[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 92 Hepatic Encephalopathy, Hepatorenal Syndrome, Hepatopulmonary Syndrome, and Systemic Complications of Liver Disease
HEPATIC ENCEPHALOPATHY
The term hepatic encephalopathy (HE) encompasses a wide array of transient and reversible neurologic and psychiatric manifestations usually found in patients with chronic liver disease and portal hypertension, but also seen in patients with acute liver failure. HE develops in 50% to 70% of patients with cirrhosis, and its occurrence is a poor prognostic indicator, with projected one- and three-year survival rates of 42% and 23%, respectively, without liver transplantation.1 Symptoms may range from mild neurologic disturbances to overt coma.2,3 HE is often triggered by an inciting event that results in a rise in the serum ammonia level. The precise underlying pathophysiologic mechanisms are not well understood, and the mainstay of therapy is the elimination of the precipitating event and excess ammonia.4 Liver transplantation generally reverses HE.
PATHOPHYSIOLOGY
A number of factors, occurring alone or in combination, have been implicated in the development of HE. These factors may differ in acute and chronic liver disease and include the production of neurotoxins, altered permeability of the blood-brain barrier, and abnormal neurotransmission (Fig. 92-1). The best-described neurotoxin involved in HE is ammonia, which is produced primarily in the colon, where bacteria metabolize proteins and other nitrogen-based products into ammonia. Enterocytes synthesize ammonia from glutamine.4–6 Once produced, ammonia enters the portal circulation and, under normal conditions, is metabolized and cleared by hepatocytes. In cirrhosis and portal hypertension, reduced hepatocyte function and portosystemic shunting contribute to increased circulating ammonia levels. Arterial hyperammonemia is observed in up to 90% of patients with HE, although serum levels are neither sensitive nor specific indicators of its presence. Increased permeability of the blood-brain barrier increases the uptake and extraction of ammonia by the cerebellum and basal ganglia.7–9 Acute hyperammonemia appears to have a direct effect on brain edema, astrocyte swelling, and the transport of neurally active compounds such as myoinositol, and thereby contributes to HE.10–12
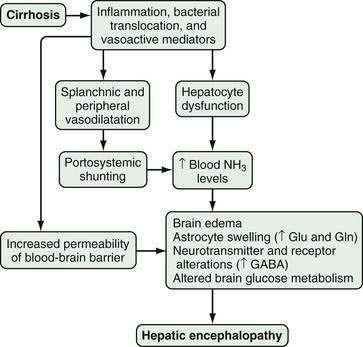
Figure 92-1. Pathophysiology of hepatic encephalopathy. GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; NH3, ammonia.
Other alterations in HE affect neuronal membrane fluidity, central nervous system (CNS) neurotransmitter expression, and neurotransmitter receptor expression and activation.13,14 The γ-aminobutyric acid (GABA)–benzodiazepine system has been the most well studied. Although CNS benzodiazepine levels and GABA receptor concentrations are unchanged in animal models of HE, increased sensitivity of the astrocyte (peripheral-type) benzodiazepine receptor enhances activation of the GABA-benzodiazepine system.15,16 This activation occurs in part through a feed-forward system in which production of neurosteroids (allopregnanolone and tetrahydrodeoxycorticosterone) by astrocytes further activates the GABAA-benzodiazepine receptor system.17,18 Other factors that influence CNS neurotransmission, including serotonin (5-hydroxytryptamine, 5-HT),19–21 nitric oxide (NO), circulating opioid peptides, manganese, and increased oxygen free radical production, have also been postulated to contribute to HE.4
Finally, hyperammonemia, particularly in acute liver failure, also increases astrocyte glutamine production via glutamine synthetase. The rise in astrocyte glutamine and glutamate concentrations contributes to factors associated with CNS dysfunction.5,22,23
CLINICAL FEATURES AND DIAGNOSIS
HE may present as a spectrum of reversible neuropsychiatric symptoms and signs, ranging from mild changes in cognition to profound coma, in patients with acute or chronic liver disease. It is often precipitated by an inciting event (e.g., gastrointestinal bleeding, electrolyte abnormalities, infections, medications, dehydration). The diagnosis of HE, therefore, requires careful consideration in the appropriate clinical situation. Occasionally, HE may be the initial presentation of chronic liver disease. Subtle findings in HE may include forgetfulness, alterations in handwriting, difficulty with driving, and reversal of the sleep-wake cycle.24,25 Overt findings may include asterixis, agitation, disinhibited behavior, seizures, and coma. Other causes of altered mental status, particularly hypoglycemia, hyponatremia, medication ingestion, and structural intracranial abnormalities resulting from coagulopathy or trauma, should be considered and rapidly excluded in patients suspected of having HE.
No specific laboratory findings indicate the presence of HE definitively. The most commonly used test to assess a patient with possible HE is the blood ammonia level. An elevation in the blood ammonia level in a patient with cirrhosis and altered mental status supports a diagnosis of HE. Blood ammonia levels may be elevated in the absence of HE, however, because of gastrointestinal bleeding or the ingestion of certain medications (e.g., diuretics, alcohol, narcotics, valproic acid).15,26,27 In addition, blood ammonia levels may be elevated in the presence of HE, even in the absence of cirrhosis and portal hypertension, in patients with metabolic disorders that influence ammonia generation or metabolism, such as urea cycle disorders (see Chapter 76) and disorders of proline metabolism (Table 92-1).28,29 Use of a tourniquet when blood is drawn and delayed processing and cooling of a blood sample may raise the blood ammonia level.15 Measurement of arterial ammonia offers no advantage over measurement of venous ammonia levels in patients with chronic liver disease.30 In patients with acute liver failure, however, elevated arterial ammonia levels (150 to 200 mg/dL or higher) may be predictive of the presence of brain edema and herniation (see Chapter 93).12,31,32
Table 92-1 Differential Diagnosis of Hyperammonemia
Of the scoring systems used to grade the severity of HE, the West Haven system, based on a scale of 0 to 4, is the most widely used in clinical practice (Table 92-2).25 Although clinically useful, the West Haven criteria are insensitive and have led to the development of standardized psychometric tests and rapid bedside mental status assessments to aid in the diagnosis of HE and facilitate research.33–37 One simple paper and pencil test, the portosystemic encephalopathy syndrome test (PSET), evaluates the patient’s attention, concentration, fine motor skills, and orientation and has been shown to be highly specific for the diagnosis of HE.33,38 The development of these tests has led to recognition of the syndrome of minimal HE, in which abnormalities are observed on testing but clinically recognizable alterations of HE are minimal or not detected. The presence of minimal HE is common in patients with cirrhosis, appears to influence the patient’s quality of life and driving ability, and confers an increased risk that overt HE will develop in the patient. Whether treatment of minimal HE confers any benefit is an area of active investigation.24,39–41
| GRADE | Impairment | |
|---|---|---|
| INTELLECTUAL FUNCTION | NEUROMUSCULAR FUNCTION | |
| 0 | Normal | Normal |
| Minimal, subclinical | Normal examination findings. Subtle changes in work or driving | Minor abnormalities of visual perception or on psychometric or number tests |
| 1 | Personality changes, attention deficits, irritability, depressed state | Tremor and incoordination |
| 2 | Changes in sleep-wake cycle, lethargy, mood and behavioral changes, cognitive dysfunction | Asterixis, ataxic gait, speech abnormalities (slow and slurred) |
| 3 | Altered level of consciousness (somnolence), confusion, disorientation, and amnesia | Muscular rigidity, nystagmus, clonus, Babinski sign, hyporeflexia |
| 4 | Stupor and coma | Oculocephalic reflex, unresponsiveness to noxious stimuli |
Modified from the West Haven Criteria; in Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: Final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002; 35:716-21.
A number of novel imaging and functional tests have been studied in the diagnosis of HE. Magnetic resonance spectroscopy (MRS) has been used to measure brain concentrations of choline and glutamine noninvasively.42 Magnetic resonance (MR) T1 mapping with partial inversion recovery (TAPIR) has been investigated as a means to measure changes in the brain quantitatively over clinically relevant measurement times.33 Whether MR-based techniques can be standardized and become practical diagnostic tests is uncertain. The critical flicker frequency test, a simple light-based test that has been used to assess cerebral cortex function in a number of disorders, has been shown to be a reliable marker of minimal HE and may become a clinically useful screening test.34–36
TREATMENT
Current treatments for HE are directed primarily toward the elimination or correction of precipitating factors (e.g., bleeding, infection, hypokalemia, medications, dehydration), reduction in elevated blood ammonia levels, and avoidance of the toxic effects of ammonia in the CNS. In the past, dietary protein restriction was considered an important component of the treatment of HE. Subsequent work, however, has suggested that limiting protein-calorie intake is not beneficial in patients with HE.43–45 Vegetable and dairy proteins are preferred to animal proteins because of a more favorable calorie-to-nitrogen ratio. Although branched-chain amino acid supplementation may improve symptoms modestly, the benefits of such supplementation are not sufficient to justify its routine use.4
Nonabsorbable disaccharides have been the cornerstone of the treatment of HE. Oral lactulose or lactitol (the latter is not available in the United States) are metabolized by colonic bacteria to byproducts that appear to have beneficial effects by causing catharsis and reducing intestinal pH, thereby inhibiting ammonia absorption.46 These agents improve symptoms in patients with acute and chronic HE when compared with placebo but do not improve psychometric test performance or mortality. The most common side effects experienced by patients who take lactulose are abdominal cramping, flatulence, diarrhea, and electrolyte imbalance. Lactulose may also be administered per rectum (as an enema) to patients who are at increased risk of aspiration, although the efficacy of enema administration has not been evaluated.
Oral antibiotics also have been used to treat HE, with the aim of modifying the intestinal flora and lowering stool pH to enhance the excretion of ammonia. Antibiotics are generally used as second-line agents after lactulose or in patients who are intolerant of nonabsorbable disaccharides. Neomycin has been approved by the U.S. Food and Drug Administration (FDA) for use in acute HE in a dose of 1 to 3 g orally every six hours for up to six days but has been used more commonly off-label to treat chronic HE in doses of 0.5 to 1 g every 12 hours, in addition to lactulose. The efficacy of neomycin in acute or chronic HE, however, is not clearly established,47 and ototoxicity and nephrotoxicity caused by neomycin have been reported, particularly in patients with preexisting renal dysfunction.4 Rifaximin has been studied and approved by the FDA for the treatment of chronic HE on the basis of the results of a multicentered, randomized, controlled trial in which the overall clinical efficacy and rate of side effects were similar in patients treated with lactitol and those treated with rifaximin.48 The usual dose is 400 mg orally three times daily. Two systematic reviews of randomized controlled trials that compared rifaximin with other therapies (nonabsorbable disaccharides and other antibiotics) for the treatment of acute or chronic HE have confirmed that the efficacy and side effect profiles are comparable.49,50 Other antibiotics, including metronidazole and vancomycin, have been reported to be effective in small trials and case series, but the data to support their use are insufficient.
In addition to antibiotics, several other agents that may modify intestinal flora and modulate ammonia generation or absorption have been evaluated as potential treatments for HE. Acarbose, an intestinal α-glucosidase inhibitor used to treat type 2 diabetes mellitus, inhibits the intestinal absorption of carbohydrates and glucose and results in their enhanced delivery to the colon. As a result, the ratio of saccharolytic to proteolytic bacterial flora is increased, and blood ammonia levels are decreased. A randomized, controlled, double-blind, crossover trial has demonstrated that acarbose improves mild HE in patients with cirrhosis and adult-onset diabetes mellitus.51 Similarly, probiotic regimens have been used to modify intestinal flora and diminish ammonia generation. Four small studies have suggested that these agents may be beneficial in humans with mild HE.40,52–55 These agents merit further evaluation and may be alternatives for patients who do not tolerate lactulose.
Strategies to enhance ammonia clearance may also be useful in the treatment of HE. Sodium benzoate, sodium phenylbutyrate, and sodium phenylacetate, all of which increase ammonia excretion in urine, are approved by the FDA for the treatment of hyperammonemia resulting from urea cycle enzyme defects and may improve HE in cirrhosis (see Chapter 76). Administration of sodium benzoate, however, results in a high sodium load, and the efficacy of this agent is not clearly established.4,56 The combination of intravenous sodium phenylacetate and sodium benzoate (Ammonul, Ucyclyd Pharma, Scottsdale, Ariz) in HE is being studied. Administration of zinc, which has been used because zinc deficiency is common in patients with cirrhosis57–59 and because zinc increases the activity of ornithine transcarbamylase, an enzyme in the urea cycle, may also improve HE; however, clear efficacy has not been established. Extracorporeal albumin dialysis using the molecular adsorbent recirculating system (MARS) has resulted in a reduction in blood ammonia levels and improvement in severe HE in patients with acute-on-chronic liver failure (see Chapter 93).60 Further studies are needed to clarify whether albumin dialysis has a role in treatment of HE. Finally, l-ornithine–l-aspartate (LOLA), a salt of the amino acids ornithine and aspartic acid that activates the urea cycle and enhances ammonia clearance, has been shown in several randomized controlled studies to improve HE compared with lactulose61–63; however, this agent is not available in the United States.
Flumazenil is a specific benzodiazepine (GABAA receptor) antagonist that has been used in patients with HE. It improves the degree of encephalopathy and electrophysiologic findings in approximately one fourth of patients with grade 3 or 4 HE. It has a short half-life and a number of potential side effects, including seizures, arrhythmias, and withdrawal symptoms, that limit its clinical usefulness.4
HEPATORENAL SYNDROME
The term hepatorenal syndrome (HRS) was first used in 1932 to describe acute kidney injury, mainly acute tubular necrosis (ATN) or interstitial nephritis, in a group of patients who had undergone biliary tract surgery.64 As pathophysiologic mechanisms were better elucidated, HRS was found to be part of a cascade of events associated with intense dilatation of the splanchnic arterial vasculature in the setting of cirrhosis or acute liver injury and resulting in profound renal arterial vasoconstriction and progressive renal failure.65 Histologically, the kidneys are normal in HRS. Function may be restored by correction of portal hypertension, liver transplantation, removal of the kidneys and transplantation of them into a noncirrhotic recipient, and, in some cases, medical therapy (see later).66–68
Acute renal dysfunction occurs in 15% to 25% of hospitalized patients with cirrhosis. Among the multiple causes of acute kidney injury, prerenal azotemia (resulting from intravascular volume depletion) is most common, accounting for 60% to 80% of cases. ATN is the second most common cause of acute kidney injury in this setting and accounts for 20% to 40% of cases.69,70 HRS appears to be an extension of the pathophysiology of prerenal azotemia and is therefore potentially reversible. The annual frequency of HRS in cirrhotic patients with ascites is roughly 8%71 and, in some reports, as high as 40%.72 HRS develops in approximately 30% of cirrhotic patients who are admitted with spontaneous bacterial peritonitis (SBP) or other infection, 25% who are hospitalized with severe alcoholic hepatitis, and 10% who require serial large-volume paracentesis.73 The observation that morbidity and mortality remain high once the syndrome is established has led to a focus on the prevention and early therapy of renal dysfunction in patients with cirrhosis.71
PATHOPHYSIOLOGY
The pathophysiology of HRS is complex and incompletely characterized. Three important components are recognized to contribute to the initiation and perpetuation of altered renal perfusion (Fig. 92-2): (1) arterial vasodilatation in the splanchnic and systemic circulation; (2) renal vasoconstriction; and (3) cardiac dysfunction.74 These components influence renal function in concert and form the basis for current therapies and preventive strategies.
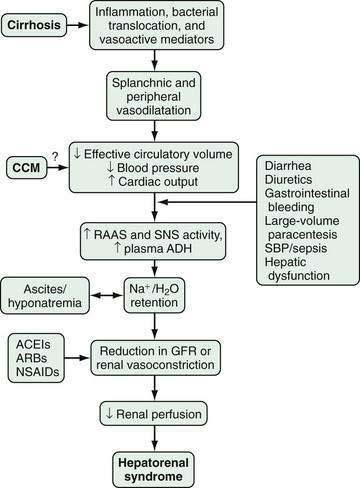
Splanchnic Arterial Vasodilatation
Splanchnic and systemic arterial vasodilation are hallmarks of the progression of portal hypertension in cirrhosis and lead to decreased effective circulating blood volume and ultimately to a decrease in blood pressure. This process is mediated by a number of endogenous substances, including NO, carbon monoxide (CO), glucagon, prostacyclin, adrenomedullin, and endogenous opiates that are released or act locally in the vasculature in response to mechanical and inflammatory signals.65,71,75,76 In the early stages of portal hypertension, compensation for the decrease in effective circulatory volume includes an increase in heart rate and cardiac output, thereby creating a hyperdynamic circulation.77 As liver disease and splanchnic vasodilatation progress, additional compensatory mechanisms are activated.
Renal Arterial Vasoconstriction
Splanchnic and systemic vasodilatation also lead to compensatory renal vasoconstriction and renal sodium and water retention. These responses are mediated by stimulation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system (RAAS), and nonosmotic release and activity of arginine vasopressin (as a result of increased secretion and decreased clearance of arginine vasopressin and apparent increased expression of vasopressin-regulated water channels), as well as intrarenal events. Although the precise intrarenal mechanisms are speculative, altered production or action of endothelins, prostaglandins, kallikreins, and F2-isoprostanes may contribute to renal vasoconstriction.72,78,79 Ultimately, the balance between vasoconstrictive responses in the kidney and systemic and splanchnic vasodilatation is lost, thus leading to a prominent increase in renal vascular resistance, decrease in renal perfusion, and reduction in the glomerular filtration rate.71,77
Cardiac Dysfunction
Impaired cardiac function also may contribute to renal hypoperfusion in HRS. In one prospective study, HRS developed in cirrhotic patients with more severe arterial vasodilatation and lower cardiac output.80 In another study of a cohort of patients who were treated for SBP, renal dysfunction (including HRS in some cases) developed in those with lower cardiac output and lower arterial pressure measurements associated with higher circulating levels of norepinephrine and renin plasma activity, despite effective treatment of the infection.81 These data demonstrate that cardiac output is impaired in patients with cirrhosis in whom HRS develops as compared with those in whom HRS does not develop, and suggests that cardiac dysfunction may be an important additional factor in the pathogenesis of HRS. The relationship between cardiac dysfunction in patients with HRS and cirrhotic cardiomyopathy (see later) has not been studied.82,83
CLINICAL FEATURES AND DIAGNOSIS
HRS is a functional disorder, and therefore diagnostic laboratory and imaging studies are not available. The diagnosis of HRS requires a high index of clinical suspicion and exclusion of other potential causes of kidney injury. The diagnostic criteria for HRS as defined by the International Ascites Club Consensus Workshop in 2007 include the following: (1) cirrhosis with ascites; (2) serum creatinine level higher than 1.5 mg/dL (133 µmol/L); (3) lack of improvement in the serum creatinine level to 1.5 mg/dL (133 µmol/L) or less after at least two days of diuretic withdrawal and volume expansion with albumin (1 g/kg of body weight/day, up to a maximum of 100 g/day); (4) absence of shock, (5) lack of current or recent treatment with nephrotoxic drugs; and (6) absence of parenchymal kidney disease as indicated by proteinuria of more than 500 mg/day, microhematuria (>50 red blood cells/high power field), or abnormal renal ultrasonographic findings (Table 92-3).74
Table 92-3 Diagnostic Criteria for Hepatorenal Syndrome*
* As defined by the International Ascites Club Consensus Workshop in 2007 (Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310-8).
