[level-membership-for-pediatrics-category]
Chapter 469 Hemostasis
The Process
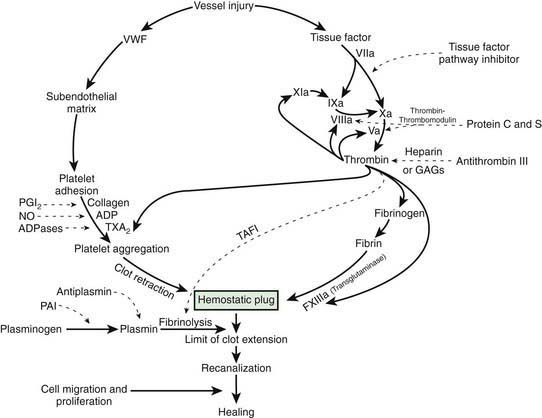
After vascular injury, vasoconstriction occurs and flowing blood comes in contact with the subendothelial matrix (Fig. 469-1). In flowing blood, when exposed to subendothelial matrix proteins, von Willebrand factor (VWF) changes conformation and provides the glue to which the platelet VWF receptor, the glycoprotein Ib complex, binds, tethering platelets to sites of injury. When the VWF receptor binds its ligand, complex signaling occurs from the outside membrane receptor to intracellular pathways, activating the platelets and triggering secretion of storage granules containing adenosine diphosphate (ADP), serotonin, and stored plasma and platelet membrane proteins. Upon activation, the platelet receptor for fibrinogen, α2bβ3, is switched on (“inside out” signaling) to bind fibrinogen and triggers the aggregation and recruitment of other platelets to form the platelet plug. Multiple physiologic agonists can trigger platelet activation and aggregation, including ADP, collagen, thrombin, and arachidonic acid. Aggregation involves the interaction of specific receptors on the platelet surface with plasma hemostatic proteins, primarily fibrinogen.
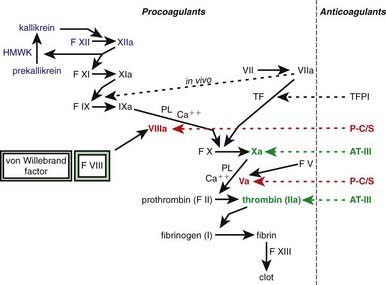
One of the subendothelial matrix proteins that are exposed after vascular injury is tissue factor. Just as exposed subendothelial matrix proteins bind VWF, exposed tissue factor binds to factor VII and activates the clotting cascade, as shown in Figure 469-2. The activated clotting factor then initiates the activation of the next sequential clotting factor in a systematic manner. Our understanding of the sequence of steps in the cascade followed assignment of the numerals for the clotting factors for the participant proteins, and thus the sequence seems “out of numerical order.” During the process of platelet activation, internalized platelet phospholipids (primarily phosphatidylserine) become externalized and interact at 2 specific, rate-limiting steps in the clotting process—those involving the cofactors factor VIII (X-ase complex) and factor V (prothrombinase complex). Both of these reactions are localized to the platelet surface and bring together the active enzyme, an activated cofactor, and the zymogen that will form the next active enzyme in the cascade. This sequence results in amplification of the process, which supplies a burst of clotting where it is physiologically needed. In vivo, autocatalysis of factor VII generates small amounts of VIIa continuously, so the system is always poised to act. Near the bottom of the cascade, the multipotent enzyme thrombin is formed. Thrombin converts fibrinogen into fibrin, activates factors V, VIII, and XI, and aggregates platelets. Activation of factor XI by thrombin amplifies further thrombin generation and contributes to inhibition of fibrinolysis. Thrombin also activates factor XIII. The stable fibrin-platelet plug is ultimately formed through clot retraction and cross linking of the fibrin clot by factor XIIIa.
