artificial pleural effusion for hepatocellular carcinoma located under the diaphragm. AJR Am J Roentgenol. 2004;183:583-588.
Koishi S, et al. Myelodysplasia in a child with Beckwith-Wiedemann syndrome previously treated for hepatoblastoma with multi-agent chemotherapy, [letter]. J Pediatr Hematol Oncol. 1996;18:419-420.
Komura E, et al. Thrombopoietin in patients with hepatoblastoma. Stem Cells. 1998;16:329-333.
Komuro H, et al. Congenital mediastinal dumbbell neuroblastoma with spontaneous regression of liver metastases. Pediatr Surg Int. 1998;14:86-88.
Kraus JA, et al. Loss of heterozygosity on chromosome 1 in human hepatoblastoma. Int J Cancer. 1996;67:467-471.
Kushner BH, et al. Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol. 1996;14:1526-1531.
Lack EE, Ornvold K. Focal nodular hyperplasia and hepatic adenoma: a review of eight cases in the pediatric age group. J Surg Oncol. 1986;33:129-135.
Lack EE, et al. Botryoid rhabdomyosarcoma of the biliary tract. Am J Surg Pathol. 1981;5:643-652.
Lack EE, et al. Hepatoblastoma: a clinical and pathologic study of 54 cases. Am J Surg Pathol. 1982;6:693-705.
Lampkin BC, et al. Solid malignancies in children and adolescents. Surg Clin North Am. 1985;65:1351-1386.
La Quaglia MP. The surgical management of metastases in pediatric cancer. Semin Pediatr Surg. 1993;2:75-82.
La Quaglia MP, et al. Prognostic factors and outcome in patients 21 years and under with colorectal carcinoma. J Pediatr Surg. 1992;27:1085-1089.
Leary DL, et al. Follow-up imaging of benign pediatric liver tumors. Pediatr Radiol. 1989;19:234-236.
Lee CL, Ko YC. Survival and distribution pattern of childhood liver cancer in Taiwan. Eur J Cancer. 1998;34:2064-2067.
Le Luyer B, et al. Use of interferon in a case of hepatic hemangioma. Arch Pediatr. 2000;7:1201-1204.
Li GC, et al. Treatment of hepatocellular carcinoma with a novel gene-viral therapeutic system CNHK300-murine endostatin. Zhonghua Yi Xue Za Zhi. 2004;84:943-948.
Liaw YF, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-1531.
Lin SM, et al. Radiofrequency ablation improves prognosis compared with ethanol injection for hepatocellular carcinoma < or = 4 cm. Gastroenterology. 2004;127:1714-1723.
Liver Cancer Study Group of Japan. Primary liver cancer in Japan: sixth report. Cancer. 1987;60:1400-1411.
Llovet JM, Beaugrand M. Hepatocellular carcinoma: present status and future prospects. J Hepatol. 2003;38(Suppl 1):S136-S149.
Magrelli A, et al. Altered microRNA expression patterns in hepatoblastoma patients. Transl Oncol. 2009;2:157-163.
Maini CL, et al. Superselective intra-arterial radiometabolic therapy with I-131 lipiodol in hepatocellular carcinoma. Clin Nucl Med. 1996;21:221-226.
Malogolowkin MH, et al. Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J Clin Oncol. 2008;26:2379-2383.
Manchester KM, et al. Establishment and characterization of a novel hepatoblastoma-derived cell line. J Pediatr Surg. 1995;30:553-558.
Mann JR, et al. Malignant hepatic tumours in children: incidence, clinical features and aetiology. Paediatr Perinat Epidemiol. 1990;4:276-289.
Mantadakis E, et al. Primary hepatic Burkitt lymphoma. Pediatr Hematol Oncol. 2008;25:331-338.
Marcellini M, et al. Adult polycystic liver disease in childhood: a case report. Ital J Surg Sci. 1986;16:217-221.
Marks J, et al. Laparoscopic liver surgery: a report on 28 patients. Surg Endosc. 1998;12:331-334.
Marsh JW, et al. Role of liver transplantation for hepatobiliary malignant disorders. Lancet Oncol. 2004;5:480-488.
Martin LW, Woodman KS. Hepatic lobectomy for hepatoblastoma in infants and children. Arch Surg. 1969;98:1-7.
