Hemorrhagic and Thrombotic Disorders
APPROACH TO A CRITICALLY ILL PATIENT WITH HEMORRHAGE OR THROMBOSIS
LABORATORY TESTS OF COAGULATION
COMPLEX THROMBOHEMORRHAGIC DISORDERS
Laboratory Tests of Coagulation
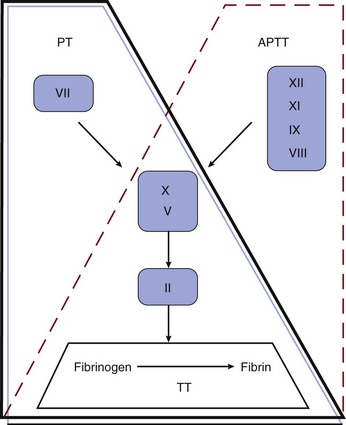
Although not reflective of the current model of coagulation, the integrity of coagulation is routinely tested by a limited set of in vitro assays (Fig. 78.1). The prothrombin time (PT) reflects the cascade of reactions traditionally called the extrinsic pathway, whereas the activated partial thromboplastin time (aPTT) reflects the intrinsic pathway. They intersect in the common pathway. The thrombin time (TT) measures the rate of conversion of fibrinogen to insoluble fibrin polymer after thrombin is added to plasma. A prolonged TT may be due to an inhibitor of thrombin (e.g., heparin, direct thrombin inhibitor [DTI]), hypofibrinogenemia or dysfibrinogenemia, fibrin degradation products (FDPs), and rarely paraproteins. The only specific coagulation factor that is routinely measured is fibrinogen. These four tests can usually localize abnormalities in the coagulation cascade.
Disorders of Platelets
Thrombocytopenia
Mechanisms and General Management
Thrombocytopenia is common in the intensive care unit (ICU). It has been estimated that 23% to 41% of patients in the ICU have a platelet count less than 100,000/µL, and 10% to 17% have a count less than 5000/µL.1 Common causes of thrombocytopenia in the ICU are shown in Box 78.1. In complex, acutely ill patients, many of these mechanisms may operate simultaneously. Severe sepsis is the most common cause of thrombocytopenia in the ICU.2
Frequently, thrombocytopenia must be managed without a specific diagnosis. Medications should be reviewed for potential offending agents.3,4 Inhibitors of platelet function should be avoided with platelet counts below 50,000/µL. If there is bleeding or if invasive procedures are anticipated, platelet transfusions should be given, unless contraindicated, to elevate the platelet count above 50,000/µL. In life-threatening situations, the goal should be a platelet count higher than 100,000/µL. In nonbleeding patients, maintenance of a platelet count above 10,000/µL (20,000/µL with fever or infection) with prophylactic transfusions is usually adequate.5
Pseudothrombocytopenia
Pseudothrombocytopenia is a laboratory artifact.6 The platelet count is factitiously lowered because of the presence of naturally occurring antibodies that cause platelet agglutination in the presence of ethylenediaminetetraacetic acid (EDTA) at room temperature. The diagnosis is suspected by finding platelet clumps on the peripheral blood smear. Repeating the platelet count with a different anticoagulant such as citrate will generally produce a normal platelet count.
Drug-Induced Thrombocytopenia
A limited number of drugs have evidence-based data to support a causal role in the development of thrombocytopenia.3,4,7,8 Medications commonly used in the ICU can cause thrombocytopenia (Box 78.2). Drug-induced thrombocytopenia most commonly occurs 7 to 21 days after exposure to the offending agent. Clinical manifestations can range from an asymptomatic decrease in platelets to life-threatening bleeding. The diagnosis is established by (1) finding a temporal relationship between starting the drug and the fall in the platelet count, (2) having no alternative diagnosis, and (3) having the platelet count recover after removal of the putative offending agent. Unfortunately, this is usually difficult to establish in the typical ICU patient. Treatment is based on removing the putative offending agent and initiating a drug of another class if possible. Though often used, steroids in general have not been shown to hasten the rate of platelet recovery. In severe thrombocytopenia with bleeding such as seen with quinine, intravenous immunoglobulin (IVIG) (1 g/kg/day for 2 days) and platelet transfusion are beneficial.
Glycoprotein IIb/IIIa Inhibitors
All platelet glycoprotein (GP) IIb/IIIa inhibitors have been associated with severe thrombocytopenia that can occur within hours of exposure (up to 2 weeks with abciximab).7 Heparin-induced thrombocytopenia (HIT) is the main differential diagnosis. Bleeding is very uncommon with HIT because of the strong prothrombotic state. Conversely, with GP IIb/IIIa inhibitor-associated thrombocytopenia, bleeding or hematoma formation may occur, especially at the site of the sheath. A platelet count less than 20,000/µL and clinical bleeding are indications for platelet transfusion. The use of IVIG and corticosteroids is not evidence based.
Immune Thrombocytopenic Purpura
Guidelines for the diagnosis and management of ITP have been developed.9,10 Indications for therapy in ITP are platelet count less than 20,000 to 30,000/µL or clinical bleeding. Corticosteroids (prednisone, 1 mg/kg/day, or pulse dexamethasone, 40 mg/day for 4 days monthly) are the usual initial therapy for ITP.9,10 Individuals with ITP in the ICU usually have severe or life-threatening bleeding. Several modalities of therapy should be used in concert to raise the platelet count in urgent situations (methylprednisolone, 1 g/day for 3 days, and IVIG, 1 g/kg/day for 2 days). Although platelets may be destroyed quickly, platelet transfusions should still be used as initial therapy. The response to platelet transfusion may improve after IVIG is given. Anti-Rh(D) IgG (WinRho), 50 to 75 µg/kg, has also been used.11 Because the dose of anti-Rh(D) must be reduced in face of anemia, its use may be problematic in a patient with severe bleeding. Anti-Rh(D) IgG is ineffective in Rh-negative patients and after splenectomy. ε-Aminocaproic acid (4-5 g IV followed by either 2-4 g IV every 4 hours or 0.5-1.0 g/hour continuous IV infusion [maximum 24 g/24 hours]) may be useful for mucosal bleeding and severe menorrhagia.
Post-transfusion Purpura
Post-transfusion purpura (PTP) is a rare condition characterized by acute, severe immune-mediated thrombocytopenia. PTP occurs in human platelet antigen-1 (HPA-1) negative individuals who receive HPA-1 positive platelets (most commonly as a contaminant in packed red blood cells [RBCs]). These individuals have previously been sensitized to HPA-1 through transfusion or pregnancy. Usually 7 to 10 days after reexposure to HPA-1, an anamnestic response occurs, resulting in a precipitous fall in the platelet count. Petechiae and bleeding are common. IVIG and plasma exchange are effective.12
Acquired Platelet Dysfunction
Medication-Induced Abnormalities
Aspirin irreversibly acetylates cyclooxygenase (COX), inhibiting platelet function for the life of the platelet (7 to 10 days). The aspirin effect can be overcome with platelet transfusion or the infusion of desmopressin (DDAVP). The effect of NSAIDs is reversible and disappears as the drug is cleared, usually within 24 to 48 hours for ibuprofen. The risk of bleeding from NSAIDs is lowest with ibuprofen and greatest with ketorolac. The thienopyridine clopidogrel tightly binds the platelet ADP P2Y12 receptor. Clopidogrel should be withheld for 5 to 7 days before elective surgery or invasive procedures.13 In an emergency, platelet transfusion can be tried, but platelet function may not be fully restored because of circulating active clopidogrel metabolites, which have a half-life of 8 hours and bind to the transfused platelets. Prasugrel is a new thienopyridine whose active metabolites have a 4-hour half-life. COX-2 inhibitors don’t affect platelet function.
