Chapter 512 Hemolytic-Uremic Syndrome
Hemolytic-uremic syndrome (HUS) is one of the most common causes of community-acquired acute kidney failure in young children. It is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and renal insufficiency (Table 512-1). HUS has clinical features in common with thrombotic thrombocytopenic purpura (TTP). The etiology and pathophysiology of the more common forms of HUS clearly delineate childhood HUS as separate from idiopathic TTP.
Table 512-1 DEFINITION OF POSTDIARRHEAL HEMOLYTIC UREMIC SYNDROME: CENTERS FOR DISEASE CONTROL AND PREVENTION, 1996
CLINICAL DESCRIPTION
Hemolytic uremic syndrome (HUS) is characterized by the acute onset of microangiopathic hemolytic anemia, renal injury, and a low platelet count. Thrombotic thrombocytopenic purpura (TTP) also is characterized by these features but can include central nervous system (CNS) involvement and fever and can have a more gradual onset. Most cases of HUS (but few cases of TTP) occur after an acute gastrointestinal illness (usually diarrheal).
LABORATORY CRITERIA FOR DIAGNOSIS
The following are both present at some time during the illness:
Note: A low platelet count can usually, but not always, be detected early in the illness, but it can then become normal or even high. If a platelet count obtained within 7 days after onset of the acute gastrointestinal illness is not <150,000/mm3, other diagnoses should be considered.
CASE CLASSIFICATION
Probable
Confirmed
COMMENT
Some investigators consider HUS and TTP to be part of a continuum of disease. Therefore, criteria for diagnosing TTP on the basis of CNS involvement and fever are not provided because cases diagnosed clinically as postdiarrheal TTP also should meet the criteria for HUS. These cases are reported as postdiarrheal HUS.
From Elliott EJ, Robins-Browne RM: Hemolytic uremic syndrome, Curr Probl Pediatr Adolesc Health Care 35:305–344, 2005.
Etiology
The various etiologies of HUS allow classification into infection-induced, genetic, medication- induced, and HUS associated with systemic diseases characterized by microvascular injury (Tables 512-2 and 512-3). The most common form of HUS is caused by toxin-producing Escherichia coli that cause prodromal acute enteritis and is commonly termed diarrhea-associated HUS. In the subcontinent of Asia and southern Africa, the shiga toxin of Shigella dysenteriae type 1 is causative, whereas in Western countries verotoxin-producing E. coli (VTEC) are the usual causes.
Table 512-2 CLASSIFICATION OF HEMOLYTIC UREMIC SYNDROME
INFECTION INDUCED
GENETIC
OTHER DISEASES ASSOCIATED WITH MICROVASCULAR INJURY
MEDICATION-INDUCED
Table 512-3 GENETIC ABNORMALITIES AND CLINICAL OUTCOME IN PATIENTS WITH ATYPICAL HEMOLYTIC-UREMIC SYNDROME
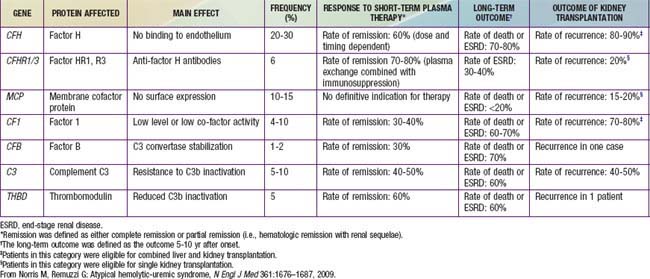
Genetic forms of HUS compose the second major category of the disease that has become clearly defined (see Tables 512-2, 512-3). Inherited deficiencies of either von Willebrand factor–cleaving protease (ADAMTS 13) or complement factor H, I, or B and defects in vitamin B12 metabolism can cause HUS. There remain familial cases transmitted in autosomal dominant or recessive patterns in which a specific genetic defect has not yet been identified. Most of the genetic forms do not have a preceding diarrhea prodrome. Genetic forms of HUS can be indolent and unremitting once they become manifest, or they can have a relapsing pattern precipitated by an infectious illness. The latter feature likely explains the association of many infectious agents with HUS, particularly in reports published before the recognition of the unique pathophysiology of VTEC and neuraminidase- producing pneumococci in causing HUS.
HUS can be superimposed on any disease associated with microvascular injury, including malignant hypertension, systemic lupus erythematosus, and antiphospholipid syndrome. It can also occur following bone marrow or solid organ transplantation and may be triggered by the use of the calcineurin inhibitors cyclosporine and tacrolimus in that setting. Several other medications can also induce HUS (see Table 512-2).
