[level-membership-for-pediatrics-category]
Chapter 84 Hemoglobinopathies
Appendix materials for this chapter are available online at http://www.expertconsult.com.
Perspective
Hemoglobinopathies are plentiful as many provide a protective effect to malaria. Although 5% of humans have hemoglobin variants, an estimated 300,000 affected homozygotes are born annually.1 Contrary to popular beliefs, hemoglobinopathies do not provide resistance to infection, but rather diminish the chance of death once infected, allowing the development of partial immunity. Although sickle cell and thalassemia trait increase fitness, homozygotes in malaria endemic areas often do not survive into their teens. Although this strong protective effect resulted in a high incidence of hemoglobinopathies in the equatorial malaria zones, the slave trade and more recent voluntary migrations has led to the worldwide distribution of these conditions.
The Globin Gene Loci
The α- and β-like genes reside within multigene loci and are transcribed at unparalleled levels in both tightly tissue specific, and developmentally specific patterns.2 This, and their involvement in human disease, has made these loci paradigms for gene regulation and pathophysiology. The β-globin gene was the first sequenced, and sickle cell was the first molecularly defined and diagnosed disease. Most forms of mutation (e.g., point, splicing, deletion, gene rearrangement) are observed in thalassemia, making these the quintessential teaching loci. This high variety and incidence of mutations result in frequent compound heterozygotes with a wide spectrum of clinical manifestations.
The five genes of the β-globin locus reside in a cluster on chromosome 11. The genes are expressed in an erythroid, and developmentally stage-specific manor; the ε, Aγ and Gγ, and δ and β genes being expressed primarily during the embryonic, fetal, and postnatal periods, respectively. At birth 95% of β-like chains are γ, with the rest being β. This ratio gradually inverts during the first year of life, explaining why phenotypes limited to the β-globin gene such as sickle cell and most β-thalassemias do not manifest until several months of age. Expression of the chromosome 16 based α-like genes differs; the embryonic ζ-gene parallels the expression of ε, but the twin α-genes are expressed from the fetal period onward. Thus α abnormalities manifest in utero, potentially with devastating consequences (e.g., hydrops fetalis). The resultant hemoglobin α-, β-heterotetramers (HbS) are developmentally expressed; embryonic: Hb Gower1 (ζ2,ε2), Hb Gower2 (α2,ε2), and Hb Portland (ζ2,γ2); fetal: HbF (fetal) (α2,γ2) and adult: HbA2 (α2,δ2) and HbA (Adult) (α2, β2) (Appendix Figure 84-A). Although many suggest HbF, with its higher oxygen affinity, evolved to aid the fetus in “stealing” oxygen from the mother, this is not supported experimentally, the advantage of HbF may in fact lie in differential transport of NO or carbon dioxide. Unfortunately, despite the intensive study, multiple Nobel prizes and the clinical importance of the globin loci, little of the molecular understanding of the loci has been translated to clinical care. The holy grail of manipulating expression from the β-locus remains elusive. Inhibiting adult sickle or thalassemic genes, while concomitantly activating expression from the normal fetal genes would alleviate most disease, obviating the need for this chapter.