Renal Failure
The hemorrhagic diathesis of renal failure is the result of metabolic derangements related to uremic toxins.14 Bleeding may worsen when the hematocrit falls below 30% due to a rheologic phenomenon in which rapidly flowing RBCs gravitate to the center of the streaming blood and force the platelets toward the vessel wall. Uremic bleeding is uncommon in the modern era of dialysis.15 For clinical bleeding, intravenous DDAVP (0.3 µg/kg given over 30 minutes) is often therapeutic.14 If the hematocrit is less than 30%, the patient should be transfused with packed RBCs.14,15 An alternative to DDAVP is cryoprecipitate (10 bags every 12 to 24 hours).14 Intravenous conjugated estrogen, 0.6 mg/kg/day for 5 days, is also effective.14 An unexpected “coagulopathy” (prolonged aPTT and TT) may develop as a result of delayed clearance of heparin after dialysis. If available, an anti–factor Xa assay will show the presence of heparin.
Complex Thrombohemorrhagic Disorders
Heparin-Induced Thrombocytopenia
Although commonly looked for, HIT is an uncommon cause of thrombocytopenia in the ICU.2,16 HIT is a paradoxical condition in which modest thrombocytopenia may be associated with devastating heparin-induced thrombocytopenia thrombosis (HITT). Thus, the entire health care team needs to be vigilant for the development of HIT in any patient receiving heparin. HIT has been associated with all types of heparin (unfractionated [UFH] and low-molecular-weight [LMWH]), at any dose, and by any route, including flushes and heparin-coated catheters.1,17 The incidence of HIT has been estimated to be less than 1% of patients in the ICU.2,16,18 HIT occurs in three time intervals. Classic HIT occurs 5 to 15 days after the initiation of heparin. Rapid-onset HIT develops hours to 1 to 2 days after heparin is started in individuals who have preformed circulating antibodies from a previous exposure to heparin, usually in the last 2 months. Classic and rapid-onset HIT may be manifested as thrombocytopenia with or without thrombosis [HIT(T)]. Delayed-onset HIT occurs an average of 12 days after the discontinuation of heparin and is manifested as isolated thrombosis. The thrombocytopenia of HIT is usually modest with an average platelet count of 50,000 to 60,000/µL.19 Severe thrombocytopenia (<10,000/µL) should suggest an alternative diagnosis. HIT may be associated with a normal platelet count if there is a baseline thrombocytosis (e.g., postoperatively). HIT may be manifested as isolated thrombocytopenia or thrombocytopenia with potentially devastating thrombosis. Venous complications include deep venous thrombosis (DVT), pulmonary embolism (PE), cerebral sinus thrombosis, infarctive adrenal hemorrhage, and skin necrosis at the heparin injection site. Arterial complications include iliofemoral artery thrombosis, digital ischemia, myocardial infarction, stroke, and mesenteric artery thrombosis.
HIT is an immune-mediated process in which the heparin–platelet factor 4 complex becomes immunogenic. Antibodies are formed that activate platelets, causing the release of thrombogenic microparticles.16,19 The antibodies can also activate monocytes and endothelial cells.
The diagnosis of HIT is clinicopathologic. HIT should be thought of in appropriate clinical situations (Box 78.3). The “4T” score has been used to predict the likelihood (pretest probability) of HIT (Table 78.1).16,20 The HEP score, devised by an expert consensus panel, has recently been validated.21 Although the decision to initiate therapy should be based on the clinical likelihood of HIT, laboratory testing should be used for the retrospective confirmation of the pretest likelihood of HIT. The complexities of HIT antibody testing are reviewed elsewhere.16,22 In general, a strongly positive HIT enzyme-linked immunosorbent assay (ELISA) and strongly positive serotonin release assay (SRA) with a high pretest probability confirm the diagnosis of HIT, whereas a negative ELISA and SRA make the diagnosis of HIT unlikely with a low pretest probability. In other situations, clinical judgment prevails in establishing or excluding a diagnosis of HIT, especially because ELISA and SRA testing from commercial labs has not been validated in clinical studies.1,16,22
Table 78.1
Determining Pretest Probability of Heparin-Induced Thrombocytopenia (HIT): The “4 Ts”
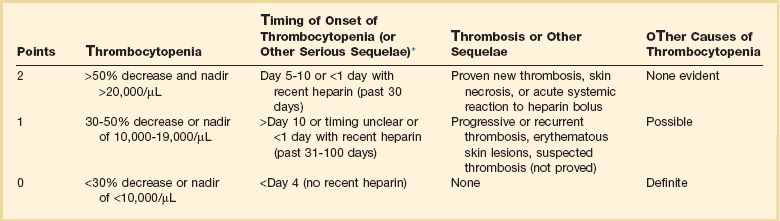
*First day of heparin exposure = day 0.
Adapted from Lo GK, Juhl D, Warkentin TE, et al: Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost 2006:4:759.
