[level-membership-for-pediatrics-category]
64 Hemolytic-Uremic Syndromes
Etiology and Pathogenesis
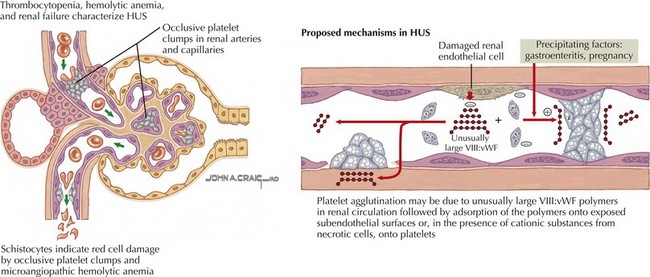
Broadly defined, HUS and TTP are syndromes whose pathologic correlate is thrombotic microangiopathy (TMA). TMA describes the microvascular occlusion that occurs most frequently within capillaries and arterioles (Figure 64-1). It is seen histologically as thrombi, endothelial cell swelling, luminal narrowing, and fibrinoid necrosis of the vessel wall. TMA results from dysfunction of the endothelial cell–platelet interface and can originate from various underlying mechanisms, which form the basis for an etiologic classification. HUS can result from infectious causes, genetic causes, or medication-related causes or in association with secondary discrete pathologic entities. TTP results from a deficiency of von Willebrand factor-cleaving protease, (vWF-cp), which can be either acquired because of the presence of an autoantibody or congenital resulting from a mutation in the ADAMTS13 gene.
Shiga Toxin–Associated Hemolytic-Uremic Syndrome
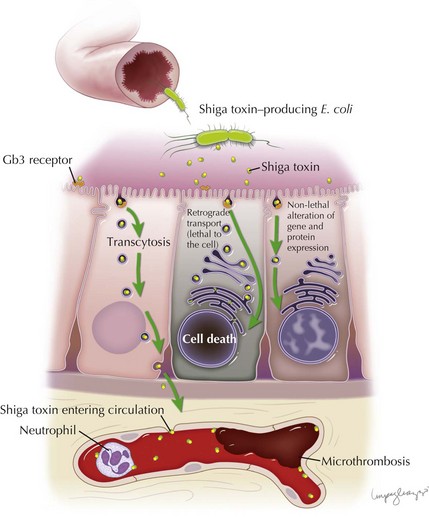
Stx is responsible for the endothelial toxicity of HUS-causing STEC, giving rise to the pathologic hallmark of TMA. It is produced in the bowel and translocated into circulation, where it can localize to the glomeruli, gastrointestinal tract, pancreas, and various other host tissues by a mechanism that has yet to be fully understood. Central to enterocyte entry and subsequent toxemia, the Stx B subunit binds to a cell surface terminal carbohydrate moiety of the globotriaosylceramide receptor (Gb3). The Gb3 receptor is a key determinant for cell sensitivity to Stx, and along with enterocytes and other cell types is present on glomerular endothelial cells, thereby targeting the toxin to the renal microvasculature. In the intestine, binding of Stx to Gb3 commences a sequence of events beginning with receptor-mediated endocytosis. Stx can follow several different pathways: (1) it can be delivered intact to the intestinal submucosa and circulation via transcytosis; (2) it can induce direct cytotoxicity by trafficking to the cell endoplasmic reticulum via the Golgi apparatus in a process known as retrograde transport; or (3) it can, in lower concentrations, alter gene and protein expression of the cell without inducing cell death. Transcytosis of toxin to the intestinal microvascular circulation is thought to give rise to the characteristic intestinal lesion that has the clinical–pathologic manifestation of bowel wall edema, thrombosis, and hemorrhage. Neutrophils localize to the intestinal mucosa during STEC infection, where they are thought to transport Stx to extraintestinal sites. They are known to bind Stx by a distinct receptor with a lower affinity than Gb3. The toxin is therefore not endocytosed, which allows it to be freely unloaded at various target sites (Figure 64-2).
Genetic Forms of Hemolytic-Uremic Syndrome
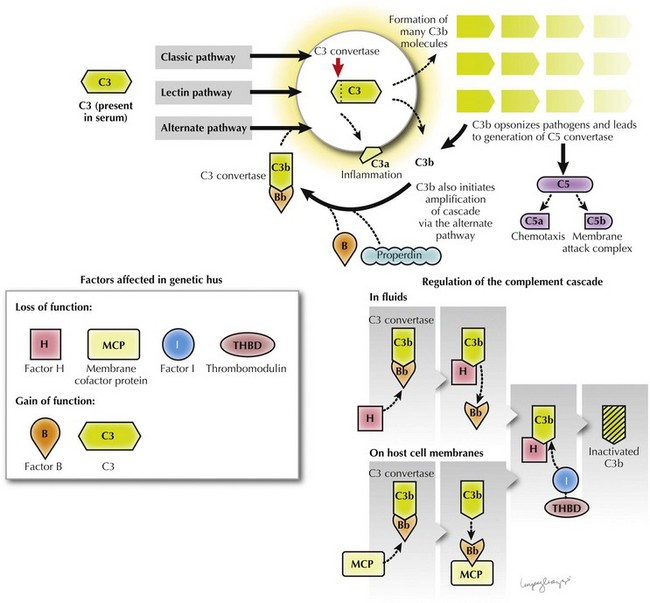
Complement component C3 was first noted in 1974 to be reduced in the serum of patients and relatives with inherited forms of NStx HUS. Low serum C3 can also occur with infection-related forms of HUS as a result of C3 consumption in the microvasculature. The low C3 in inherited HUS is persistent and reflects abnormalities that give rise to hyperactivation of the complement cascade. The complement cascade is a key component of the innate immune system comprising groups of plasma and membrane-bound zymogen proteins that follow one of three activation pathways: classical, lectin, or alternative. These pathways converge with the generation of C3 convertase enzymes that convert C3 into C3a and C3b (see Figure 64-2). C3b is necessary for opsonization and generation of C5 convertase, which in turn is necessary for chemotaxis (C5a) and the generation of a membrane attack complex (C5b). This process is tightly regulated by specific regulatory proteins to prevent nonspecific destruction of host cells (Figure 64-3).
Common Renal Pathology
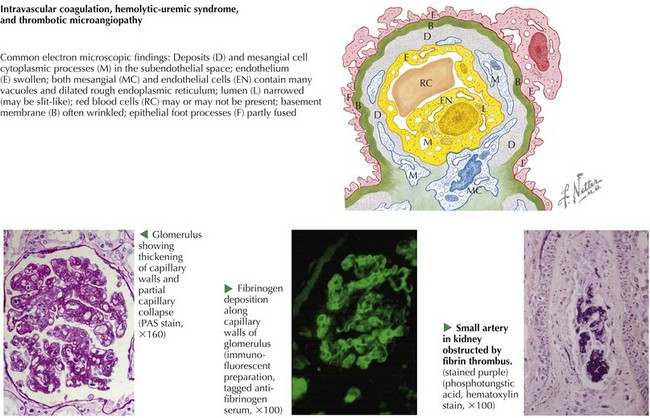
The common “endotheliopathy” that results from various etiologic forms of HUS produces the histopathologic lesion of glomerular TMA (Figure 64-4). Thrombosis affects the glomerular capillaries and can extend proximally into the afferent arterioles, even rarely into interlobular arteries. Extensive thrombosis into the arteries may produce shrunken, ischemic glomeruli or cortical infarct, which can be responsible for severe hypertension. More commonly, the glomeruli have thickened capillary walls, luminal obstruction or thrombosis, and endothelial cell swelling. The glomeruli may appear large. In more severe cases of HUS, patchy cortical necrosis can be seen. Occasionally, necrosis can be more diffuse and affect the entire superficial cortex. It is important to note that whereas fibrin- and RBC-rich thrombi are seen in Stx and pneumococcal HUS, platelet thrombi are seen in TTP. In TTP, the platelet-dominant thrombi affect the heart, pancreas, kidney, adrenal glands, and central nervous system (CNS) in order of increasing severity. A renal biopsy is rarely necessary in the diagnosis of HUS or TTP.
Constantinescu AR, Bitzan M, Weiss L, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43:976-982.
Copelovitch L, Kaplan BS. Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol. 2008;23:1951-1956.
Copelovitch L, Kaplan BS. The thrombotic microangiopathies. Pediatr Nephrol. 2008;23:1761-1767.
Loirat C, Taylor CM. Hemolytic uremic syndrome. In: Avner E, Harmon W, Niaudet P, editors. Pediatric Nephrology. ed 5. Philadelphia, Lippincott: Williams & Wilkins; 2004:887-915.
Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035-1050.
Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073-1086.
Trachtman H, Kaplan B. Hemolytic uremic syndrome: Stx-HUS. In: Kaplan B, Meyers K, editors. Pediatric Nephrology and Urology: The Requisites. Philadelphia: Elsevier Mosby; 2005:203-208.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
64 Hemolytic-Uremic Syndromes
Etiology and Pathogenesis
Broadly defined, HUS and TTP are syndromes whose pathologic correlate is thrombotic microangiopathy (TMA). TMA describes the microvascular occlusion that occurs most frequently within capillaries and arterioles (Figure 64-1). It is seen histologically as thrombi, endothelial cell swelling, luminal narrowing, and fibrinoid necrosis of the vessel wall. TMA results from dysfunction of the endothelial cell–platelet interface and can originate from various underlying mechanisms, which form the basis for an etiologic classification. HUS can result from infectious causes, genetic causes, or medication-related causes or in association with secondary discrete pathologic entities. TTP results from a deficiency of von Willebrand factor-cleaving protease, (vWF-cp), which can be either acquired because of the presence of an autoantibody or congenital resulting from a mutation in the ADAMTS13 gene.
Shiga Toxin–Associated Hemolytic-Uremic Syndrome
Stx is responsible for the endothelial toxicity of HUS-causing STEC, giving rise to the pathologic hallmark of TMA. It is produced in the bowel and translocated into circulation, where it can localize to the glomeruli, gastrointestinal tract, pancreas, and various other host tissues by a mechanism that has yet to be fully understood. Central to enterocyte entry and subsequent toxemia, the Stx B subunit binds to a cell surface terminal carbohydrate moiety of the globotriaosylceramide receptor (Gb3). The Gb3 receptor is a key determinant for cell sensitivity to Stx, and along with enterocytes and other cell types is present on glomerular endothelial cells, thereby targeting the toxin to the renal microvasculature. In the intestine, binding of Stx to Gb3 commences a sequence of events beginning with receptor-mediated endocytosis. Stx can follow several different pathways: (1) it can be delivered intact to the intestinal submucosa and circulation via transcytosis; (2) it can induce direct cytotoxicity by trafficking to the cell endoplasmic reticulum via the Golgi apparatus in a process known as retrograde transport; or (3) it can, in lower concentrations, alter gene and protein expression of the cell without inducing cell death. Transcytosis of toxin to the intestinal microvascular circulation is thought to give rise to the characteristic intestinal lesion that has the clinical–pathologic manifestation of bowel wall edema, thrombosis, and hemorrhage. Neutrophils localize to the intestinal mucosa during STEC infection, where they are thought to transport Stx to extraintestinal sites. They are known to bind Stx by a distinct receptor with a lower affinity than Gb3. The toxin is therefore not endocytosed, which allows it to be freely unloaded at various target sites (Figure 64-2).
[/not-level-membership-for-pediatrics-category]