Sickle Cell Disease
Molecular Description and Epidemiology
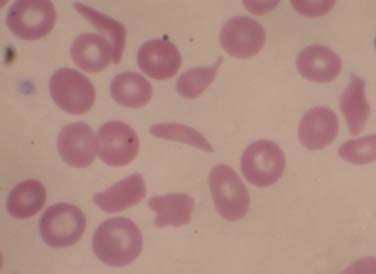
Sickle cell disease (SCD) is a group of single gene, autosomal recessive disorders common in people originating from specific regions of Africa or India and in those of Hispanic descent. SCD encompasses a group of disorders characterized by the presence of a single nucleotide change in the adult β-globin gene, and a second abnormal allele allowing the sickle hemoglobin to polymerize. Substitution of an adenine (A) for a thymidine (T) in codon 6 results in replacement of a hydrophilic glutamic acid residue with a hydrophobic valine residue. The allosteric changes in hemoglobin structure occurring with deoxygenation leads to exposure of a destabilizing valine-containing pocket. The high concentration of hemoglobin in erythrocytes (MCHC) allows for the alignment of these pockets, leading to polymerization and transition to the classic sickle cell morphology (Figure 84-1) and the initiation of downstream events leading to pain and chronic end-organ damage. The presence of HbA in sickle trait, or HbF in the newborn, attenuate polymerization, diminishing the clinical consequences of HbS. In the United States, approximately 1 in 14 and 1 in 400 African Americans have sickle trait and SCD, respectively, with approximately 100,000 cases and approximately 1000 new births per year (in contrast to approximately 1 million births per year in Africa). With increased ethnic mixing, compound heterozygous forms of SCD are increasingly being observed (e.g., S/β-thalassemia or HbS/E). Sadly, though simple public health measures have dramatically decreased mortality rates in the United States, the countries with the highest incidence of SCD often lack such resources and children with SCD often do not reach their teens.
Sickle Cell Trait
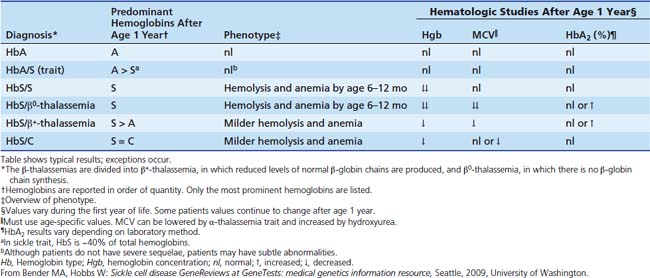
Once the presence of HbS has been confirmed, the differential diagnosis consists of severe forms, less severe forms, and carrier states. Sickle cell trait (SCT) is distinguished from SCD by the presence of a normal β-globin allele and more HbA than HbS (typically 40% HbS) (Table 84-1). SCT does not result in the classic spectrum of sickle-related complications or alter red cell indices, but can lead to clinical abnormalities. Impaired renal concentrating abilities and intermittent microhematuria is common. Although of unclear clinical significance, this damage may interact with other pathophysiology in the African-American population (e.g., diabetes), explaining the high risk of chronic renal disease. In addition, there may be an increased risk of venous thromboembolism.3 Most controversial is the concern that extremes of physical exertion, dehydration, and/or altitude may induce sickle cell vaso-occlusive events in some individuals with sickle cell trait.4 This is particularly relevant to student athletes for whom no formal activity restrictions are recommended, but empirically, aggressive hydration during extremes of physical exertion is generally recommended.
Spectrum of Sickle Cell Disease Genotypes
Although homozygous sickle cell (Hb S/S, often called sickle cell anemia) is most common, it is essential to be aware of additional genotypes that result in a similar spectrum of disease (see Table 84-1). That said, once in the intensive care unit (ICU), the form of SCD plays little role in decision-making. In HbS/β+-thalassemia and HbSβ0-thalassemia, the lower the expression of β-like chains from the thalassemic allele, the lower the hemoglobin concentration (Hgb) and MCV, and the higher the percentage of HbS and polymerization, resulting in a more severe clinical phenotype. In contrast, when HbS is expressed in conjunction with HbC, HbD, or Hb O-Arab or several other variants, near normal amounts of hemoglobin are made, but these forms are less able to attenuate HbS polymerization than HbA and result in milder sickle syndromes.