Virtually all procoagulant proteins are balanced by an anticoagulant protein that regulates or inhibits procoagulant function. Four clinically important, naturally occurring anticoagulants regulate the extension of the clotting process. They are antithrombin III (AT-III), protein C, protein S, and tissue factor pathway inhibitor (TFPI). AT-III is a serine protease inhibitor that regulates factor Xa and thrombin primarily and factors IXa, XIa, and XIIa to a lesser extent. When thrombin in flowing blood encounters intact endothelium, thrombin binds to thrombomodulin, its endothelial receptor. The thrombin-thrombomodulin complex then converts protein C into activated protein C. In the presence of the cofactor protein S, activated protein C proteolyses and inactivates factor Va and factor VIIIa. Inactivated factor Va is, in fact, a functional anticoagulant that inhibits clotting. TFPI limits activation of factor X by factor VIIa and tissue factor and shifts the activation site of tissue factor and factor VIIa to that of factor IX (see Figs. 469-1 and 469-2).
469.1 Clinical and Laboratory Evaluation of Hemostasis
Laboratory Tests
Prothrombin Time and Activated Partial Thromboplastin Time
Because clotting factors were named in the order of discovery, they do not necessarily reflect the sequential order of activation (Table 469-1). In fact, factors III, IV, and VI were not subsequently found to be independent proteins; thus, these terms are no longer used. The dual mechanisms of activating clotting have been termed the intrinsic (surface activation) and extrinsic (tissue factor–mediated) pathways. Study of the hemostatic mechanism is further complicated in that the interactions in vivo may use different pathways from those studied in clinical laboratory testing. PT measures the activation of clotting by tissue factor (thromboplastin) in the presence of calcium. Addition of tissue factor causes a burst of factor VIIa generation. The tissue factor–factor VIIa complex activates factor X. Whether factor X is activated by the intrinsic or the extrinsic pathway, factor Xa on the platelet phospholipid surface complexes with factor V and calcium (the “prothrombinase” complex) to activate prothrombin to thrombin (also referred to as factor IIa). Once thrombin is generated, fibrinogen is converted to a fibrin clot, the end-point of the reaction (see Fig. 469-2). PT is not prolonged with deficiencies of factors VIII, IX, XI, and XII. In most laboratories, the normal PT value is 10-13 sec. PT has been standardized using the International Normalized Ratio (INR) so that values can be compared from 1 laboratory or instrument to another. This ratio is used to determine similar degrees of anticoagulation with warfarin (Coumadin)–like medications.
| CLOTTING FACTOR | SYNONYM | DISORDER |
|---|---|---|
| I | Fibrinogen | Congenital deficiency (afibrinogenemia) or dysfunction (dysfibrinogenemia) |
| II | Prothrombin | Congenital deficiency or dysfunction |
| V | Labile factor, proaccelerin | Congenital deficiency (parahemophilia) |
| VII | Stable factor or proconvertin | Congenital deficiency |
| VIII | Antihemophilic factor | Congenital deficiency is hemophilia A (classic hemophilia) |
| IX | Christmas factor | Congenital deficiency is hemophilia B |
| X | Stuart-Prower factor | Congenital deficiency |
| XI | Plasma thromboplastin antecedent | Congenital deficiency, sometimes referred to as hemophilia C |
| XII | Hageman factor | Congenital deficiency is not associated with clinical symptoms |
| XIII | Fibrin-stabilizing factor | Congenital deficiency |
Mixing Studies
If there is unexplained prolongation of PT, PTT, or thrombin time, a mixing study is usually performed. Normal plasma is added to the patient’s plasma, and the PT or PTT is repeated. Correction of PT or PTT by 1 : 1 mixing with normal plasma suggests deficiency of a clotting factor, because a 50% level of individual clotting proteins is sufficient to produce normal PT or PTT. If the clotting time is not corrected or only partially corrected, an inhibitor is usually present. An inhibitor of clotting may be either a chemical similar to heparin that delays coagulation or an antibody directed against a specific clotting factor or the phospholipid used in clotting tests. In the inpatient setting, the most common cause of this finding is heparin contamination of the sample. The presence of heparin in the sample can be ruled in or out with the use of thrombin time and reptilase time, as noted earlier. If the mixing study is not corrected or if its result becomes more prolonged and the patient has clinical bleeding, an inhibitor of a specific clotting factor (antibody directed against the factor), most commonly factor VIII, factor IX, or factor XI, may be present. If the patient has no bleeding symptoms and both PTT and the mixing study are prolonged, a lupus-like anticoagulant (Chapter 476) is often present. Patients with these findings usually have a long PTT, do not bleed, and may have a clinical predisposition to excessive clotting.
Clotting Factor Assays
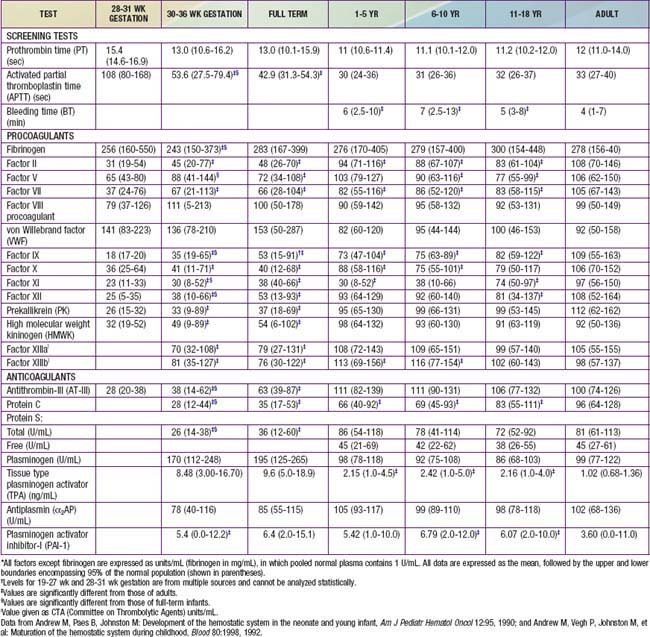
Each of the clotting factors can be measured in the clinical laboratory using individual factor–deficient plasmas. For most clotting factors, activity is measured against pooled normal plasma or against a standard, by which 100% activity is expressed as 100 IU/dL. By definition, 1 international unit (IU) of each factor is defined as that amount in 1 mL of normal plasma referenced against a standard established by the World Health Organization (WHO). For most clotting factors, the normal range is 50-150 IU/dL (50-150%) (Table 469-2).
Developmental Hemostasis
The normal newborn infant has reduced levels of most procoagulants and anticoagulants (see Table 469-1). In general, there is a more marked abnormality in the preterm infant. Although major differences exist in the normal ranges for newborn and preterm infants, these ranges vary greatly among laboratories. During gestation, there is progressive maturation and increase of the clotting factors synthesized by the liver. The extremely premature infant has prolonged PT and PTT values as well as a marked reduction in anticoagulant protein levels (protein C, protein S, and AT-III). Levels of fibrinogen, factors V and VIII, VWF, and platelets are near-normal throughout the later stages of gestation (Chapter 97.4). Because protein C and protein S are physiologically reduced, the normal factors V and VIII are not balanced with their regulatory proteins. In contrast, the physiologic deficiency of vitamin K–dependent procoagulant proteins (factors II, VII, IX, and X) is partially balanced by the physiologic reduction of AT-III. The net effect is that newborns (especially premature infants) are at increased risk for complications of bleeding, clotting, or both.
Cantor AB. Developmental hemostasis: relevance to newborns and infants. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:147-191.
Colman RW, Clowes AW, George JN, et al. Overview of hemostasis. In: Colman RW, Marder VJ, Clowes AW, et al, editors. Hemostasis and thrombosis: basic principles and clinical practice. ed 5. Lippincott: Williams and Wilkins; 2006:3-16.
Harrison P. The role of PFA-100 testing in the investigation and management of haemostatic defects in children and adults. Br J Haematol. 2005;130:3-10.
Lippi G, Franchini M, Montagnana M, et al. Coagulation testing in pediatric patients: the young are not just miniature adults. Semin Thromb Hemost. 2007;33:816-820.
Parmar N, Albisetti M, Berry LR, Chan AK. The fibrinolytic system in newborns and children. Clin Lab. 2006;52:115-124. erratum in Clin Lab 2006;52:324
Rajpurkar M, Lusher J. Clinical and laboratory approach to the patient with bleeding. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1449-1462.
Saxonhouse MA, Manco-Johnson MJ. The evaluation and management of neonatal coagulation disorders. Semin Perinatol. 2009;33:52-65.
Sugar NF, Taylor JA, Feldman KW. Bruises in infants and toddlers: those who don’t cruise rarely bruise. Arch Pediatr Adolesc Med. 1999;153:399-403.
Mann KG, Brummel-Ziedens K. Blood coagulation. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1399-1424.
Newman PK, Newman DK. Platelets and the vessel wall. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1379-1399.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 469 Hemostasis
The Process
After vascular injury, vasoconstriction occurs and flowing blood comes in contact with the subendothelial matrix (Fig. 469-1). In flowing blood, when exposed to subendothelial matrix proteins, von Willebrand factor (VWF) changes conformation and provides the glue to which the platelet VWF receptor, the glycoprotein Ib complex, binds, tethering platelets to sites of injury. When the VWF receptor binds its ligand, complex signaling occurs from the outside membrane receptor to intracellular pathways, activating the platelets and triggering secretion of storage granules containing adenosine diphosphate (ADP), serotonin, and stored plasma and platelet membrane proteins. Upon activation, the platelet receptor for fibrinogen, α2bβ3, is switched on (“inside out” signaling) to bind fibrinogen and triggers the aggregation and recruitment of other platelets to form the platelet plug. Multiple physiologic agonists can trigger platelet activation and aggregation, including ADP, collagen, thrombin, and arachidonic acid. Aggregation involves the interaction of specific receptors on the platelet surface with plasma hemostatic proteins, primarily fibrinogen.
One of the subendothelial matrix proteins that are exposed after vascular injury is tissue factor. Just as exposed subendothelial matrix proteins bind VWF, exposed tissue factor binds to factor VII and activates the clotting cascade, as shown in Figure 469-2. The activated clotting factor then initiates the activation of the next sequential clotting factor in a systematic manner. Our understanding of the sequence of steps in the cascade followed assignment of the numerals for the clotting factors for the participant proteins, and thus the sequence seems “out of numerical order.” During the process of platelet activation, internalized platelet phospholipids (primarily phosphatidylserine) become externalized and interact at 2 specific, rate-limiting steps in the clotting process—those involving the cofactors factor VIII (X-ase complex) and factor V (prothrombinase complex). Both of these reactions are localized to the platelet surface and bring together the active enzyme, an activated cofactor, and the zymogen that will form the next active enzyme in the cascade. This sequence results in amplification of the process, which supplies a burst of clotting where it is physiologically needed. In vivo, autocatalysis of factor VII generates small amounts of VIIa continuously, so the system is always poised to act. Near the bottom of the cascade, the multipotent enzyme thrombin is formed. Thrombin converts fibrinogen into fibrin, activates factors V, VIII, and XI, and aggregates platelets. Activation of factor XI by thrombin amplifies further thrombin generation and contributes to inhibition of fibrinolysis. Thrombin also activates factor XIII. The stable fibrin-platelet plug is ultimately formed through clot retraction and cross linking of the fibrin clot by factor XIIIa.
Virtually all procoagulant proteins are balanced by an anticoagulant protein that regulates or inhibits procoagulant function. Four clinically important, naturally occurring anticoagulants regulate the extension of the clotting process. They are antithrombin III (AT-III), protein C, protein S, and tissue factor pathway inhibitor (TFPI). AT-III is a serine protease inhibitor that regulates factor Xa and thrombin primarily and factors IXa, XIa, and XIIa to a lesser extent. When thrombin in flowing blood encounters intact endothelium, thrombin binds to thrombomodulin, its endothelial receptor. The thrombin-thrombomodulin complex then converts protein C into activated protein C. In the presence of the cofactor protein S, activated protein C proteolyses and inactivates factor Va and factor VIIIa. Inactivated factor Va is, in fact, a functional anticoagulant that inhibits clotting. TFPI limits activation of factor X by factor VIIa and tissue factor and shifts the activation site of tissue factor and factor VIIa to that of factor IX (see Figs. 469-1 and 469-2).