Once the diagnosis of HIT is thought to be likely, all heparin should be discontinued. Heparin-coated pulmonary artery catheters should be replaced with noncoated catheters. Catheters for dialysis or apheresis should be locked with 4% citrate or tissue plasminogen activator. The patient’s chart and bedside should be labeled heparin allergy. Platelet transfusions should be avoided except for life-threatening bleeding, given the anecdotal observations of acute thrombosis occurring after platelet transfusion. A patient with life- or limb-threatening arterial thrombosis should be evaluated for surgical intervention. Anticoagulation with a DTI (Table 78.2) should be initiated in all patients unless contraindicated. LMWH is contraindicated in HIT caused by UFH. Conversion to warfarin can be considered after a minimum of 5 days of DTI therapy if the platelet count has returned to normal (suggesting that the process has cooled off and the patient is no longer hyperprothrombotic) and no future invasive procedures are planned. Simply discontinuing heparin therapy and not starting a DTI is inappropriate because occult thrombosis may have already developed.23 Starting or continuing warfarin monotherapy is also contraindicated because of the risk of venous limb gangrene.24 If warfarin has been given at the time HIT is diagnosed, vitamin K should be given to replenish proteins C and S. If invasive procedures are needed (e.g., tracheostomy, pacemaker), it is best to delay them, if medically safe, until the platelet count is normal to minimize the risk of developing thrombosis during the time that DTI therapy is withheld. Inferior vena cava (IVC) filters should be avoided because of the risk of vena cava thrombosis. Patients with active HIT may need percutaneous coronary intervention (PCI). Argatroban has been approved by the Food and Drug Administration (FDA) for use during PCI in patients with HIT.25 Though not FDA approved, bivalirudin has been used safely in this situation.26 In a patient who has active HIT or persistent HIT antibodies and who needs open heart surgery, medical management is recommended until the antibody becomes negative. If urgent surgery is needed, bivalirudin is most commonly used.27 For the patient with a past history of HIT who needs open heart surgery and is currently HIT antibody negative, heparin can be used during bypass and a DTI started as soon as it is surgically safe postoperatively.28
Table 78.2
Parenteral Anticoagulant Doses
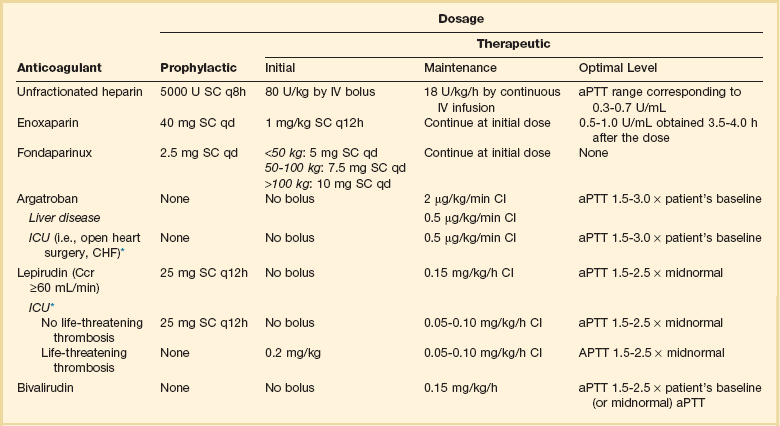
*Suggested dose modification in ICU patients.
Data from Selleng K, Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia in intensive care patients. Crit Care Med 2007;35:1165-1176.
Thrombotic Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura (TTP) is a relatively rare disorder whose hallmarks are thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and neurologic dysfunction. The diagnosis of TTP should be considered in any patient with unexplained thrombocytopenia and MAHA.29 TTP is the clinical manifestation of a heterogeneous group of underlying disorders driven by different pathophysiologic processes. Although most cases of TTP are idiopathic, TTP may be associated with exposure to drugs (e.g., ticlopidine, clopidogrel, quinine, mitomycin C, gemcitabine, cyclosporine), pregnancy, HIV infection, and bone marrow transplantation.30 The morphologic hallmark of TTP is hyaline thrombi in precapillary arterioles composed primarily of platelets. Classic TTP is due to an immune-mediated deficiency of the metalloproteinase ADAMTS13, which cleaves ultralarge multimers of von Willebrand factor (vWF) into smaller forms that are less platelet reactive.31 Deficiency of this enzyme allows ultralarge forms of vWF that are normally sequestered in the endothelium to bind platelets and form microthrombi in the circulation. Conversely, ADAMTS13 activity is normal in TTP associated with bone marrow transplantation and the hemolytic-uremic syndrome.30,31 Organ dysfunction is due to microvascular thrombosis. Commonly involved organs are the brain, kidneys, heart, and pancreas. It is critical to establish the diagnosis quickly because TTP can be rapidly fatal if not properly treated.
Once the diagnosis of TTP is thought to be likely, therapy should be initiated quickly because of the proclivity of the disease to progress rapidly. All patients should initially be treated in the ICU. Platelet transfusions should be avoided except for life-threatening bleeding because of anecdotal reports of acute decompensation after platelet transfusions. A large-bore catheter will need to be placed, even in the face of severe thrombocytopenia, because the mainstay and only evidence-based component of therapy for TTP is plasma exchange.32,32a Most commonly, 1.5 plasma volumes are exchanged daily with FFP. A clue that TTP is the correct diagnosis is the color of the plasma removed from the first plasmapheresis. A red or brown color suggests free hemoglobin from intravascular hemolysis. If plasma exchange cannot be initiated in a timely manner, infusion of 30 mL/kg of FFP daily can be a temporizing maneuver.32 Corticosteroids (e.g., prednisone, 1 mg/kg/day) are commonly used as well.32a The use of antiplatelet agents, which may increase the risk of bleeding, and vincristine is controversial.33 In patients who do not respond to initial therapy, rituximab is an attractive second-line therapy.30,34
Hemolytic-Uremic Syndrome
Hemolytic-uremic syndrome (HUS), the triad of thrombocytopenia, MAHA, and acute kidney injury, is most commonly due to infection with Shiga toxin–producing Escherichia coli in children.35 Adult atypical HUS (aHUS) is not Shiga toxin associated. It may be idiopathic or associated with calcineurin inhibitors (cyclosporin, tacrolimus), pregnancy, and HIV. aHUS is associated with low C3 and is a complement deposition disease. Mutations in complement factor H (CFH) are found in a minority of cases. Although not as efficacious as when used for TTP, plasma exchange is used for aHUS. The anti-C5 antibody eculizumab (Soliris) has been FDA approved for the treatment of aHUS.36 Plasma exchange should be initiated while waiting to obtain eculizumab. Supplemental eculizumab dosing is required after each plasma exchange.37 Patients need to receive quadravalent meningococcal vaccine before starting eculizumab.
Disseminated Intravascular Coagulation
DIC is always a manifestation of a severe underlying pathologic process (Box 78.4). The final common pathway is the generation of thrombin, which produces microthrombi that can lead to organ dysfunction.38 In the process, natural anticoagulants are consumed and fibrinolysis is activated. Circulating FDPs, along with the consumption of platelets and coagulation factors, lead to a bleeding diathesis. The balance between these competing processes results in the clinical manifestations in any particular individual.
There are no strict, evidence-based laboratory criteria for the diagnosis of DIC.38 The diagnosis is based on identifying an underlying predisposition, the clinical features, and laboratory testing. The most common laboratory features of DIC are thrombocytopenia, elevated FDP or D-dimers, and decreased fibrinogen. The PT and aPTT may or may not be prolonged, depending on the sensitivity of the assay, degree of baseline elevation of coagulation factors as acute-phase reactants, the rate of consumption of coagulation factors, and the amount of FDPs released. An absolute decrease in fibrinogen is very suggestive of DIC. However, fibrinogen activity in the normal range may still be consistent with DIC because fibrinogen is an acute-phase reactant and its level may be inappropriately low for the underlying physiologic state.39 Serial measurement of fibrinogen is often useful. Schistocytes are seen in half the cases. Although the natural anticoagulants antithrombin (AT) (previously called AT-III) and proteins C and S are decreased, and thrombin-antithrombin complexes are increased, their levels usually cannot be measured in real time and are of limited clinical value.