Natural History
The clinical manifestations of SCD result from intermittent episodes of vascular occlusion leading to tissue ischemia/reperfusion injury and variable degrees of hemolysis, both of which contribute to multiorgan dysfunction. The severity of disease manifestations varies widely in individuals with the same genotype and is affected by (1) known genetic modifiers, e.g., α-thalassemia that lowers the MCHC; (2) genetic modifiers of additional pathways contributing to the pathophysiology of SCD (below); (3) behavioral and environmental factors, (e.g., family and community support, racial disparities and access to health care); and (4) psychological factors, (e.g., coping with chronic disease, recurrent pain, and a high rate of depression). The spectrum of manifestations changes with age, and is beautifully described in Gill et al.5 Previously, median survival in the United States for those with SCD was 42 years for men and age 48 years for women6; however, considerable improvements in longevity have been suggested.7,8 The main causes of death are infection, acute chest syndrome, pulmonary artery hypertension, and cerebrovascular events.9,10 What is not fully appreciated by pediatricians is that this increased lifespan is often associated with significant morbidity related to chronic organ damage.
Pathophysiology
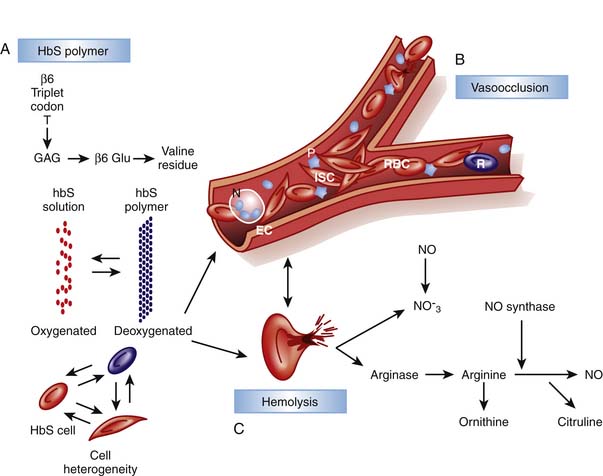
Although unable to fully provide the elegant details of the rapidly evolving insights into the multifaceted pathophysiology of SCD, this chapter outlines key pathways with direct relevance to management basics. While the initial era of understanding pathophysiology focused on HbS polymerization and red cell “sickling,” this was followed by an awareness of the critical role of endothelial interactions and inflammation and later, of hemolysis and perturbations of NO metabolism (Figure 84-2).11–14
Red Cells, Inflammation, and the Endothelium
Crystallization leads to deformation of erythrocytes and oxidative damage to cell membrane proteins, leading to the activation of pathways contributing to the stimulation of white cells and inflammation, endothelial damage and the initiation of the clotting cascade and platelet aggregation. Membrane damage triggers movement of phosphatidylserine to the outer leaf of the red cell membrane where it acts as a substrate for factor V and VIII binding, promoting the clotting cascade. Reticulocytes and sickled cells expose increased adherence proteins, slowing flow and increasing CTT and enhancing binding to other cell types. Binding to platelets leads to platelet activation and aggregation, whereas binding to neutrophils stimulates the oxidative burst, damaging endothelium and exposing tissue factor, further promoting coagulation, platelet aggregation, and clot formation. In addition, bound white cells release cytokines, increasing inflammatory cell recruitment and division, perpetuating the process. Thus, sickle cell represents an activated, inflammatory state in which the CTT is easily compromised. This increases the likelihood of additional cells sickling and blood flow becoming further compromised, resulting in ischemic damage, inflammation, and propagation of the cycle. This activation and integration of multiple pathways explains the tremendous clinical heterogeneity observed in people with the same hemoglobin genotype, as well as why patients can show rapid clinical decline. The clinical importance of these paths in SCD is re-enforced by an elevated white blood cell (WBC) count being a risk factor for pain episodes, acute chest syndrome, and early death.6,15 This also explains why infection and trauma are frequent triggers for sickle related pain, and why granulocyte-colony stimulating factor (G-CSF) has led to death in SCD.16 Therapeutically, this points to the importance of nonsteroidal anti-inflammatory drugs (NSAIDs) in affecting the underlying pathophysiology as well as providing analgesia, and why clinical response to hydroxyurea is best correlated with a decrease in WBC.17,18 Finally the identification of these interacting pathways identifies potential targets for new therapies affecting inflammation and cell-endothelial interactions.