Diagnosis and Differential Diagnosis
The diagnosis is made by the combination of microangiopathic hemolytic anemia with schistocytes, thrombocytopenia, and some degree of kidney involvement (see Table 512-1). The anemia, mild at presentation, rapidly progresses. Thrombocytopenia is an invariable finding in the acute phase, with platelet counts usually 20,000-100,000/mm3. Partial thromboplastin and prothrombin times are usually normal. The Coombs test is negative, with the exception of pneumococci-induced HUS, where the Coombs test is usually positive. Leukocytosis is present and significant. Urinalysis typically shows microscopic hematuria and low-grade proteinuria. The renal insufficiency can vary from mild elevations in serum BUN and creatinine to acute, anuric kidney failure.
Prognosis and Treatment
The acute prognosis, with careful supportive care, for diarrhea-associated HUS has <5% mortality in most major medical centers. Half of the patients require dialysis support during the acute phase of the disease. Most recover renal function completely, but of surviving patients, 5% remain dependent on dialysis, and up to 20-30% are left with some level of chronic renal insufficiency. The prognosis for HUS not associated with diarrhea is more severe. Pneumococci-associated HUS causes increased patient morbidity, with mortality reported as 20%. The familial, genetic forms of HUS can be insidiously progressive or relapsing diseases and have a poor prognosis (see Table 512-3). Identification of specific factor deficiencies in some of these genetic forms provides opportunity for specific replacement therapy to improve outcomes.
Bender JM, Ampofo K, Byington CL, et al. Epidemiology of Streptococcus pneumoniae–induced hemolytic uremic syndrome in Utah children. Pediatr Infect Dis J. 2010;29(8):712-716.
Centers for Disease Control and Prevention. Escherichia coli O157:H7 infection associated with drinking raw milk—Washington and Oregon, November-December, 2005. MMWR Morb Mortal Wkly Rep. 2007;56:165-167.
Centers for Disease control and Prevention. Laboratory-confirmed non-O157 shiga toxin–producing Escherichia coli—Connecticut, 2000–2005. MMWR Morb Mortal Wkly Rep. 2007;56:29-32.
Centers for Disease Control and Prevention. Two multistate outbreaks of Shiga toxin–producing Escherichia coli infections linked to beef from a single slaughter facility—United States, 2008. MMWR. 2010;59(18):557-560.
Clark WF, Sontrop JM, Macnab JJ, et al. Long term risk for hypertension, renal impairment, and cardiovascular disease after gastroenteritis from drinking water contaminated with Escherichia coli O157:H7: a prospective cohort study. BMJ. 2010;341:1089.
Copelovitch L, Kaplan BS. Streptococcus pneumoniae–associated hemolytic uremic syndrome. Pediatr Nephrol. 2008;23:1951-1956.
Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345-356.
Garg AX, Salvadori M, Moist LM, et al. Renal prognosis of toxigenic E. coli infection. Kidney Int Suppl. 2009;112:S38-S41.
Hee Lee B, Kwak Heon, Il Shin J, et al. Atypical hemolytic uremic syndrome associated with complement factor H autoantibodies and CFHR1/CFHR3 deficiency. Pediatr Res. 2009;66:336-340.
Iijima K, Kamioka I, Nozu K. Management of diarrhea-associated hemolytic uremic syndrome in children. Clin Exp Nephrol. 2008;12:16-29.
Michael M, Elliott EJ, Ridley GF, et al: Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura (review), Cochrane Database Syst Rev (1):CD003595, 2009.
Norris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676-1687.
Oakes RS, Siegler RL, McReynolds MA, et al. Predictors of fatality in post-diarrheal hemolytic uremic syndrome. Pediatrics. 2006;117:1656-1662.
Pennington H. Escherichia coil O157. Lancet. 2010;376:1428-1434.
Piercefield EW, Bradley KK, Coffman RL, et al. Hemolytic uremic syndrome after an Escherichia coli O111 outbreak. Arch Intern Med. 2010;170(18):1656-1663.
Tarr PI. Shiga toxin–associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: distinct mechanisms of pathogenesis. Kidney Int Suppl. 2009;112:S29-S32.
Tarr PI, Gordon CA, Chandler WL. Shiga-toxin–producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073-1086.
Waters AM, Kerecuk L, Luk D, et al. Hemolytic uremic syndrome associated with invasive pneumococcal disease: the United Kingdom experience. J Pediatr. 2007;151:140-144.
Wong CS, Jelacic S, Habeeb RL, et al. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342:1930-1936.