The International Society of Thrombosis and Haemostasis (ISTH) has developed a scoring system for the identification of overt DIC using four commonly available tests (platelets, prothrombin time, FDPs such as D-dimer or soluble fibrin monomer, and fibrinogen) (Table 78.3), which has been prospectively validated.40,41 It is important to remember that all the laboratory abnormalities seen in DIC can be caused by other disease processes. Vitamin K deficiency,42 a heparin-contaminated blood sample, or a lupus anticoagulant can cause a coagulopathy, whereas elevations in FDPs can be seen with metastatic cancer or can be found after surgery or trauma. Other microangiopathies cause thrombocytopenia and RBC fragmentation. Liver failure can produce a constellation of laboratory abnormalities indistinguishable from those observed in DIC.43
Table 78.3
| Test | Score |
| Platelet count (per µL) | |
| ≥100,000 | 0 |
| 50,000-99,999 | 1 |
| <50,000 | 2 |
| D-dimer (µg/mL) | |
| ≤0.39 (upper limit of normal) | 0 |
| 0.40-4.0 | 2 |
| >4.0 | 3 |
| Prothrombin time prolongation (s) | |
| ≤3 | 0 |
| >3 but <6 | 1 |
| ≥6 | 2 |
| Fibrinogen (mg/dL) | |
| >100 | 0 |
| <100 | 1 |
Original ISTH score: (platelets + D-dimer + PT + fibrinogen) ≥ 5 signifies overt DIC.
Modified ISTH score: (platelets + D-dimer + PT) ≥ 5 signifies overt DIC in severe sepsis.
ISTH, International Society of Thrombosis and Haemostasis; PT, prothrombin time.
Adapted from Taylor FB, Toh CH, Hoots WK, et al: Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 2001;86:1327; and Dhainaut J-F, Yan SB, Joyce DE, et al: Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost 2003;2:1924.
Other treatment modalities have been tried in DIC. The use of AT concentrates showed no significant decrease in mortality rate.44 Activated protein C (drotrecogin alfa) used for the treatment of sepsis has been withdrawn from the U.S. market. Because of the intense fibrinolytic activity in DIC, antifibrinolytic agents (ε-aminocaproic acid and tranexamic acid) have been tried in an attempt to reduce bleeding. In general, failure rates have been quite high, and serious thromboses have occurred because the lysis of diffuse microthrombi was suppressed.
Disorders of Hemostasis
Coagulation Factor Abnormalities
Acquired Deficiencies of Procoagulants
Vitamin K Deficiency
Because factor VII has the shortest half-life (approximately 4 hours), early vitamin K deficiency is manifested by an isolated prolongation of the PT. As factors II, IX, and X become depleted, the aPTT will become prolonged. Vitamin K deficiency is common in critically ill patients and may result in serious bleeding.42 Treatment of vitamin K deficiency depends on the clinical situation. In a nonbleeding patient, vitamin K should be given enterally if the gut is working. Subcutaneous vitamin K should be avoided if the gut is working because of its erratic absorption, especially in face of impaired tissue perfusion and because anaphylactic reactions have been reported with subcutaneous vitamin K.45 In a bleeding patient, FFP will correct the coagulopathy most quickly. In critically ill patients with an urgent need for vitamin K replacement, phytonadione may be administered intravenously if the benefits are thought to outweigh the risk of anaphylaxis. Vitamin K should be given no faster than 1 mg/minute with cardiorespiratory monitoring because anaphylactic reactions can occur even at this slow infusion rate.45 One milligram of vitamin K twice a week can prevent vitamin K deficiency.
Liver Disease
The perturbation of hemostasis in liver disease is complex. All hemostatic factors except for vWF and plasminogen activator inhibitor-1 are synthesized in the liver. Loss of the ability to synthesize factor VII is disproportionately greater in patients with liver disease than in those receiving warfarin. Thus, the bleeding risk of a prolonged international normalized ratio (INR) from liver disease is not as great as the same INR with warfarin therapy.46 As outlined later, changes in factor VIII/vWF, and decreased synthesis of natural anticoagulants and fibrinolytic proteins, may help keep the patient with liver disease in hemostatic balance,43 making the autoanticoagulation of liver disease promulgated among house staff a myth.
Although hepatic dysfunction is most commonly associated with bleeding, the risk of thrombosis has been underappreciated.43 Synthesis of hemostatic proteins becomes impaired when the albumin falls below 2.5 g/dL. Similar to vitamin K deficiency, the earliest abnormality is a reduction in factor VII. In more severe liver disease, the other coagulation factors become deficient. Factor V and fibrinogen do not usually decrease significantly until the liver failure is severe. In contrast, the activities of factor VIII and vWF are normal or elevated. AT, proteins C and S, and plasminogen are decreased. Hepatic clearance of activated procoagulants and FDP is reduced. Thrombocytopenia may develop due to decreased thrombopoietin production or splenic sequestration. Advanced liver disease may also be complicated by vitamin K deficiency from malnutrition and decreased vitamin K absorption due to cholestasis.
A variety of approaches have been used to treat the coagulopathy of liver disease. If the patient is not bleeding, a trial of vitamin K can be given for 3 days to see if the coagulopathy corrects. Because all patients with liver disease have some degree of biliary obstruction and fat malabsorption, the subcutaneous route is preferred over oral administration. Although the risk of anaphylaxis with intravenous vitamin K is low, it is not generally recommended empirically in a patient with liver disease because of the low likelihood that vitamin K will completely correct the coagulopathy.45 If bleeding is a problem, FFP should be administered. Because the relationship between the levels of coagulation factors and the PT/PTT is nonlinear, the greater the prolongation of the PT and PTT, the greater the chance of correction with FFP. However, it is difficult to normalize the PT.47 On average, a unit of FFP increases all coagulation factors by 6% to 7%. A typical patient with the coagulopathy of liver disease may have approximately 10% to 15% of the normal level of coagulation factors. If a factor level of 50% of normal is needed for hemostasis, 6 units of FFP would be expected to correct the bleeding diathesis. The PT and PTT may still be prolonged, however, depending upon the sensitivities of PT and PTT assays. The PT (and aPTT) should be checked immediately after the FFP is administered. Given the short half-life of factor VII, waiting until the next morning’s routine blood draw will produce a prolongation of at least the PT and give the impression that the coagulopathy is uncorrectable.
Massive Transfusion
Massive volume replacement (>1 blood volume in less than 24 hours) may not give the liver sufficient time to replace coagulation factors. In a hemodynamically stable patient, coagulation studies should be followed serially and FFP infused when a coagulopathy develops. Conversely, exsanguinating patients should be managed with balanced transfusion of RBC : FFP:platelets in a ratio of 1 : 1 : 1 (either 1 unit of single donor or 6 units or random donor platelets).48
Acquired Circulating Inhibitors
Factor VIII Inhibitors
The clinical manifestations of factor VIII inhibitors can range from an unexplained prolongation of the aPTT to severe bleeding. The aPTT is markedly prolonged with a normal BT, PT, TT, and fibrinogen. The mixing study does not correct. Further evaluation includes factor VIII, IX, XI, and XII activities and a lupus anticoagulant panel; hematomas should not be drained except in severe circumstances (e.g., compartment syndrome) because of the risk for bleeding, even with replacement therapy. Treatment options depend on the severity of the inhibitor. A low-titer inhibitor (<5 Bethesda units [BU]) may be treated with factor VIII infusion (200 U/kg every 8 to 12 hours). For a high-titer antibody (>5 BU), the treatment options are recombinant human factor VIIa (rH-VIIa; NovoSeven) and virally inactivated concentrates containing activated coagulation factors (FEIBA [factor eight inhibitor bypass activity]). The dose of rH-VIIa is 90 µg/kg, which can be repeated every 2 hours until the bleeding stops. rH-VIIa carries a risk of thrombotic and ischemic events even in individuals with severe coagulopathy.49 Coagulation studies are not used to adjust the dose. The PT always falls below the lower end of normal. The FEIBA dose is 50 to 100 U/kg, which is repeated every 6 to 12 hours (maximum daily dose 200 U/kg/day due to thrombotic risk). A total of 500 units of heparin can be added to each bag to prevent infusion phlebitis. The clinical response to FEIBA is monitored.