Several aspects of the diagnosis of HRS deserve emphasis. First, in patients with no prior evidence or history of renal impairment, the diagnostic criteria for HRS include an increase in the serum creatinine level by 50% above baseline to a level higher than 1.5 mg/dL (133 µmol/L).71 Although this definition is standardized, a subset of patients with cirrhosis and end-stage liver disease have a profound decrease in muscle mass and urea synthesis that may in turn result in reduced serum creatinine and blood urea nitrogen levels, thereby potentially delaying recognition of HRS.73,84 Second, many medications, most notably diuretics, lactulose, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and nonsteroidal anti-inflammatory drugs, may influence intravascular volume status and renal perfusion and should be identified expeditiously and discontinued in the setting of acute renal dysfunction. Third, even though SBP may not be accompanied by obvious symptoms and signs, HRS may develop in as many as 20% of affected patients.85 Therefore, a low threshold for evaluating cirrhotic patients with ascites for the presence of SBP is required.
CLASSIFICATION
HRS has traditionally been classified into two types on the basis of clinical characteristics and prognosis (types 1 and 2).74 Some authors have advocated expanding the classification to include patients who are not encompassed within the current framework (i.e., type 3 HRS).72
Type 1 HRS presents as a rapidly progressive form of renal dysfunction. Typically, the serum creatinine level doubles to a value higher than 2.5 mg/dL in a period of two weeks or less. Type 1 HRS is often triggered by an inciting event that causes a rapid decline in the hemodynamic parameters that maintain renal homeostasis in cirrhotic patients.70 The most common triggers include severe bacterial infections,86–88 gastrointestinal bleeding, surgical procedures, and acute liver injury.74,89,90 SBP is the main bacterial infection that predisposes cirrhotic patients to develop HRS. Other bacterial infections have less of an impact on the development of the syndrome, unless sepsis is present or the response to antibiotic therapy is poor.71,87,91 Patients who exhibit high levels of inflammatory response markers and patients with severe circulatory depression prior to the onset of infection are most susceptible to the development of HRS. Some degree of adrenal insufficiency has been found in 80% of patients with septic shock in whom HRS develops, and administration of glucocorticoids may improve survival.92
Type 2 HRS is a more slowly progressing entity compared with type 1 HRS but still carries a median survival of only approximately six months. Type 2 HRS is observed in patients with severe ascites (diuretic resistant) and is characterized by serum creatinine levels lower than 2.5 mg/dL. The degree of arterial hypotension and circulatory dysfunction is less than that seen with type 1 HRS. Type 1 HRS may develop in patients with type 2 HRS following a triggering event.71,72
Many patients with cirrhosis and portal hypertension also have underlying chronic kidney disease, which complicates recognition of HRS, even in the presence of underlying pathophysiologic mechanisms that favor the development of HRS. Whether these patients should be considered to have a unique form of HRS (type 3) and whether they should be treated in a fashion similar to that for other patients with HRS have not been clearly defined.72
PREVENTION AND TREATMENT
The high mortality rate of HRS underscores the importance of prevention. In particular, intravascular volume depletion (resulting from overdiuresis, diarrhea caused by lactulose, gastrointestinal bleeding from gastroesophageal varices, or large-volume paracentesis without colloid administration), nephrotoxins (e.g., nonsteroidal anti-inflammatory drugs, nephrotoxic antibiotics), and infection (SBP, bacteremia) should be avoided or addressed if present. Specific guidelines address the primary and secondary prophylaxis of variceal bleeding (see Chapter 90), administration of colloid (albumin) to patients with a rising serum creatinine level after a large-volume paracentesis and in the presence of SBP (see Chapter 91), and prophylactic administration of antibiotics to patients at high risk of SBP or other infections because of hospitalization for gastrointestinal bleeding (see Chapters 19 and 90).85,86,89,91 Routine invasive hemodynamic monitoring of cirrhotic patients with a rising serum creatinine level does not have a clear benefit and is not recommended.
The concept that treatment of HRS is possible and may improve survival has emerged since 2000. Current options include medical therapies, transjugular intrahepatic portosystemic shunt (TIPS) placement, and liver transplantation. Medical therapies for HRS are directed toward reversing the underlying splanchnic and systemic vasodilatation with vasoconstrictors and increasing effective circulatory volume with the use of colloid. Such treatment is used increasingly as a temporizing measure until definitive treatment for liver disease (liver transplantation) or portal hypertension (TIPS) is undertaken, or until an acute process (SBP, gastrointestinal bleeding) has been reversed (Table 92-4).93,94
Table 92-4 Management of Hepatorenal Syndrome
ACEIs, angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; IV, intravenous; MAP, mean arterial pressure; NSAIDs, nonsteroidal anti-inflammatory drugs; SBP, spontaneous bacterial peritonitis.
* Not available in the United States.
Data from references 69,74,85,86,89,91,93–94,97–124.
Medical Therapy
Terlipressin is an intravenously administered selective vasopressin 1 receptor agonist vasoconstrictor that has been used in Europe and is under review by the FDA for the treatment of type 1 HRS.95,96 It has been evaluated in approximately 330 patients included in four randomized, controlled trials and two meta-analyses.69,97–109 In two multicenter studies of patients with type 1 HRS, terlipressin (given in two different doses in the two studies) in combination with albumin improved serum creatinine levels relative to albumin alone (30% to 43% vs. 8% to 13%), although survival was not significantly different in the two groups.106,107 In addition, in one study,106 terlipressin was associated with a significantly increased rate of cardiovascular complications compared with albumin alone; therefore, this agent should only be used with close monitoring. Finally, the response to terlipressin in patients with type 1 HRS appeared to be better in patients with less severe renal dysfunction at baseline, thus supporting the early initiation of therapy. These studies indicate that administration of terlipressin in combination with albumin can improve renal function in HRS, although the optimal dose and duration of therapy are not defined.
Midodrine, an orally administered α1-adrenergic agonist, and octreotide, a somatostatin analog that inhibits endogenous vasodilators,110,111 have been used in combination with albumin for type 1 HRS in three small nonrandomized studies. In two studies, treatment with midodrine, titrated to cause a rise in mean arterial blood pressure, was associated with improved serum creatinine levels and improved survival compared with no treatment and was associated with few major side effects.112–114 This regimen has the advantage of ease of administration and appears to have a favorable safety profile; however, its efficacy has not been established in randomized controlled trials.
Norepinephrine, a widely available intravenously administered α1-adrenergic agonist, in combination with albumin, has been proposed as an alternative to terlipressin on the basis of two small pilot studies.115,116 In one study of 22 patients with type 1 or 2 HRS, norepinephrine appeared to be as effective and safe as terlipressin. Significant cardiovascular side effects, however, have been reported with the use of norepinephrine to treat HRS, and whether the efficacy and safety of norepinephrine are similar to those of terlipressin have not been fully defined.
Radiologic and Surgical Therapy
Transjugular Intrahepatic Portosystemic Shunt
Insertion of a transjugular intrahepatic portosystemic shunt (TIPS) creates a portosystemic shunt and lowers portal venous pressure, thereby decreasing venous pooling in the splanchnic circulation and increasing venous return. TIPS is effective for the treatment of diuretic-resistant ascites, a precursor to type 2 HRS.117 Four pilot studies have evaluated the use of TIPS in nontransplant candidates with type 1 or 2 HRS.118–121 In these studies, serum creatinine levels declined, sodium excretion increased, and neurohumoral responses improved after TIPS, although survival was not affected. The major benefit was seen in patients with type 2 HRS. In one study, the use of midodrine, octreotide, and albumin followed by TIPS appeared to be effective in a small cohort of patients with type 1 HRS. An important limitation of the use of TIPS in HRS is the potential to worsen hepatic function. Therefore, important questions regarding the safety and benefit of TIPS in HRS remain.
Liver Transplantation
Liver transplantation is the only therapeutic modality that has the potential to reverse both liver dysfunction and HRS and should be considered in any patient found to have HRS.65,66,71,122 Rates of postoperative complications and in-hospital mortality are higher in patients transplanted with HRS than in those without HRS,75,122 and up to 35% of patients with HRS require long-term renal replacement therapy.66 Still, the three-year survival rate of patients transplanted with HRS is approximately 60% compared with 70% to 80% for patients transplanted without HRS. The duration and degree of renal dysfunction preoperatively may be independent predictors of survival, and patients who require hemodialysis carry a mortality risk that is 1.77 times higher than that of patients who do not need dialysis.122–124 In one study, patients with HRS who responded to treatment with a vasopressin analog prior to liver transplantation had outcomes similar to those of patients who underwent liver transplantation without HRS,94 a finding that supports the use of such therapy as a bridge to liver transplantation. Larger trials are needed to confirm this observation.
Other Therapies
Extracorporeal albumin dialysis with MARS is an experimental therapeutic modality that enhances the removal of water-soluble and albumin-binding toxins from the circulation.125 The results of one small randomized trial have supported the use of MARS to improve serum creatinine levels and survival rates in patients with HRS, although larger studies are needed to confirm these findings.126,127 Several other vasoconstrictive medical therapies, including dopamine and octreotide in combination with albumin, have not improved the outcome of HRS, and in one study use of a nonselective endothelin receptor antagonist to inhibit intrarenal vasoconstriction in HRS proved deleterious.95
HEPATOPULMONARY SYNDROME AND PORTOPULMONARY HYPERTENSION
Cirrhosis and portal hypertension are accompanied by alterations in the vascular beds of multiple organ systems. In the pulmonary circulation, two distinct clinical entities, termed the hepatopulmonary syndrome (HPS) and portopulmonary hypertension (PPH), have been recognized. HPS occurs when pulmonary microvascular alterations impair gas exchange and is found in up to 30% of patients evaluated for liver transplantation.128–130 PPH occurs when vasoconstriction and remodeling in resistance vessels increase pulmonary arterial pressures and is found in as many as 5% of patients with cirrhosis. The mechanisms whereby these two entities develop are incompletely characterized, although they occur in similar clinical settings and may share pathogenic pathways. The presence of HPS or PPH increases mortality in affected patients. No effective medical therapies are available for HPS, although liver transplantation reverses the syndrome in most patients. Medical therapies that improve pulmonary hemodynamics in patients with PPH have become available, but the specific role of liver transplantation in PPH is not clearly defined.131–138
PATHOPHYSIOLOGY
Hepatopulmonary Syndrome
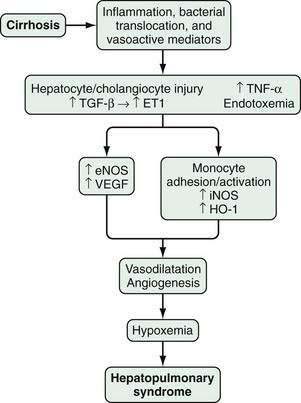
HPS is characterized by microvascular alterations and dilatation in the precapillary and capillary pulmonary arterial circulation. In human HPS, the production of vasodilatory substances within the pulmonary vasculature, most notably NO, is increased. Although increased circulating and pulmonary NO levels appear to be features of human HPS, improvement in oxygenation in response to acute inhibition of NO is variable,139–142 and HPS may take more than one year to resolve after liver transplantation in advanced cases.135 These findings suggest that other vasoactive mediators or angiogenesis in the pulmonary microvasculature may contribute to vascular alterations and hypoxemia in human HPS.
In experimental HPS induced by bile duct ligation in the rat, pulmonary NO overproduction has also been observed and is triggered by a series of pathophysiologic events. During the onset of pulmonary vascular alterations, increased biliary production and release of endothelin-1143 in conjunction with shear stress induces pulmonary microvascular endothelin-B receptor overexpression, which leads in turn to endothelin-1–mediated endothelial NO synthase (eNOS)-derived NO production.144,145 As HPS progresses, tumor necrosis factor-α (TNF-α) levels rise as a result of bacterial translocation, which leads to adherence of macrophages in the pulmonary microvasculature and inducible NO synthase (iNOS)–derived NO production and heme oxygenase-1–derived carbon monoxide production.146–150 Endothelin receptor antagonists and inhibition of NOS, bacterial translocation, TNF-α, and heme oxygenase all improve experimental HPS.148–151
Studies have found that experimental HPS is accompanied by enhanced pulmonary vascular endothelial growth factor (VEGF) expression and angiogenesis.151 Both antiangiogenic peptides and pentoxifylline, a phosphodiesterase inhibitor with TNF-α antagonist properties, have been shown to inhibit angiogenesis and decrease the severity of HPS. Two small studies of pentoxifylline in human HPS have reported conflicting results, and the role of angiogenesis and angiogenesis inhibition in human HPS has yet to be defined (Fig. 92-3).152–155
Portopulmonary Hypertension
The mechanisms whereby PPH develops are poorly understood. Histologically, PPH shares the characteristic features of other forms of pulmonary arterial hypertension (PAH): medial proliferation and hypertrophy, plexiform arteriopathy, and in situ vascular thrombosis.156–158 The precise role of portal hypertension in this process is not clear, however, and whether PPH shares pathophysiologic mechanisms with PAH is unknown. Studies have found PPH, like PAH, to be more common in women than men.159 In addition, endothelin-1 levels are increased and thought to contribute to vascular alterations in PAH, a finding that has also been observed in cirrhotic patients with PPH compared with cirrhotic patients without PPH.160 By contrast, genetic polymorphisms in serotonin metabolism, which appear to increase vascular tone in a subset of patients with PAH, have not been found in patients with PPH.161 Nevertheless, the observation that therapies used for PAH also appear to be effective for PPH supports the notion that downstream effector mechanisms are similar in the two disorders.
CLINICAL FEATURES AND DIAGNOSIS
Hepatopulmonary Syndrome
HPS is defined as a widened age-corrected alveolar-arterial oxygen gradient (AaPo2) on room air in the presence or absence of hypoxemia (AaPo2 = 15 mm Hg, or 20 mm Hg in patients older than 64 years) as a result of intrapulmonary vasodilation. HPS can be graded, according to a task force, on the basis of the degree of hypoxemia, as follows: mild (Pao2 ≥ 80 mm Hg); moderate (Pao2 = 61 to 80 mm Hg), severe (Pao2 = 50 to 60 mm Hg), and very severe (Pao2 < 50 mm Hg).162 The frequency of HPS ranges from 10% to 35% in patients with cirrhosis who undergo evaluation for liver transplantation.128,131,136 In addition, HPS has been found to increase mortality significantly in patients with cirrhosis. Moreover, survival of patients with HPS after liver transplantation is diminished compared with those without HPS, particularly when hypoxemia is severe.163
Patients with HPS present most commonly with respiratory complaints in the setting of chronic liver disease. Other specific pulmonary abnormalities may accompany certain liver diseases and cause respiratory symptoms. These abnormalities include panacinar emphysema in patients with α1-antitrypsin deficiency, pulmonary granulomas and interstitial fibrosis in those with primary biliary cirrhosis and sarcoidosis, and characteristic lung disease in patients with cystic fibrosis. Occasionally, HPS may be the initial manifestation of cirrhosis, and it also may be found in the setting of noncirrhotic and posthepatitic portal hypertension, ischemic hepatitis, and chronic hepatitis in the absence of confirmed cirrhosis. A syndrome similar to HPS has been described in children with congenital abnormalities that divert hepatic blood from the pulmonary circulation.164–166
Classic clinical manifestations of HPS include platypnea (dyspnea worsened by an erect position and improved by a supine position), orthodeoxia (exacerbation of hypoxia and hypoxemia in an upright position), an insidious onset and slow progression of dyspnea, clubbing, and distal cyanosis.128,131,167 Although clubbing and hypoxemia (partial pressure of oxygen < 60 mm Hg) in patients with liver disease in the absence of intrinsic cardiopulmonary disease are highly suggestive of HPS,131 other clinical features are not reliable for detecting HPS, and many patients, particularly those with early HPS, are asymptomatic or have symptoms only on exertion. Cough has also been described as a presenting symptom of HPS.168 In most studies, the presence or severity of HPS does not appear to correlate with the degree of hepatic dysfunction.135
The diagnosis of HPS requires a high degree of clinical suspicion, measurement of arterial blood gases, detection of intrapulmonary shunting, and exclusion of intrinsic cardiopulmonary disease as the cause of hypoxemia. The most sensitive test for the diagnosis of intrapulmonary shunting is contrast echocardiography,169 which is performed by peripheral intravenous injection of agitated saline to produce microbubbles that are visualized during transthoracic echocardiography. Normally, microbubbles travel from the right ventricle to the lungs, where they are absorbed and do not reach the left ventricle. In patients with intracardiac shunting, microbubbles reach the left ventricle early (within one to three cardiac cycles after injection). In patients with intrapulmonary shunting, microbubbles reach the left ventricle in a delayed fashion (three to six cardiac cycles after injection). Up to 60% of patients with cirrhosis have intrapulmonary vasodilatation on contrast echocardiography, but only a subset of patients have sufficient vasodilatation to cause abnormal arterial blood gas results and HPS.