Hemolysis and Nitric Oxide Homeostasis
In contrast to the vaso-occlusive complications that correlate with a high WBC and diminished HbF, recently a subset of complications including pulmonary hypertension, skin ulcers, and priapism were found to correlate with increased lactate dehydrogenase and bilirubin and reticulocyte count.12–14 This defines a hemolytic phenotype arising from disturbances in NO hemostasis, and as described at the end of this section, this may be responsible for the inverse association of hemolysis and pain. Hemoglobin accelerates destruction of NO several thousand-fold. Normally laminar flow positions red cells centrally in vessels sequestering hemoglobin away from the endothelium where NO is synthesized and utilized. Hemolysis releases hemoglobin (Fe++) that is now free to diffuse and reduce NO to nitrate with resultant formation of methemoglobin (Fe+++). In addition, hemolysis releases erythrocyte arginase, resulting in arginine depletion, the precursor for NO synthesis. Thus chronic hemolysis leads to decreased NO synthesis and increased destruction, resulting in altered NO hemostasis and increased tone of small vessels, slowing flow and increasing sickling. Optimal management of hemolysis related pathophysiology remains uncertain. Hemolysis prevention with hydroxyurea is appropriate and well accepted, and agents to prevent cellular dehydration (e.g., Mg++ or ICA-17043) are being investigated, but neither is appropriate for acute management. NO metabolism is directly modified with class 5 phosphodiesterase inhibitors (e.g., sildenafil) or by administering NO or related metabolites, and both approaches are being actively investigated for both acute and chronic use. Endogenous sulfur compounds may be tightly integrated with NO pathways, thus a therapeutic role of sulfur compounds may emerge.
In an exciting new paradigm for understanding SCD pain suggested by Gladwin and colleagues,19 NO has been found to affect the processing of nociceptive signals, that may explain why patients with more severe hemolytic disease have fewer vaso-occlusive events (VOE). NO and resultant cGMP signaling increase the hyperexcitability of nociceptive neurons.20 Thus as hemolysis depletes NO, it potentially attenuates pain signaling. Conversely, this may explain why SCD patients with fewer markers of hemolysis have more VOE. The implications of this biochemical pathway on pain management in SCD and other states of high hemolysis such as cardiac bypass are not known (see “Pain” below).
Clinical Manifestations
Although a summary of clinical problems and management are listed in the following section, the reader is referred to the National Heart Lung and Blood Institute standard of care guidelines21 for more details: www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf.
Pain
See Appendix Table 84-A for a detailed care plan, and Appendix Figures 84-B, C, and D for overviews of pain management.22 Vaso-occlusive events, defined as the acute onset of severe pain secondary to ischemic tissue injury, are a hallmark of SCD. The complexities of SCD pain are best summarized by Shapiro and Ballas: “Vaso-occlusion is a physiological process, but the resultant pain is a biopsychosocial phenomenon. Psychosocial issues such as coping skills, social context, personality, mood, and interactions with the health care system mingle with the biologic factors and contribute to the expression of the illness.”23 SCD pain can be acute, recurrent and/or chronic, and is complicated by co-existing chronic disease as well as social and racial overlays. The need for aggressive and rapid treatment is critical and well documented in guidelines of the American Pain Society and British Society of Hematology and should be consulted.24,25 Although aggressive intervention is essential for humane care and physiologic improvements (e.g., improving respiratory mechanics when having rib infarct pain), too often racial attitudes and concerns of drug seeking prevent sufficient delivery to patients in excruciating pain.26 Pain is the most common reason for hospitalization in SCD. The impact of pain is extremely heterogenous in SCD, likely because of differences in genetic background affecting the pathways described above, environment and psychosocial and behavioral variables. In one cooperative study, 80% of patients sought care once or less per year, whereas 5% of patients accounted for one third of hospitalizations.27 Recent daily pain diary assessments suggest these prior studies underestimate the routine pain many experience.28 Risk factors for pain include a high Hgb, high WBC, coexisting alpha thalassemia, and increased levels of cell free DNA, whereas HbF is protective.27,29,30
Pathophysiology, Diagnosis, and Presentation
Pain can start spontaneously but common precipitating factors relate to the pathophysiology and include weather changes (especially cold), dehydration, inflammation, and infections, all of which increase CTT. Occlusion results in ischemic/reperfusion injury and the release of multiple inflammatory mediators that activate nociceptors, evoking a pain response. Pain may involve any part of the body though most commonly affecting the back and long bones. The unpredictability of the pain contributes to the psychological component of pain, potentially interfering with coping mechanisms.31 Recurrent and chronic pain contribute to a decreased quality of life, decreased function, depression and suicidal ideations.28,32,33 Diagnosis is based on a qualitative description, typically biting or gnawing, throbbing, and fatiguing; and quantitative description, that must be trusted (Figure 84-E). The description is essential to help rule out other etiologies of pain, because no physical exam, laboratory, or radiographic studies can confirm or negate this subjective complaint. Physical findings can include swelling, warmth, erythema and tenderness, but exams can be normal and critically, it must be re-enforced that patients can appear without distress. The role of laboratory and radiologic studies is to rule out other etiologies of pain; particularly important for sickle mesenteric pain that must be distinguished from other causes of abdominal pain (e.g., ultrasound to rule out appendicitis or cholecystitis, or tapping a joint to rule out infectious bone or joint processes). Serial quantitative assessments with a developmentally stage specific pain scale is standard of care, though fraught with problems.
Management
See Appendix Table 84-A for a detailed care plan, and Appendix Figures 84-B to 84-D for overviews of pain management.22 An effective management strategy addresses the underlying tissue damage, nociception, history of pain episodes, and doses of medications required to achieve acceptable analgesia, baseline pain medications, history of tolerance, and how people process pain and coexisting depression. Although many are tempted to begin with analgesic medications, there are several other key components to management, such as the following: (1) environmental manipulation. A dark, calm, quiet environment with supportive interactions supports pain reduction. (2) Complementary methods. Many benefit from distraction, prayer, biofeedback, self-hypnosis and similar modalities. (3) Addressing pathophysiology and triggers. Fluids to maintain euvolemia, warmth and NSAIDs decrease CTT and promote conditions less likely to result in vaso-occlusion. (4) Adjuncts. Physical therapy, ambulation, and incentive spirometry should be encouraged to maintain blood flow and prevent atelectasis and acute chest syndrome (ACS). Additional adjuvants such as massage and acupuncture have been tried. After these have been addressed, providers may focus on analgesic medications.
Patients with SCD are the experts in their own pain management. Ideally patients and staff have agreed on individualized, predefined guidelines for pain management taking into account the patient’s degree of opiate tolerance, and side effect profile, and these are documented online or on a patient identification card. Early initiation of NSAIDs is essential, although the choice of NSAID is controversial (e.g., ibuprofen is a better anti-inflammatory, whereas ketoralac is a better analgesic). Notably, half of children will have resolution of pain with intravenous ketorolac.34 For severe episodes, rapid and repeated doses of opiates (dosed every 20 minutes), followed by transition to patient controlled analgesia (if developmentally appropriate) is the accepted standard.35 Although no consensus on the opiate of choice exists, many controversies remain. There are conflicting reports regarding an increased incidence of acute chest syndrome with morphine, with some suggesting decreased rates when nalbuphine is used.36,37 Many avoid meperidine due to concerns of increased seizures and neuroexcitation.38 The goal of opiate use is to balance providing sufficient pain relief to allow one to rest and perform incentive spirometry, while avoiding oversedation, which contributes to the development of pulmonary processes. Certain subsets of patients may benefit from regional anesthesia including epidural,39 dexmedetomidine,40 clonidine, gabapentin, ketamine, and intranasal diamorphine41; although few data exist evaluating their efficacy. One additional promising agent is low molecular weight heparin, which targets the pro-thrombotic and inflammatory state in SCD, Notably a recent trial reported fewer days of severe pain and length of stay with this intervention.42
As mentioned in the discussion of the pathophysiology of SCD, hemolysis leads to depletion of NO and this may attenuate nociceptive signaling through modulation of cGMP signaling.19 This has substantial implications for SCD where, as described in the following section, modulators of NO metabolism are being increasingly employed therapeutically. Expected outcomes are not obvious as additional signaling pathways modulate cGMP activity, and NO and cGMP production can decrease pain under some conditions.20 Some of this complexity is explained by the differential effects of modulating distinct sets of NO synthase genes and proteins.20 Thus, ultimately agents that specifically modulate distinct arms of these pathways in specific regions of the body (e.g., periphery versus central nervous system) will be required to extract the full clinical benefits of NO metabolism manipulation for SCD while maintaining the benefit of NO deletion on chronic pain.
Sepsis
See Appendix Figure 84-F for a detailed care plan.22 Sepsis is a major cause of morbidity and mortality in SCD. Strikingly, in the 1970s, 20% of SCD patients in the United States died before age 6 years, primarily from sepsis with encapsulated organisms, particularly Streptococcus pneumoniae.5 Many patients were diagnosed with SCD when presenting with sepsis, and diagnosed families often did not appreciate that fever in a toddler was an emergency. This led to newborn screening programs and the development of comprehensive care programs focused on education and immunizations. As expected, this did not affect the incidence of sepsis, but deaths from sepsis plummeted,43,44 and further decreased with the initiation of prophylactic penicillin in those younger than 6.45 Widespread use of pneumococcal and Hib vaccines further decreased severe pneumococcal disease to 2.3 events per 100 patient-years, with ages 6 to 36 months being the highest risk group.46,47
Pathophysiology and Etiology
The developmental timing of sepsis risk correlates with decreased splenic function because of infarcts leading to functional asplenia, which can be observed as early as 3 months. The resultant defects in cellular immunity, the alternate complement pathway and decrease in memory cells and opsonizing antibodies reduces clearance of encapsulated organisms. Together these lead to a lessened response to the 23-valent polysaccharide vaccine requiring routine reimmunization and results in a 100 to 400 times increased risk of bacteremia with encapsulated pathogens. SCD patients have shown a strong response to the conjugated PCV-7 vaccine, resulting in a two-thirds reduction in invasive pneumococcal disease.48 Serotypes not covered by the PCV-7 are increasing in prevalence, prompting the development of the PCV-13 vaccine. Although S. pneumoniae accounts for the majority of invasive infections other major pathogens include Haemophilus influenzae type b, Escherichia coli urosepsis, and Salmonella and Staphylococcus osteomyelitis.49
Management
See Figure 84-F for a detailed care plan.22 Although the pathophysiology, diagnosis, and management of sepsis are presented in Chapter 103, several aspects are unique to SCD, including (1) a focus on prevention with aggressive vaccinations with PCV-13, the conjugated 23-valent vaccine (and recurrent boosters from falling titers), Hib, and meningococcus50; (2) prophylactic penicillin given through age 6 years; (3) the critical role of family and provider education in seeking medical care and parenteral antibiotics emergently for fevers higher than 38.3° C. Pneumococcus has doubling time of 20 minutes; thus a 2-hour delay can result in a 50-fold increase in bacterial burden; (4) antibiotic choice should be driven by clinical presentation, but for empiric therapy ceftriaxone is preferred over cefotaxime and cefuroxime due to increasing resistance of pneumococcus; and (5) possible transfusion if cardiovascularly unstable. Evolving care practices will need to account for (1) the influence of hydroxyurea, (2) increasing antibiotic resistance, and (3) the recent release of the PCV-13 vaccine. (Hydroxyurea can result in regrowth of splenic tissue, and early use in young children may prevent autoinfarction, but how this correlates with preservation of immune function and resistance to encapsulated organisms is unknown.)