Martinez F, et al. Rhabdomyosarcoma of the biliary tree: the case for aggressive surgery. J Pediatr Surg. 1982;17:508-511.
Mascarello JT, Krous HF. Second report of a translocation involving 19q13.4 in a mesenchymal hamartoma of the liver. Cancer Genet Cytogenet. 1992;58:141-142.
Mazuryk M, et al. Benefit of aggressive multimodality therapy with autologous stem cell support for intra-abdominal desmoplastic small round cell tumor. Bone Marrow Transplant. 1998;21:961-963.
McClusky DA3rd, et al. Hepatic surgery and hepatic surgical anatomy: historical partners in progress. World J Surg. 1997;21:330-342.
Medary I, et al. Kinetics of primary tumor regression with chemotherapy: implications for the timing of surgery. Ann Surg Oncol. 1996;3:521-525.
Meinders AJ, et al. Mesenchymal hamartoma of the liver: failed management by marsupialization. J Pediatr Gastroenterol Nutr. 1998;26:353-355.
Meng F, Henson R, Patel T. Chemotherapeutic stress selectively activates NF-kappa B-dependent AKT and VEGF expression in liver cancer-derived endothelial cells. Am J Physiol Cell Physiol. 2007;293:C749-C760.
Meyers R. Tumors of the liver in children. Surg Oncol. 2007;16:195-203.
Meyers R, et al. Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg. 2007;42:2050-2056.
Meyers RL, et al. Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2009;53:1016-1022.
Mills AE. Undifferentiated primary hepatic non-Hodgkin’s lymphoma in childhood. Am J Surg Pathol. 1988;12:721-726.
Moore L, et al. Hepatocellular carcinoma following neonatal hepatitis. Pediatr Pathol Lab Med. 1997;17:601-610.
Morotti RA, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol. 2006;30:962-968.
Morrow CE, et al. Hepatic resection for secondary neoplasms. Surgery. 1982;92:610-614.
Mulliken JB. Diagnosis and natural history of hemangiomas. In: Mulliken, JB, Young, AE. Vascular Birthmarks: Hemangiomas and Malformations. Philadelphia: Saunders; 1988:41-62.
Muraji T, et al. The prognostic implication of hypercholesterolemia in infants and children with hepatoblastoma. J Pediatr Surg. 1985;20:228-230.
Nagaraj HS, et al. Rhabdomyosarcoma of the bile ducts. J Pediatr Surg. 1977;12:1071-1074.
Nagorney DM. Benign hepatic tumors: focal nodular hyperplasia and hepatocellular adenoma. World J Surg. 1995;19:13-18.
Napoli VM, Campbell WGJr. Hepatoblastoma in infant sister and brother. Cancer. 1977;39:2647-2650.
National Cancer Institute. SEER Cancer Statistics Review, 1973-1992: Tables and Graphs, National Cancer Institute. NIH Publication No. 96-2789. Bethesda, MD: National Institutes of Health; 1995.
Newman KD, et al. Malignant mesenchymoma of the liver in children. J Pediatr Surg. 1989;24:781-783.
Ng KK, et al. Three-dimensional magnetic resonance cholangiopancreatography for evaluation of obstructive jaundice. J Formos Med Assoc. 1997;96:586-592.
Ni YH, et al. Hepatocellular carcinoma in childhood: clinical manifestations and prognosis. Cancer. 1997;68:1737-1741.
Niemela M, et al. Interferon alpha-2a therapy in 18 hemangioblastomas. Clin Cancer Res. 2001;7:510-516.
Noronha R, Gonzalez-Crussi F. Hepatic angiosarcoma in childhood: a case report and review of the literature. Am J Surg Pathol. 1984;8:863-871.
Norton KI, et al. Leiomyosarcoma of the kidney in an HIV-infected child. Pediatr Radiol. 1997;27:557-558.
Oda H, et al. Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res. 1996;56:3320-3323.
Ohama K, et al. Alpha-fetoprotein (AFP) levels in normal children. Eur J Pediatr Surg. 1997;7:267-269.
Ohnuma N, et al. The role of magnetic resonance imaging for treatment in children with malignant solid tumor. Nippon Geka Gakkai Zasshi. 1991;92:1143-1146.
Ordonez NG. Desmoplastic small round cell tumor: I. A histopathologic study of 39 cases with emphasis on unusual histological patterns. Am J Surg Pathol. 1998;22:1303-1313.
Orozco H, et al. Undifferentiated (embryonal) sarcoma of the liver: report of a case. Rev Invest Clin. 1991;43:255-258.
Ortega JA, et al. Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Children’s Cancer Study Group. J Clin Oncol. 1991;9:2167-2176.
Ortega JA, et al. Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: a report from the Children’s Cancer Group and the Pediatric Oncology Group. J Clin Oncol. 2000;18:2665-2675.
O’Sullivan MJ, et al. Undifferentiated embryonal sarcoma with unusual features arising within mesenchymal hamartoma of the liver: report of a case and review of the literature. Pediatr Dev Pathol. 2001;4:482-489.
Otte JB. Progress in the surgical treatment of malignant liver tumors in children. Cancer Treat Rev. 2010;36:360-371.
Otte J, et al. Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer. 2004;42:74-83.
Otte JB, de Ville de Goyet J, Reding R. Liver transplantation for hepatoblastoma: indications and contraindications in the modern era. Pediatr Transplant. 2005;9:557-565.
Pang R, Poon R. Angiogenesis and antiangiogenic therapy in hepatocellular carcinoma. Cancer Lett. 2006;242:151-167.
Parada LA, et al. Cytogenetic abnormalities and clonal evolution in an adult hepatoblastoma. Am J Surg Pathol. 1997;21:1381-1386.
Passmore SJ, et al. Prolonged survival following multiple thoracotomies for metastatic hepatoblastoma. Med Pediatr Oncol. 1995;24:58-60.
Patterson K, Kapur SP, Chandra RS. Hepatocellular carcinoma in a noncirrhotic infant after prolonged parenteral nutrition. J Pediatr. 1985;106:797-800.
Perera MT, et al. Embryonal rhabdomyosarcoma of the ampulla of Vater in early childhood: report of a case and review of literature. J Pediatr Surg. 2009;44:e9-e11.
Perilongo G, et al. Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer. 2000;89:1845-1853.
Perisic VN, et al. Cholestasis caused by biliary botryoid sarcoma. Eur J Pediatr Surg. 1991;1:242-243.
Pham TH, et al. Outcomes of primary liver cancer in children: an appraisal of experience. J Pediatr Surg. 2007;42:834-839.
Pichlmayr R, et al. Role of liver transplantation in the treatment of unresectable liver cancer. World J Surg. 1995;19:807-813.
Pinna AD, et al. Treatment of fibrolamellar hepatoma with subtotal hepatectomy or transplantation. Hepatology. 1997;26:877-883.
Powers C, et al. Primary liver neoplasms: MR imaging with pathologic correlation. Radiographics. 1994;14:459-482.
Prasad VK, et al. Hepatic focal nodular hyperplasia in infant antenatally exposed to steroids, [letter]. Lancet. 1995;346:371.
Probst-Cousin S, et al. Malignant peripheral nerve sheath tumor with extensive miliary metastases: a case report. Gen Diagn Pathol. 1997;142:357-360.
Pul N, Pul M. Congenital solitary nonparasitic cyst of the liver in infancy and childhood. J Pediatr Gastroenterol Nutr. 1995;21:461-462.
Ramani K, et al. Novel gene delivery to liver cells using engineered virosomes. FEBS Lett. 1997;404:164-168.
Rao PS, et al. Multicystic kidney in association with hepatoblastoma: a case report. Jpn J Surg. 1989;19:583-585.
Raut CP, et al. Significant long-term survival after radiofrequency ablation of unresectable hepatocellular carcinoma in patients with cirrhosis. Ann Surg Oncol. 2005;12:616-628.
Ravindra KV. Long-term survival after spontaneous rupture of a malignant rhabdoid tumor of the liver. J Pediatr Surg. 2002;37:1488-1490.
Reyes J, et al. Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr. 2000;136:795-804.
Reynolds M. Conversion of unresectable to resectable hepatoblastoma and long-term follow-up study. World J Surg. 1995;19:814-816.
Reynolds M. Current status of liver tumors in children. Semin Pediatr Surg. 2001;10:140-145.
Reynolds P, et al. Birth characteristics and hepatoblastoma risk in young children. Cancer. 2004;100:1070-1076.
Rodriguez E, et al. Abnormalities of 2q: a common genetic link between rhabdomyosarcoma and hepatoblastoma. Genes Chromosomes Cancer. 1991;3:122-127.
Rocourt DV, et al. Contemporary management of benign hepatic adenoma using percutaneous radiofrequency ablation. J Pediatr Surg. 2006;41:1149-1152.
Roebuck DJ, et al. Hepatobiliary rhabdomyosarcoma in children: diagnostic radiology. Pediatr Radiol. 1998;28:101-108.
Roebuck DJ, et al. 2005 PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol. 2007;37:123-132.
Rojiani AM, et al. Hepatic hemangioblastoma: an unusual presentation in a patient with von Hippel–Lindau disease. Am J Surg Pathol. 1991;15:81-86.
Ross JS, et al. Primary hepatic leiomyosarcoma in a child with the acquired immunodeficiency syndrome. Hum Pathol. 1992;23:69-72.
Ruck P, Xiao JC. Stem-like cells in hepatoblastoma. Med Pediatr Oncol. 2002;39:504-507.
Ruymann FB, et al. Rhabdomyosarcoma of the biliary tree in childhood: a report from the intergroup rhabdomyosarcoma study. Cancer. 1985;56:575-581.
Ryu M, et al. Therapeutic results of resection, transcatheter arterial embolization and percutaneous transhepatic ethanol injection in 3225 patients with hepatocellular carcinoma: a retrospective multicenter study. Jpn J Clin Oncol. 1997;27:251-257.
Saito Y, et al. Anesthetic management and metabolic control of a patient with von Gierke’s disease (glycogen storage disease type I) associated with hepatic adenoma. Masui. 1984;33:1395-1399.
Sakatoku H, et al. Focal nodular hyperplasia in an adolescent with glycogen storage disease type I with mesocaval shunt operation in childhood: a case report and review of the literature. Acta Paediatr Jpn. 1996;38:172-175.
Saltzman DA, et al. Aggressive metastasectomy for pulmonic sarcomatous metastases: a follow-up study. Am J Surg. 1993;166:543-547.
Samama G, et al. Laparoscopic anatomical hepatic resection: report of four left lobectomies for solid tumors. Surg Endosc. 1998;12:76-78.
Santambrogio R, et al. Safety and efficacy of laparoscopic radiofrequency ablation of hepatocellular carcinoma in patients with liver cirrhosis. Surg Endosc. 2003;17:1826-1832.
Sanz N, et al. Rhabdomyosarcoma of the biliary tree. Pediatr Surg Int. 1997;12:200-201.
Sari N, et al. Infantile hepatic hemangioendothelioma with elevated serum alpha-fetoprotein. Pediatr Hematol Oncol. 2006;23:639-647.
Scaglioni PP, et al. Recent advances in the molecular biology of hepatitis B virus. Baillieres Clin Gastroenterol. 1996;10:207-225.
Schaller RTJr, et al. The advantages of hemodilution anesthesia for major liver resection in children. J Pediatr Surg. 1984;19:705-710.
Schnater JM, et al. Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer. 2002;94:1111-1120.
Schneid H, et al. The Beckwith-Wiedemann syndrome phenotype and the risk of cancer. Med Pediatr Oncol. 1997;28:411-415.
Schwarz RE, et al. Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol. 1998;5:416-422.
Selby DM, et al. Angiosarcoma of the liver in childhood: a clinicopathologic and follow-up study of 10 cases. Pediatr Pathol. 1992;12:485-498.
Shamberger RC, et al. Long-term hepatic regeneration and function in infants and children following liver resection. J Am Coll Surg. 1996;182:515-519.
Shapiro RS, et al. Case report: calcified liver metastases from osteosarcoma. J Comput Assist Tomogr. 1988;12:196-198.
Shimada H, et al. Pathology of fatal rhabdomyosarcoma: report from Intergroup Rhabdomyosarcoma Study (IRS-I and IRS-II). Cancer. 1987;59:459-465.
Simms LA, et al. Genetic mosaicism at the insulin locus in liver associated with childhood hepatoblastoma. Genes Chromosomes Cancer. 1995;13:72-73.
Spunt SL, et al. Aggressive surgery is unwarranted for biliary tract rhabdomyosarcoma. J Pediatr Surg. 2000;35:309-316.
Srouji MN, et al. Mesenchymal hamartoma of the liver in infants. Cancer. 1978;42:2483-2489.
Stocker JT. Hepatoblastoma. Semin Diagn Pathol. 1994;11:136-143.
Stringer MD, et al. Improved outcome for children with hepatoblastoma. Br J Surg. 1995;82:386-391.
Su WT, et al. Hepatic metastasectomy in children. Cancer. 2007;109:2089-2092.
Superina R, Bilik R. Results of liver transplantation in children with unresectable liver tumors. J Pediatr Surg. 1996;31:835-839.
Surendran N, et al. Hepatoblastoma in siblings. J Pediatr Surg. 1989;24:1169-1171.
Swarts S, et al. Significance of extra copies of chromosome 20 and the long arm of chromosome 2 in hepatoblastoma. Cancer Genet Cytogenet. 1996;91:65-67.
Taat F, et al. Hepatoblastoma in a girl with biliary atresia: coincidence or co-incidence. Pediatr Blood Cancer. 2004;43:603-605.
Tagge EP, et al. Resection, including transplantation, for hepatoblastoma and hepatocellular carcinoma: impact on survival. J Pediatr Surg. 1992;27:292-296.
Takvorian P, et al. Hepatic rupture after puncture biopsy: value of embolization—apropos of a case with hepatoblastoma. Pediatrie (Bucur). 1988;43:531-533.
Tamano S, et al. Histogenesis and the role of p53 and K-ras mutations in hepatocarcinogenesis by glyceryl trinitrate (nitroglycerin) in male F344 rats. Carcinogenesis. 1996;17:2477-2486.
Terris B, et al. Interphase cytogenetic analysis reveals numerical chromosome aberrations in large liver cell dysplasia. J Hepatol. 1997;27:313-319.
Theegarten D, et al. Mixed malignant germ cell tumour of the liver. Virchows Arch. 1998;433:93-96.
Thomas D, et al. Familial hepatoblastoma and APC gene mutations: renewed call for molecular research. Eur J Cancer. 2003;39:2200-2204.
Thomas PR, et al. Prognostic implications of hepatic adhesion, invasion, and metastases at diagnosis of Wilms’ tumor. The National Wilms’ Tumor Study Group. Cancer. 1991;68:2486-2488.
Tomlinson GE, et al. Cytogenetic evaluation of a large series of hepatoblastomas: numerical abnormalities with recurring aberrations involving 1q12-q21. Genes Chromosomes Cancer. 2005;44:177-184.
Torre W, Rodriguez-Spiteri N, Sierrasesumaga L. Current role for resection of thoracic metastases in children and young adults: do we need different strategies for this population? Thorac Cardiovasc Surg. 2004;52:90-95.
Toyosaka A, et al. Pathologic and radiographic studies of intrahepatic metastasis in hepatocellular carcinoma: the role of efferent vessels. HPB Surg. 1996;10:97-103.
Tozzi MC, et al. The hepatic malignant mesenchymoma: a case report. Eur J Pediatr. 1992;151:488-491.
Tsai SY, et al. Hepatoblastoma in an infant with Beckwith-Wiedemann syndrome. J Formos Med Assoc. 1996;95:180-183.
Tsao K, et al. Fetal therapy for giant hepatic cysts. J Pediatr Surg. 2002;37:E31.
Tsuchida Y, et al. The role of subfractionation of alpha-fetoprotein in the treatment of pediatric surgical patients. J Pediatr Surg. 1997;32:514-517.
Tsunoda Y, et al. Non-alpha-fetoprotein-producing anaplastic hepatoblastoma cell line. In Vitro Cell Dev Biol Anim. 1996;32:194-196. (letter)
Tung C, et al. Rapid production of interleukin-2-secreting tumor cells by herpes simplex virus–mediated gene transfer: implications for autologous vaccine production. Hum Gene Ther. 1996;7:2217-2224.
Van Tornout JM, et al. Rate and magnitude of decline in alphafetoprotein (AFP) levels in treated children with unresectable or metastatic hepatoblastoma (HB) are indicators of outcome: a report from the Children’s Cancer Group, [abstract]. Proc Annu Meet Am Soc Clin Oncol. 1993;12:A1408.
Van Tornout JM, et al. Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children’s Cancer Group. J Clin Oncol. 1997;15:1190-1197.
Vaughan WG, et al. Favorable outcome in children with Beckwith-Wiedemann syndrome and intraabdominal malignant tumors. J Pediatr Surg. 1995;30:1042-1044.
Vetter D, et al. Hepatic undifferentiated (or embryonal) sarcoma: diagnostic and therapeutic problems apropos of botryoid rhabdomyosarcoma. Gastroenterol Clin Biol. 1989;13:98-103.
von Schweinitz D, et al. Production of interleukin-1 beta and interleukin-6 in hepatoblastoma. Int J Cancer. 1993;53:728-734.
von Schweinitz D, et al. Clinico-pathological criteria with prognostic relevance in hepatoblastoma. Eur J Cancer. 1994;30A:1052-1058.
von Schweinitz D, et al. Results of the HB-89 study in treatment of malignant epithelial liver tumors in childhood and concept of a new HB-94 protocol. Klin Paediatr. 1994;206:282-288.
von Schweinitz D, et al. Extramedullary hematopoiesis and intratumoral production of cytokines in childhood hepatoblastoma. Pediatr Res. 1995;38:555-563.
von Schweinitz D, et al. Complete resection before development of drug resistance is essential for survival from advanced hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg. 1995;30:845-852.
von Schweinitz D, et al. Liver tumors in neonates and very young infants: diagnostic pitfalls and therapeutic problems. Eur J Pediatr Surg. 1995;5:72-76.
von Schweinitz D, et al. Hepatocyte growth factor in pediatric hepatoblastoma. Langenbecks Arch Chir Suppl Kongressbd. 1997;114:37-40.
von Schweinitz D, et al. Prognostic factors and staging systems in childhood hepatoblastoma. Int J Cancer. 1997;74:593-599.
Vujanic GM, et al. Rhabdoid tumour of the kidney: a clinicopathological study of 22 patients from the International Society of Paediatric Oncology (SIOP) nephroblastoma file. Histopathology. 1996;28:333-340.
Walhof CM, et al. Half-life of alpha-fetoprotein in patients with a teratoma, endodermal sinus tumor, or hepatoblastoma. Pediatr Hematol Oncol. 1988;5:217-227.
Wang JN, et al. Cardiac tumors in infants and children. Acta Paediatr Taiwan. 2003;44:215-219.
Wegmann W, et al. [Liver cell carcinoma as a late complication of Alagille syndrome (arterio-hepatic dysplasia)]. Leber Magen Darm. 1996;26:157-158. 161-163
Weinberg AG, Finegold MJ. Primary hepatic tumors of childhood. Hum Pathol. 1983;14:512-537.
Weitman S, et al. Pediatric phase II cancer chemotherapy trials: a Pediatric Oncology Group study. J Pediatr Hematol Oncol. 1997;19:187-191.
Westaby D, et al. Androgen-related primary hepatic tumors in non-Fanconi patients. Cancer. 1983;51:1947-1952.
Wheatley JM, et al. Liver regeneration in children after major hepatectomy for malignancy-evaluation using a computer-aided technique of volume measurement. J Surg Res. 1996;61:183-189.
White FV, et al. Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. Am J Surg Pathol. 1999;23:249-256.
Wong KK, et al. The use of positron emission tomography in detecting hepatoblastoma recurrence: a cautionary tale. J Pediatr Surg. 2004;39:1779-1781.
Yedibela S, et al. Undifferentiated, embryonal sarcoma as a rare cause of spontaneous liver rupture in adults. Chirurg. 2000;71:101-105.
Yen JB, et al. Hepatic mesenchymal hamartoma. J Paediatr Child Health. 2003;39:632-634.
Yokomori K, et al. Complete disappearance of unresectable hepatoblastoma by continuous infusion therapy through hepatic artery. J Pediatr Surg. 1991;26:844-846.
Yuri T, et al. Malignant rhabdoid tumor of the liver: case report and literature review. Pathol Int. 2004;54:623-629.
Zampieri N, et al. Botryoid rhabdomyosarcoma of the biliary tract in children: a unique case report. Eur J Cancer Care (Engl). 2006;15:463-466.
Zatkova A, et al. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer. 2004;39:126-137.