Inherited Deficiencies of Procoagulants
von Willebrand Disease
vWD is inherited in an autosomal dominant manner with partial penetrance. The clinical features, diagnosis, and subclassification of vWD are reviewed elsewhere.50
Factor VIII Deficiency
For major surgery, closed-head trauma, major trauma, or life-threatening hemorrhage, the factor VIII level should be raised to 100% and then maintained continuously above 50% for the first 5 to 7 days and above 40% for an additional 5 to 7 days. rH-VIII is the treatment of choice because it has no risk of viral infection. One unit of factor VIII per kilogram should raise the plasma factor VIII activity by 2%. Thus, the initial dose is 50 U/kg followed by 25 U/kg every 8 hours. Peak and trough factor VIII levels should be obtained after the first dose and at least daily. In an emergency situation if rH-VIII is not available, virally inactivated factor VIII would be the treatment of choice. The dosing schema is similar to that of rH-VIII. In an emergency situation in a smaller facility that does not stock hemostatic factors on site, cryoprecipitate can be given every 8 hours until the patient can be transferred. Similar to vWD, DDAVP can be given for mild or moderate factor VIII deficiency in known DDAVP responders. As the hemophilia population ages, coronary artery disease (CAD) is becoming an increasing problem. Individuals with hemophilia A should be supported through indicated cardiac procedures and surgery.51
Factor XI Deficiency
Factor XI deficiency is an autosomal recessive disorder most commonly seen in Ashkenazi Jews. The bleeding history is variable, ranging from menorrhagia to bleeding with delivery or surgery. Therapy depends on the clinical situation.52 For severe deficiency, FFP (10 to 20 mL/kg followed by 3 to 6 mL/kg every 6 hours) is used. The goal is factor XI activity greater than 40% to 45%. Individuals with factor XI deficiency may be seen in the ICU because factor XI deficiency is not protective against coronary artery disease. These individuals may require emergency cardiac catheterization and coronary intervention.53 Clopidogrel has been given safely after coronary stenting.54
Venous Thromboembolism
Venous thromboembolism (VTE) is common in critically ill patients. Twenty-nine percent of medical ICU patients have been found to have asymptomatic lower extremity DVT, although 5.8% and 4.2% have symptomatic DVT and PE, respectively.55 Most patients admitted to the ICU have multiple risk factors including recent surgery, trauma, sepsis, malignancy, immobilization, stroke, advanced age, heart or respiratory failure, previous VTE, and indwelling catheters.55–57
Deep Venous Thrombosis Prophylaxis
All patients admitted to the ICU should be evaluated for their risk for DVT. The perceived risk of thrombosis and bleeding needs to be assessed in each ICU patient.55 In medical patients, options are LMWH and UFH.55 In medical patients with a high bleeding risk, mechanical prophylaxis (graded compression stockings and intermittent pneumatic compression devices) is recommended.55 Beyond the size limitations of this chapter, specific recommendations for DVT prophylaxis are available for trauma, abdominal/pelvic, thoracic, cardiac, and neurologic surgery.58
Therapy for Venous Thromboembolism
In the noncritical care setting, VTE is routinely treated with at least 5 days of UFH, LMWH, or fondaparinux (see Table 78.2), followed by warfarin.59 In the ICU, intravenous UFH is the safest mode of anticoagulation give its rapid reversabilty for procedures or bleeding. LMWH has the disadvantages of a longer half-life and being only partially reversed with protamine. As discussed in the section on anticoagulation, fondaparinux should not be used routinely in the ICU. ICU patients should not be transitioned to warfarin given their risk of bleeding and frequent need for invasive procedures. Warfarin is hard to regulate due to frequent drug interactions (especially with antibiotics) and erratic oral intake. Warfarin may stick to feeding tubes.60 Warfarin is absorbed in the upper small bowel and may not be absorbed if given via J-tube.61
Inferior Vena Cava Filter
Approximately 80% of PE occur as a result of the lower extremity DVT. IVC filter placement is indicated in two situations: (1) acute DVT and a contraindication to anticoagulation (e.g., recent intracranial hemorrhage or stroke, recent neurologic or ophthalmologic surgery, active gastrointestinal hemorrhage, cerebral metastasis with a high risk of bleeding [e.g., melanoma, renal cell, seminoma]62), and (2) recurrent PE despite therapeutic anticoagulation. Filters may have some benefit in face of PE causing hypotension.59 IVC filters are not indicated for PE without DVT. IVC filters do not eliminate all risk of PE and do not decrease mortality rates.59 There is an approximate 10% risk of thrombosis at the filter insertion site.59
Anticoagulants
A variety of parenteral and oral anticoagulants acting through different mechanisms are commercially available with many more in development. Commonly used dosing schema are shown in Table 78.2.
Heparin, Heparin Derivatives, and Heparinoid
Heparin
Heparin is a glycosaminoglycan that anticoagulates blood by augmenting the activity of AT.63 Complexes of heparin and AT inhibit thrombin, factor Xa, and other procoagulant proteases. The first five sugars of heparin bind to AT. The additional sugars determine the protein binding properties and pharmacokinetics of UFH and LMWH.63
Unfractionated Heparin
The goal of therapeutic heparin is to maintain the aPTT in a range that corresponds to a heparin level of 0.3 to 0.7 U/mL.63 Given the marked variability in aPTT response to heparin because of differences in reagents and laboratory equipment, this range needs to be determined in each laboratory. Because some hospitals do not have a heparin protocol, an adaptation of the Raschke nomogram is shown in Table 78.4.63,64 An aPTT ratio of 1.5 to 2.5 times the control value correlates very poorly with the anti-Xa heparin assay and should not be used. The dose of heparin is weight based. No adjustments are needed for obesity, hepatic dysfunction, or renal impairment. The hemoglobin and platelet count should be monitored daily as surveillance for occult bleeding and HIT. Overanticoagulation with heparin can usually be managed by simply interrupting the infusion. With serious bleeding, heparin can be reversed immediately with intravenous protamine sulfate.63 One milligram of protamine will neutralize 100 units of heparin. If bleeding occurs during the constant infusion, the amount of heparin given during the previous 2 hours should be used for the calculation. Thus, a patient bleeding after a 5000-unit heparin bolus will need 50 mg of protamine, whereas a patient receiving a 1250 U/hour infusion will need only 25 mg of protamine. Protamine should be given slowly over 1 to 3 minutes to minimize the risk for hypotension and bradycardia. The risk for anaphylaxis is increased in those who have received protamine zinc insulin or who have had a vasectomy.
Table 78.4
| Anti–Factor Xa (U/mL) | aPTT |
| Initial dose | 80 U/kg bolus, then 18 U/kg/hour |
| <0.15 | 80 U/kg bolus, increase by 4 U/kg/hour |
| 0.15-0.29 | 40 U/kg bolus, increase by 2 U/kg/hour |
| 0.30-0.70 | No change |
| 0.71-0.85 | Decrease infusion by 2 U/kg/hour |
| >0.85 | Hold 1 hour, decrease infusion by 3 U/kg/hour |
aPTT, activated partial thromboplastin time.
Adapted from Raschke RA, Reilly BM, Guidry JR, et al: The weight based heparin dosing nomogram compared with a “standard care” nomogram: A randomized controlled trial. Ann Intern Med 1993;119:874.
Low-Molecular-Weight Heparin
The properties of LMWH vary significantly from those of UFH.63 The anti-Xa : anti-IIa ratio of LMWH is approximately 4 : 1. Thus, LMWH does not significantly affect the aPTT. There is little protein binding, allowing for predictable renal excretion. The properties of the available LMWHs (enoxaparin, dalteparin, tinzaparin) are different. Their kinetics and dosing are not interchangeable.63 In a normal-sized adult (50 to 150 kg) with normal renal function, monitoring is not necessary. If monitoring is necessary (weight outside the usual range, impaired renal function, pregnancy), anti-Xa activity is measured 3.5 to 4.0 hours after an enoxaparin dose. The therapeutic range for enoxaparin (1 mg/kg every 12 hours) is 0.5 to 1.0 U/mL. LMWH is only 60% reversible by protamine.63 If bleeding occurs within 8 hours of a LMWH injection, 1 mg of protamine per 100 anti-Xa units should be given. Because of the kinetics of LMWH, a second protamine dose of 0.5 mg per 100 anti-Xa units should be given if the bleeding persists.63
Pentasaccharide
Fondaparinux (Arixtra) is a synthetic analog of the first five sugars of heparin. Due to its small size, it is a pure factor Xa antagonist. Fondaparinux is approved for treatment of DVT and PE and for DVT prophylaxis in orthopedic and abdominal surgery. Although not FDA approved, fondaparinux has been used off-label for the prophylaxis and treatment of HIT(T).16 As a result of its 18- to 21-hour half-life, its use in the ICU is limited because it is not rapidly cleared in a patient population that needs frequent procedures, that has a high risk of bleeding, and often has renal impairment (contraindicated with creatine clearance [Ccr] < 30 mL/minute). It does not prolong the PT, aPTT, or TT. It can be measured in the anti-Xa assay calibrated for fondaparinux. It is not reversed by protamine. Bleeding due to pentasaccharide should be supported by transfusion. rH-VIIa can be tried.
Heparinoid
Danaparoid is a mixture of sulfated glycosaminoglycans, distinct from heparin, that produces an anticoagulant effect primarily by enhancing the activity of AT. Danaparoid is primarily used in the setting of HIT. Dosing schema are available for most clinical situations.65 If monitoring is necessary, such as during renal failure, anti-Xa activity must be determined. Though no longer available in the United States, danaparoid is available in Canada and Europe.
Direct Thrombin Inhibitors
Argatroban is an arginine analog with a 40-minute half-life. It is metabolized in the liver, and its half-life increases to around 140 minutes with hepatic dysfunction. Because of its short half-life, an initial bolus is not needed. Although argatroban is monitored by the aPTT, it has a significant effect on the PT.66 Argatroban monotherapy will approximately double the INR and can markedly prolong the INR with warfarin cotherapy. As long as the aPTT is therapeutic, this exaggerated increase in INR does not increase the bleeding risk.
Hirudin is a family of recombinant proteins originally extracted from the salivary glands of leeches.67 Lepirudin is used subcutaneously or intravenously. The half-life of intravenous lepirudin is 80 to 180 minutes but extends to days with severe renal failure. Although lepirudin can be removed by hemofiltration, the filters are not approved for use in the United States.68 It should not be used in those with sulfite sensitivity. Approximately 40% of individuals exposed to lepirudin will develop antilepirudin antibodies, which will impair drug excretion and which may cause anaphylaxis on reexposure. Lepirudin is no longer produced for the European Union.
Bivalirudin is a reversible synthetic analog of hirudin. Bivalirudine has the advantage of a 25-minute half-life. Although it is cleared primarily by intravascular proteolysis, the dose must be reduced with renal dysfunction. Although not FDA approved, bivalirudin is used off-label in many centers for systemic anticoagulation for HIT(T).69 It is also used off-label for bypass surgery in individuals with HIT.27
Dabigatran (Pradaxa) is the first FDA approved oral DTI. It is approved for prevention of stroke and systemic embolization with nonvalvular atrial fibrillation.70 It should be used with extreme caution in the elderly, especially because of the risk of bleeding due to overestimation of the Ccr and risk of trauma in the infirm. The dose is 150 mg twice a day (if Ccr > 30 mL/minute). Although not FDA approved, dabigatran has shown efficacy for the treatment of VTE.71 It binds free and clot bound thrombin, and is primarily cleared via the kidneys with a half-life of 12 to 17 hours.72 Drugs that are metabolized via P-glycoprotein may alter metabolism of dabigatran, which is a prodrug. There is no standardized laboratory test to measure the dabigatran level. Thus, in a patient who is having bleeding or thrombotic events while taking dabigatran, there is no test to tell whether the event may be related to over- or underanticoagulation. Given its sensitivity, a normal TT suggests that there is no dabigatran present. How long dabigatran should be discontinued before procedures depends upon the renal function and the bleeding risk of the procedure.72 Dabigatran has no antidote. In the bleeding patient, in addition to supportive care and transfusion, the use of activated charcoal (if within 2 hours of ingestion), FEIBA, rH-VIIA, and dialysis have been suggested. Although antifibrinolytics have been shown to decrease bleeding after trauma,73 they should be used with trepidation with FEIBA and rH-VIIa because of the risk of thrombosis.
Oral Factor Xa Inhibitors74
Apixiban
Apixiban (Eliquis), 5 mg twice a day, has been shown to be superior to warfarin for stroke prevention in atrial fibrillation and is under FDA review.75 Its elimination is fecal and renal with an 8- to 15-hour half-life. Platelet GP and strong CYP3A4 inhibitors significantly increase its plasma concentration.
Rivaroxaban
Rivaroxaban (Xarelto), 20 mg daily, is FDA approved for the prevention of stroke and systemic embolization with nonvalvular atrial fibrillation, and doses of 10 mg daily have been approved for the prevention of VTE with knee and hip replacement surgery.76 Although not FDA approved, it has been shown to be efficacious for the treatment of DVT and PE.77 It has a 5- to 9-hour half-life (9-13 hours in the elderly) and is metabolized primarily via the cytochrome system. Combined platelet GP and strong CYP3A4 inhibitors may significantly increase the plasma concentration of rivaroxaban. It should be avoided in face of significant hepatic dysfunction, and dose reduction is required for a Ccr less than 50 mL/minute. It should be discontinued at least 24 hours before surgery. Rivaroxaban activity can be measured by the anti-Xa assay used to monitor heparins. Unfortunately, at the time of this writing, standards to calibrate the assay are not available in the United States. Prolongation of the INR by rivaroxaban has been reversed with the use of PCC in healthy volunteers.78 There are no data on the efficacy of PCC in bleeding patients. Rivaroxaban is not dialyzable.
Warfarin
The response to warfarin is variable and is affected by many factors common in the ICU, such as poor nutrition (vitamin K deficiency), liver dysfunction, and coadministration of medications that affect warfarin pharmacokinetics. It is not uncommon for patients to become over-anticoagulated with warfarin. An approach to warfarin reversal is shown in Box 78.5.79,80
References
1. Napolitano, LM, Warkentin, TE, Almahameed, A, et al. Heparin-induced thrombocytopenia in the critical care setting: Diagnosis and management. Crit Care Med. 2006; 34:2898–2911.
2. Greinacher, A, Selleng, K. Thrombocytopenia in the intensive care unit patient. Hematol Am Soc Hematol Educ Program. 2010; 2010:135–143.
3. George, JN, Raskob, GE, Shah, SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med. 1998; 129:886–890.
4. George, JN. Platelets on the Web. Update available at www.ouhsc.edu/platelets/index.html.
5. Blajchman, MM, Schlicter, SJ, Heddle, NM, Murphy, MF. New strategies for the optimal use of platelet transfusions. Hematology Am Soc Hematol Educ Program. 2008; 2008:198–204.
6. Bizzaro, N. EDTA-dependent pseudothrombocytopenia: A clinical and epidemiological study of 112 cases, with 10-year follow-up. Am J Hematol. 1995; 50:103–109.
7. Huxtable, LM, Tafreshi, MJ, Rakkar, ANS. Frequency and management of thrombocytopenia with the glycoprotein IIb/IIIa receptor antagonists. Am J Cardiol. 2006; 97:426–429.
8. Von Drygalski, A, Curtis, BR, Bougie, DW, et al. Vancomycin-induced immune thrombocytopenia. N Engl J Med. 2007; 356:904–910.
9. Provan, D, Stasi, R, Newland, AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010; 115:168–186.
10. Neunert, C, Lim, W, Crowther, M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011; 117:4190–4207.
11. Newman, GC, Novoa, MV, Fodero, EM, et al. A dose of 75 µg/kg/d of IV anti-D increases the platelet count more rapidly and for a longer period of time than does 50 µg/kg/d in adults with immune thrombocytopenic purpura (ITP). Br J Haematol. 2001; 112:1076–1078.
12. McCrae, KR, Herman, JH. Posttransfusion purpura: Two unusual cases and a literature review. Am J Hematol. 1996; 52:205–211.
13. Douketis, JD, Spyropoulos, AC, Spencer, FA, et al. Perioperative management of anticoagulant therapy: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e326S–e350S.
14. Hedges, SJ, Dehoney, SB, Hooper, JS, et al. Evidence-based treatment recommendations for uremic bleeding. Nat Clin Pract Nephrol. 2007; 3:138–153.
15. Fernandez, F, Goudable, C, Sie, P, et al. Low hematocrit and prolonged bleeding time in uraemic patients: Effect of red cell transfusions. Br J Haematol. 1985; 59:139–148.
16. Linkins, L-A, Dans, AL, Moores, LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e495S–e530S.
17. Laster, J, Silver, D. Heparin coated catheters and heparin-induced thrombocytopenia. J Vasc Surg. 1988; 7:667–672.
18. Selleng, K, Warkentin, TE, Greinacher, A. Heparin-induced thrombocytopenia in intensive care patients. Crit Care Med. 2007; 35:1165–1176.
19. Warkentin, TE. Heparin-induced thrombocytopenia: Pathogenesis and management. Br J Haematol. 2003; 116:535–555.
20. Lo, GK, Juhl, D, Warkentin, TE, et al. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006; 4:759–765.
21. Cuker, A, Arepally, G, Crowther, MA, et al. The HIT Expert Probability (HEP) score: A novel pre-test probability model for heparin-induced thrombocytopenia based on broad expert opinion. J Thromb Haemost. 2010; 8:2642–2650.
22. Warkentin, TE, Sheppard, J-AL. Testing for heparin-induced thrombocytopenia antibodies. Trans Med Rev. 2006; 20:259–272.
23. Wallis, DE, Workman, DK, Lewis, BE, et al. Failure of early heparin cessation as treatment for heparin-induced thrombocytopenia. Am J Med. 1999; 106:629–635.
24. Warkentin, TE, Elavathil, LJ, Hayward, CPM, et al. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Intern Med. 1997; 127:804–812.
25. Lewis, BE, Maathai, WH, Jr., Cohen, M, et al. Argatroban anticoagulation during percutaneous coronary artery intervention in patients with heparin-induced thrombocytopenia. Cathet Cardiovasc Intervent. 2002; 57:177–184.
26. Mahaffey, KW, Lewis, BE, Wildermann, NM, et al. Anticoagulant therapy with bivalirudin to assist in the performance of percutaneous coronary intervention in patients with heparin-induced thrombocytopenia (ATBAT study): Main results. J Invasive Cardiol. 2003; 15:611–616.
27. Greinacher, A. The use of direct thrombin inhibitors in cardiovascular surgery in patients with heparin-induced thrombocytopenia. Semin Thromb Haemost. 2004; 30:315–327.
28. Warkentin, TE, Kelton, JG. Temporal aspects of heparin induced thrombocytopenia. N Engl J Med. 2001; 344:1286–1292.
29. George, JN. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006; 354:1927–1935.
30. Sadler JE, Poncz M: Antibody mediated thrombotic disorders: Idiopathic thrombocytopenic purpura and heparin induced thrombocytopenia. In Kaushansky K, Lichtman MA, Beutler E, et al (eds): Williams Hematology, 8th ed. New York, McGraw Hill, ••, pp 2163–2184
30a. George, JN. How I treat thrombotic thrombocytopenic purpura. Blood. 2010; 116:4060–4069.
31. Moake, JL. Thrombotic microangiopathies. N Engl J Med. 2002; 347:589–600.
32. Rock, GA, Shumak, KH, Buskard, NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med. 1991; 325:393–397.
33. Ziman, A, Mitri, M, Klapper, E, et al. Combination vincristine and plasma exchange as initial therapy in patients with thrombotic thrombocytopenic purpura: One institution’s experience and review of the literature. Transfusion. 2005; 45:41–49.
34. Jasti, S, Coyle, T, Gentile, T, et al. Rituximab as an adjunct to plasma exchange in TTP: A report of 12 cases and review of literature. J Clin Apheresis. 2008; 23:151.
35. Norris, M, Remuzzi, G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009; 361:1676–1687.
36. Ohanian, M, Cable, C, Halka, K. Eculizumab safely reverses neurologic impairment and eliminates need for dialysis in severe atypical hemolytic uremic syndrome. Clin Pharmacol Adv Applications. 2011; 3:5–12.
37. Alexion Pharmaceuticals. Soliris. Available at http://soliris.net/ahus/HCP/Dosing_and_Administration.html.
38. Marder, VJ, Feinstein, DI, Colman, RW, et al. Consumptive thrombohemorrhagic disorders. In: Colman RW, Marder VJ, Clowes AW, et al, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:1571–1600.
39. Spero, AJ, Lewis, JH, Hasiba, U. DIC: Findings in 346 patients. Thromb Haemost. 1980; 43:28–33.
40. Taylor, FB, Toh, C-H, Hoots, WK, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001; 86:1327–1330.
41. Bakhtiari, K, Meijers, JC, de Jonge, E, et al. Prospective validation of the international society of thrombosis and haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004; 32:2416.
42. Alperin, JB. Coagulopathy caused by vitamin K deficiency in critically ill, hospitalized patients. JAMA. 1987; 258:1916–1919.
43. Lisman, T, Porte, RJ. Rebalanced hemostasis in patients with liver disease: Evidence and clinical consequences. Blood. 2010; 116:878–885.
44. Warren, BL, Eid, A, Singer, P, et al. Caring for the critically ill patient. High-dose anti-thrombin III in severe sepsis: A randomized controlled trial. JAMA. 2001; 286:1869–1878.
45. Fiore, LD, Scola, MA, Cantillon, CE, et al. Anaphylactoid reactions to vitamin K. J Thromb Thrombolysis. 2001; 11:175–183.
46. Deitcher, SR. Interpretation of the international normalized ratio (INR) in patients with liver disease. Lancet. 2002; 359:47–48.
47. Abdel-Wahab, OL, Healey, B, Dzik, WH. Effect of fresh-frozen plasma transfusion on prothrombin time and bleeding in patients with mild coagulation abnormalities. Transfusion. 2006; 46:1279–1285.
48. Holcomb, JB. Optimal use of blood products in severely injured trauma patients. Hematol Am Soc Hematol Educ Program. 2010; 2010:465–469.
49. Abshire, T, Kenet, G. Recombinant factor VIIa: Review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004; 2:899–909.
50. Nichols, WL, Hultin, MB, James, AH, et al. von Willebrand disease (VWD): Evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008; 14:171–232.
51. Konkle, BA, Kessler, C, Aledort, L, et al. Emerging clinical concerns in the ageing haemophilia patient. Haemophilia. 2009; 15:1197–1209.
52. Bolton-Maggs, P. Factor XI deficiency—Resolving the enigma? Hematol Am Soc Hematol Educ Program. 2009; 2009:97–105.
53. Salomon, O, Stenberg, DM, Dardick, R, et al. Inherited factor XI deficiency confers no protection against myocardial infarction. J Thromb Haemost. 2003; 1:658–661.
54. Lee, SH, Jeong, MH, Sohn, S, et al. Successful management of a patient with factor XI deficiency and unstable angina by percutaneous coronary intervention. Korean Circ J. 2005; 35:860–863.
55. Kahn, SR, Lim, W, Dunn, AS, et al. Prevention of VTE in nonsurgical patients: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e195S–e226S.
56. Geerts, W, Cook, D, Selby, R, et al. Venous thromboembolism and its prevention in critical care. J Crit Care. 2002; 17:95–104.
57. Joynt, GM, Kew, J, Gomersall, CD, et al. Deep venous thrombosis caused by femoral venous catheters in critically ill adult patients. Chest. 2000; 117:178–183.
58. Gould, MK, Garcia, DA, Wren, SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e227s–e277s.
59. Kearon, C, Akl, EA, Comerota, AJ, et al. Antithrombotic therapy for VTE disease: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e419S–e494S.
60. Klang, M, Graham, D, McLymont, V. Warfarin bioavailability with feeding tubes and enteral formula. J Parent Ent Nutr. 2010; 34:300–304.
61. Lutomski, DM, LaFrance, RT, Bower, RH, et al. Warfarin absorption after massive small bowel resection. Am J Gastroenterol. 1985; 80:99–102.
62. Gerber, DE, Grossman, SA, Streiff, MB. Management of venous thromboembolism in patients with primary and metastatic brain tumors. J Clin Oncol. 2006; 24:1310–1318.
63. Garcia, DA, Baglin, TP, Weitz, JI, et al. Parenteral anticoagulants: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e24S–e43S.
64. Raschke, RA, Reilly, BM, Guidry, JR, et al. The weight based heparin dosing nomogram compared with a “standard care” nomogram: A randomized controlled trial. Ann Intern Med. 1993; 119:874–881.
65. Wilde, MI, Markham, A. Danaparoid: A review of its pharmacology and clinical use in the management of heparin-induced thrombocytopenia. Drugs. 1997; 54:903–924.
66. Hursting, MJ, Alford, KL, Becker, J-CP, et al. Novastan (brand of argatroban): A small-molecule, direct thrombin inhibitor. Semin Thromb Hemost. 1997; 23:503–516.
67. Greinacher, A, Janssens, U, Berg, G, et al. Lepirudin (recombinant hirudin) for parenteral anticoagulation in patients with heparin-induced thrombocytopenia. Circulation. 1999; 100:587–593.
68. Fischer, K-G, van de Loo, A, Bohler, J. Recombinant hirudin (lepirudin) as anticoagulant in intensive care patients treated with continuous hemodialysis. Kidney Int. 1999; 56(Suppl 72):S46–S50.
69. Lee, P, Smith, R, Deal, EN, et al. Comparison of bivalirudin and argatroban for the management of heparin-induced thrombocytopenia. Pharmacotherapy. 2010; 30:1229–1238.
70. Connolly, SJ, Ezekowitz, MD, Yusuf, S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009; 361:1139–1151.
71. Schulman, S, Kearon, C, Kakkar, AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009; 361:2342–2352.
72. van Ryn, J, Stanfier, J, Haertter, J, et al. Dabigatran etexilate—A novel, reversible, oral direct thrombin inhibitor: Interpretation of coagulation assays and reversal of anticoagulant activity. J Thromb Haemost. 2010; 103:1116–1127.
73. CRASH-2 Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): A randomized, placebo-controlled trial. Lancet. 2010; 376:23–32.
74. Weitz, JI, Eikelboom, JW, Samama, MM. New antithrombotic drugs: Antithombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e44s–e88s.
75. Granger, CB, Alexander, JH, McMurray, JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011; 365:981–992.
76. Patel, MR, Mahaffey, KW, Garg, J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011; 365:883–891.
77. The EINSTEIN-PE Investigators, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012; 366(14):1287–1297.
78. Eerenberg, ES, Kamphuisen, PW, Sjipkens, MK, et al. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: A randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011; 124(14):1573–1579.
79. Ansell, J, Hirsh, J, Hylek, E, et al. Pharmacology and management of the vitamin K antagonists, 8th ed. ACCP evidence-based clinical practice guidelines. Chest. 2008; 133(Suppl):160S–198S.
80. Ageno, W, Gallus, AS, Wittkowsky, A, et al. Oral anticoagluant therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed. ACCP evidence-based clinical practice guidelines. Chest. 2012; 141:e44S–e88S.