In patients with pulmonary symptoms and hypoxemia who are found to have intrapulmonary shunting, intrinsic cardiopulmonary disease should be excluded. Chest radiography or computed tomography (CT) and pulmonary function tests are generally performed in patients being considered for liver transplantation. If potentially reversible cardiopulmonary disorders are detected, treatment is undertaken, and the assessment of oxygenation is repeated. In addition to its use in the evaluation of patients for pulmonary disease, high-resolution chest CT has been reported to detect peripheral pulmonary vascular dilatations in a small cohort of patients with HPS.139,170 The diagnostic usefulness of chest CT for the diagnosis of HPS, however, is not defined. In the small subset of patients found to have advanced hypoxemia (Pao2 < 60 mm Hg) and both intrapulmonary shunting and significant cardiopulmonary disease, a technetium-labeled macroaggregated albumin scan may confirm that HPS is contributing to the gas exchange abnormalities. In this test, the shunting of intravenously administered 99mTc-labeled macroaggregated albumin (20 µm in diameter) through the pulmonary vasculature is quantified by measuring activity and uptake in both the lungs and brain.169,171,172 In one study, the scan result was positive (>6% shunt fraction) in patients with HPS and a Pao2 of <60 mm Hg, but not in patients with chronic obstructive pulmonary disease and a similar degree of hypoxemia.171
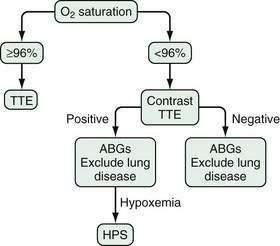
Screening algorithms for HPS have been evaluated, particularly in patients who are candidates for liver transplantation. One validated approach uses pulse oximetry screening (Po2 < 96%) to detect patients with a Pao2 < 70 mm Hg (sensitivity 100%, specificity 88%; Fig. 92-4). This subgroup of patients (~30% of all liver transplantation candidates) is then targeted for arterial blood gas analysis, contrast echocardiography, and evaluation for intrinsic cardiopulmonary disease, thereby limiting full evaluation for HPS to those patients likely to have at least moderate HPS.173,174 Currently, however, no practice guidelines for screening for HPS in liver transplantation candidates have been developed.
Portopulmonary Hypertension
PPH is defined as the development of PAH in the setting of portal hypertension. The diagnostic criteria for PPH include the presence of PAH as defined by the World Health Organization: mean arterial pulmonary pressure (mPAP) > 25 mm Hg at rest or 30 mm Hg with exercise; pulmonary capillary wedge pressure < 15 mm Hg; and pulmonary vascular resistance > 240 dynes • s • cm−5) occurring in the setting of portal hypertension (splenomegaly, thrombocytopenia, portosystemic shunts, or portal vein hemodynamic abnormalities).129,175,176 PPH is generally graded according to the degree of elevation in mPAP, which correlates with the mortality risk associated with liver transplantation and influences decisions regarding therapy.172 Mild PPH (mPAP = 25 to 35 mm Hg) is not associated with an increased operative risk for liver transplantation and may not require medical therapy. Moderate PPH (mPAP = 35 to 50 mm Hg) is associated with an increased operative risk for liver transplantation and is an indication for medical therapy. Severe PPH (mPAP > 50 mm Hg) has a prohibitive operative mortality and is managed with medical therapy. PPH has been found in as many as 6% of cirrhotic patients who are evaluated for liver transplantation, and outcomes are worse than in cirrhotic patients without PPH.
The most common symptom associated with PPH is exertional dyspnea; other nonspecific symptoms such as orthopnea, fatigue, chest pressure, syncope, edema, and lightheadedness also may occur.176 Characteristic physical examination features of PAH, including an elevated jugular venous pressure, loud second pulmonic heart sound, murmur of tricuspid regurgitation, and lower extremity edema, have been reported but are not sufficiently sensitive nor specific to be diagnostically useful. In cirrhotic patients, peripheral edema out of proportion to the degree of ascites should prompt consideration of right ventricular dysfunction secondary to pulmonary hypertension. In a number of screening studies, most cirrhotic patients with significant PPH were asymptomatic.133,177
The diagnosis of PPH warrants a high degree of clinical suspicion, and all patients considered for liver transplantation as well as patients with suggestive symptoms or physical findings should be evaluated for PPH. Transthoracic echocardiography is the recommended screening test because it evaluates right-sided cardiac function and allows an estimation of right ventricular systolic pressure by evaluating the tricuspid regurgitant jet.178 In addition, other causes of elevated right-sided cardiac pressures, including secondary pulmonary hypertension, volume overload, and a hyperdynamic circulation, should be considered and assessed. Methods for estimating right ventricular systolic pressure vary among centers but, in general, in the absence of significant pulmonary artery stenosis, an estimated right ventricular systolic pressure higher than 40 mm Hg or the presence of right ventricular abnormalities support further evaluation for PPH. The absence of both these echocardiographic findings essentially excludes PPH from the differential diagnosis.133 In all patients found to have echocardiographic features suggestive of PPH, pulmonary artery catheterization should be performed to establish the diagnosis and assess the severity of PPH. Findings on pulmonary artery catheterization are useful for distinguishing volume overload and a hyperdynamic circulation from PPH.
TREATMENT
Hepatopulmonary Syndrome
Treatment options for HPS are limited. Currently, no established medical therapies exist, although case reports and small case series have suggested that some treatments may improve oxygenation. Therefore, patients with well-preserved hepatic synthetic function who have hypoxemia are generally treated symptomatically until oxygenation worsens sufficiently to permit listing for liver transplantation based on an exception to the Model for End-stage Liver Disease (MELD) score (see Chapter 95). Liver transplantation reverses HPS in most affected patients, although mortality after liver transplantation appears to be higher in patients with HPS than in those without HPS.163,179–181
Medical Therapy
A number of agents have been tried in patients with HPS empirically or on the basis of data from experimental models in uncontrolled trials, case series, and case reports. Among the compounds studied are garlic preparations, pentoxifylline, aspirin, N-acetylcysteine, glucocorticoids, antimetabolites (cyclophosphamide), antibiotics (norfloxacin), somatostatin, and almitrine (Duxil, a respiratory stimulant).139,172,182–184 Of these agents, only two have been evaluated in small clinical studies. An open-label trial of a garlic preparation reported an increase in Pao2 of approximately 10 mm Hg in 40% of patients when used for periods of at least six months.182 Two small studies have evaluated pentoxifylline as a treatment for HPS; one found improvement in oxygenation, and the other observed dose-limiting gastrointestinal toxicity and no clinical improvement.185,186
Interventional Radiologic Therapy
Two radiologic techniques—TIPS,187 to lower portal pressure, and pulmonary angiography with embolization, to occlude areas of intrapulmonary shunting,172 have been reported to be useful in HPS. These invasive approaches have been described only in case reports or small retrospective analyses, and whether either treatment reliably improves oxygenation is not clear. Therefore, placement of a TIPS specifically to treat HPS in the absence of other indications is not recommended.187,188 Pulmonary angiography has been considered in patients with severe hypoxemia to identify focal arteriovenous shunting that might be diminished with embolization. Little evidence to support such an approach is available, however, and arteriovenous shunting sufficient to cause hypoxemia and amenable to embolization may be detected by high-resolution CT.172
Liver Transplantation
Liver transplantation reverses HPS in as many as 80% of affected patients.139 Hypoxemia may persist after transplantation, however, particularly when severe before transplantation, and may require more than one year to resolve. Mortality also appears to be higher in patients with HPS who undergo liver transplantation than in patients transplanted without HPS, although this observation has not been completely uniform.181 The increase in postoperative mortality is related in part to the severity of HPS, with one prospective study demonstrating that patients with profound hypoxemia (Pao2 < 50 mm Hg) and marked intrapulmonary shunting (shunt fraction > 20%) had a marked increase in mortality.135,136,163,179 In addition, unique complications such as transient worsening of hypoxemia following liver transplantation, the development of pulmonary hypertension, and embolic cerebrovascular events have been observed and may contribute to adverse postoperative outcomes. On the basis of the relationship between the severity of hypoxemia and poor outcomes in nontransplanted patients with HPS, MELD exception points may be given to patients with HPS and a resting Pao2 of <60 mm Hg, thereby increasing their priority status for transplantation (see Chapter 95).179–181
Portopulmonary Hypertension
Medical Therapy
The therapy of PPH is largely empirical. In general, diuretics are used for volume overload, supplemental oxygen is provided if hypoxemia is present, and anticoagulation may be appropriate in the subset of patients with PPH who do not have hepatic decompensation, marked coagulopathy, or gastroesophageal varices. In one study, withdrawal of β-adrenergic blocker therapy improved right-sided cardiac function in patients with PPH, and some clinicians advocate treating varices with band ligation if feasible and withdrawing medical therapy for varices in patients with PPH.189
Despite data from trials in patients with PAH, no randomized controlled studies have evaluated vasodilator therapy in patients with PPH. Nevertheless, case reports and series have evaluated prostacyclin analogs, several of which require complex intravenous administration (e.g., epoprostenol, iloprost, treprostinil),138,177,190,191 oral endothelin receptor antagonists including bosentan (a mixed endothelin-A and -B receptor antagonist),192–194 ambrisentan (an endothelin-A blocker),65 and oral phosphodiesterase-5 inhibitors (e.g., sildenafil).195–197 Bosentan hepatotoxicity has been described uncommonly and is potentially mediated by inhibitory effects on the canalicular bile salt export protein. Close monitoring of liver biochemical test levels and use only in patients with well-compensated liver disease is appropriate. Use of ambrisentan in patients with PPH is increasing, although few data about its efficacy are available. Sildenafil generally has been well tolerated in patients with PPH. The magnitude of improvement in pulmonary arterial pressures has been modest with oral agents, and some clinicians use endothelin receptor antagonists and phosphodiesterase-5 inhibitors in combination, although the beneficial effects are limited.
Liver Transplantation
Traditionally, moderate to severe PPH has been a contraindication to liver transplantation, particularly when the mPAP is higher than 50 mm Hg.198 With the availability of agents to lower pulmonary pressures, consideration has been given to improving pulmonary hemodynamics with medical therapy followed by liver transplantation to attempt to resolve PPH.199 Three case series have described such an approach.190,200,201 Although results have varied among centers, some patients with PPH appear to have resolved after liver transplantation. Whether liver transplantation is an effective treatment for PPH and whether subgroups of patients may derive the specific benefit from such an approach have not been clarified.
CIRRHOTIC CARDIOMYOPATHY
Although systemic hemodynamic changes in cirrhotic patients have been observed for more than 100 years, not until the 1950s were patients with alcoholic cirrhosis found to have a so-called hyperdynamic circulation—decreased arterial blood pressure, decreased peripheral resistance, and increased cardiac output.202 Later studies described vascular hyporesponsiveness to vasoconstrictors in cirrhotic patients that was attributed to effects of alcohol on the heart and termed alcoholic cardiomyopathy.203 Subsequently, specific vascular and cardiac abnormalities have been found in human and animal models of cirrhosis, independent of alcohol ingestion, and have led to the concept that cirrhosis itself triggers cardiac dysfunction; this has been termed cirrhotic cardiomyopathy (CCM).204
PATHOPHYSIOLOGY
Although diagnostic criteria for CCM have not been established, three major pathophysiologic features have been observed: (1) structural and functional ventricular abnormalities; (2) an abnormal ventricular response in the presence of pharmacologic, physiologic, or surgical stress; and (3) cardiac electrophysiologic abnormalities (Fig. 92-5).83,204–207
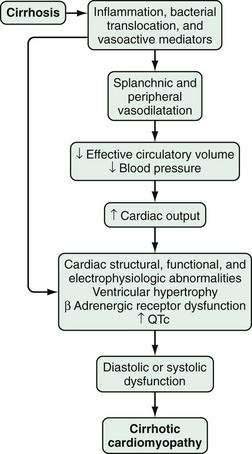
The major structural and functional ventricular abnormalities, found in histologic and echocardiographic studies, are left ventricular hypertrophy and diastolic dysfunction.83,208–211 The structural changes have been attributed to a hypertrophic response to the hyperdynamic circulation. Diastolic alterations may be precipitated or worsened by the presence of a significant amount of ascites.212,213 In addition, impaired systolic function and histologic injury to cardiomyocytes have been observed.214 Whether the severity of hepatic synthetic dysfunction correlates with the degree of cardiac dysfunction is not clear.215 The impaired ventricular response to stress and exercise observed in human and animal models of cirrhosis83,204,208,210 has been attributed to impaired β-adrenergic signaling pathways that lead to subnormal chronotropic and contractile responses,216–218 as well as to cardiomyocyte dysfunction resulting from overproduction of NO, carbon monoxide, and endocannabinoids.219–224 Electrophysiologic abnormalities, most notably prolongation of the QT interval (QTc), have also been observed in patients with CCM.225,226 The degree of prolongation of the QTc appears to correlate with the severity of liver disease and may contribute to the dissociation between electrical and mechanical events in the heart and to cardiac dysfunction.227 These abnormalities appear to improve after liver transplantation.
CLINICAL FEATURES AND DIAGNOSIS
Because CCM typically becomes clinically detectable under circumstances of stress, the diagnosis is difficult to make and the process may not be recognized. Overt cardiac dysfunction may occur after common clinical interventions in cirrhosis. For example, a prospective multicenter analysis has shown that more than 10% of patients who undergo TIPS placement for refractory ascites exhibit signs of heart failure when compared with patients treated by repeated large-volume paracentesis.228 Subsequent studies have revealed that both the severity of liver disease (as measured by the MELD score) and diastolic dysfunction 28 days after the procedure are independent predictors of death in patients with cirrhosis who are treated with a TIPS.229 Similarly, in a case series of patients who underwent liver transplantation, 47% were found to have radiologic evidence of pulmonary edema within the first 24 hours after transplantation. Volume replacement was considered unlikely to be the sole culprit, suggesting that cardiac dysfunction may have played a role.230,231 Finally, in prospective studies, a lower cardiac output has been found in patients in whom HRS developed than in those in whom HRS did not develop, thereby supporting the role of cardiac dysfunction in HRS.80,81
No precise diagnostic criteria for CCM have been established, because no baseline clinical, imaging, or biochemical findings have been found to predict the development of overt cardiac dysfunction under stress definitively.232–236 The presence of QTc prolongation and echocardiographic evidence of diastolic dysfunction are readily detected but do not appear to correlate with the risk of development of cardiac dysfunction under stress. Serum levels of markers of cardiac dysfunction—brain natriuretic peptide, atrial natriuretic peptide, and troponin I—are elevated in patients with cirrhosis and appear to correlate with QTc prolongation, diastolic dysfunction, and the severity of liver disease. Evaluation of cardiac function under stress conditions with echocardiography or ventriculography has been reported, and cardiac function may be impaired but does not correlate clearly with subsequent clinical cardiac dysfunction. Further work is needed to define the role of serum markers and stress testing in the diagnosis of CCM.235,236 In cirrhotic patients found to have evidence of congestive heart failure, alternative causes of cardiac dysfunction, including coronary artery disease, valvular abnormalities, and other causes of cardiomyopathy, should be excluded.
TREATMENT
Therapy of volume overload in patients suspected of having CCM includes standard supportive measures and diuresis.237 Use of preload and afterload reducing agents should be considered with caution, because these agents may worsen hypotension in the setting of underlying systemic vasodilatation.37,204 Inotropic and chronotropic agents do not appear to be of benefit.205 In patients without overt heart failure but with electrocardiographic or echocardiographic features consistent with CCM, chronic use of aldosterone antagonists has been shown to improve echocardiographic features marginally,237,238 and short-term administration of a noncardioselective β-adrenergic antagonist has been shown to improve QTc prolongation.239 Whether these agents prevent or ameliorate cardiac dysfunction in response to stress is unknown.
The effects of liver transplantation on cardiac abnormalities in patients with cirrhosis have not been fully characterized. A small study has demonstrated that resting echocardiographic and stress radionuclide ventriculographic abnormalities found in patients with cirrhosis were improved or reversed after liver transplantation.212,240 Further data are needed to determine whether CCM reverses after liver transplantation.
ENDOCRINE DYSFUNCTION
ADRENAL INSUFFICIENCY
The recognition that adrenal insufficiency worsens outcomes in sepsis, a syndrome that shares physiologic abnormalities seen in liver failure, has led to evaluation of adrenal dysfunction in patients with liver disease. Five studies have demonstrated the presence of relative adrenal insufficiency in up to 69% of critically ill patients with liver disease.241–245 The presence of relative adrenal insufficiency was associated with greater hemodynamic instability and increased mortality. In one study,245 glucocorticoid therapy improved survival, but, in two others,242,243 such therapy was associated with increased mortality secondary to nosocomial and opportunistic infections. Inadequate hepatic cholesterol production as a result of liver disease may predispose patients to impaired adrenal cortisol synthesis from cholesterol during stress. Currently, no standardized diagnostic criteria have been developed for relative adrenal insufficiency, and whether a subset of critically ill patients with liver disease will benefit from glucocorticoid treatment remains an area of active investigation.
GONADAL DYSFUNCTION
Historical data have suggested a high frequency (70% to 80%) of central and peripheral hypogonadism in cirrhotic patients. Hypogonadism in this setting is associated with a decreased concentration of free or bioavailable testosterone in direct proportion to the degree of liver dysfunction.246,247 Sex hormone-binding globulin (SHBG), which binds testosterone and 17β-estradiol in serum, has a lower affinity for estrogens than testosterone. In patients with cirrhosis, elevated concentrations of SHBG (with a resulting shift in the balance of hormones in favor of estrogens) and a decreased production of dehydroepiandrosterone sulfate (a precursor of androgenic hormones) may account for the “feminization syndrome” seen in male patients with cirrhosis.247–249 Acute and chronic alcohol consumption is responsible for direct toxic effects on Leydig cells and for alterations of the hypothalamic-pituitary-gonadal axis. These alterations include a decrease in the serum concentration of luteinizing hormone and decreased responsiveness to gonadotropin-releasing hormone.250,251 Spironolactone, which frequently is used to treat fluid overload, causes painful gynecomastia by displacing androgen from its receptor and binding protein and by increasing estradiol production and testosterone clearance.252 Topical testosterone appears to improve muscle strength and survival in patients with chronic allograft failure after liver transplantation, but whether testosterone supplementation improves the symptoms and signs related to hypogonadism in pretransplantation cirrhotic patients is unknown.253
THYROID DYSFUNCTION
A number of thyroid abnormalities, including increased thyroid volume and decreased serum levels of free triiodothyronine, have been found in patients with cirrhosis. These alterations appear to correlate with the severity of liver disease, and the presence of thyroid disease may be a predictor of decreased survival.254–257 In patients with hepatitis C and autoimmune liver disease, the incidence of hypothyroidism and autoimmune thyroid disease is increased.258–260 The detection of hepatitis C virus in thyroid cells raises the possibility of direct viral cytotoxicity.261 Interferon-based treatments for viral hepatitis may cause thyroiditis and have been implicated in both hyper- and hypothyroidism in 10% to 15% of treated patients (see Chapter 79).262 Whether routine screening for thyroid disease influences survival or quality of life is unknown.
BONE DISEASE
The frequency of osteoporosis among patients with all causes of chronic liver disease ranges from 12% to 55%.263 Potential risk factors include cholestasis, a maternal history of hip fracture, progression of liver disease, alcohol consumption, lower body mass index, oral glucocorticoid use for more than three months, and older patient age. Women with primary biliary cirrhosis have a four-fold higher risk of developing osteoporosis and a two-fold higher risk of bone fractures than age-matched controls.264 Ursodeoxycholic acid treatment does not influence bone density in patients with primary biliary cirrhosis,265 but lower glucocorticoid use and better nutrition appear to decrease the frequency of osteoporosis.266 Bone mineral density is an appropriate screening tool for osteoporosis in patients with cirrhosis (especially primary biliary cirrhosis and primary sclerosing cholangitis) and those who require more than three months of glucocorticoid therapy.266–268 Treatment of osteoporosis in chronic liver disease is based on studies of postmenopausal women and is an area of ongoing investigation. The use of calcium and vitamin D and of therapeutic bisphosponates improves bone mineral density and appears not to have significant side effects (see Chapter 89).268,269
COAGULATION DISORDERS
Cirrhosis is well recognized to be associated with a bleeding diathesis because of the presence of a prolonged prothrombin time and thrombocytopenia. Studies in the 2000s have suggested, however, that the interplay among abnormalities in both pro- and anticoagulant factors may result in increased bleeding and hypercoagulability. The precise mechanisms for these clinical events are not fully characterized.270 In cirrhosis, the progressive loss of hepatocytes leads to decreased synthesis of procoagulant factors, including vitamin K–dependent factors (II, VII, IX, X), factor V, and factor XI. The severity of clotting abnormalities, measured by the prothrombin time (PT), activated partial thromboplastin time (aPTT), and international normalized ratio (INR) increase as liver disease progresses,271 and the increase in INR predicts survival.272 A prolonged PT alone is not generally considered to be a major risk factor for spontaneous bleeding in patients with cirrhosis but does increase the severity of bleeding when it occurs.273 Common practice is to administer fresh frozen plasma (FFP), vitamin K, and occasionally recombinant factor VIIa to correct coagulopathy in patients with chronic liver disease, particularly in the setting of bleeding or prior to invasive procedures.274 Despite current practice, however, clinical evidence that vitamin K, FFP, or recombinant factor VIIa administration reduces the severity of variceal bleeding, for example, is not strong.275–277 In addition, the volume of FFP (>6 units) required to achieve a clinically significant reduction in the PT in patients with cirrhosis has been associated with a significant increase in the risk of acute lung injury and volume overload.278,279 In one trial of patients who underwent laparoscopic liver biopsy, recombinant factor VIIa therapy normalized the PT, and no patients required blood transfusions at the time of biopsy.280 Nevertheless, no placebo-controlled trial of recombinant factor VIIa in cirrhotic patients undergoing invasive procedures has been performed. One concern about use of the INR in patients with cirrhosis is the substantial interlaboratory variability with use of the standard international sensitivity index (ISI) to normalize varying sensitivities of thromboplastin reagents.281 This variability may result in as much as a 25% difference in mean INR for a single patient sample assayed with different reagents. Adjusting the ISI calibration by using samples from cirrhotic patients appears to eliminate this variation but is time-consuming and not widely used.282 Therefore, use of the INR to predict bleeding risk and to calculate the MELD score for organ allocation priority has limitations.
Thrombocytopenia is also a common feature in cirrhotic patients with portal hypertension. It is associated with the presence of hypersplenism, but decreased hepatic thrombopoietin synthesis and, in some cases, direct bone marrow toxicity (e.g., from alcohol or hepatitis C virus) also play a role.283 In addition to quantitative abnormalities, platelet thrombin generation is impaired, particularly at platelet counts below 50,000/mL, and may contribute to impaired clot formation.284 The presence of thrombocytopenia, however, does not appear to increase the risk of bleeding in patients with cirrhosis. The data on whether the administration of platelets influences the bleeding risk in patients who undergo invasive procedures or decreases blood loss or transfusion requirements in patients with variceal hemorrhage are limited. Nevertheless, common clinical practice is to administer platelets to achieve a platelet count of approximately 50,000/mL prior to invasive procedures and in the setting of active bleeding.278,279 Although impaired platelet function, as assessed by measuring the bleeding time, is commonly observed in patients with cirrhosis, neither prolongation of the bleeding time nor its correction with desmopressin administration influence the risk of bleeding.285,286
Dysfibrinogenemia, reflected by elevated circulating levels of D-dimer and fibrinogen degradation products, and by prolongation of the clot lysis time, is seen in up to 46% of cirrhotic patients.287,288 These abnormalities result from altered production of activators and inhibitors of fibrinolysis, activation of the coagulation cascade by endotoxemia, and decreased clearance of fibrinolytic proteins in the setting of hepatic synthetic dysfunction. Dysfibrinogenemia becomes more severe as liver disease progresses and may progress to overt disseminated intravascular coagulation, further increasing the risk of bleeding. Results of tests for D-dimer and fibrinogen degradation products are variable in cirrhosis, but no uniformly accepted standard for assessing dysfibrinogenemia is available. Antihyperfibrinolytic therapy with compounds such as ε-aminocaproic acid has been used to prevent blood loss in stable cirrhotic patients and during liver transplantation, but evidence to support such an approach is limited.289,290 In addition to effects that predispose to bleeding, hepatic synthetic dysfunction in patients with cirrhosis also impairs the production of endogenous anticoagulant proteins, including protein C, protein S, antithrombin, tissue plasminogen activator, and thrombomodulin.291 These abnormalities may result in hypercoagulability and a risk for thrombosis. For example, portal vein thrombosis is a well-recognized complication of cirrhosis, and case-control studies have found up to a two-fold increased risk of thromboembolic events (deep venous thrombosis and pulmonary embolism) in patients with cirrhosis and noncirrhotic liver disease compared with controls.292–294 Whether a subset of these patients has underlying inherited causes of hypercoagulability that contribute to thrombosis has not been fully defined.295 In some studies, anticoagulation has appeared to be safe and of benefit for patients with noncirrhotic, nonmalignant portal vein thrombosis associated with gastroesophageal varices, but the role of anticoagulation therapy in cirrhotic portal vein thrombosis remains unclear.296 The potential role of hypercoagulability in the progression of liver disease and in some complications of cirrhosis (e.g., HRS, PPH) are areas for future investigation.297
Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28:59-69. (Ref 205.)
Bass NM. Review article: The current pharmacological therapies for hepatic encephalopathy. Aliment Pharmacol Ther. 2007;25(Suppl 1):23-31. (Ref 43.)
Esrailian E, Pantangco ER, Kyulo NL, et al. Octreotide/Midodrine therapy significantly improves renal function and 30-day survival in patients with type 1 hepatorenal syndrome. Dig Dis Sci. 2007;52:742-8. (Ref 114.)
Gaskari SA, Honar H, Lee SS. Therapy insight: Cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3:329-37. (Ref 210.)
Martin-Llahi M, Pépin M-N, Guevara M, et al. Terlipressin and albumin vs. albumin in patients with cirrhosis and hepatorenal syndrome: A randomized study. Gastroenterology. 2008;134:1352-9. (Ref 106.)
Mas A, Rodes J, Sunyer L, et al. Comparison of rifaximin and lactitol in the treatment of acute hepatic encephalopathy: Results of a randomized, double-blind, double-dummy, controlled clinical trial. J Hepatol. 2003;38:51-8. (Ref 48.)
Møller S, Henriksen JH. Cardiovascular complications of cirrhosis. Gut. 2008;57:268-78. (Ref 206.)
Palma DT, Fallon MB. The hepatopulmonary syndrome. J Hepatol. 2006;45:617-25. (Ref 139.)
Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome—a liver-induced vascular disorder. N Engl J Med. 2008;358:2378-87. (Ref 172.)
Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310-18. (Ref 74.)
Sanyal AJ, Boyer T, Garcia-Tsao G, et al. A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology. 2008;134:1360-8. (Ref 107.)
Swanson K, Wiesner R, Krowka M. Natural history of hepatopulmonary syndrome: Impact of liver transplantation. Hepatology. 2005;41:1122-9. (Ref 135.)
Torregrosa M, Genesca J, Gonzalez A, et al. Role of Doppler echocardiography in the assessment of portopulmonary hypertension in liver transplantation candidates. Transplantation. 2001;71:572-4. (Ref 134.)
Wong F, Pantea L, Sniderman K. Midodrine, octreotide, albumin, and TIPS in selected patients with cirrhosis and type 1 hepatorenal syndrome. Hepatology. 2004;40:55-64. (Ref 118.)
1. Bustamante J, Rimola A, Ventura PJ, et al. Prognostic significance of hepatic encephalopathy in patients with cirrhosis. J Hepatol. 1999;30:890-5.
2. Amodio P, Montagnese S, Gatta A, et al. Characteristic of minimal hepatic encephalopathy. Metab Brain Dis. 2004;19:253-65.
3. Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: Final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-21.
4. Dbouk N, McGuire BM. Hepatic encephalopathy: A review of its pathophysiology and treatment. Curr Treat Options Gastroenterol. 2006;9:464-74.
5. Albrecht J, Sonnewald U, Waagepertersen HS, et al. Glutamine in the central nervous system: Function and dysfunction. Front Biosci. 2007;12:332-43.
6. Shawcross DL, Olde Damink SW, Butterworth RF, et al. Ammonia and hepatic encephalopathy: The more things change, the more they remain the same. Metab Brain Dis. 2005;20:169-79.
7. Lockwood AH. Blood ammonia levels and hepatic encephalopathy. Metab Brain Dis. 2004;19:345-54.
8. Ahl B, Weissenborn K, van den Hoff J, et al. Regional differences in cerebral blood flow and cerebral ammonia metabolism in patients with cirrhosis. Hepatology. 2004;40:73-9.
9. Sathyasaikumar KV, Swapna I, Reddy PV, et al. Fulminant hepatic failure in rats induces oxidative stress differentially in cerebral cortex, cerebellum and pons medulla. Neurochem Res. 2007;32:517-24.
10. Jover R, Rodrigo R, Felipo V, et al. Brain edema and inflammatory activation in bile duct ligated rats with diet-induced hyperammonemia: A model of hepatic encephalopathy in cirrhosis. Hepatology. 2006;43:1257.
11. Cordoba J, Blei AT. Brain edema and hepatic encephalopathy. Semin Liver Dis. 1996;16:271-80.
12. Kundra A, Jain A, Banga A, et al. Evaluation of plasma ammonia levels in patients with acute liver failure and chronic liver disease and its correlation with the severity of hepatic encephalopathy and clinical features of raised intracranial pressure. Clin Biochem. 2005;38:696-9.
13. Swapna I, Kumar KV, Reddy PV, et al. Phospholipid and cholesterol alterations accompany structural disarray in myelin membrane of rates with hepatic encephalopathy induced by thioacetamide. Neurochem Int. 2006;49:238-44.
14. Swapna I, Sathyasaikumar KV, Murthy ChR, et al. Changes in cerebral membrane lipid composition and fluidity during thioacetamide-induced hepatic encephalopathy. J Neurochem. 2006;98:1899-907.
15. Ferenci P. Clinical manifestations and diagnosis of hepatic encephalopathy. Available at http://www.uptodate.com/patients/content/topic.do?topicKey=~YWDuul/31599/rR
16. Butterworht RF. The astrocytic (“peripheral-type”) benzodiazepine receptor: Role in the pathogenesis of portal-systemic encephalopathy. Neurochem Int. 2000;36:411-16.
17. Ahboucha S, Butterworth RF. The neurosteroid system: An emerging therapeutic target for hepatic encephalopathy. Metab Brain Dis. 2007;22:291-308.
18. Ahboucha S, Pomier-Layrargues G, Mamer O, et al. Increased levels of pregnenolone and its neuroactive metabolite allopregnanolone in autopsied brain tissue from cirrhotic patients who died in hepatic coma. Neurochem Int. 2006;49:372-8.
19. Lozeva-Thomas V. Serotonin brain circuits with a focus on hepatic encephalopathy. Metab Brain Dis. 2004;19:413-20.
20. Lozeva V, Montgomery JA, Tuomisto L, et al. Increased brain serotonin turnover correlates with the degree of shunting and hyperammonemia in rats following variable portal vein stenosis. J Hepatol. 2004;40:742-8.
21. Michalak A, Chatauret N, Butterworth RF. Evidence for a serotonin transporter deficit in experimental acute liver failure. Neurochem Int. 2001;38:163-8.
22. Verma A, Saraswat VA, Radha Krishna Y, et al. In vivo 1H magnetic resonance spectroscopy-derived metabolite variations between acute-on-chronic liver failure and acute liver failure. Liver Int. 2008;28:1095-103.
23. Hertz L, Kala G. Energy metabolism in brain cells: Effects of elevated ammonia concentrations. Metab Brain Dis. 2007;22:199-218.
24. Dhiman RK, Chawla YK. Minimal hepatic encephalopathy: Time to recognize and treat. Trop Gastroenterol. 2008;29:6-12.
25. Prasad S, Dhiman RK, Duseja A, et al. Lactulose improves cognitive functions and health-related quality of life in patients with cirrhosis who have minimal hepatic encephalopathy. Hepatology. 2007;45:549-59.
26. Kim JM, Ryu WS, Hwang YH, et al. Aggravation of ataxia due to acetazolamide induced hyperammonaemia in episodic ataxia. J Hepatol. 2007;47:245-52.
27. Mehndiratta MM, Mehndiratta P, Phul P, et al. Valproate induced nonhepatic hyperammonaemic encephalopathy (VNHE)—a study from tertiary care referral university hospital, north India. J Pak Med Assoc. 2008;58:627-31.
28. Mitsubuchi H, Nakamura K, Matsumoto S, et al. Inborn errors of proline metabolism. J Nutr. 2008;138:2016S-20S.
29. Summar ML, Dobbelaere D, Brusilow S, et al. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420-5.
30. Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188-93.
31. Bernal W, Hall C, Karvellas CJ, et al. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844-52.
32. O’Beirne JP, Chouhan M, Hughes RD. The role of infection and inflammation in the pathogenesis of hepatic encephalopathy and cerebral edema in acute liver failure. Nat Clin Pract Gastroenterol Hepatol. 2006;3:118-19.
33. Shah NJ, Neeb H, Zaitsev M, et al. Quantitative T1 mapping of hepatic encephalopathy using magnetic resonance imaging. Hepatology. 2003;38:1219-26.
34. Kircheis G, Wettstein M, Timmermann L, et al. Critical flicker frequency for quantification of low-grade hepatic encephalopathy. Hepatology. 2002;35:357-66.
35. Sharma P, Sharma BC, Puri V, et al. Critical flicker frequency: Diagnostic tool for minimal hepatic encephalopathy. J Hepatol. 2007;47:67-73.
36. Romero-Gómez M, Córdoba J, Jover R, et al. Value of the critical flicker frequency in patients with minimal hepatic encephalopathy. Hepatology. 2007;45:879-85.
37. Kircheis G, Fleig WE, Görtelmeyer R, et al. Assessment of low-grade hepatic encephalopathy: A critical analysis. Hepatology. 2007;47:642-50.
38. Weissenborn K, Ennen JC, Schomerus H, et al. Neuropsychological characterization of hepatic encephalopathy. J Hepatol. 2001;34:768-73.
39. Bajaj JS. Management options for minimal hepatic encephalopathy. Expert Rev Gastroenterol Hepatol. 2008;2:785-90.
40. Bajaj JS, Saeian K, Christensen KM, et al. Probiotic yogurt for the treatment of minimal hepatic encephalopathy. Am J Gastroenterol. 2008;103:1707-15.
41. Bajaj JS. Minimal hepatic encephalopathy matters in daily life. World J Gastroenterol. 2008;21:3609-15.
42. Grover VP, Dresner MA, Forton DM, et al. Current and future applications of magnetic resonance imaging and spectroscopy of the brain in hepatic encephalopathy. World J Gastroenterol. 2006;21:2969-78.
43. Bass NM. Review article: The current pharmacological therapies for hepatic encephalopathy. Aliment Pharmacol Ther. 2007;25(Suppl 1):23-31.
44. Als-Nielsen B, Koretz RL, Kjaergard LL, et al. Branched-chain amino acids for hepatic encephalopathy. Cochrane Database Syst Rev 2003; (2):CD001939.
45. Cordoba J, Lopez-Hellin J, Planas M, et al. Normal protein diet for episodic hepatic encephalopathy: Results of a randomized study. J Hepatol. 2004;41:38-43.
46. Als-Nielsen B, Gluud LL, Gluud C. Nonabsorbable disaccharides for hepatic encephalopathy. Cochrane Database Syst Rev 2004; (2):CD003044.
47. Rothenberg ME, Keefe EB. Antibiotics in the management of hepatic encephalopathy: An evidence-based review. Rev Gastroenterol Disord. 2005;5(Suppl 3):S26-S35.
48. Mas A, Rodes J, Sunyer L, et al. Comparison of rifaximin and lactitol in the treatment of acute hepatic encephalopathy: Results of a randomized, double-blind, double-dummy, controlled clinical trial. J Hepatol. 2003;38:51-8.
49. Lawrence KR, Klee JA. Rifaximin for the treatment of hepatic encephalopathy. Pharmacotherapy. 2008;28:1019-32.
50. Jiang Q, Jiang XH, Zheng MH, et al. Rifaximin versus nonabsorbable disaccharides in the management of hepatic encephalopathy: A meta-analysis. Eur J Gastroenterol Hepatol. 2008;20:1064-70.
51. Gentile S, Guarino G, Romano M, et al. A randomized controlled trial of acarbose in hepatic encephalopathy. Clin Gastroenterol Hepatol. 2005;3:184-91.
52. Sheth AA, Garcia-Tsao G. Probiotics and liver disease. J Clin Grastroenterol. 2008;42(Suppl 2):S80-4.
53. Sharma P, Sharma BC, Puri V, et al. An open-label randomized controlled trial of lactulose and probiotics in the treatment of minimal hepatic encephalopathy. Eur J Gastroenterol Hepatol. 2008;20:506-11.
54. Malaguarnera M, Greco F, Barone G, et al. Bifidobacterium longum with fructo-oligosaccharide (FOS) treatment in minimal hepatic encephalopathy: A randomized, double-blind, placebo-controlled study. Dig Dis Sci. 2007;52:3259-65.
55. Liu Q, Duan ZP, Ha DK, et al. Symbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology. 2004;39:1441-9.
56. Efrati C, Masini A, Merli M, et al. Effect of sodium benzoate on blood ammonia response to oral glutamine challenge in cirrhotic patients: A note of caution. Am J Gastroenterol. 2000;95:3574-8.
57. Shaposhnikova NA, Drozdov VN, Petrakov AV, et al. Effect of zinc deficiency on effectiveness of the treatment of liver encephalopathy in patients with liver cirrhoses. Eksp Klin Gastroenterol. 2007;164:46-50.
58. Chetri K, Choudhuri G. Role of trace elements in hepatic encephalopathy: Zinc and manganese. Indian J Gastroenterol. 2003;22(Suppl 2):S28-30.
59. Rahelic D, Kujundzic M, Romic Z, et al. Serum concentration of zinc, copper, manganese and magnesium in patients with liver cirrhosis. Coll Antropol. 2006;30:523-8.
60. Hassanein TI, Tofteng F, Brown RSJr, et al. Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology. 2007;46:1853-62.
61. Stadlbauer V, Tauss J, Portugaller HR, et al. Hepatic encephalopathy following transjugular intrahepatic portosystemic shunt (TIPS): Management with l-ornithine–l-aspartate and stent reduction. Metab Brain Dis. 2007;22:45-50.
62. Reesa CJ, Opponga K, Al Mardinia H, et al. Effect of l-ornithine–l-aspartate on patients with and without TIPS undergoing glutamine challenge: A double-blind, placebo-controlled trial. Gut. 2000;47:571-4.
63. Poo JL, Góngora J, Sánchez-Avila F, et al. Efficacy of oral l-ornithine–l-aspartate in cirrhotic patients with hyperammonemic hepatic encephalopathy. Results of a randomized, lactulose-controlled study. Ann Hepatol. 2006;5:281-8.
64. Helvig FC, Schutz CB. A liver and kidney syndrome: Clinical, pathological, and experimental studies. Surg Gynecol Obstet. 1932;55:570-82.
65. Angeli P, Merkel C. Pathogenesis and management of hepatorenal syndrome in patients with cirrhosis. J Hepatol. 2008;48:S93-103.
66. Marik PE, Wood K, Starzl TE. The course of type 1 hepatorenal syndrome post liver transplantation. Nephrol Dial Transplant. 2006;21:478-82.
67. Shusterman B, Mchedishvili G, Rosner MH. Outcomes for hepatorenal syndrome and acute kidney injury in patients undergoing liver transplantation: A single center experience. Transplant Proc. 2007;39:1496-500.
68. Arroyo V. The liver and the kidney: Mutual clearance or mixed intoxication. Contrib Nephrol. 2007;156:17-23.
69. Moreau R, Durand F, Poynard T, et al. Terlipressin in patients with cirrhosis and type 1 hepatorenal syndrome: A retrospective multicenter study. Gastroenterology. 2002;122:923-30.
70. Moreau R, Lebrec D. Acute renal failure in patients with cirrhosis: Perspective in the age of MELD. Hepatology. 2003;37:233-43.
71. Arroyo V, Fernandez J, Ginès P. Pathogenesis and treatment of hepatorenal syndrome. Semin Liver Dis. 2008;28:81-95.
72. Munoz SJ. The hepatorenal syndrome. Med Clin N Am. 2008;92:813-37.
73. Moreau R, Lebrec D. Diagnosis and treatment of acute renal failure in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21:111-23.
74. Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310-18.
75. Bolognesi M, Sacerdoti D, Piva A, et al. Carbon monoxide-mediated activation of large conductance calcium-activated potassium channel contributes to mesenteric vasodilation in cirrhotic rats. J Pharmacol Exp Ther. 2007;321:187-94.
76. Ro J, Claria J, To-Figueras J, et al. Endogenous cannabinoids: A new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology. 2002;122:85-93.
77. Fernandez-Seara J, Prieto J, Quiroga J, et al. Systemic and regional hemodynamics in patients with liver cirrhosis and ascites with and without functional renal failure. Gastroenterology. 1989;97:1304-12.
78. Salerno F, Gerbes A, Ginès P, et al. Authors’ response. Gut. 2008;57:139-40.
79. Neuhoffer W, Pittrow D. Role of endothelin receptor antagonis in renal disesase. Eur J Clin Invest. 2006;36(Suppl 3):78-88.
80. Ruiz-del-Arbol L, Monescillo A, Arocena C, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439-47.
81. Ruiz-del-Arbol L, Urman J, Fernández J, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2003;38:1210-18.
82. Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28:59-69.
83. Baik SK, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Orphanet J Rare Dis. 2007;27:2-15.
84. Sherman DS, Fish DN, Teitelbaum I. Assessing renal function in cirrhotic patients: Problems and pitfalls. Am J Kidney Dis. 2003;41:269-78.
85. Ginès P, Guevara M, Arroyo V, et al. Hepatorenal syndrome. Lancet. 2003;29:1819-27.
86. Arroyo V, Terra C, Ginès P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol. 2007;46:935-46.
87. Terra C, Guevara M, Torre A, et al. Renal failure in patients with cirrhosis and sepsis unrelated to spontaneous bacterial peritonitis: Value of MELD score. Gastroenterology. 2005;129:1944-53.
88. Fasolato S, Angeli P, Dallangnese L, et al. Renal failure and bacterial infections in patients with cirrhosis: Epidemiology and clinical features. Hepatology. 2007;45:223-9.
89. Cardenas A, Ginès P, Uriz J, et al. Renal failure after gastrointestinal bleeding in cirrhosis: Incidence, clinical course, predictive factors, and short term prognosis. Hepatology. 2001;34:671-6.
90. Hampel H, Bynum GD, Zamora E, et al. Risk factors for the development of renal dysfunction in hospitalized patients with cirrhosis. Am J Gastroenterol. 2001;96:2206-10.
91. Fernandez J, Manteagudo J, Bargallo X, et al. A randomized unblended pilot study comparing albumin versus hydroxyethyl starch in spontaneous bacterial peritonitis. Hepatology. 2005;42:627-34.
92. Fernández J, Escorsell A, Zabalza M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival. Hepatology. 2006;44:1288-95.
93. Turban S, Tuluvath PJ, Atta MG. Hepatorenal syndrome. World J Gastroenterol. 2007;13:4046-55.
94. Restuccia T, Ortega R, Guevara M, et al. Effects of treatment of hepatorenal syndrome before transplantation on posttransplantation outcome. A case-control study. J Hepatol. 2004;40:140-6.
95. . Ikaria acquires North American rights to lucassin from orphan therapeutics—no currently approved drugs to treat hepatorenal syndrome type 1 in U.S., Sept. 2, 2008. Available at http://www.ikaria.com/media/news/press_2008_2_sept_LUCASSIN_Acquisition.pdf.
96. Treschan TA, Peters J. The vasopressin system: Physiology and clinical strategies. Anesthesia. 2006;105:599-612.
97. Halimi C, Bonnard P, Bernard B, et al. Effect of terlipressin (Glypressin) on hepatorenal syndrome in cirrhotic patients: Results of a multicentre pilot study. Eur J Gastroenterol Hepatol. 2002;14:153-8.
98. Duhamel C, Mauillon J, Berkelmans J, et al. Hepatorenal syndrome in cirrhotic patients: Terlipressin is a safe and efficient treatment; propranolol and digitalic treatments: Precipitating and preventing factors? Am J Gastroenterol. 2000;95:2984-5.
99. Colle I, Durand F, Pessione F, et al. Clinical course, predictive factors and prognosos in patients with cirrhosis and type-1 hepatorenal syndrome treated with terlipressin: A retrospective analysis. J Gastroenterol Hepatol. 2002;17:882-7.
100. Danalioglu A, Cakaloglu Y, Karaca C, et al. Terlipressin and albumin combination treatment in hepatorenal syndrome. Hepatogastroenterology. 2003;50(Suppl 2):ccciii-v.
101. Mulkay JP, Louis H, Donckier V, et al. Long-term terlipressin administration improves renal function in cirrhotic patients with type 1 hepatorenal syndrome: A pilot study. Acta Gastroenterol Belg. 2001;64:15-19.
102. Angeli P, Guarda S, Fasolato S, et al. Switch therapy with ciprofloxacin vs. intravenous ceftazidime in the treatment of spontaneous bacterial peritonitis in patients with cirrhosis: Similar efficacy at lower cost. Aliment Pharmacol Ther. 2006;23:75-84.
103. Uriz J, Ginès P, Cardenas A, et al. Terlipressin plus albumin infusion: An effective and safe therapy of hepatorenal syndrome. J Hepatol. 2000;33:43-48.
104. Ortega R, Ginès P, Uriz J, et al. Terlipressin therapy with and without albumin for patients with hepatorenal syndrome: Results of a prospective nonrandomized study. Hepatology. 2002;36:941-8.
105. Solanki P, Chawla A, Garg R, et al. Beneficial effects of terlipressin in hepatorenal syndrome: A prospective, randomized placebo-controlled clinical trial. J Gastroenterol Hepatol. 2003;18:152-6.
106. Martin-Llahi M, Pépin M-N, Guevara M, et al. Terlipressin and albumin vs. albumin in patients with cirrhosis and hepatorenal syndrome: A randomized study. Gastroenterology. 2008;134:1352-9.
107. Sanyal AJ, Boyer T, Garcia-Tsao G, et al. A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology. 2008;134:1360-8.
108. Fabrizi F, Dixit V, Martin P. Meta-analysis: Terlipressin therapy for the hepatorenal syndrome. Aliment Pharmacol Ther. 2006;24:935-44.
109. Gluud LL, Kjaer MS, Christensen E. Terlipressin for hepatorenal syndrome. Cochrane Database Syst Rev 2006; (4):CD005162.
110. Panteris V, Karamanolis DG. The puzzle of somatostatin: Action, receptors, analogues and therapy. Hepatogastroenterology. 2005;52:1771-81.
111. Schmidt LE, Ring-Larsen H. Vasoconstrictor therapy for hepatorenal syndrome in liver cirrhosis. Curr Pharm Des. 2006;12:4637-47.
112. Angeli P, Volpin R, Gerunda G, et al. Reversal of type 1 hepatorenal syndrome with the administration of midodrine and octreotide. Hepatology. 1999;29:1690-7.
113. Esrailian E, Runyon BA. Alcoholic cirrhosis–associated hepatorenal syndrome treated with vasoactive agents. Nat Clin Pract Nephrol. 2006;2:169-72.
114. Esrailian E, Pantangco ER, Kyulo NL, et al. Octreotide/Midodrine therapy significantly improves renal function and 30-day survival in patients with type 1 hepatorenal syndrome. Dig Dis Sci. 2007;52:742-8.
115. Sharma P, Kumar A, Sharma BC, et al. An open label, pilot, randomized controlled trial of noradrenaline versus terlipressin in the treatment of type 1 hepatorenal syndrome and predictors of response. Am J Gastroenterol. 2008;103:1689-97.
116. Alessandria C, Ottobrelli A, Debernardi-Venon W, et al. Noradrenalin vs. terlipressin in patients with hepatorenal syndrome: A prospective, randomized, unblinded, pilot study. J Hepatol. 2007;47:499-505.
117. Boyer TD. Transjugular intrahepatic portosystemic shunt in the management of complications of portal hypertension. Curr Gastroenterol Rep. 2008;10:30-5.
118. Wong F, Pantea L, Sniderman K. Midodrine, octreotide, albumin, and TIPS in selected patients with cirrhosis and type 1 hepatorenal syndrome. Hepatology. 2004;40:55-64.
119. Testino G, Ferro C, Sumberaz A, et al. Type-2 hepatorenal syndrome and refractory ascites: Role of transjugular intrahepatic portosystemic stent-shunt in eighteen patients with advanced cirrhosis and awaiting orthotopic liver transplantation. Hepatogastroenterology. 2003;50:1753-5.
120. Brensing KA, Textor J, Perz J, et al. Long term outcome after transjugular intrahepatic portosystemic stent-shunt in non-transplant cirrhotics with hepatorenal syndrome: A phase II study. Gut. 2000;47:288-95.
121. Guevara M, Ginès P, Bandi JC, et al. Transjugular intrahepatic portosystemic shunt in hepatorenal syndrome: Effects on renal function and vasoactive systems. Hepatology. 1998;28:416-22.
122. Nair S, Verma S, Tuluvath PJ. Pretransplant renal function predicts survival in patients undergoing orthotopic liver transplantation. Hepatology. 2002;35:1179-85.
123. Gonwa TA, McBride MA, Anderson K, et al. Continued influence of preoperative renal function on outcome of orthotopic liver transplant (OLTX) in the US: Where will MELD lead us? Am J Transplant. 2006;6:2651-9.
124. Campbell MS, Kotylar DS, Brensinger CM, et al. Renal function after orthotopic liver transplantation is predicted by duration of pretransplantation creatinine elevation. Liver Transpl. 2005;11:1048-55.
125. Abe T, Shono M, Kodama T, et al. Extracorporeal albumin dialysis. Ther Apher Dial. 2004;8:217-22.
126. Mitzner SR, Klammt S, Peszynski P, et al. Improvement of multiple organ functions in hepatorenal syndrome during albumin dialysis with the molecular adsorbent recirculating system. Ther Apher. 2001;5:417-22.
127. Mitzner SR, Stange J, Klammt S, et al. Improvement of hepatorenal syndrome with extracorporeal albumin dialysis MARS: Results of a prospective, randomized, controlled clinical trial. Liver Transpl. 2000;6:277-86.
128. Fallon M, Abrams G. Pulmonary dysfunction in chronic liver disease. Hepatology. 2000;32:859-65.
129. Budhiraja R, Hassoun PM. Portopulmonary hypertension: A tale of two circulations. Chest. 2003;123:562-76.
130. Ferreira PP, Camara EJN, Paula RLP, et al. Prevalence of hepatopulmonary syndrome in patients with decompensated chronic liver disease and its impact on short-term survival. Arq Gastroenterol. 2008;45:34-7.
131. Martinez G, Barbera J, Visa J, et al. Hepatopulmonary syndrome in candidates for liver transplantation. J Hepatol. 2001;34:651-7.
132. Schenk P, Fuhrmann V, Madl C, et al. Hepatopulmonary syndrome: Prevalence and predictive value of various cutoffs for arterial oxygenation and their clinical consequences. Gut. 2002;51:853-9.
133. Colle I, Moreau R, Godinho E, et al. Portopulmonary hypertension in candidates for liver transplantation: Diagnosis at evaluation comparing doppler echocardiography with cardiac catheterization and incidence on the waiting list. Hepatology. 2003;37:401-7.
134. Torregrosa M, Genesca J, Gonzalez A, et al. Role of Doppler echocardiography in the assessment of portopulmonary hypertension in liver transplantation candidates. Transplantation. 2001;71:572-4.
135. Swanson K, Wiesner R, Krowka M. Natural history of hepatopulmonary syndrome: Impact of liver transplantation. Hepatology. 2005;41:1122-9.
136. Schenk P, Schoniger-Hekele M, Furhmann V, et al. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology. 2003;125:1042-52.
137. Kawut S, Taichman D, Ahya V, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107-11.
138. Swanson KL, McGoon MD, Krowka MJ. Survival in portopulmonary hypertension. Am J Respir Crit Care Med. 2003;167:A683.
139. Palma DT, Fallon MB. The hepatopulmonary syndrome. J Hepatol. 2006;45:617-25.
140. Brussino L, Bucca C, Morello M, et al. Effect on dyspnoea and hypoxaemia of inhaled NG-nitro-l-arginine methyl ester in hepatopulmonary syndrome. Lancet. 2003;362:43-4.
141. Schenk P, Madl C, Rezale-Majd S, et al. Methylene blue improves the hepatopulmonary syndrome. Ann Intern Med. 2000;133:701-6.
142. Gomez F, Barbera JA, Roca J, et al. Effects of nebulized NG-nitro-l-arginine methyl ester in patients with hepatopulmonary syndrome. Hepatology. 2006;43:1084-91.
143. Luo B, Tang L, Wang Z, et al. Cholangiocyte endothelin 1 and transforming growth factor β1 production in rat experimental hepatopulmonary syndrome. Gastroenterology. 2005;129:682-95.
144. Ling Y, Zhang J, Luo B, et al. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of experimental hepatopulmonary syndrome. Hepatology. 2004;39:1593-602.
145. Luo B, Liu L, Tang L, et al. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: A potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556-63.
146. Rabiller A, Nunes H, Lebrec D, et al. Prevention of gram-negative translocation reduces the severity of hepatopulmonary syndrome. Am J Respir Crit Care Med. 2002;166:514-17.
147. Zhang HY, Han DW, Ai-Rong S, et al. Intestinal endotoxemia plays a central role in development of hepatopulmonary syndrome in a cirrhotic rat model induced by multiple pathogenic factors. World J Gastroenterol. 2007;21:6385-95.
148. Zhang HY, Han DW, Zhao ZF, et al. Multiple pathogenic factor-induced complications of cirrhosis in rats: A new model of hepatopulmonary syndrome with intestinal endotoxemia. World J Gastroenterol. 2007;13:3500-7.
149. Arguedas M, Drake B, Kapoor A, et al. Carboxyhemoglobin levels in cirrhotic patients with and without hepatopulmonary syndrome. Gastroenterology. 2005;128:328-33.
150. Zhang J, Ling Y, Luo B, et al. Analysis of pulmonary heme oxygenase-1 and nitric oxide synthase alterations in experimental hepatopulmonary syndrome. Gastroenterology. 2003;125:1441-51.
151. Zhang J, Luo B, Tang L, et al. Pulmonary angiogenesis in a rat model of hepatopulmonary syndrome. Gastroenterology. 2009;136:1070-80.
152. Alvaro D, Mancino MG, Glaser S, et al. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology. 2007;132:415-31.
153. Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270-82.
154. Cramer T, Schuppan D, Bauer M, et al. Hepatocyte growth factor and c-Met expression in rat and human liver fibrosis. Liver Int. 2004;24:335-44.
155. Gaudio E, Onori P, Franchitto A, et al. Vascularization of the intrahepatic biliary tree and its role in the regulation of cholangiocyte growth. In: Alpini G, Alvaro D, LeSage G, et al, editors. The pathophysiology of biliary epithelia. Georgetown, Tex: Landes Biosciences; 2004:41.
156. Homsi J, Daud AI. Spectrum of activity and mechanism of action of VEGF/PDGF inhibitors. Cancer Control. 2007;14:285-94.
157. Ono M, Sawa Y, Matsumoto K, et al. In vivo gene transfecton with hepatocyte growth factor via the pulmonary artery induce angiogenesis in the rat lung. Circulation. 2002;106(Suppl 1):I264-9.
158. Assaad AM, Kawut SM, Arcasoy SM, et al. Platelet-derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest. 2007;113:850-5.
159. Castaño G, Sookoian S. Female sex and autoimmune hepatitis and the risk of portopulmonary hypertension. Hepatology. 2008;48:2090.
160. Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut. 2003;52:1355-62.
161. Kawut SM, Al-Naamani N, Agerstrand C, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest. 2009;135:752-9.
162. Rodriguez-Roisin R, Krowka MJ, Herve P, et al. ERS Task Force Pulmonary-Hepatic Vascular Disorders Scientific Committee. Eur Respir J. 2004;24:861-80.
163. Arguedas M, Abrams GA, Krowka MJ, et al. Prospective evaluation of outcomes and predictors of mortality in patients with hepatopulmonary syndrome undergoing liver transplantation. Hepatology. 2003;37:192-7.
164. Fuhrmann V, Madl C, Mueller C, et al. Hepatopulmonary syndrome in patients with hypoxic hepatitis. Gastroenterology. 2006;131:69-75.
165. Mahmoodi E, Kianifar HR, Partovi S, et al. Intrapulmonary arteriovenous shunt in children with chronic liver disease: Clinical features, laboratory data and outcome. Indian J Gastroenterol. 2008;27:16-18.
166. Kim JS, Kim KM, Ko JK, et al. Intrapulmonary shunt in the course of pediatric liver transplantation. Transplant Proc. 2008;40:2512-4.
167. Gomez F, Martinez-Palli G, Barbera J, et al. Gas exchange mechanism of orthodeoxia in hepatopulmonary syndrome. Hepatology. 2004;40:660-6.
168. Sindhu S, Ramesh P, Juneja R, et al. Hepatopulmonary syndrome, an unusual cause of hypoxemia. Indian J Pediatr. 2007;74:1127-9.
169. Abrams GA, Jaffe CC, Hoffer PB, et al. Diagnostic utility of contrast echocardiography and lung perfusion scan in patients with hepatopulmonary syndrome. Gastroenterology. 1995;109:1283-8.
170. Lee KN, Lee HJ, Shin WW, et al. Hypoxemia and liver cirrhosis (hepatopulmonary syndrome) in eight patients: Comparison of the central and peripheral pulmonary vasculature. Radiology. 1999;211:549-53.
171. Abrams GA, Nanda NC, Dubovsky EV, et al. Use of macroaggregated albumin lung perfusion scan to diagnose hepatopulmonary syndrome: A new approach. Gastroenterology. 1998;114:305-10.
172. Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome—a liver-induced vascular disorder. N Engl J Med. 2008;358:2378-87.
173. Palma DT, Philips GM, Arguedas MR, et al. Oxygen desaturation during sleep in hepatopulmonary syndrome. Hepatology. 2008;47:1257-63.
174. Abrams GA, Sanders MK, Fallon MB. Utility of pulse oximetry in the detection of arterial hypoxemia in liver transplant candidates. Liver Transpl. 2002;8:391-6.
175. Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:S5-S12.
176. Krowka MJ. Portopulmonary hypertension: Diagnostic advances and caveats. Liver Transpl. 2003;9:1336-7.
177. Swanson KL, Krowka MJ. Screen for portopulmonary hypertension, especially in liver transplant candidates. Cleve Clin J Med. 2008;75:121-36.
178. Murray KF, Carrithers RLJr. AASLD Practice Guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005:1-26.
179. Fallon MB, Mulligan DC, Gish RG, et al. Model for end-stage liver disease (MELD): Exception for hepatopulmonary syndrome. Liver Transpl. 2006;12:S105-S7.
180. Krowka MJ, Fallon MB. Liver transplantation for hepatopulmonary syndrome (HPS): What is the MESSAGE? Am J Transplant. 2008;8:911-12.
181. Sulleman BM, Hunslcker LG, Katz DA, et al. OPTN policy regarding prioritization of patients with hepatopulmonary syndrome: Does it provide equitable organ allocation. Am J Transpl. 2008;8:954-64.
182. Abrams GA, Fallon MB. Treatment of hepatopulmonary syndrome with Allium sativum (garlic): A pilot trial. J Clin Gastroenterol. 1998;27:232-5.
183. Anel RM, Sheagren JN. Novel presentation and approach to management of hepatopulmonary syndrome with use of antimicrobial agents. Clin Infect Dis. 2001;32:E131-6.
184. Varcelino R, Tieppo J, Simoes Dias A, et al. N-Acetylcysteine effects on genotoxic and oxidative stress parameters in cirrhotic rats with hepatopulmonary syndrome. Basic Clin Pharmacol Toxicol. 2008;102:370-6.
185. Tanikella R, Philips GM, Faulk DK, et al. Pilot study of pentoxifylline in hepatopulmonary syndrome. Liver Transpl. 2008;14:1199-203.
186. Zhang J, Ling Y, Tang L, et al. Pentoxifylline attenuation of experimental hepatopulmonary syndrome. J Appl Physiol. 2007;102:949-55.
187. Chevalier P, Novelli L, Montamedi JP, et al. Hepatopulmonary syndrome successfully treated with transjugular intrahepatic portosystemic shunt: A three-year follow-up. J Vasc Interv Radiol. 2004;15:647-8.
188. Lasch HM, Fried MW, Zacks SL, et al. Use of transjugular intrahepatic portosystemic shunt as a bridge to liver transplantation in a patient with severe hepatopulmonary syndrome. Liver Transp. 2001;7:147-9.
189. Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology. 2006;130:120-6.
190. Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: A single-center series. Am J Transplant. 2006;6:2177-82.
191. Kähler CM, Graziadei I, Wiedermann CJ, et al. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr. 2000;112:637-40.
192. Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor antagonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol. 2004;2:1039-42.
193. Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation. 2004;77:1774-5.
194. Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: A potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223-31.
195. Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther. 2002;71:398-402.
196. Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant. 2004;10:945-50.
197. Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J. 2006;28:563-7.
198. Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: A report of the multicenter liver transplant database. Liver Transplant. 2004;10:174-82.
199. Krowka MJ, Fallon MB, Mullingan DC, et al. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transpl. 2006;12:S114-16.
200. Fix OK, Bass NM, De Marco T, et al. Long-term follow-up of portopulmonary hypertension: Effect of treatment with epoprostenol. Liver Transpl. 2007;13:875-85.
201. Ashfaq M, Chinnakotla S, Rogers L, et al. The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant. 2007;7:1258-64.
202. Kowalski HJ, Abelmann WH. The cardiac output at rest in Laennec’s cirrhosis. J Clin Invest. 1953;32:1025-33.
203. Regan TJ, Levinson GE, Oldewur A, et al. Ventricular function in noncardiacs with alcoholic fatty liver: Role of ethanol in the production of cardiomyopathy. J Clin Invest. 1969;48:397-407.
204. Lee RF, Glenn TK, Lee SS. Cardiac dysfunction in cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21:125-40.
205. Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28:59-69.
206. Møller S, Henriksen JH. Cardiovascular complications of cirrhosis. Gut. 2008;57:268-78.
207. Milani A, Zaccaria R, Bombardieri G, et al. Cirrhotic cardiomyopathy. Dig Liver Dis. 2007;39:507-15.
208. Kelbaek H, Eriksen J, Brynjolf I, et al. Cardiac performance in patients with asymptomatic alcoholic cirrhosis of the liver. Am J Cardiol. 1984;54:852-5.
209. Finucci G, Desideri A, Sacerdoti D, et al. Left ventricular diastolic function in liver cirrhosis. Sacnd J Gastroenterol. 1996;31:279-84.
210. Gaskari SA, Honar H, Lee SS. Therapy insight: Cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3:329-37.
211. Alexander J, Mishra P, Desai N, et al. Cirrhotic cardiomyopathy: Indian scenario. J Gastroenterol Hepatol. 2007;22:395-9.
212. Torregrosa M, Aguade S, Dos L, et al. Cardiac alterations in cirrhosis: Reversibility after liver transplantation. J Hepatol. 2005;42:68-74.
213. Pozzi M, Carugo S, Boari G, et al. Evidence of functional and structural cardiac abnormalities in cirrhotic patients with and without ascites. Hepatology. 1997;26:1131-7.
214. Møller S, Henriksen JH. Cardiovascular dysfunction in cirrhosis. Pathophysiological evidence of a cirrhotic cardiomyopathy. Scand J Gastroenterol. 2001;36:785-94.
215. Pozzi M, Redaelli E, Ratti L, et al. Time-course of diastolic dysfunction in different stages of chronic HCV related liver diseases. Minerva Gastroenterol Dietol. 2005;51:179-86.
216. Bal JS, Thuluvath PJ. Prolongation of QTc interval: Relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23:243-8.
217. Ma Z, Zhang Y, Huet PM, et al. Differential effects of jaundice and cirrhosis on β-adrenoreceptor signaling in three rat models of cirrhotic cardiomyopathy. J Hepatol. 1999;30:485-91.
218. Lee SS, Marty J, Mantz J, et al. Desensitization of myocardial β-adrenergic receptors in cirrhotic rats. Hepatology. 1990;12:481-5.
219. Batkai S, Jarai Z, Wagner JA, et al. Endocannabinoids acting as vascular Cb1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827-32.
220. Ros J, Claria J, To-Figueras J, et al. Endogenous cannabinoids: A new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology. 2002;122:85-93.
221. Domenicali M, Ros J, Fernandez-Varo G, et al. Increased anandamide induced relaxation in mesenteric arteries of cirrhotic rats: Role of cannabinoid and vanilloid receptors. Gut. 2005;54:522-7.
222. Gaskari SA, Liu H, Moezi L, et al. Role of endocannabinoid in the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Br J Pharmacol. 2005;146:315-23.
223. Liu H, Ma Z, Lee SS. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology. 2000;118:937-44.
224. Liu H, Song D, Lee SS. Role of heme oxygenase–carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G68-G74.
225. Finucci G, Lunardi F, Sacerdoti D, et al. Q-T interval prolongation in liver cirrhosis. Reversibility after orthotopic liver transplantation. Jpn Heart J. 1998;39:321-9.
226. Bernardi M, Calandra S, Colantoni A, et al. Q-T interval prolongation in cirrhosis: Prevalence, relationship with severity, and etiology of the disease and possible pathogenic factors. Hepatology. 1998;27:28-34.
227. Henriksen JH, Fuglsang S, Bendtsen F, et al. Dyssynchronous electrical and mechanical systole in patients with cirrhosis. J Hepatol. 2002;36:513-20.
228. Gines P, Uriz J, Calahorra B, et al. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrosis. Gastroenterology. 2002;123:1839-47.
229. Cazzaniga M, Salerno F, Pagnozzi G, et al. Diastolic dysfunction is associated with poor survival in cirrhotic patients with transjugular intrahepatic portosystemic shunt. Gut. 2007;56:869-75.
230. Snowden CP, Hughes T, Rose J, et al. Pulmonary edema in patients after liver transplantation. Liver Transpl. 2000;6:466-70.
231. Therapondos G, Flapan AD, Plevris JN, et al. Cardiac morbidity and mortality related to orthotopic liver transplantation. Liver Transpl. 2004;10:1441-53.
232. Wong F, Siu S, Liu P, et al. Brain natriuretic peptide: Is it a predictor of cardiomyopathy in cirrhosis. Clin Sci. 2001;101:621-8.
233. Henriksen JH, Gotze JP, Fuglsang S, et al. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: Relation to cardiovascular dysfunction and severity of disease. Gut. 2003;52:1511-17.
234. Pateron D, Beyne P, Laperche T, et al. Elevated circulating cardiac troponin I in patients with cirrhosis. Hepatology. 1999;29:640-3.
235. Donovan CL, Marcovitz PA, Punch JD, et al. Two-dimensional and dobutamine stress echocardiography in the preoperative assessment of patients with end-stage liver disease prior to orthotopic liver transplantation. Transplantation. 1996;61:1180-8.
236. Mohamed R, Forsey PR, Davies MK, et al. Effect of liver transplantation on QT interval prolongation and autonomic dysfunction in end-stage liver disease. Hepatology. 1996;23:1128-34.
237. Liu H, Song D, Lee SS. Cirrhotic cardiomyopathy. Gastreoenterol Clin Biol. 2002;26:842-7.
238. Pozzi M, Grassi G, Ratti L, et al. Cardiac, neuroadrenergic, and portal hemodynamic effects of prolonged aldosterone blockade in post viral child A cirrhosis. Am J Gastroenterol. 2005;100:1110-16.
239. Henriksen JH, Bendtsen F, Hansen EF, et al. Acute non-selective beta-adrenergic blockade reduces prolonged frequency-adjusted Q-T interval (QTc) in patients with cirrhosis. J Hepatol. 2004;40:239-46.
240. Liu H, Lee SS. What happens to cirrhotic cardiomyopathy after liver transplantation? Hepatology. 2005;42:1203-5.
241. Harry R, Auzinger G, Wendon J. The clinical importance of adrenal insufficiency in acute hepatic dysfunction. Hepatology. 2002;36:395-402.
242. Harry R, Auzinger G, Wendon J. The effects of supraphysiologic doses of corticosteroids in hypotensive liver failure. Liver Int. 2003;23:71-7.
243. Marik PE. Adrenal-exhaustion syndrome in patients with liver disease. Intensive Care Med. 2006;32:275-80.
244. Tsai MH, Peng YS, Cheng YC, et al. Adrenal insufficiency in patients with cirrhosis, severe sepsis and septic shock. Hepatology. 2006;43:673-81.
245. Fernandez J, Escorsell A, Zabalza M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival. Hepatology. 2006;44:1288-95.
246. Karagiannis A, Harsoulis F. Gonadal dysfunction in systemic diseases. Eur J Endocrinol. 2005;152:501-13.
247. Luppa PB, Thaler M, Schulte-Frohlinde E, et al. Unchanged androgen-binding properties of sex hormone-binding globulin in male patients with liver cirrhosis. Clin Chem Lab Med. 2006;44:967-73.
248. Ahbouch S, Pomier-Layrargues G, Vincent C, et al. Reduced plasma dehydroepiandrosterone sulfate levels are significantly correlated with fatigue severity in patients with primary biliary cirrhosis. Neurochem Int. 2008;52:569-74.
249. Charlton M, Angulo P, Chalasani N, et al. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology. 2008;47:484-92.
250. Frias J, Torres JM, Miranda MT, et al. Effects of acute alcohol intoxication on pituitary-gonadal axis hormones, pituitary-adrenal axis hormones, beta-endorphins and prolactin in human adults of both sexes. Alcohol Alcohol. 2002;37:169-73.
251. Hasselblatt M, Dieg-Hartig C, Hufner M, et al. Persistent disturbance of the hypothalamic-pituitary-gonadal axis in abstinent alcoholic men. Alcohol Alcohol. 2003;38:239-42.
252. Prisant LM, Chin E. Gynecomastia and hypertension. J Clin Hypertens. 2005;7:245-8.
253. Neff GW, O’Brien CB, Shire NJ, et al. Topical testosterone treatment for chronic allograft failure in liver transplant recipients with recurrent hepatitis C virus. Transplant Proc. 2004;36:3071-4.
254. Spadaro L, Bolognesi M, Pierbon A, et al. Alterations in thyroid Doppler arterial resistance indices, volume and hormones in cirrhosis: Relationship with splanchnic haemodynamics. Ultrasound Med Biol. 2004;30:19-25.
255. Kayacetin E, Kisakol G, Kaya A. Low serum total thyroxine and thriiodothyronine in patients with hepatic encephalopathy due to non-alcoholic cirrhosis. Swiss Med Wkly. 2003;133:210-13.
256. Rodríguez-Torres M, Ríos-Bedoya CF, Ortiz-Lasanta G, et al. Thyroid dysfunction (TD) among chronic hepatitis C patients with mild and severe hepatic fibrosis. Ann Hepatol. 2008;7:72-7.
257. Caregaro L, Alberino F, Amodio P, et al. Nutritional and prognostic significance of serum hypothyroxinemia in hospitalized patients with liver cirrhosis. J Hepatol. 1998;28:115-21.
258. Antonelli A, Ferri C, Fallahi P, et al. Thyroid disorders in chronic hepatitis C virus infection. Thyroid. 2006;16:563-72.
259. Antonelli A, Ferri C, Pampana A, et al. Thyroid disorders in chronic hepatitis C. Am J Med. 2004;117:10-13.
260. Antonelli A, Ferri C, Ferrari SM, et al. Endocrine manifestations of hepatitis C virus infection. Nat Clin Pract Endocrinol Metab. 2009;5:26-34.
261. Bartolomé J, Rodríguez-Iñigo E, Quadros P, et al. Detection of hepatitis C virus in thyroid tissue from patients with chronic HCV infection. J Med Virol. 2008;80:1588-94.
262. Tarantino G, Gagliardi G, Conca P. Do thyroid abnormalities detected in patients treated for HCV-related chronic hepatitis persist? Int J Immunopathol Pharmacol. 2008;21:467-9.
263. Collier J. Bone disorders in chronic liver disease. Hepatology. 2007;46:1271-8.
264. Guanabens N, Pares A, Ros I, et al. Severity of cholestasis and advanced histological stage but not menopausal status are the major risk factors for osteoporosis in primary biliary cirrhosis. J Hepatol. 2005;42:573-7.
265. Lindor KD, Janes CH, Crippin JS, et al. Bone disease in primary biliary cirrhosis: Does ursodeoxycholic acid make a difference? Hepatology. 1995;21:389-92.
266. Guichelaar MMJ, Kendall R, Schmoll J, et al. Bone mineral density before and after OLT; long-term follow-up and predictive factors. Liver Transpl. 2006;12:1390-402.
267. Leslie WD, Bernstein CN, Leboff MS. American Gastroenterological Association Clinical Practice Committee: AGA technical review on osteoporosis in hepatic disorders. Gastroenterology. 2003;125:941-66.
268. Collier J, Ninkovic M, Compston JE. Guidelines on the management of osteoporosis associated with chronic liver disease. Gut. 2002;50(Suppl 1):1-9.
269. Pares A, Guanabens N. Treatment of bone disorders in liver disease. J Hepatol. 2006;45:445-53.
270. Caldwell SH, Sanyal AJ. Coagulation and hemostasis in liver disease: Controversies and advances. Guest editors. Clin Liver Dis, 13, 2009:1-166
271. Rodriguez-Inigo E, Bartolome J, Quiroga JA, et al. Expression of factor VII in the liver of patients with liver disease: Correlations with the disease severity and impairment in the hemostasis. Blood Coagul Fibrinolysis. 2001;12:193-9.
272. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-70.
273. Dell’era A, Bosch J. Review article: The relevance of portal pressure and other risk factors in acute gastro-oesophageal variceal bleeding. Aliment Pharmacol Ther. 2004;20(Suppl 3):8-15.
274. Kujovich JL. Hemostatic defects in end-stage liver disease. Crit Care Clin. 2005;21:563-87.
275. Marti-Carvajal AJ, Cortés-Jofré M, Martí-Peña AJ. Vitamin K for upper gastrointestinal bleeding in patients with liver diseases. Cochrane Database Syst Rev 2008; (3):CD004792.
276. Youssef WI, Salazar F, Dasarathy S, et al. Role of fresh frozen plasma infusion in correction of coagulopathy of chronic liver disease: A dual phase study. Am J Gastroenterol. 2003;98:1391-4.
277. Bosch J, Thabut D, Albillos A, et al. Recombinant factor VIIa for variceal bleeding in patients with advanced cirrhosis: A randomized, controlled trial. Hepatology. 2008;47:1604-14.
278. Khan H, Belser J, Yilmaz M, et al. Fresh-frozen plasma and platelet transfusions are associated with development of acute lung injury in critically ill medical patients. Chest. 2007;131:1308-14.
279. Eder AF, Herron R, Strupp A, et al. Transfusion-related acute lung injury surveillance (2003-2005) and the potential impact of the selective use of plasma from male donors in the American Red Cross. Transfusion. 2007;47:599-607.
280. Jeffers L, Chalasani N, Balart L, et al. Safety and efficacy of recombinant factor VIIa in patients with liver disease undergoing laparoscopic liver biopsy. Gastroenterology. 2002;123:118-26.
281. Arjal R, Trotter JF. International normalized ratio of prothrombin time in the model for end-stage liver disease score: An unreliable measure. Clin Liver Dis. 2009;13:67-71.
282. Tripodi A, Chantarangkul V, Primignani M, et al. The international normalized ratio calibrated for cirrhosis (INRliver) normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology. 2007;46:528-34.
283. Hugenholtz GGC, Porte RJ, Lisman T. The platelet and platelet function testing in liver disease. Clin Liver Dis. 2009;13:11-20.
284. Tripodi A, Primignani M, Chantarangkul V, et al. Thrombin generation in patients with cirrhosis: The role of platelets. Hepatology. 2006;44:440-5.
285. Wong AY, Irwin MG, Hui TW, et al. Desmopressin does not decrease blood loss and transfusion requirements in patients undergoing hepatectomy. Can J Anaesth. 2003;50:14-20.
286. Pivalizza EG, Warters RD, Gebhard R. Desmopressin before liver transplantation. Can J Anaesth. 2003;50:748-9.
287. Hu KQ, Yu AS, Tyyagura L, et al. Hyperfibrinolytic activity in hospitalized cirrhotic patients in a referral liver unit. Am J Gastroenterol. 2001;96:1581-6.
288. Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: Reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol. 2007;46:727-33.
289. Gunawan B, Runyon B. The efficacy and safety of epsilon-aminocaproic acid treatment in patients with cirrhosis and hyperfibrinolysis. Aliment Pharmacol Ther. 2006;23:115-20.
290. Kang Y, Lewis JH, Navalgund A, et al. Epsilon-aminocaproic acid for treatment of fibrinolysis during liver transplantation. Anesthesiology. 1987;66:766-73.
291. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol. 2002;37:280-7.
292. Søgaard KK, Horváth-Puhó E, Grønbaek H, et al. Risk of venous thromboembolism in patients with liver disease: A nationwide population-based case-control study. Am J Gastroenterol. 2009;104:96-101.
293. Northup PG, McMahon MM, Ruhl AP, et al. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am J Gastroenterol. 2006;101:1524-8.
294. Garcia-Fuster MJ, Abadilla N, Fabia MJ, et al. Venous thromboembolism and liver cirrhosis. Rev Esp Enferm Dig. 2008;100:259-62.
295. Amitrano L, Brancaccio V, Guardascione MA, et al. Inherited coagulation disorders in cirrhotic patients with portal vein thrombosis. Hepatology. 2000;31:345-8.
296. Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: Risk and benefit of anticoagulant therapy. Gastroenterology. 2001;120:490-7.
297. Northup PG, Sundaram V, Fallon MB, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6:2-9.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 92 Hepatic Encephalopathy, Hepatorenal Syndrome, Hepatopulmonary Syndrome, and Systemic Complications of Liver Disease
HEPATIC ENCEPHALOPATHY
The term hepatic encephalopathy (HE) encompasses a wide array of transient and reversible neurologic and psychiatric manifestations usually found in patients with chronic liver disease and portal hypertension, but also seen in patients with acute liver failure. HE develops in 50% to 70% of patients with cirrhosis, and its occurrence is a poor prognostic indicator, with projected one- and three-year survival rates of 42% and 23%, respectively, without liver transplantation.1 Symptoms may range from mild neurologic disturbances to overt coma.2,3 HE is often triggered by an inciting event that results in a rise in the serum ammonia level. The precise underlying pathophysiologic mechanisms are not well understood, and the mainstay of therapy is the elimination of the precipitating event and excess ammonia.4 Liver transplantation generally reverses HE.
PATHOPHYSIOLOGY
A number of factors, occurring alone or in combination, have been implicated in the development of HE. These factors may differ in acute and chronic liver disease and include the production of neurotoxins, altered permeability of the blood-brain barrier, and abnormal neurotransmission (Fig. 92-1). The best-described neurotoxin involved in HE is ammonia, which is produced primarily in the colon, where bacteria metabolize proteins and other nitrogen-based products into ammonia. Enterocytes synthesize ammonia from glutamine.4–6 Once produced, ammonia enters the portal circulation and, under normal conditions, is metabolized and cleared by hepatocytes. In cirrhosis and portal hypertension, reduced hepatocyte function and portosystemic shunting contribute to increased circulating ammonia levels. Arterial hyperammonemia is observed in up to 90% of patients with HE, although serum levels are neither sensitive nor specific indicators of its presence. Increased permeability of the blood-brain barrier increases the uptake and extraction of ammonia by the cerebellum and basal ganglia.7–9 Acute hyperammonemia appears to have a direct effect on brain edema, astrocyte swelling, and the transport of neurally active compounds such as myoinositol, and thereby contributes to HE.10–12
Figure 92-1. Pathophysiology of hepatic encephalopathy. GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; NH3, ammonia.
Other alterations in HE affect neuronal membrane fluidity, central nervous system (CNS) neurotransmitter expression, and neurotransmitter receptor expression and activation.13,14 The γ-aminobutyric acid (GABA)–benzodiazepine system has been the most well studied. Although CNS benzodiazepine levels and GABA receptor concentrations are unchanged in animal models of HE, increased sensitivity of the astrocyte (peripheral-type) benzodiazepine receptor enhances activation of the GABA-benzodiazepine system.15,16 This activation occurs in part through a feed-forward system in which production of neurosteroids (allopregnanolone and tetrahydrodeoxycorticosterone) by astrocytes further activates the GABAA-benzodiazepine receptor system.17,18 Other factors that influence CNS neurotransmission, including serotonin (5-hydroxytryptamine, 5-HT),19–21 nitric oxide (NO), circulating opioid peptides, manganese, and increased oxygen free radical production, have also been postulated to contribute to HE.4
Finally, hyperammonemia, particularly in acute liver failure, also increases astrocyte glutamine production via glutamine synthetase. The rise in astrocyte glutamine and glutamate concentrations contributes to factors associated with CNS dysfunction.5,22,23
CLINICAL FEATURES AND DIAGNOSIS
HE may present as a spectrum of reversible neuropsychiatric symptoms and signs, ranging from mild changes in cognition to profound coma, in patients with acute or chronic liver disease. It is often precipitated by an inciting event (e.g., gastrointestinal bleeding, electrolyte abnormalities, infections, medications, dehydration). The diagnosis of HE, therefore, requires careful consideration in the appropriate clinical situation. Occasionally, HE may be the initial presentation of chronic liver disease. Subtle findings in HE may include forgetfulness, alterations in handwriting, difficulty with driving, and reversal of the sleep-wake cycle.24,25 Overt findings may include asterixis, agitation, disinhibited behavior, seizures, and coma. Other causes of altered mental status, particularly hypoglycemia, hyponatremia, medication ingestion, and structural intracranial abnormalities resulting from coagulopathy or trauma, should be considered and rapidly excluded in patients suspected of having HE.
No specific laboratory findings indicate the presence of HE definitively. The most commonly used test to assess a patient with possible HE is the blood ammonia level. An elevation in the blood ammonia level in a patient with cirrhosis and altered mental status supports a diagnosis of HE. Blood ammonia levels may be elevated in the absence of HE, however, because of gastrointestinal bleeding or the ingestion of certain medications (e.g., diuretics, alcohol, narcotics, valproic acid).15,26,27 In addition, blood ammonia levels may be elevated in the presence of HE, even in the absence of cirrhosis and portal hypertension, in patients with metabolic disorders that influence ammonia generation or metabolism, such as urea cycle disorders (see Chapter 76) and disorders of proline metabolism (Table 92-1).28,29 Use of a tourniquet when blood is drawn and delayed processing and cooling of a blood sample may raise the blood ammonia level.15 Measurement of arterial ammonia offers no advantage over measurement of venous ammonia levels in patients with chronic liver disease.30 In patients with acute liver failure, however, elevated arterial ammonia levels (150 to 200 mg/dL or higher) may be predictive of the presence of brain edema and herniation (see Chapter 93).12,31,32
Table 92-1 Differential Diagnosis of Hyperammonemia
Of the scoring systems used to grade the severity of HE, the West Haven system, based on a scale of 0 to 4, is the most widely used in clinical practice (Table 92-2).25 Although clinically useful, the West Haven criteria are insensitive and have led to the development of standardized psychometric tests and rapid bedside mental status assessments to aid in the diagnosis of HE and facilitate research.33–37 One simple paper and pencil test, the portosystemic encephalopathy syndrome test (PSET), evaluates the patient’s attention, concentration, fine motor skills, and orientation and has been shown to be highly specific for the diagnosis of HE.33,38 The development of these tests has led to recognition of the syndrome of minimal HE, in which abnormalities are observed on testing but clinically recognizable alterations of HE are minimal or not detected. The presence of minimal HE is common in patients with cirrhosis, appears to influence the patient’s quality of life and driving ability, and confers an increased risk that overt HE will develop in the patient. Whether treatment of minimal HE confers any benefit is an area of active investigation.24,39–41
| GRADE | Impairment | |
|---|---|---|
| INTELLECTUAL FUNCTION | NEUROMUSCULAR FUNCTION | |
| 0 | Normal | Normal |
| Minimal, subclinical | Normal examination findings. Subtle changes in work or driving | Minor abnormalities of visual perception or on psychometric or number tests |
| 1 | Personality changes, attention deficits, irritability, depressed state | Tremor and incoordination |
| 2 | Changes in sleep-wake cycle, lethargy, mood and behavioral changes, cognitive dysfunction | Asterixis, ataxic gait, speech abnormalities (slow and slurred) |
| 3 | Altered level of consciousness (somnolence), confusion, disorientation, and amnesia | Muscular rigidity, nystagmus, clonus, Babinski sign, hyporeflexia |
| 4 | Stupor and coma | Oculocephalic reflex, unresponsiveness to noxious stimuli |
Modified from the West Haven Criteria; in Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: Final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002; 35:716-21.
A number of novel imaging and functional tests have been studied in the diagnosis of HE. Magnetic resonance spectroscopy (MRS) has been used to measure brain concentrations of choline and glutamine noninvasively.42 Magnetic resonance (MR) T1 mapping with partial inversion recovery (TAPIR) has been investigated as a means to measure changes in the brain quantitatively over clinically relevant measurement times.33 Whether MR-based techniques can be standardized and become practical diagnostic tests is uncertain. The critical flicker frequency test, a simple light-based test that has been used to assess cerebral cortex function in a number of disorders, has been shown to be a reliable marker of minimal HE and may become a clinically useful screening test.34–36
TREATMENT
Current treatments for HE are directed primarily toward the elimination or correction of precipitating factors (e.g., bleeding, infection, hypokalemia, medications, dehydration), reduction in elevated blood ammonia levels, and avoidance of the toxic effects of ammonia in the CNS. In the past, dietary protein restriction was considered an important component of the treatment of HE. Subsequent work, however, has suggested that limiting protein-calorie intake is not beneficial in patients with HE.43–45 Vegetable and dairy proteins are preferred to animal proteins because of a more favorable calorie-to-nitrogen ratio. Although branched-chain amino acid supplementation may improve symptoms modestly, the benefits of such supplementation are not sufficient to justify its routine use.4
Nonabsorbable disaccharides have been the cornerstone of the treatment of HE. Oral lactulose or lactitol (the latter is not available in the United States) are metabolized by colonic bacteria to byproducts that appear to have beneficial effects by causing catharsis and reducing intestinal pH, thereby inhibiting ammonia absorption.46 These agents improve symptoms in patients with acute and chronic HE when compared with placebo but do not improve psychometric test performance or mortality. The most common side effects experienced by patients who take lactulose are abdominal cramping, flatulence, diarrhea, and electrolyte imbalance. Lactulose may also be administered per rectum (as an enema) to patients who are at increased risk of aspiration, although the efficacy of enema administration has not been evaluated.
Oral antibiotics also have been used to treat HE, with the aim of modifying the intestinal flora and lowering stool pH to enhance the excretion of ammonia. Antibiotics are generally used as second-line agents after lactulose or in patients who are intolerant of nonabsorbable disaccharides. Neomycin has been approved by the U.S. Food and Drug Administration (FDA) for use in acute HE in a dose of 1 to 3 g orally every six hours for up to six days but has been used more commonly off-label to treat chronic HE in doses of 0.5 to 1 g every 12 hours, in addition to lactulose. The efficacy of neomycin in acute or chronic HE, however, is not clearly established,47 and ototoxicity and nephrotoxicity caused by neomycin have been reported, particularly in patients with preexisting renal dysfunction.4 Rifaximin has been studied and approved by the FDA for the treatment of chronic HE on the basis of the results of a multicentered, randomized, controlled trial in which the overall clinical efficacy and rate of side effects were similar in patients treated with lactitol and those treated with rifaximin.48 The usual dose is 400 mg orally three times daily. Two systematic reviews of randomized controlled trials that compared rifaximin with other therapies (nonabsorbable disaccharides and other antibiotics) for the treatment of acute or chronic HE have confirmed that the efficacy and side effect profiles are comparable.49,50 Other antibiotics, including metronidazole and vancomycin, have been reported to be effective in small trials and case series, but the data to support their use are insufficient.
In addition to antibiotics, several other agents that may modify intestinal flora and modulate ammonia generation or absorption have been evaluated as potential treatments for HE. Acarbose, an intestinal α-glucosidase inhibitor used to treat type 2 diabetes mellitus, inhibits the intestinal absorption of carbohydrates and glucose and results in their enhanced delivery to the colon. As a result, the ratio of saccharolytic to proteolytic bacterial flora is increased, and blood ammonia levels are decreased. A randomized, controlled, double-blind, crossover trial has demonstrated that acarbose improves mild HE in patients with cirrhosis and adult-onset diabetes mellitus.51 Similarly, probiotic regimens have been used to modify intestinal flora and diminish ammonia generation. Four small studies have suggested that these agents may be beneficial in humans with mild HE.40,52–55 These agents merit further evaluation and may be alternatives for patients who do not tolerate lactulose.
Strategies to enhance ammonia clearance may also be useful in the treatment of HE. Sodium benzoate, sodium phenylbutyrate, and sodium phenylacetate, all of which increase ammonia excretion in urine, are approved by the FDA for the treatment of hyperammonemia resulting from urea cycle enzyme defects and may improve HE in cirrhosis (see Chapter 76). Administration of sodium benzoate, however, results in a high sodium load, and the efficacy of this agent is not clearly established.4,56 The combination of intravenous sodium phenylacetate and sodium benzoate (Ammonul, Ucyclyd Pharma, Scottsdale, Ariz) in HE is being studied. Administration of zinc, which has been used because zinc deficiency is common in patients with cirrhosis57–59 and because zinc increases the activity of ornithine transcarbamylase, an enzyme in the urea cycle, may also improve HE; however, clear efficacy has not been established. Extracorporeal albumin dialysis using the molecular adsorbent recirculating system (MARS) has resulted in a reduction in blood ammonia levels and improvement in severe HE in patients with acute-on-chronic liver failure (see Chapter 93).60 Further studies are needed to clarify whether albumin dialysis has a role in treatment of HE. Finally, l-ornithine–l-aspartate (LOLA), a salt of the amino acids ornithine and aspartic acid that activates the urea cycle and enhances ammonia clearance, has been shown in several randomized controlled studies to improve HE compared with lactulose61–63; however, this agent is not available in the United States.
Flumazenil is a specific benzodiazepine (GABAA receptor) antagonist that has been used in patients with HE. It improves the degree of encephalopathy and electrophysiologic findings in approximately one fourth of patients with grade 3 or 4 HE. It has a short half-life and a number of potential side effects, including seizures, arrhythmias, and withdrawal symptoms, that limit its clinical usefulness.4
HEPATORENAL SYNDROME
The term hepatorenal syndrome (HRS) was first used in 1932 to describe acute kidney injury, mainly acute tubular necrosis (ATN) or interstitial nephritis, in a group of patients who had undergone biliary tract surgery.64 As pathophysiologic mechanisms were better elucidated, HRS was found to be part of a cascade of events associated with intense dilatation of the splanchnic arterial vasculature in the setting of cirrhosis or acute liver injury and resulting in profound renal arterial vasoconstriction and progressive renal failure.65 Histologically, the kidneys are normal in HRS. Function may be restored by correction of portal hypertension, liver transplantation, removal of the kidneys and transplantation of them into a noncirrhotic recipient, and, in some cases, medical therapy (see later).66–68
Acute renal dysfunction occurs in 15% to 25% of hospitalized patients with cirrhosis. Among the multiple causes of acute kidney injury, prerenal azotemia (resulting from intravascular volume depletion) is most common, accounting for 60% to 80% of cases. ATN is the second most common cause of acute kidney injury in this setting and accounts for 20% to 40% of cases.69,70 HRS appears to be an extension of the pathophysiology of prerenal azotemia and is therefore potentially reversible. The annual frequency of HRS in cirrhotic patients with ascites is roughly 8%71 and, in some reports, as high as 40%.72 HRS develops in approximately 30% of cirrhotic patients who are admitted with spontaneous bacterial peritonitis (SBP) or other infection, 25% who are hospitalized with severe alcoholic hepatitis, and 10% who require serial large-volume paracentesis.73 The observation that morbidity and mortality remain high once the syndrome is established has led to a focus on the prevention and early therapy of renal dysfunction in patients with cirrhosis.71
PATHOPHYSIOLOGY
The pathophysiology of HRS is complex and incompletely characterized. Three important components are recognized to contribute to the initiation and perpetuation of altered renal perfusion (Fig. 92-2): (1) arterial vasodilatation in the splanchnic and systemic circulation; (2) renal vasoconstriction; and (3) cardiac dysfunction.74 These components influence renal function in concert and form the basis for current therapies and preventive strategies.
Splanchnic Arterial Vasodilatation
Splanchnic and systemic arterial vasodilation are hallmarks of the progression of portal hypertension in cirrhosis and lead to decreased effective circulating blood volume and ultimately to a decrease in blood pressure. This process is mediated by a number of endogenous substances, including NO, carbon monoxide (CO), glucagon, prostacyclin, adrenomedullin, and endogenous opiates that are released or act locally in the vasculature in response to mechanical and inflammatory signals.65,71,75,76 In the early stages of portal hypertension, compensation for the decrease in effective circulatory volume includes an increase in heart rate and cardiac output, thereby creating a hyperdynamic circulation.77 As liver disease and splanchnic vasodilatation progress, additional compensatory mechanisms are activated.
Renal Arterial Vasoconstriction
Splanchnic and systemic vasodilatation also lead to compensatory renal vasoconstriction and renal sodium and water retention. These responses are mediated by stimulation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system (RAAS), and nonosmotic release and activity of arginine vasopressin (as a result of increased secretion and decreased clearance of arginine vasopressin and apparent increased expression of vasopressin-regulated water channels), as well as intrarenal events. Although the precise intrarenal mechanisms are speculative, altered production or action of endothelins, prostaglandins, kallikreins, and F2-isoprostanes may contribute to renal vasoconstriction.72,78,79 Ultimately, the balance between vasoconstrictive responses in the kidney and systemic and splanchnic vasodilatation is lost, thus leading to a prominent increase in renal vascular resistance, decrease in renal perfusion, and reduction in the glomerular filtration rate.71,77
Cardiac Dysfunction
Impaired cardiac function also may contribute to renal hypoperfusion in HRS. In one prospective study, HRS developed in cirrhotic patients with more severe arterial vasodilatation and lower cardiac output.80 In another study of a cohort of patients who were treated for SBP, renal dysfunction (including HRS in some cases) developed in those with lower cardiac output and lower arterial pressure measurements associated with higher circulating levels of norepinephrine and renin plasma activity, despite effective treatment of the infection.81 These data demonstrate that cardiac output is impaired in patients with cirrhosis in whom HRS develops as compared with those in whom HRS does not develop, and suggests that cardiac dysfunction may be an important additional factor in the pathogenesis of HRS. The relationship between cardiac dysfunction in patients with HRS and cirrhotic cardiomyopathy (see later) has not been studied.82,83
CLINICAL FEATURES AND DIAGNOSIS
HRS is a functional disorder, and therefore diagnostic laboratory and imaging studies are not available. The diagnosis of HRS requires a high index of clinical suspicion and exclusion of other potential causes of kidney injury. The diagnostic criteria for HRS as defined by the International Ascites Club Consensus Workshop in 2007 include the following: (1) cirrhosis with ascites; (2) serum creatinine level higher than 1.5 mg/dL (133 µmol/L); (3) lack of improvement in the serum creatinine level to 1.5 mg/dL (133 µmol/L) or less after at least two days of diuretic withdrawal and volume expansion with albumin (1 g/kg of body weight/day, up to a maximum of 100 g/day); (4) absence of shock, (5) lack of current or recent treatment with nephrotoxic drugs; and (6) absence of parenchymal kidney disease as indicated by proteinuria of more than 500 mg/day, microhematuria (>50 red blood cells/high power field), or abnormal renal ultrasonographic findings (Table 92-3).74
Table 92-3 Diagnostic Criteria for Hepatorenal Syndrome*
* As defined by the International Ascites Club Consensus Workshop in 2007 (Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310-8).
Several aspects of the diagnosis of HRS deserve emphasis. First, in patients with no prior evidence or history of renal impairment, the diagnostic criteria for HRS include an increase in the serum creatinine level by 50% above baseline to a level higher than 1.5 mg/dL (133 µmol/L).71 Although this definition is standardized, a subset of patients with cirrhosis and end-stage liver disease have a profound decrease in muscle mass and urea synthesis that may in turn result in reduced serum creatinine and blood urea nitrogen levels, thereby potentially delaying recognition of HRS.73,84 Second, many medications, most notably diuretics, lactulose, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and nonsteroidal anti-inflammatory drugs, may influence intravascular volume status and renal perfusion and should be identified expeditiously and discontinued in the setting of acute renal dysfunction. Third, even though SBP may not be accompanied by obvious symptoms and signs, HRS may develop in as many as 20% of affected patients.85 Therefore, a low threshold for evaluating cirrhotic patients with ascites for the presence of SBP is required.
CLASSIFICATION
HRS has traditionally been classified into two types on the basis of clinical characteristics and prognosis (types 1 and 2).74 Some authors have advocated expanding the classification to include patients who are not encompassed within the current framework (i.e., type 3 HRS).72
Type 1 HRS presents as a rapidly progressive form of renal dysfunction. Typically, the serum creatinine level doubles to a value higher than 2.5 mg/dL in a period of two weeks or less. Type 1 HRS is often triggered by an inciting event that causes a rapid decline in the hemodynamic parameters that maintain renal homeostasis in cirrhotic patients.70 The most common triggers include severe bacterial infections,86–88 gastrointestinal bleeding, surgical procedures, and acute liver injury.74,89,90 SBP is the main bacterial infection that predisposes cirrhotic patients to develop HRS. Other bacterial infections have less of an impact on the development of the syndrome, unless sepsis is present or the response to antibiotic therapy is poor.71,87,91 Patients who exhibit high levels of inflammatory response markers and patients with severe circulatory depression prior to the onset of infection are most susceptible to the development of HRS. Some degree of adrenal insufficiency has been found in 80% of patients with septic shock in whom HRS develops, and administration of glucocorticoids may improve survival.92
Type 2 HRS is a more slowly progressing entity compared with type 1 HRS but still carries a median survival of only approximately six months. Type 2 HRS is observed in patients with severe ascites (diuretic resistant) and is characterized by serum creatinine levels lower than 2.5 mg/dL. The degree of arterial hypotension and circulatory dysfunction is less than that seen with type 1 HRS. Type 1 HRS may develop in patients with type 2 HRS following a triggering event.71,72
Many patients with cirrhosis and portal hypertension also have underlying chronic kidney disease, which complicates recognition of HRS, even in the presence of underlying pathophysiologic mechanisms that favor the development of HRS. Whether these patients should be considered to have a unique form of HRS (type 3) and whether they should be treated in a fashion similar to that for other patients with HRS have not been clearly defined.72
PREVENTION AND TREATMENT
The high mortality rate of HRS underscores the importance of prevention. In particular, intravascular volume depletion (resulting from overdiuresis, diarrhea caused by lactulose, gastrointestinal bleeding from gastroesophageal varices, or large-volume paracentesis without colloid administration), nephrotoxins (e.g., nonsteroidal anti-inflammatory drugs, nephrotoxic antibiotics), and infection (SBP, bacteremia) should be avoided or addressed if present. Specific guidelines address the primary and secondary prophylaxis of variceal bleeding (see Chapter 90), administration of colloid (albumin) to patients with a rising serum creatinine level after a large-volume paracentesis and in the presence of SBP (see Chapter 91), and prophylactic administration of antibiotics to patients at high risk of SBP or other infections because of hospitalization for gastrointestinal bleeding (see Chapters 19 and 90).85,86,89,91 Routine invasive hemodynamic monitoring of cirrhotic patients with a rising serum creatinine level does not have a clear benefit and is not recommended.
The concept that treatment of HRS is possible and may improve survival has emerged since 2000. Current options include medical therapies, transjugular intrahepatic portosystemic shunt (TIPS) placement, and liver transplantation. Medical therapies for HRS are directed toward reversing the underlying splanchnic and systemic vasodilatation with vasoconstrictors and increasing effective circulatory volume with the use of colloid. Such treatment is used increasingly as a temporizing measure until definitive treatment for liver disease (liver transplantation) or portal hypertension (TIPS) is undertaken, or until an acute process (SBP, gastrointestinal bleeding) has been reversed (Table 92-4).93,94
Table 92-4 Management of Hepatorenal Syndrome
ACEIs, angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; IV, intravenous; MAP, mean arterial pressure; NSAIDs, nonsteroidal anti-inflammatory drugs; SBP, spontaneous bacterial peritonitis.
* Not available in the United States.
Data from references 69,74,85,86,89,91,93–94,97–124.
Medical Therapy
Terlipressin is an intravenously administered selective vasopressin 1 receptor agonist vasoconstrictor that has been used in Europe and is under review by the FDA for the treatment of type 1 HRS.95,96 It has been evaluated in approximately 330 patients included in four randomized, controlled trials and two meta-analyses.69,97–109 In two multicenter studies of patients with type 1 HRS, terlipressin (given in two different doses in the two studies) in combination with albumin improved serum creatinine levels relative to albumin alone (30% to 43% vs. 8% to 13%), although survival was not significantly different in the two groups.106,107 In addition, in one study,106 terlipressin was associated with a significantly increased rate of cardiovascular complications compared with albumin alone; therefore, this agent should only be used with close monitoring. Finally, the response to terlipressin in patients with type 1 HRS appeared to be better in patients with less severe renal dysfunction at baseline, thus supporting the early initiation of therapy. These studies indicate that administration of terlipressin in combination with albumin can improve renal function in HRS, although the optimal dose and duration of therapy are not defined.
Midodrine, an orally administered α1-adrenergic agonist, and octreotide, a somatostatin analog that inhibits endogenous vasodilators,110,111 have been used in combination with albumin for type 1 HRS in three small nonrandomized studies. In two studies, treatment with midodrine, titrated to cause a rise in mean arterial blood pressure, was associated with improved serum creatinine levels and improved survival compared with no treatment and was associated with few major side effects.112–114 This regimen has the advantage of ease of administration and appears to have a favorable safety profile; however, its efficacy has not been established in randomized controlled trials.
Norepinephrine, a widely available intravenously administered α1-adrenergic agonist, in combination with albumin, has been proposed as an alternative to terlipressin on the basis of two small pilot studies.115,116 In one study of 22 patients with type 1 or 2 HRS, norepinephrine appeared to be as effective and safe as terlipressin. Significant cardiovascular side effects, however, have been reported with the use of norepinephrine to treat HRS, and whether the efficacy and safety of norepinephrine are similar to those of terlipressin have not been fully defined.
Radiologic and Surgical Therapy
Transjugular Intrahepatic Portosystemic Shunt
Insertion of a transjugular intrahepatic portosystemic shunt (TIPS) creates a portosystemic shunt and lowers portal venous pressure, thereby decreasing venous pooling in the splanchnic circulation and increasing venous return. TIPS is effective for the treatment of diuretic-resistant ascites, a precursor to type 2 HRS.117 Four pilot studies have evaluated the use of TIPS in nontransplant candidates with type 1 or 2 HRS.118–121 In these studies, serum creatinine levels declined, sodium excretion increased, and neurohumoral responses improved after TIPS, although survival was not affected. The major benefit was seen in patients with type 2 HRS. In one study, the use of midodrine, octreotide, and albumin followed by TIPS appeared to be effective in a small cohort of patients with type 1 HRS. An important limitation of the use of TIPS in HRS is the potential to worsen hepatic function. Therefore, important questions regarding the safety and benefit of TIPS in HRS remain.
Liver Transplantation
Liver transplantation is the only therapeutic modality that has the potential to reverse both liver dysfunction and HRS and should be considered in any patient found to have HRS.65,66,71,122 Rates of postoperative complications and in-hospital mortality are higher in patients transplanted with HRS than in those without HRS,75,122 and up to 35% of patients with HRS require long-term renal replacement therapy.66 Still, the three-year survival rate of patients transplanted with HRS is approximately 60% compared with 70% to 80% for patients transplanted without HRS. The duration and degree of renal dysfunction preoperatively may be independent predictors of survival, and patients who require hemodialysis carry a mortality risk that is 1.77 times higher than that of patients who do not need dialysis.122–124 In one study, patients with HRS who responded to treatment with a vasopressin analog prior to liver transplantation had outcomes similar to those of patients who underwent liver transplantation without HRS,94 a finding that supports the use of such therapy as a bridge to liver transplantation. Larger trials are needed to confirm this observation.
Other Therapies
Extracorporeal albumin dialysis with MARS is an experimental therapeutic modality that enhances the removal of water-soluble and albumin-binding toxins from the circulation.125 The results of one small randomized trial have supported the use of MARS to improve serum creatinine levels and survival rates in patients with HRS, although larger studies are needed to confirm these findings.126,127 Several other vasoconstrictive medical therapies, including dopamine and octreotide in combination with albumin, have not improved the outcome of HRS, and in one study use of a nonselective endothelin receptor antagonist to inhibit intrarenal vasoconstriction in HRS proved deleterious.95
HEPATOPULMONARY SYNDROME AND PORTOPULMONARY HYPERTENSION
Cirrhosis and portal hypertension are accompanied by alterations in the vascular beds of multiple organ systems. In the pulmonary circulation, two distinct clinical entities, termed the hepatopulmonary syndrome (HPS) and portopulmonary hypertension (PPH), have been recognized. HPS occurs when pulmonary microvascular alterations impair gas exchange and is found in up to 30% of patients evaluated for liver transplantation.128–130 PPH occurs when vasoconstriction and remodeling in resistance vessels increase pulmonary arterial pressures and is found in as many as 5% of patients with cirrhosis. The mechanisms whereby these two entities develop are incompletely characterized, although they occur in similar clinical settings and may share pathogenic pathways. The presence of HPS or PPH increases mortality in affected patients. No effective medical therapies are available for HPS, although liver transplantation reverses the syndrome in most patients. Medical therapies that improve pulmonary hemodynamics in patients with PPH have become available, but the specific role of liver transplantation in PPH is not clearly defined.131–138
PATHOPHYSIOLOGY
Hepatopulmonary Syndrome
HPS is characterized by microvascular alterations and dilatation in the precapillary and capillary pulmonary arterial circulation. In human HPS, the production of vasodilatory substances within the pulmonary vasculature, most notably NO, is increased. Although increased circulating and pulmonary NO levels appear to be features of human HPS, improvement in oxygenation in response to acute inhibition of NO is variable,139–142 and HPS may take more than one year to resolve after liver transplantation in advanced cases.135 These findings suggest that other vasoactive mediators or angiogenesis in the pulmonary microvasculature may contribute to vascular alterations and hypoxemia in human HPS.
In experimental HPS induced by bile duct ligation in the rat, pulmonary NO overproduction has also been observed and is triggered by a series of pathophysiologic events. During the onset of pulmonary vascular alterations, increased biliary production and release of endothelin-1143 in conjunction with shear stress induces pulmonary microvascular endothelin-B receptor overexpression, which leads in turn to endothelin-1–mediated endothelial NO synthase (eNOS)-derived NO production.144,145 As HPS progresses, tumor necrosis factor-α (TNF-α) levels rise as a result of bacterial translocation, which leads to adherence of macrophages in the pulmonary microvasculature and inducible NO synthase (iNOS)–derived NO production and heme oxygenase-1–derived carbon monoxide production.146–150 Endothelin receptor antagonists and inhibition of NOS, bacterial translocation, TNF-α, and heme oxygenase all improve experimental HPS.148–151
Studies have found that experimental HPS is accompanied by enhanced pulmonary vascular endothelial growth factor (VEGF) expression and angiogenesis.151 Both antiangiogenic peptides and pentoxifylline, a phosphodiesterase inhibitor with TNF-α antagonist properties, have been shown to inhibit angiogenesis and decrease the severity of HPS. Two small studies of pentoxifylline in human HPS have reported conflicting results, and the role of angiogenesis and angiogenesis inhibition in human HPS has yet to be defined (Fig. 92-3).152–155
Portopulmonary Hypertension
The mechanisms whereby PPH develops are poorly understood. Histologically, PPH shares the characteristic features of other forms of pulmonary arterial hypertension (PAH): medial proliferation and hypertrophy, plexiform arteriopathy, and in situ vascular thrombosis.156–158 The precise role of portal hypertension in this process is not clear, however, and whether PPH shares pathophysiologic mechanisms with PAH is unknown. Studies have found PPH, like PAH, to be more common in women than men.159
[/not-level-membership-for-gastroenterology-and-hepatology-category]