Acute Chest Syndrome
See Appendix Table 84-B for a detailed care plan.22 ACS is clinically heterogeneous and its definitions vary in the literature, making comparisons of studies difficult. The most general definition is a new nonatelectatic infiltrate on chest radiograph in a patient with SCD, though others add requirements for fever or respiratory symptoms (Figure 84-3). ACS is the second leading cause of hospitalization in SCD. ACS can progress in hours resulting in a mortality rate of 1% in children (vs. 4% in adults), with most deaths occurring in children younger than age 3 years.51 ACS accounts for 25% of deaths.51 Risk factors include a history of asthma, high baseline Hgb and low HbF. Factors associated with mortality include a prior episode of ACS, development of respiratory failure within 48 hours of presentation, sepsis, and simultaneous presentation with pain.51 Etiologies vary by age and included bacterial infections with typical and atypical organisms, viral infections, fat emboli, and pulmonary hemorrhage and multiple etiologic factors are often present (see also Appendix Figure 84-G).52
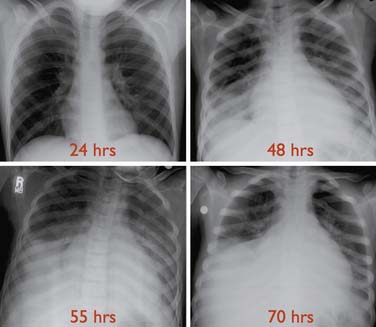
Presentation
ACS frequently occurs 2 to 3 days into a VOE. Consistent with the variety of etiologies, presenting signs and symptoms are variable and vary with age (e.g., young children are more likely to present with fever while older adolescents are more likely to present with chest pain).51 Notably, no single or pattern of signs and symptoms predicts ACS, and up to 35% of patients will have a normal pulmonary exam.51 Therefore there should always be a low threshold to obtain a chest radiograph. Infiltrates are most common in the lower lobes, and pleural effusions are more common in adults.51,52 Leukocytosis and significant drops in hemoglobin and platelets are common.51 Significant morbidity is associated with ACS including pneumothorax and empyema and 14% of patients developed respiratory failure in one study.52 Risk factors for respiratory failure include (1) extensive lobar involvement, (2) platelet count less than 199,000/µL and (3) history of cardiac disease.52
Management
See Appendix Table 84-B for a detailed care plan.22 Management of ACS can be addressed in stages; initial conservative interventions are followed by transfusion for clinical decline or if severely ill and finally biphasic positive airway pressure (BiPAP) or ventilatory support if needed. Initial care should consist of (1) antibiotics: a cephalosporin to cover encapsulated organisms, particularly S. pneumoniae, and a macrolide because Mycoplasma and Chlamydia are the most common infectious pathogens; (2) oxygen to maintain saturations more than 94%; (3) judicious fluid resuscitation to maintain euvolemia while avoiding overly aggressive fluid resuscitation that may worsen cardiac and respiratory status due to a combination of severe anemia and decreased cardiac function; and (4) prevention of atelectasis. Incentive spirometry (IS) has been shown to prevent progression, and chest physiotherapy and ambulation may be of benefit.53 Although IS is often limited by chest pain and patient compliance, use of a positive expiratory pressure (PEP) device has been shown to be as effective and better tolerated.54 The use of BiPAP or continuous positive airway pressure (CPAP) as a preventive measure is untested but appropriate as early intervention may prevent the need for intubation. (5) Opiates should be carefully titrated to minimize splinting and allow IS, while minimizing respiratory depression (small opiate boluses before IS may be helpful). Pain relief can improve respiratory mechanics, improving clinical status. (6) Bronchodilators are indicated as asthma is common in SCD and its presence increases the risk of ACS, and a subset of patients respond independent of documented wheezing.55 (7) The use of corticosteroids remains controversial. