101 Hepatic Encephalopathy
Hepatic encephalopathy encompasses a spectrum of neuropsychiatric abnormalities that occur in patients with liver disease in the absence of other brain disease.1–2 The spectrum includes personality changes, impaired mental function, motor abnormalities (asterixis, tremor, hyperventilation, hyperactive reflexes), and altered consciousness. A consensus panel of experts proposed classification of hepatic encephalopathy into type A, associated with acute liver failure; type B, associated with portal-systemic bypass without intrinsic liver disease; and type C, associated with chronic liver disease.3
The encephalopathy accompanying acute hepatic failure (type A) is commonly associated with cerebral edema and increased intracranial pressure (ICP), exhibits abrupt onset with a short prodrome and rapid progression, and often ends with the death of the patient.4–6 Patients sequentially experience drowsiness, delirium, agitation or convulsions, decerebrate rigidity, unresponsiveness, and deep coma within a comparatively short period of time, usually hours to days. Irreversible neurologic damage may occur as a result of brain ischemia or herniation. Patients who develop coma in the setting of acute liver failure have a grave prognosis; fewer than 20% survive without hepatic transplantation.7
In patients with chronic liver disease, encephalopathy (type C) develops insidiously and often is heralded by a change in mental or behavioral status. Encephalopathy may be episodic, persistent, or minimal and subclinical.3 Episodes are sporadic, characterized by exacerbations and remissions, and generally are precipitated by inciting events.1–2 Although the initial manifestation of portosystemic encephalopathy (PSE) is usually a subtle change in mentation, neurologic dysfunction may progress and be classified according to confusion, lethargy, and even coma (Table 101-1).8 Neurologic signs vary and fluctuate but usually include asterixis, hyperreflexia, clonus, and an extensor plantar response. Causes of PSE may not always be apparent, but azotemia, sepsis, gastrointestinal (GI) bleeding, dehydration, electrolyte imbalances, and sedatives are frequent precipitants (Box 101-1). In some patients, chronic encephalopathy may not be clinically obvious, but only detectable by psychometric testing. By these tests, about two-thirds of cirrhotic patients with portal hypertension have unsuspected subclinical hepatic encephalopathy.9–12 Patients who undergo portal-systemic shunt or bypass, either surgical or transjugular intrahepatic portosystemic shunt (TIPS), often develop encephalopathy (type B) which is similar to the encephalopathy experienced by patients with chronic liver disease.
TABLE 101-1 Stages of Encephalopathy in Chronic Liver Disease
| Stage | Clinical Signs |
|---|---|
| Stage I | Mental slowness, euphoria or anxiety, shortened attention span, impaired calculating ability |
| Stage II | Lethargy or apathy, inappropriate behavior, personality change, more obvious problems with calculations |
| Stage III | Lethargic, somnolent, marked confusion and disorientation, but responds to verbal stimuli |
| Stage IV | Coma, patient may or may not respond to noxious stimuli |
Patients with chronic liver disease rarely, if ever, demonstrate cerebral edema, regardless of the stage of encephalopathy.
 General Principals
General Principals
No single abnormality of hepatic or neurologic metabolism adequately explains all of the clinical, biochemical, physiologic, or experimental findings of encephalopathy occurring in patients or animal models.1–26 Abnormalities of multiple neurotransmitters including glutamate, γ-aminobutyric acid (GABA), dopamine, serotonin, and opioids have been described, and plasma levels of a wide array of potential neurotoxins (ammonia [NH3], GABA, short-chain fatty acids, methanethiols) are increased (Box 101-2).13–14 Despite this seeming confusion, several lines of investigation focus on NH3 as a key factor in the pathogenesis of hepatic encephalopathy. Ammonia accumulation deranges glutamate and glutamine metabolism in the central nervous system (CNS) and alters the metabolism of GABA and its function as an inhibitory neurotransmitter. In addition, benzodiazepine receptors in the CNS are physically linked to GABA receptors. The latter finding provides an explanation for the increased sensitivity of patients with liver disease to the sedative and hypnotic effects of benzodiazepines and a rationale for use of benzodiazepine antagonists in the treatment of PSE. Hepatic encephalopathy occurring in the setting of either acute liver failure or chronic liver disease is also associated with marked changes in CNS glial cells on neuropathologic examination. Encephalopathy of acute liver failure is characterized by astrocytic swelling, but chronic encephalopathy is characterized by Alzheimer type II astrocytosis.15
Cerebral Blood Flow
In acute liver failure, the brain is potentially subject to hypoxic injury due to complications such as systemic arterial hypotension, respiratory failure, and reduction in cerebral blood flow that accompanies cerebral edema and intracranial hypertension. Therapy is often directed at optimal oxygenation, maintenance of cerebral perfusion pressure (goal > 40 mm Hg), and reduction of ICP (goal < 20 mm Hg).4,9 Paradoxically, increases in cerebral blood flow, however, may aggravate cerebral edema and worsen neurologic damage. In humans with acute liver failure, cerebral blood flow has been measured primarily by the xenon-133 washout technique.16–20 These data suggest that cerebral blood flow is initially relatively low but then increases with increasing blood concentration of NH3, which decreases cerebral arteriolar tone.
Cerebral Glucose and Oxygen Metabolism
Brain energy metabolism is unique in that glucose is the only substrate under normal physiologic conditions, and its uptake and utilization by the brain is independent of insulin.21–25 Under stress, the brain can utilize β-hydroxybutyrate and acetoacetate. Ammonia accumulation during hepatic failure in humans or in experimental models of hyperammonemia is associated with altered cerebral glucose metabolism. In early acute liver failure, prior to onset of intracranial hypertension, cerebral glucose metabolism and cerebral oxygen consumption are proportionately diminished.21 There is no evidence of cerebral hypoxia, implying that the reduced glucose and oxygen utilization reflect diminished metabolic demand by the brain at this early stage. Cerebral lactate uptake is increased despite sufficient glucose delivery and preserved oxidative metabolism. Acute short-term mechanical ventilation, resulting in moderate reduction in PCO2 and cerebral blood flow, does not adversely affect oxidative brain metabolism. Thus, prior to the development of intracranial hypertension, cerebral glucose and oxygen metabolism are reduced, but these changes are consistent with normal aerobic metabolism and physiologic regulation. After development of intracranial hypertension, oxygen metabolism remains reduced, but measurements of cerebral glucose utilization vary from reduced rates to increased rates, and glycolysis may be accelerated.22–2325 These findings suggest that progression of acute liver failure and development of intracranial hypertension are associated with relative cerebral hypoxia and a switch to anaerobic metabolism.
Ammonia Hypothesis
The ammonia hypothesis states that the major mechanism of hepatic encephalopathy is excessive accumulation of NH3, which induces neuronal metabolic derangements but also promotes astrocytic swelling.26 In addition, NH3 perturbs cerebral nitric oxide metabolism, which can mediate some of the effects.27 Studies using positron-emission tomography (PET) and magnetic resonance spectroscopy have demonstrated an increase in cerebral metabolic rate and increased permeability of the blood-brain barrier for NH3.28–34 Blood NH3 originates mainly from four sources: intrahepatic deamination of amino acids, extrahepatic metabolism of nucleotides, gut metabolism of glutamine, and bacterial degradation of intestinal protein and urea.35 More than 50% of blood NH3 is derived from the latter source. NH3 is normally metabolized by the liver to either urea or glutamine by the actions of carbamoyl-phosphate synthetase I (the initiating enzyme of the urea cycle) and glutamine synthetase, respectively. Patients with hepatic failure have impaired NH3 metabolism related to a reduction in liver metabolism and an increase in portal-systemic shunting. As a result, an elevation in blood NH3 concentration is a characteristic feature of severely impaired hepatic function.
Certain clinical and experimental observations link the increase in blood NH3 concentration to hepatic encephalopathy.13,15,36–37 Hyperammonemia and elevated concentrations of NH3 within the cerebrospinal fluid (CSF) are features of acute and chronic hepatic encephalopathy, Reye syndrome, deficiencies of urea cycle enzymes, and sodium valproate toxicity. In cirrhotics or patients with portocaval shunts, ingestion of NH3-generating substances (proteins, amino acids, urea, ammonium salts) may precipitate encephalopathy. In animal models, chronic administration of ammonium salts results in Alzheimer type II astrocytosis, a change indistinguishable from that observed in patients with chronic hepatic encephalopathy.15
Glutamine-Glutamatergic Neurotransmitter System
The glutamatergic excitatory neurotransmitter system in the CNS is markedly altered in patients with both acute and chronic liver disease and in animal models of hepatic encephalopathy.6,26–27 CNS astrocytes are a major regulatory cell in the glutamatergic system.15 Normally the astrocyte avidly takes up excess glutamate from the synaptic cleft (against a 3000-10,000 fold concentration gradient), an important function that terminates glutamate-induced neuroexcitation. Once glutamate is taken up by the astrocyte, it is metabolized to glutamine via the action of glutamine synthetase, which utilizes blood-derived NH3 (Figure 101-1). The hyperammonemia of liver failure favors the formation of glutamine but also impairs the release of glutamine from the astrocyte. The accumulation of osmotically active glutamine in the astrocyte is associated with cell swelling. Normally, glutamine is actively extruded from the astrocyte and then taken up by presynaptic nerve terminals for conversion back to glutamate and subsequent utilization in neurotransmission. Under the conditions of liver failure and hyperammonemia, glutamate uptake into neurons and astrocytes is diminished, and glutamate accumulates in the extracellular fluid. Clinically, levels of glutamine and glutamate increase in CSF fluid during hyperammonemic states, and CSF concentrations of glutamine correlate loosely with the stage of encephalopathy. In animal models of acute encephalopathy, blockade of glutamine production by an inhibitor of glutamine synthetase, methionine sulfoximine, decreases cerebral edema and reduces astrocyte swelling.14 Production of NH3 from the intestine is reduced by orally administered nonabsorbable antibiotics such as rifaximin and neomycin, as well as nonabsorbable disaccharides including lactulose, lactitol, and lactose (in lactase-deficient patients). These treatments lower plasma NH3 concentration and improve subjective and objective measures of encephalopathy.
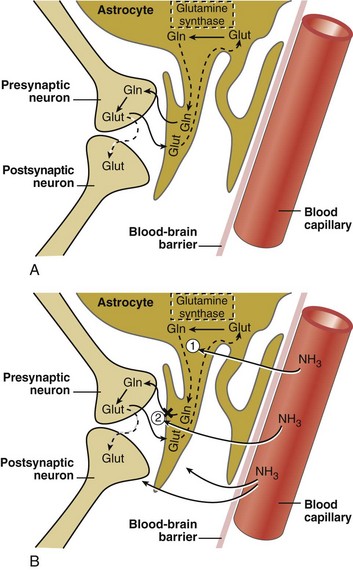
Other clinical and experimental observations refute the link between NH3 and hepatic encephalopathy. Blood levels of NH3 are elevated in cirrhotic patients regardless of the presence or absence of encephalopathy. Some patients with hepatic encephalopathy have normal blood levels of NH3. The grade of hepatic encephalopathy does not correlate with the blood concentration of NH3. Seizures and hyperexcitability are commonly observed in animal models of NH3 intoxication and in human congenital hyperammonemia but are rarely observed in patients with chronic hepatic encephalopathy. Administration of ammonium chloride to cirrhotic patients induces a mild hyperkinesis but fails to exacerbate typical chronic encephalopathy.13
γ-Aminobutyric Acid-Benzodiazepine Receptor Hypothesis
GABA is an inhibitory neurotransmitter found throughout the CNS.38 The GABA hypothesis states that an excess of GABA or increased sensitivity to GABA is responsible for hepatic encephalopathy.14,38–39 Observations in rabbits with galactosamine-induced hepatic failure provided the initial support for this hypothesis. GABA originates from the intestine, and plasma levels increase in hepatic failure due to inadequate hepatic extraction. During acute liver failure, the blood-brain barrier becomes more permeable, and increased amounts of GABA enter the CNS. Once in the brain, GABA binds to its receptor to produce neuroinhibition and clinical encephalopathy. A key component to understanding the relationship of GABA and benzodiazepines was the recognition that the GABA receptor was tightly linked and modulated by the benzodiazepine receptor.40–43 Binding of benzodiazepines to the benzodiazepine receptor induces a conformational change in the GABA receptor enhancing the binding of GABA and neuroinhibition. Activation of the GABA receptor opens a chloride channel, the third component of the GABA receptor complex (Figure 101-2). In summary, GABA-induced inhibition of neurotransmission is enhanced by binding of benzodiazepines or related compounds to the benzodiazepine receptor, increasing the number of GABA receptors, increasing the activity of the GABA receptor, or opening the GABA-associated chloride channel.
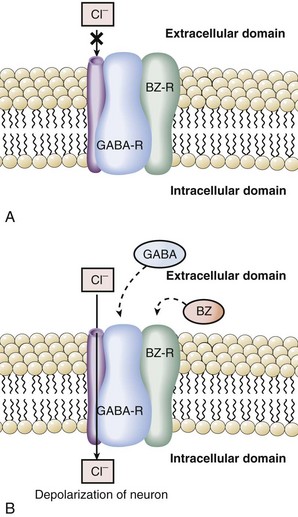
The GABA hypothesis predicts that benzodiazepines would increase the severity of hepatic encephalopathy, and benzodiazepine antagonists such as flumazenil might ameliorate hepatic encephalopathy. Clinical experience clearly suggests that cirrhotic patients, especially those with encephalopathy, are particularly sensitive to the amnesic and sedative effects of benzodiazepines. In our experience, use of benzodiazepines and other sedative/hypnotics is a common reason for exacerbations of hepatic encephalopathy. Recent studies have demonstrated that patients with hepatic encephalopathy have increased plasma levels of benzodiazepines or “natural” benzodiazepine-like compounds that then may act as “false neurotransmitters.”44–47 Some have suggested that patients with hepatic encephalopathy are particularly sensitive to GABA neuroinhibition, owing to an increase in background benzodiazepine stimulation of the GABA receptor.
Taurine
Taurine is an inhibitory neurotransmitter which is increased in brains of animal models of experimental hepatic encephalopathy and in the CSF of primates with encephalopathy secondary to portocaval shunts. Plasma levels of taurine are greatest in patients with the greatest degrees of encephalopathy, suggesting that this inhibitory neurotransmitter may be involved in hepatic encephalopathy.48 Additional neurotransmitters that may be altered in hepatic encephalopathy include endogenous opioids and melatonin.
Methanethiols
Interest in methanethiol as a potential neurotoxin began with the finding of methanethiol in the urine of a patient with fetor hepaticus. Subsequently, levels of methanethiol, 4-methylthio-2-oxobutyrate, and methanethiol-mixed disulfides were found to be elevated in the plasma of cirrhotic patients.49 It was suggested that these compounds may exacerbate the toxic effects of NH3 and short-chain fatty acids. However, blood levels of methanethiols are similar in deeply comatose patients and those with only mild cerebral dysfunction, and there is little correlation between grade of encephalopathy blood levels of methanethiol.
Manganese, Zinc
The liver is responsible for manganese excretion, and liver disease is associated with manganese accumulation. Magnetic resonance imaging (MRI) studies of the brain in patients with cirrhosis reveal pallidal hyperintensity on T1-weighted images (Figure 101-3), which correlates with the presence of extrapyramidal signs and symptoms and blood levels of manganese.50 Manganese concentrations in the globus pallidus are markedly increased in autopsy studies of cirrhotics who died in hepatic coma. PET imaging reveals reduced cerebral glucose utilization in these areas. Such findings suggest a relationship between manganese, brain hypometabolism, and some of the neuropsychiatric and motor abnormalities of hepatic encephalopathy.
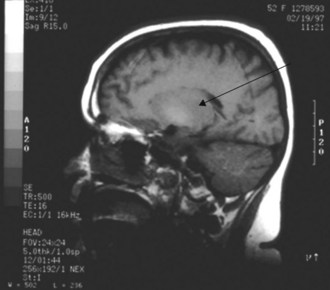
Zinc deficiency is common in long-standing cirrhosis. Its importance to the pathogenesis of hepatic encephalopathy is unknown, and three randomized controlled trials of zinc supplementation have yielded conflicting results as regards improvement in encephalopathy (one positive, two negative).51–53
 Encephalopathy in the Setting of Acute Hepatic Failure
Encephalopathy in the Setting of Acute Hepatic Failure
Definition
Acute liver failure is defined by the development of coagulopathy (prothrombin time [PT] >20 sec prolonged with an international normalized ratio [INR] >1.5) in a patient with acute hepatitis who lacks underlying chronic liver disease (exception in Wilson’s disease).54–56 Patients with acute liver failure usually have extreme elevations of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) with the initial injury (1000 to 5000 IU/L), often are jaundiced, and exhibit constitutional symptoms. They are at risk for encephalopathy, although most recover uneventfully. Progressive hepatic encephalopathy is a poor prognostic sign and signals the need for hepatic transplantation.
Etiology
There are approximately 2000 cases of acute liver failure in the United States each year.57,58 Among the most common causes are acetaminophen toxicity, other types of acute drug toxicity, and hepatitis A and B virus (HAV, HBV) infection. However, the second leading diagnostic category for fulminant hepatic failure (FHF) is cryptogenic, cause unknown (Table 101-2). Recent data obtained since 1998 indicate that over 50% of cases of FHF in the United States are due to acetaminophen overdosage (38%) or idiosyncratic drug reactions (~14%).58 Many cases of acetaminophen-induced liver failure are due to “therapeutic misadventure” due to self-administration of as little as 4 grams/day over several days in the setting of fasting and alcohol use. Sporadic cases of FHF due to both cocaine and Ecstasy have recently been described. FHF from mushroom poisoning occasionally occurs with inexperienced amateur mushroom fanciers. Infiltration of the liver with rapid progression of tumor growth can lead to FHF and has been described for cases of metastatic breast carcinoma, lymphoma, and melanoma. Biopsy of the liver is required to establish the latter diagnoses. FHF also may occur in the third trimester of pregnancy, related to acute fatty liver of pregnancy, HELLP syndrome, or disseminated herpes infection.
| Acetaminophen | 20% |
| Cryptogenic | 15% |
| Non-acetaminophen drug toxicity | 12% |
| Hepatitis B | 10% |
| Hepatitis A | 7% |
| Autoimmune hepatitis | 6% |
| Wilson’s disease | 6% |
| Miscellaneous* | 24% |
* Budd-Chiari syndrome, herpes simplex, paramyxovirus, Epstein-Barr virus, amanita poisoning, ischemia, malignant infiltration.
Adapted from Schiodt FV, Atillasoy E, Shakil O et al. Etiologic factors and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999;5:29-34.
Prognosis
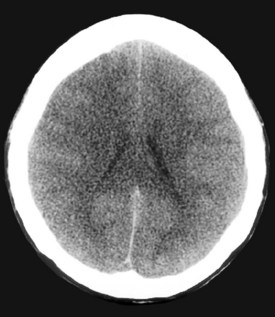
A major determinant of prognosis is the level of encephalopathy (see Table 101-1). Patients with acute liver failure who have progressed to higher stages of encephalopathy (stage III or IV) have the worst prognosis. Glasgow Coma Scale is useful in assessing need for transplantation.59 Cerebral edema on computed tomography (CT) scan of the brain is a late feature of progressive encephalopathy (Figure 101-4). Additional clinical features that indicate a poor prognosis include metabolic acidosis, renal failure, severe jaundice, or markedly prolonged prothrombin time.56,59–60 The likelihood of survival varies with the cause of acute liver injury. Patients with acetaminophen overdose have a relatively favorable outcome, and over 50% survive.61 Patients with fulminant HAV and HBV infection have an intermediate prognosis, and 30% to 50%62 survive. In contrast, patients with a fulminant presentation of Wilson’s disease or severe sporadic non-A, non-B, non-C hepatitis have a survival of less than 10%.63
General Clinical Management
All patients should be placed on needle (HBV, NANBV) and stool (HAV) precautions but not in isolation. Gloves should be worn when handling biological specimens, and specimens should be clearly labeled (Hepatitis Patient). All used instruments should be autoclaved or appropriately disposed. FHF due to acute viral hepatitis is not reversible by antiviral therapy, although some advocate use of lamivudine or related drugs for severe acute HBV. Corticosteroids are contraindicated, as they may increase the risk of developing chronic hepatitis and are ineffective in treatment of encephalopathy or cerebral edema in this setting. Removal of the offending drug, toxin, or alcohol is the mainstay of therapy of drug-induced and alcoholic hepatitis, respectively. N-acetylcysteine is an effective primary intervention for hepatic injury related to acetaminophen64 and is currently under investigation in the treatment of FHF due to other etiologies (Box 101-3).
Box 101-3
Use of N-Acetylcysteine in Treatment of Acetaminophen Overdose
Oral Dosing Schedule
Intravenous Dosing Schedule (Limited Availability in Research Centers)
Infection is a leading comorbidity in patients with acute liver failure.56–60 Blood, urine, and sputum should be cultured frequently (even in absence of fever or other signs of infection) and antibiotic therapy instituted only for positive cultures and directed against specific organisms. Fever is not a feature of most forms of acute liver injury and if present usually signifies concurrent infection. Febrile patients should be cultured immediately and treated empirically with antibiotics. The most common sources of infection are the respiratory tract, the urinary tract, and line sepsis. Currently, we use vancomycin with a broad-spectrum cephalosporin such as cefotaxime or a fluoroquinolone such as levofloxacin as initial treatment, and then tailor antibiotic use once results of cultures are known.
 Use of Intracranial Pressure Monitoring
Use of Intracranial Pressure Monitoring
Advantages
The use of ICP monitoring for managing high-grade encephalopathy in acute liver failure remains controversial. Several studies advocate ICP monitoring for its ability to provide important prognostic information about neurologic recovery after hepatic transplantation and in managing intracranial hypertension (ICH) while awaiting liver transplantation.65–67 Therefore, ICP monitoring could be considered for patients with high-grade encephalopathy (stage III or IV) who are waiting on the transplant list. Also, centers could consider ICP monitoring in nontransplant candidates if there is a reasonable likelihood of spontaneous recovery, such as in acetaminophen-induced acute liver failure. In addition to helping with the management of ICH, ICP monitoring also assists with close monitoring during the crucial perioperative period of liver transplantation, since it has been found that there is a transient increase in ICP that can last for about 12 hours postoperatively, after the dissection of native liver and the reperfusion of the graft.68
Disadvantages
However, the use of ICP monitoring may lead to severe complications such as intracranial hemorrhage in these already critically ill patients. In addition, several nonrandomized studies have failed to demonstrate an improvement in survival with the use of ICP monitoring.65–67 One prospective study of 92 out of 332 patients with acute liver failure, high-grade encephalopathy, and ICP monitoring found a 10.3% rate of intracranial hemorrhage.67 However, nearly half of the cases of intracranial bleeding were incidental radiographic findings without clinical consequence. Regardless of ICP use, 30-day survival post transplant was approximately 85%. Other reports confirm that bleeding complications from the placement of ICP monitoring devices in patients with acute liver failure are mostly mild and without clinical significance.65–6769 Use of ICP monitoring remains controversial, but experts agree that these devices should not be used in patients with mild hepatic encephalopathy (grades I or II) or in patients with evidence of cerebral herniation, hypotension, or imminent death.
 Management of Encephalopathy and Intracranial Hypertension
Management of Encephalopathy and Intracranial Hypertension
Encephalopathy is a hallmark of acute liver failure. It is also observed in patients with underlying chronic liver disease who sustain superimposed acute liver injury. The encephalopathy of acute hepatic failure is related to both metabolic factors, such as progressive elevation in blood NH3 concentration, and cerebral edema. Progressively worsening encephalopathy is an ominous clinical feature; development of grade III or IV encephalopathy may herald the death of the patient due to cerebral edema, increased ICP, and central herniation of the brain. Efforts to control the encephalopathy of acute liver failure are directed at preventing or resolving cerebral edema (Box 101-4).16,67,70–73 Because emerging evidence suggests that NH3 may play a role in the development of cerebral edema, we recommend that administration of protein should be limited to less than 40 grams/d in adults, and lactulose (20-40 g/d in divided doses) should be administered enterally to purge the bowel. However, one must exercise caution when using lactulose in the setting of FHF; dosing should be monitored carefully and adjusted to avoid excessive diarrhea and alterations in electrolytes and volume depletion. If oral (PO) lactulose is given simultaneously with intravenous (IV) mannitol, marked deficits of free water can develop, inducing severe hypernatremia. Although one recent study suggested that infusion of hypertonic (3%) saline to maintain serum sodium concentration between 145 and 155 mEq/L is beneficial,74 rapid shifts in sodium concentration have been associated with central pontine myelinolysis. Further discussion of hypertonic saline is provided later in this chapter. Administration of terlipressin or vasopressin may worsen intracranial hypertension and should be avoided.75
Box 101-4
Measures Used to Monitor and Control Cerebral Edema Due to Fulminant Hepatic Failure
Second-Line Therapies For Treatment Of Mannitol-Refractory Intracranial Hypertension
Hypothermia in ALF
Therapeutic hypothermia or intentional reduction of body core temperature has been increasingly used to treat hypoxic brain injury after cardiac arrest as well as in the traumatic setting. Animal models of acute liver failure suggest that hypothermia may be effective in the prevention of cerebral edema.76–78 Several case reports suggest that hypothermia can be used as an effective form of supportive therapy while awaiting liver transplantation.
A series of cases reported by Jalan et al. demonstrated that use of hypothermia in 38 patients with acute liver failure and uncontrolled ICH reduced cerebral edema and ICP.24,79–83 In these studies, patients had high-grade hepatic encephalopathy (grades III or IV) and ICP above 25 mm Hg despite two doses of mannitol. Body temperature reduction to 32°C to 33°C for 8 to 14 hours decreased mean ICP from 45 to 16 mm Hg, and cerebral blood flow dropped from 103 to 44 mL/100 g/min. Cerebral perfusion pressure increased from 45 to 70 mm Hg. Unfortunately, those who were not deemed transplant candidates died after rewarming. Later cases showed similar findings and support the notion that hypothermia provides some neurologic benefits in transplant candidates. Although these studies show potential benefit, a randomized controlled trial to further validate the use of hypothermia is still needed to define its role.
Hypertonic Saline in ALF
Another possible method to treat refractory ICH during acute liver failure is hypertonic saline. There has been great concern for the use of hypertonic saline because of the potential consequence of osmotic shifts across the blood-brain barrier (BBB). However, a presumed advantage for hypertonic saline to treat ICH is a higher osmotic reflection coefficient across the BBB.84–86 Thus, hypertonic saline could potentially lead to edema reduction, lower ICPs, and better perfusion by developing a higher osmotic gradient in the cerebral vascular compartment. In a randomized placebo-controlled study, Murphy et al. studied the effect of hypertonic saline infusion on ICP and clinical outcomes among ICU patients with acute liver failure.74 Thirty patients were treated with hypertonic saline 30% (5-20 mL/hour) to maintain serum sodium levels at 145 to 155 mmol/L. After 24 to 72 hours, ICP was significantly lower in the treated group (P = 0.04) compared to the standard care group. However, high osmolar loads and continuous hemofiltration were required, and mortality was similar among the treated and standard care groups. The use of hypertonic saline in the management of ICH in acute liver failure warrants further investigation.
L-Ornithine-L-Aspartate (LOLA)
As already stated earlier in the chapter, NH3 is thought to be a key neurotoxin in acute liver failure. Higher blood levels of NH3 have been correlated with higher mortality, higher grades of hepatic encephalopathy, increased frequency of ICH, and severe complications. L-Ornithine-L-aspartate (LOLA) may be useful for both acute and chronic hepatic encephalopathy. This compound salt has been found to reduce NH3 levels by increasing NH3 disposal through enhanced peripheral metabolism.87 LOLA increases the activity of hepatic urea cycle enzymes and also increases the rate of glutamine production within skeletal muscle.
The largest randomized controlled trial to test the efficacy of LOLA in high-grade encephalopathy on patients with acute liver failure was performed by Acharya et al.87; 201 patients with acute liver failure were randomized to either standard of care or LOLA (30 g daily infusion for 3 days). The primary endpoint, mortality, was not improved by LOLA (RR 1.27, P = 0.204). In a multivariate analysis, only blood NH3 level was an independent predictor of survival, and this parameter also was not significantly affected by treatment with LOLA. Accordingly, LOLA is not currently recommended in the management of severe encephalopathy from acute liver failure.
Other Options For Refractory Ich
Other potential therapies that can be considered in acute liver failure patients with refractory ICH include pentobarbital or thiopental and indomethacin. By inducing a comatose state and reducing cerebral edema, barbiturates such as pentobarbital (3-5 mg/kg IV load, then 1-3 mg/kg/h infusion) or thiopental (5-10 mg/kg load, then 3-5 mg/kg/h infusion) have been shown to have some benefit in refractory ICH.65–6788 However, severe side effects such as arterial hypotension, hypokalemia, and prolonged coma limit their use. These medications also often require coadministration of vasopressors in order to maintain cerebral perfusion pressure above 50 mm Hg, and require a provider to have prior experience using these drugs. Indomethacin (dosed at 25 mg IV over 1 minute) also has the potential to cause an acute decrease in ICP and an acute increase in CPP by causing cerebral vasoconstriction. Therefore, these medications may also play a role as second- or third-line options for patients with persistent refractory ICH.65,89–90
Experimental Therapies
Albumin Dialysis
Stange et al. recently reported use of an extracorporeal liver assist device based on albumin dialysis (MARS) in 26 patients with chronic liver disease who had either acute or chronic liver failure.91 The treatments lowered plasma bilirubin and bile acid levels, but the effect on clinical outcome was unclear: nine patients with advanced liver disease died within an average of 15 days, but the remainder survived and were thought to have benefited. Further studies will be needed to define benefit and overall utility.
Bioartificial Liver
Demetriou et al. reported the results of multicenter randomized controlled trial of a porcine hepatocyte-based BAL in 171 patients with acute liver failure (n = 147) and primary nonfunction after liver transplantation (n = 24).92 In both the group as a whole and the subgroup of acute liver failure, 30-day patient survival was slightly, but insignificantly, higher in the BAL group (entire cohort: 71% versus 62%, P = NS; acute liver failure: 73% versus 59%, P = NS). A major confounding variable in this study was the overwhelmingly positive effect of hepatic transplantation. Transplanted patients (55% of cohort) experienced a 70% reduction in relative risk (RR) of death (P < .0001). Additional analysis of survival in the subgroup of patients with acute liver failure, after controlling for impact of transplantation, suggested survival benefit for BAL-treated patients (RR death = 0.56, P = 0.048). Although this initial report is encouraging, additional studies will be needed to determine the efficacy and role of BAL in treatment of acute liver failure.
Hepatocyte Transplantation
The principals guiding use of hepatocyte transplantation are similar to those of the bioartificial liver: provide support during a period of critical need so the patient can be bridged to recovery or transplantation.93–95 One potential advantage of hepatocyte transplantation is the ability of liver stem cells to regenerate, raising the potential for repopulation of a dying or dead liver by allogeneic donor hepatocytes. The latter theoretical consideration has not been proven in humans with acute liver failure. Experience with hepatocyte transplantation in FHF is limited. Our center reported the outcome of six patients who were not candidates for liver transplantation due to active substance abuse or prohibitive underlying medical illness, and one patient listed for transplantation who had disseminated herpes infection. Despite a suggestion of improvement in neurologic status after hepatocyte transplantation, all seven died. Currently, hepatocyte transplantation for acute liver failure should be viewed as unproven and experimental.
Liver Transplantation
Liver transplantation is the only treatment that has been proven to improve survival in patients with acute liver failure and grade III or IV encephalopathy.7 Survival without transplantation is 10% to 20%. Survival increases to 60% to 80% with liver transplantation. In the study of BAL by Demetriou et al. noted above, the survival of patients with acute liver failure who underwent hepatic transplantation was 92%.92
Resolution of Cerebral Edema
At some stage, cerebral edema is irreversible and patients, despite transplantation, will experience brain death or massive irreversible brain injury.96–97 Risk of irreversible neurologic injury is greatest in those with CPP less than 40 mm Hg for more than 4 hours. Lesser increases in ICP may be associated with neurologic injury, but usually the cerebral edema resolves in the posttransplant period, and complete or partial neurologic recovery may be expected. In most cases of acute liver failure, all the manifestations of the neurologic illness (cerebral edema, encephalopathy, coma) totally reverse without sequelae following successful hepatic transplantation. One complication, central pontine myelinolysis, may occur in the absence of cerebral edema and may be related to fluctuations in plasma sodium during resuscitative measures in the ICU, such as IV fluids, transfusions, antibiotics, sedatives, narcotics, invasive procedures, and ventilatory support. Central pontine myelinolysis may result in significant neurologic impairment, requiring prolonged support and rehabilitation (physical therapy, speech therapy). Despite the serious nature of central pontine myelinolysis , significant neurologic recovery can occur.98
Living Donor Liver Transplantation
Donor safety is a major concern in the performance of living donor liver transplantation (LDLT). Current statistics suggest that donor mortality is approximately 0.13% for adult-to-pediatric cases and 0.25% for adult-to-adult cases.99 This procedure should be used with caution and only performed by centers with extensive experience and expertise in hepatic transplantation and liver resection.
Experience with LDLT for acute liver failure is limited worldwide. Japan has reported the largest number of LDLT cases for acute liver failure, largely because more than 99% of all liver transplants in Japan are from living donors.100 In the largest series of LDLT for acute liver failure, Lee et al. report patient survival as 82.3% at 1 and 5 years after transplantation for 57 recipients.101 These results are similar to those of cadaveric transplantation for acute liver failure and also demonstrate the durability of living donor allografts.
 Encephalopathy in the Setting of Chronic Liver Disease
Encephalopathy in the Setting of Chronic Liver Disease
Patients with cirrhosis of any cause and chronic PSE may present with a host of neuropsychiatric symptoms, ranging from subtle changes in mental status to coma.1–3 Fetor hepaticus is common but not invariable. Asterixis, the “flapping tremor,” is due to involuntary intermittent relaxation of sustained motor activity, but is less specific than fetor hepaticus for hepatic encephalopathy and is usually only present during the late stages of encephalopathy. Asterixis is most easily elicited with the patient’s arm outstretched, fingers separated, and wrists hyperextended. Although reported, cerebral edema rarely occurs in patients with encephalopathy in the setting of chronic liver disease. As the patient recovers from hepatic encephalopathy, asterixis and other manifestations of encephalopathy disappear.
Risk Factors And Precipitating Events: Implications For Diagnostic Testing And Treatment
Surgical Shunt Procedure or TIPS
Hepatic encephalopathy is a common complication of portal diversion following surgical portal-systemic shunts or TIPS (see Table 101-3).102–103 Predictors of post-shunt encephalopathy include pre-shunt encephalopathy, severe liver disease (Child-Pugh score > 10 or Model-for-End-Stage-Liver-Disease [MELD] score > 15), poor clearance of indocyanine green and lidocaine, and elderly age. The mechanisms of hepatic encephalopathy after placement of a portal-systemic shunt include: lack of compensatory dilatation of the hepatic artery, lack of perfusion of the liver via the portal vein, and reduction in hepatocyte function. Clinically apparent encephalopathy after placement of a shunt usually responds to medical treatment (low-protein diet, lactulose, neomycin). In rare circumstances, narrowing of the shunt with a flow-reducing stent or occlusion of the shunt may be necessary to control encephalopathy.104
TABLE 101-3 Neuropsychiatric Tests Used to Evaluate Hepatic Encephalopathy
| Cerebral Function | Test |
|---|---|
| Learning and delayed recall | Story Memory Test |
| Figure Memory Test | |
| Concentration | Digit Vigilance Test |
| Fine motor coordination | Grooved Pegboard |
| Sequential procedures | Trail Making Test |
| Problem solving | Wisconsin Card Sorting Test |
| Attention | WAIS-R* Digit Symbol Subtest |
| Vocabulary | WAIS-R Vocabulary Subtest |
| Verbal fluency skills | Controlled Oral Word Association |
| Animal Naming | |
| Auditory comprehension | Complex Material |
| Visual-spatial analysis | WAIS-R Block Design Subtest |
| Psychological function | MMPI-2† |
Diagnosis
The diagnosis of PSE is based upon clinical suspicion in patients with chronic liver disease, and the impression is confirmed by resolution following medical therapy. Occasionally it may be necessary to employ additional testing to confirm the diagnosis of PSE. Additional testing is particularly useful when encephalopathy is the primary clinical manifestation of otherwise unsuspected liver disease, or if the manifestations of encephalopathy are predominantly a change in behavior or an unusual neurologic syndrome (seizures, focal neurologic deficits). Rarely, cerebral edema complicates chronic liver disease.105
Radiologic Imaging
Standard CT scans or nuclear brain scans exhibit little or no specific distinguishing features, although loss of cortical volume may be common in patients with Laënnec’s cirrhosis and chronic encephalopathy. CT may be used to document cerebral edema or to exclude CNS complications such as tumor, infection, or hemorrhage. MRI imaging studies have revealed a few features relatively unique to hepatic encephalopathy. One feature, hyperintensity on T1-weighted images of the globus pallidus (see Figure 101-3), correlates with (extrapyramidal) motor disorders and excess accumulation of manganese.
Neuropsychiatric Testing
In general, neuropsychiatric testing is used primarily to follow efficacy of treatment. A battery of tests is employed to distinguish hepatic encephalopathy and organic brain syndrome from other causes of encephalopathy and underlying psychiatric disease. These tests are itemized in Table 101-3. Poor performance on number connection tests correlates reasonably well with severity of encephalopathy and Child-Pugh and/or MELD classification.
Therapeutic Options
Traditional treatment for hepatic encephalopathy has included a protein-restricted diet of 40 grams or less per day.106–108 However, cirrhotic patients often develop severe muscle wasting; thus, in patients with advanced disease, unnecessary protein restriction might further worsen the poor nutritional state. Most hepatologists currently avoid use of protein restriction in management of chronic hepatic encephalopathy.
Branched-Chain Amino Acids
Early studies demonstrated that cirrhotic patients had an increase in aromatic amino acids and a decrease in branched-chain amino acids (BCAAs) in blood samples. Subsequent clinical work suggested that patients with the greatest imbalance in plasma amino acids were more likely to be encephalopathic and to experience early and higher mortality. For this reason, there have been at least 14 controlled trials of the use of BCAAs in the treatment of cirrhotic patients with chronic encephalopathy. A recent well-controlled trial suggested efficacy.109–110 However, results of these trials have been inconsistent, and separate meta-analyses yielded opposite conclusions regarding efficacy.108,111 In addition, BCAA preparations are much more expensive than standard amino acid supplements. A trial of BCAA might be considered in patients who develop encephalopathy on standard protein diets and manifest protein-calorie malnutrition. BCAA supplements may allow adequate protein intake in this select group of patients without increasing the frequency of attacks of encephalopathy.
Lactulose
One of the most successful treatments for hepatic encephalopathy is lactulose, a nonabsorbable disaccharide which is fermented by bacteria in the intestine to yield acetic, butyric, propionic, and lactic acids.112–116 The fermentation of lactulose produces an acidic milieu that alters the composition of the bacterial flora, lowers colonic pH, and produces an osmotic diarrhea. Each of these effects may be responsible for the ameliorative effects of lactulose on hepatic encephalopathy. Changing the composition of the bacterial flora may alter the metabolism of fecal contents and reduce the production of toxins, NH3, and methanethiols that are responsible for the encephalopathy. The acidic luminal milieu creates an environment capable of trapping NH3:
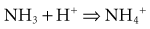
Neomycin
Neomycin is highly nephrotoxic and should never be given IV or parenterally. Orally administered neomycin, on the other hand, is poorly absorbed and has a limited entrance to the circulation and is therefore much less nephrotoxic. The goal of therapy with oral neomycin is to alter the bacterial composition of the colonic flora. The major advantage of neomycin over lactulose is that it does not cause diarrhea. The main disadvantage is that despite its poor absorption, some neomycin does gain entry to the body which can contribute to nephrotoxicity. We recommend use of neomycin in patients who are intolerant of lactulose (usually due to diarrhea). Also, we may add neomycin to a lactulose-based regimen to improve efficacy for controlling encephalopathy.8,117–119 Some have recommended that neomycin be given in short courses lasting only 2 to 8 weeks.
Metronidazole
Studies have demonstrated that oral metronidazole (500 mg to 1.5 g/d given for 1 week) was well tolerated, safe (no obvious neurotoxicity), and as effective as neomycin or lactulose in controlling encephalopathy. Others have not observed similar efficacy and have measured little effect of metronidazole on blood NH3 levels.119 The advantages of metronidazole are that it does not cause diarrhea and it is not nephrotoxic. A disadvantage is that many patients complain of epigastric discomfort with its use (poor compliance with long-term treatment). Maintenance therapy can be expected to cause peripheral neuropathy (already a problem in patients with advanced liver disease), and metronidazole has been reported to cause the “disulfiram reaction” when alcohol is consumed. The physician prescribing metronidazole to cirrhotic patients also should be aware that this drug undergoes extensive hepatic metabolism. One study of cirrhotics with encephalopathy revealed a threefold reduction in hepatic elimination and maintenance of therapeutic levels with as little as 500 mg given every 24 to 48 hours.
Helicobacter pylori
Published reports have suggested that Helicobacter pylori infections might increase blood NH3 levels and precipitate hepatic encephalopathy in patients with cirrhosis. Controlled trials of interventions to eradicate H. pylori have failed to confirm this initial observation.120–122
Benzodiazepine Antagonists
There have been several randomized controlled trials of short-term administration of flumazenil in the treatment of hepatic encephalopathy.123–131 In some studies, flumazenil was superior to placebo in improving the grade of encephalopathy; 30% to 60% of encephalopathic patients improved after administration of flumazenil, and EEG changes paralleled this improvement. In other studies, flumazenil was no better than placebo in ameliorating the symptoms of encephalopathy, and EEGs did not improve. A recent meta-analysis suggested benefit of flumazenil over placebo.132 Flumazenil has a limited role in the treatment of hepatic encephalopathy, and additional trials of larger numbers of subjects with varying grades of encephalopathy are needed.
LOLA
Although studies using LOLA in acute liver failure did not find significant changes in survival, other studies investigated its potential use in hepatic encephalopathy from chronic liver disease. One prospective study, which used eight Child-Pugh B or C patients without baseline hepatic encephalopathy and seven cirrhotic patients with TIPS, tested to see if LOLA dosing could prevent or reduce glutamine-induced hepatic encephalopathy. After each subject was given two doses of glutamine (20-gram challenges), each subject was randomly selected to receive either 5 g of IV LOLA once or placebo infusion. Blood NH3 levels, psychometric tests, and electroencephalography were tested in both groups, and LOLA was found to provide significant benefit of reducing encephalopathy symptoms when compared to placebo in non-TIPS patients, as well as minimizing the negative psychometric measurements after glutamine challenge.133 A meta-analysis was also performed to analyze the effects by LOLA on chronic hepatic encephalopathy. The meta-analysis included three randomized trials which pooled 212 patients. This analysis found an overall significant effect on improvement of chronic hepatic encephalopathy symptoms (RR 1.89; 95% CI: 1.32-2.71, P = 0.0005). However, most benefit was seen in the grade I or II patients (RR 1.87, 95% CI: 1.30-2.68, P = 0.0007).134
Rifaximin
Rifaximin is the newest agent developed for the treatment of hepatic encephalopathy. It is a nonabsorbable antibiotic derivative of rifamycin with broad antimicrobial activity.135 In comparison studies of rifaximin with lactulose, patients treated with rifaximin showed greater improvement in the degree of hepatic encephalopathy, had lower NH3 levels, and had lower PSE index scores.136 A recent randomized controlled trial of 299 patients demonstrated reduction of recurrent hepatic encephalopathy episodes (HR 0.42, 95% CI: 0.28-0.64, P < 0.001) and number of hospitalizations due to hepatic encephalopathy (HR 0.5, 95% CI: 0.29-0.87, P = 0.01).135,137–138 Rifaximin is likely to become a first-line agent for the treatment of chronic hepatic encephalopathy.
Hepatic Transplantation
The development of encephalopathy in a patient with chronic liver disease indicates severe portal-systemic shunting and hepatic dysfunction. The prognosis for patients who develop this complication is grim; one recent study indicated that the 1-year survival rate is 42% and the 3-year survival rate is 23%.139 In addition, there are numerous comorbidities in encephalopathic patients, including inability to continue gainful employment, poor function at home, nursing strains on spouse or family, inability to drive a vehicle, and inability to handle personal finances. Although medical therapies can ameliorate the major symptoms of encephalopathy, they rarely are effective enough to return the patient to full function. Often the patient with encephalopathy is at risk for other life-threatening complications of liver disease such as variceal hemorrhage and spontaneous bacterial peritonitis. Posttransplant 1-year survival rates are 80% to 85%, doubling 1-year survival rates for patients with hepatic encephalopathy without a transplant. For all the above reasons, any patient with hepatic encephalopathy should be considered for hepatic transplantation.
 Summary
SummaryVaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11:1581-1589.
Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology. 2009;136:2159-2168.
Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362:1071-1081.
Critical paper to demonstrate efficacy of rifaximin to treat hepatic encephalopathy.
Bustamante J, Rimola A, Ventura PJ, et al. Prognostic significance of hepatic encephalopathy in patients with cirrhosis. J Hepatol. 1999;30:890-895.
Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
Most recent definition of the different grades of hepatic encephalopathy.
1 Blei AT, Cordoba J. Hepatic Encephalopathy. Am J Gastroenterol. 2001;96:1968-1976.
2 Lockwood AH. Hepatic encephalopathy. Neurol Clin. 2002;20:241-246. vii
3 Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
4 Blei AT. Brain edema in acute liver failure. Crit Care Clin. 2008;24:99-114. ix
5 Larsen FS, Wendon J. Prevention and management of brain edema in patients with acute liver failure. Liver Transpl. 2008;14(Suppl 2)):S90-S96.
6 Vaquero J, Butterworth RF. Mechanisms of brain edema in acute liver failure and impact of novel therapeutic interventions. Neurol Res. 2007;29:683-690.
7 Liou IW, Larson AM. Role of liver transplantation in acute liver failure. Semin Liver Dis. 2008;28:201-209.
8 Conn HO, Lieberthal MM. The hepatic coma syndromes and lactulose. Baltimore: Williams & Wilkins; 1979.
9 Das A, Dhiman RK, Saraswat VA, Verma M, Naik SR. Prevalence and natural history of subclinical hepatic encephalopathy in cirrhosis. J Gastroenterol Hepatol. 2001;16:531-535.
10 Groeneweg M, Moerland W, Quero JC, Hop WC, Krabbe PF, Schalm SW. Screening of subclinical hepatic encephalopathy. J Hepatol. 2000;32:748-753.
11 Stewart CA, Smith GE. Minimal hepatic encephalopathy. Nat Clin Pract Gastroenterol Hepatol. 2007;4:677-685.
12 Romero-Gomez M, Boza F, Garcia-Valdecasas MS, Garcia E, Aguilar-Reina J. Subclinical hepatic encephalopathy predicts the development of overt hepatic encephalopathy. Am J Gastroenterol. 2001;96:2718-2723.
13 Vogels BA, van Steynen B, Maas MA, Jorning GG, Chamuleau RA. The effects of ammonia and portal-systemic shunting on brain metabolism, neurotransmission and intracranial hypertension in hyperammonaemia-induced encephalopathy. J Hepatol. 1997;26:387-395.
14 Cauli O, Rodrigo R, Llansola M, et al. Glutamatergic and GABAergic neurotransmission and neuronal circuits in hepatic encephalopathy. Metab Brain Dis. 2009;24:69-80.
15 Norenberg MD, Jayakumar AR, Rama Rao KV, Panickar KS. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219-234.
16 Bass NM. Monitoring and treatment of intracranial hypertension. Liver Transpl. 2000:S21-S26.
17 Strauss GI, Knudsen GM, Kondrup J, Moller K, Larsen FS. Cerebral metabolism of ammonia and amino acids in patients with fulminant hepatic failure. Gastroenterology. 2001;121:1109-1119.
18 Madsen PL, Holm S, Herning M, Lassen NA. Average blood flow and oxygen uptake in the human brain during resting wakefulness: a critical appraisal of the Kety-Schmidt technique. J Cereb Blood Flow Metab. 1993;13:646-655.
19 Strauss GI, Moller K, Holm S, Sperling B, Knudsen GM, Larsen FS. Transcranial Doppler sonography and internal jugular bulb saturation during hyperventilation in patients with fulminant hepatic failure. Liver Transpl. 2001;7:352-358.
20 Aggarwal S, Obrist W, Yonas H, et al. Cerebral hemodynamic and metabolic profiles in fulminant hepatic failure: relationship to outcome. Liver Transpl. 2005;11:1353-1360.
21 Strauss GI, Moller K, Larsen FS, Kondrup J, Knudsen GM. Cerebral glucose and oxygen metabolism in patients with fulminant hepatic failure. Liver Transpl. 2003;9:1244-1252.
22 Larsen FS, Ejlersen E, Clemmesen JO, Kirkegaard P, Hansen BA. Preservation of cerebral oxidative metabolism in fulminant hepatic failure: an autoregulation study. Liver Transpl Surg. 1996;2:348-353.
23 Vaquero J, Chung C, Blei AT. Cerebral blood flow in acute liver failure: a finding in search of a mechanism. Metab Brain Dis. 2004;19:177-194.
24 Jalan R, Olde Damink SW, Deutz NE, Hayes PC, Lee A. Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology. 2004;127:1338-1346.
25 Tofteng F, Jorgensen L, Hansen BA, Ott P, Kondrup J, Larsen FS. Cerebral microdialysis in patients with fulminant hepatic failure. Hepatology. 2002;36:1333-1340.
26 Norenberg MD, Rama Rao KV, Jayakumar AR. Signaling factors in the mechanism of ammonia neurotoxicity. Metab Brain Dis. 2009;24:103-117.
27 Rose C, Felipo V. Limited capacity for ammonia removal by brain in chronic liver failure: potential role of nitric oxide. Metab Brain Dis. 2005;20:275-283.
28 Balata S, Olde Damink SW, Ferguson K, et al. Induced hyperammonemia alters neuropsychology, brain MR spectroscopy and magnetization transfer in cirrhosis. Hepatology. 2003;37:931-939.
29 Laubenberger J, Haussinger D, Bayer S, Gufler H, Hennig J, Langer M. Proton magnetic resonance spectroscopy of the brain in symptomatic and asymptomatic patients with liver cirrhosis. Gastroenterology. 1997;112:1610-1616.
30 Grover VP, Dresner MA, Forton DM, et al. Current and future applications of magnetic resonance imaging and spectroscopy of the brain in hepatic encephalopathy. World J Gastroenterol. 2006;12:2969-2978.
31 McConnell JR, Antonson DL, Ong CS, et al. Proton spectroscopy of brain glutamine in acute liver failure. Hepatology. 1995;22:69-74.
32 Ross BD, Jacobson S, Villamil F, et al. Subclinical hepatic encephalopathy: proton MR spectroscopic abnormalities. Radiology. 1994;193:457-463.
33 Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology. 1994;107:1475-1480.
34 Cordoba J, Sanpedro F, Alonso J, Rovira A. 1H magnetic resonance in the study of hepatic encephalopathy in humans. Metab Brain Dis. 2002;17:415-429.
35 Huizenga JR, Gips CH, Tangerman A. The contribution of various organs to ammonia formation: a review of factors determining the arterial ammonia concentration. Ann Clin Biochem. 1996;33(Pt 1):23-30.
36 Huizenga JR, van Dam GM, Gips CH. Arterial ammonia with Blood Ammonia Checker II and with indophenol reaction to assess presence of hepatic encephalopathy. Clin Chim Acta. 1996;252:73-82.
37 Huizenga JR, Gips CH, Conn HO, Jansen PL. Determination of ammonia in ear-lobe capillary blood is an alternative to arterial blood ammonia. Clin Chim Acta. 1995;239:65-70.
38 Butterworth RF. Neuroactive amino acids in hepatic encephalopathy. Metab Brain Dis. 1996;11:165-173.
39 Knudsen GM, Schmidt J, Almdal T, Paulson OB, Vilstrup H. Passage of amino acids and glucose across the blood-brain barrier in patients with hepatic encephalopathy. Hepatology. 1993;17:987-992.
40 Ahboucha S, Butterworth RF. Role of endogenous benzodiazepine ligands and their GABA-A-associated receptors in hepatic encephalopathy. Metab Brain Dis. 2005;20:425-437.
41 Basile AS, Harrison PM, Hughes RD, et al. Relationship between plasma benzodiazepine receptor ligand concentrations and severity of hepatic encephalopathy. Hepatology. 1994;19:112-121.
42 Wilkinson SP. GABA, benzodiazepines and hepatic encephalopathy. Eur J Gastroenterol Hepatol. 1995;7:323-324.
43 Macdonald GA, Frey KA, Agranoff BW, et al. Cerebral benzodiazepine receptor binding in vivo in patients with recurrent hepatic encephalopathy. Hepatology. 1997;26:277-282.
44 Zeneroli ML, Venturini I, Avallone R, et al. Hepatic encephalopathy in liver transplant recipients precipitated by benzodiazepines present in transfused blood. Transplantation. 1996;62:764-767.
45 Ahboucha S, Pomier-Layrargues G, Butterworth RF. Increased brain concentrations of endogenous (non-benzodiazepine) GABA-A receptor ligands in human hepatic encephalopathy. Metab Brain Dis. 2004;19:241-251.
46 Medina JH, Pena C, Piva M, et al. Benzodiazepines in the brain. Their origin and possible biological roles. Mol Neurobiol. 1992;6:377-386.
47 Baraldi M, Avallone R, Corsi L, Venturini I, Baraldi C, Zeneroli ML. Natural endogenous ligands for benzodiazepine receptors in hepatic encephalopathy. Metab Brain Dis. 2009;24:81-93.
48 Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis. 2006;23:512-521.
49 Blom HJ, Ferenci P, Grimm G, Yap SH, Tangerman A. The role of methanethiol in the pathogenesis of hepatic encephalopathy. Hepatology. 1991;13:445-454.
50 Barron TF, Devenyi AG, Mamourian AC. Symptomatic manganese neurotoxicity in a patient with chronic liver disease: correlation of clinical symptoms with MRI findings. Pediatr Neurol. 1994;10:145-148.
51 Bresci G, Parisi G, Banti S. Management of hepatic encephalopathy with oral zinc supplementation: a long-term treatment. Eur J Med. 1993;2:414-416.
52 Riggio O, Ariosto F, Merli M, et al. Short-term oral zinc supplementation does not improve chronic hepatic encephalopathy. Results of a double-blind crossover trial. Dig Dis Sci. 1991;36:1204-1208.
53 Reding P, Duchateau J, Bataille C. Oral zinc supplementation improves hepatic encephalopathy. Results of a randomised controlled trial. Lancet. 1984;2:493-495.
54 Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol. 2009;6:542-553.
55 Lee WM, Squires RHJr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401-1415.
56 1st Edition ed. Lee WM, Williams R, editors. Acute Liver Failure. Cambridge, UK: Cambridge University Press, 1997.
57 Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142-152.
58 Lee WM. Acute Liver Failure in Clinical Perspectives in Gastroenterology, AGA, March-April, 2001, p. 101-10.
59 Riordan SM, Williams R. Use and validation of selection criteria for liver transplantation in acute liver failure. Liver Transpl. 2000;6:170-173.
60 Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transpl. 2000;6:163-169.
61 Pathikonda M, Munoz SJ. Acute liver failure. Ann Hepatol. 2010;9:7-14.
62 Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol. 2003;98:448-453.
63 Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am. 2008;92:761-794. viii
64 Schiodt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med. 1997;337:1112-1117.
65 Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;35:2498-2508.
66 Keays RT, Alexander GJ, Williams R. The safety and value of extradural intracranial pressure monitors in fulminant hepatic failure. J Hepatol. 1993;18:205-209.
67 Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11:1581-1589.
68 Keays R, Potter D, O’Grady J, Peachey T, Alexander G, Williams R. Intracranial and cerebral perfusion pressure changes before, during and immediately after orthotopic liver transplantation for fulminant hepatic failure. Q J Med. 1991;79:425-433.
69 Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet. 1993;341:157-158.
70 Wendon J, Lee W. Encephalopathy and cerebral edema in the setting of acute liver failure: pathogenesis and management. Neurocrit Care. 2008;9:97-102.
71 Wijdicks EF, Plevak DJ, Rakela J, Wiesner RH. Clinical and radiologic features of cerebral edema in fulminant hepatic failure. Mayo Clin Proc. 1995;70:119-124.
72 Detry O, Arkadopoulos N, Ting P, et al. Intracranial pressure during liver transplantation for fulminant hepatic failure. Transplantation. 1999;67:767-770.
73 Lidofsky SD, Bass NM, Prager MC, et al. Intracranial pressure monitoring and liver transplantation for fulminant hepatic failure. Hepatology. 1992;16:1-7.
74 Murphy N, Auzinger G, Bernel W, Wendon J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39:464-470.
75 Shawcross DL, Davies NA, Mookerjee RP, et al. Worsening of cerebral hyperemia by the administration of terlipressin in acute liver failure with severe encephalopathy. Hepatology. 2004;39:471-475.
76 Traber P, DalCanto M, Ganger D, Blei AT. Effect of body temperature on brain edema and encephalopathy in the rat after hepatic devascularization. Gastroenterology. 1989;96:885-891.
77 Cordoba J, Crespin J, Gottstein J, Blei AT. Mild hypothermia modifies ammonia-induced brain edema in rats after portacaval anastomosis. Gastroenterology. 1999;116:686-693.
78 Rose C, Michalak A, Pannunzio M, Chatauret N, Rambaldi A, Butterworth RF. Mild hypothermia delays the onset of coma and prevents brain edema and extracellular brain glutamate accumulation in rats with acute liver failure. Hepatology. 2000;31:872-877.
79 Vaquero J, Butterworth RF. Mild hypothermia for the treatment of acute liver failure—what are we waiting for? Nat Clin Pract Gastroenterol Hepatol. 2007;4:528-529.
80 Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet. 1999;354:1164-1168.
81 Jalan R, Olde Damink SW, Deutz NE, Hayes PC, Lee A. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001;34:50-54.
82 Jalan R, Olde Damink SW, Deutz NE, et al. Moderate hypothermia prevents cerebral hyperemia and increase in intracranial pressure in patients undergoing liver transplantation for acute liver failure. Transplantation. 2003;75:2034-2039.
83 Jalan R. Intracranial hypertension in acute liver failure: pathophysiological basis of rational management. Semin Liver Dis. 2003;23:271-282.
84 Himmelseher S. Hypertonic saline solutions for treatment of intracranial hypertension. Curr Opin Anaesthesiol. 2007;20:414-426.
85 Qureshi AI, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301-3313.
86 Suarez JI. Hypertonic saline for cerebral edema and elevated intracranial pressure. Cleve Clin J Med. 2004;71(Suppl 1)):S9-13.
87 Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology. 2009;136:2159-2168.
88 Forbes A, Alexander GJ, O’Grady JG, et al. Thiopental infusion in the treatment of intracranial hypertension complicating fulminant hepatic failure. Hepatology. 1989;10:306-310.
89 Clemmesen JO, Hansen BA, Larsen FS. Indomethacin normalizes intracranial pressure in acute liver failure: a twenty-three-year-old woman treated with indomethacin. Hepatology. 1997;26:1423-1425.
90 Tofteng F, Larsen FS. The effect of indomethacin on intracranial pressure, cerebral perfusion and extracellular lactate and glutamate concentrations in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2004;24:798-804.
91 Stange J, Mitzner SR, Klammt S, et al. Liver support by extracorporeal blood purification: a clinical observation. Liver Transpl. 2000;6:603-613.
92 Demetriou AA, Brown RSJr, Busuttil RW, et al. Prospective, randomized, multicenter, controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg. 2004;239:660-667. discussion 7-70
93 Fox IJ, Chowdhury JR. Hepatocyte transplantation. Am J Transplant. 2004;4(Suppl 6)):7-13.
94 Tsiaoussis J, Newsome PN, Nelson LJ, Hayes PC, Plevris JN. Which hepatocyte will it be? Hepatocyte choice for bioartificial liver support systems. Liver Transpl. 2001;7:2-10.
95 Bilir BM, Guinette D, Karrer F, et al. Hepatocyte transplantation in acute liver failure. Liver Transpl. 2000;6:32-40.
96 Ardizzone G, Arrigo A, Schellino MM, et al. Neurological complications of liver cirrhosis and orthotopic liver transplant. Transplant Proc. 2006;38:789-792.
97 Blanco R, De Girolami U, Jenkins RL, Khettry U. Neuropathology of liver transplantation. Clin Neuropathol. 1995;14:109-117.
98 Bennet J. Recovery from Central pontine myelinolysis in patients with hepatic failure and orthotopic liver transplantation: Section of Hepatology, Depts. of Medicine, Surgery, and Anesthesiology, Univ. of CO School of Med., Denver. Hepatology. 1995;22:A394-A.
99 Trotter JF, Wachs M, Everson GT, Kam I. Adult-to-adult transplantation of the right hepatic lobe from a living donor. N Engl J Med. 2002;346:1074-1082.
100 Akamatsu N, Sugawara Y. Acute liver failure and living donor liver transplantation. Hepatol Res. 2008;38:S60-S71.
101 Lee SG, Ahn CS, Kim KH. Which types of graft to use in patients with acute liver failure? (A) Auxiliary liver transplant (B) Living donor liver transplantation (C) The whole liver. (B) I prefer living donor liver transplantation. J Hepatol. 2007;46:574-578.
102 Riggio O, Angeloni S, Salvatori FM, et al. Incidence, natural history, and risk factors of hepatic encephalopathy after transjugular intrahepatic portosystemic shunt with polytetrafluoroethylene-covered stent grafts. Am J Gastroenterol. 2008;103:2738-2746.
103 Boyer TD. Transjugular intrahepatic portosystemic shunt: current status. Gastroenterology. 2003;124:1700-1710.
104 Madoff DC, Wallace MJ, Ahrar K, Saxon RR. TIPS-related hepatic encephalopathy: management options with novel endovascular techniques. Radiographics. 2004;24:21-36. discussion-7
105 Donovan JP, Schafer DF, Shaw BWJr, Sorrell MF. Cerebral oedema and increased intracranial pressure in chronic liver disease. Lancet. 1998;351:719-721.
106 Morgan TR, Moritz TE, Mendenhall CL, Haas R. Protein consumption and hepatic encephalopathy in alcoholic hepatitis. VA Cooperative Study Group #275. J Am Coll Nutr. 1995;14:152-158.
107 Bianchi GP, Marchesini G, Fabbri A, et al. Vegetable versus animal protein diet in cirrhotic patients with chronic encephalopathy. A randomized cross-over comparison. J Intern Med. 1993;233:385-392.
108 Wright G, Jalan R. Management of hepatic encephalopathy in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21:95-110.
109 Marchesini G, Bianchi G, Merli M, et al. Nutritional supplementation with branched-chain amino acids in advanced cirrhosis: a double-blind, randomized trial. Gastroenterology. 2003;124:1792-1801.
110 Charlton M. Branched-chain amino acid-enriched supplements as therapy for liver disease: Rasputin lives. Gastroenterology. 2003;124:1980-1982.
111 Marchesini G, Marzocchi R, Noia M, Bianchi G. Branched-chain amino acid supplementation in patients with liver diseases. J Nutr. 2005;135:1596S-1601S.
112 Weber FLJr. Lactulose and combination therapy of hepatic encephalopathy: the role of the intestinal microflora. Dig Dis. 1996;14(Suppl 1)):53-63.
113 Ferenci P. Treatment of hepatic encephalopathy in patients with cirrhosis of the liver. Dig Dis. 1996;14(Suppl 1)):40-52.
114 Horsmans Y, Solbreux PM, Daenens C, Desager JP, Geubel AP. Lactulose improves psychometric testing in cirrhotic patients with subclinical encephalopathy. Aliment Pharmacol Ther. 1997;11:165-170.
115 Blanc P, Daures JP, Rouillon JM, et al. Lactitol or lactulose in the treatment of chronic hepatic encephalopathy: results of a meta-analysis. Hepatology. 1992;15:222-228.
116 Dhiman RK, Sawhney MS, Chawla YK, Das G, Ram S, Dilawari JB. Efficacy of lactulose in cirrhotic patients with subclinical hepatic encephalopathy. Dig Dis Sci. 2000;45:1549-1552.
117 Festi D, Vestito A, Mazzella G, Roda E, Colecchia A. Management of hepatic encephalopathy: focus on antibiotic therapy. Digestion. 2006;73(Suppl 1)):94-101.
118 Strauss E, Tramote R, Silva EP, et al. Double-blind randomized clinical trial comparing neomycin and placebo in the treatment of exogenous hepatic encephalopathy. Hepatogastroenterology. 1992;39:542-545.
119 Alexander T, Thomas K, Cherian AM, Kanakasabapathy. Effect of three antibacterial drugs in lowering blood & stool ammonia production in hepatic encephalopathy. Indian J Med Res. 1992;96:292-296.
120 Huber M, Rossle M, Siegerstetter V, et al. Helicobacter pylori infection does not correlate with plasma ammonia concentration and hepatic encephalopathy in patients with cirrhosis. Hepatogastroenterology. 2001;48:541-544.
121 Abdel-Hady H, Zaki A, Badra G, et al. Helicobacter pylori infection in hepatic encephalopathy: Relationship to plasma endotoxins and blood ammonia. Hepatol Res. 2007;37:1026-1033.
122 Scotiniotis IA, Lucey MR, Metz DC. Helicobacter pylori infection is not associated with subclinical hepatic encephalopathy in stable cirrhotic patients. Dig Dis Sci. 2001;46:2744-2751.
123 Pomier-Layrargues G, Giguere JF, Lavoie J, et al. Flumazenil in cirrhotic patients in hepatic coma: a randomized double-blind placebo-controlled crossover trial. Hepatology. 1994;19:32-37.
124 Gyr K, Meier R, Haussler J, et al. Evaluation of the efficacy and safety of flumazenil in the treatment of portal systemic encephalopathy: a double blind, randomised, placebo controlled multicentre study. Gut. 1996;39:319-324.
125 Groeneweg M, Gyr K, Amrein R, et al. Effect of flumazenil on the electroencephalogram of patients with portosystemic encephalopathy. Results of a double blind, randomised, placebo-controlled multicentre trial. Electroencephalogr Clin Neurophysiol. 1996;98:29-34.
126 Van der Rijt CC, Schalm SW, Meulstee J, Stijnen T. Flumazenil therapy for hepatic encephalopathy. A double-blind cross over study. Gastroenterol Clin Biol. 1995;19:572-580.
127 Hoffman EJ, Warren EW. Flumazenil: a benzodiazepine antagonist. Clin Pharm. 1993;12:641-656. quiz 99-701
128 Howard CD, Seifert CF. Flumazenil in the treatment of hepatic encephalopathy. Ann Pharmacother. 1993;27:46-48.
129 Gooday R, Hayes PC, Bzeizi K, O’Carroll RE. Benzodiazepine receptor antagonism improves reaction time in latent hepatic encephalopathy. Psychopharmacology (Berl). 1995;119:295-298.
130 Cadranel JF, el Younsi M, Pidoux B, et al. Flumazenil therapy for hepatic encephalopathy in cirrhotic patients: a double-blind pragmatic randomized, placebo study. Eur J Gastroenterol Hepatol. 1995;7:325-329.
131 Devictor D, Tahiri C, Lanchier C, Navelet Y, Durand P, Rousset A. Flumazenil in the treatment of hepatic encephalopathy in children with fulminant liver failure. Intensive Care Med. 1995;21:253-256.
132 Goulenok C, Bernard B, Cadranel JF, et al. Flumazenil vs. placebo in hepatic encephalopathy in patients with cirrhosis: a meta-analysis. Aliment Pharmacol Ther. 2002;16:361-372.
133 Rees CJ, Oppong K, Al Mardini H, Hudson M, Record CO. Effect of L-ornithine-L-aspartate on patients with and without TIPS undergoing glutamine challenge: a double blind, placebo controlled trial. Gut. 2000;47:571-574.
134 Jiang Q, Jiang XH, Zheng MH, Chen YP. L-Ornithine-L-aspartate in the management of hepatic encephalopathy: a meta-analysis. J Gastroenterol Hepatol. 2009;24:9-14.
135 Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362:1071-1081.
136 Mas A, Rodes J, Sunyer L, et al. Comparison of rifaximin and lactitol in the treatment of acute hepatic encephalopathy: results of a randomized, double-blind, double-dummy, controlled clinical trial. J Hepatol. 2003;38:51-58.
137 Lawrence KR, Klee JA. Rifaximin for the treatment of hepatic encephalopathy. Pharmacotherapy. 2008;28:1019-1032.
138 Romero-Gomez M. Pharmacotherapy of hepatic encephalopathy in cirrhosis. Expert Opin Pharmacother. 2010;11:1317-1327.
139 Bustamante J, Rimola A, Ventura PJ, et al. Prognostic significance of hepatic encephalopathy in patients with cirrhosis. J Hepatol. 1999;30:890-895.
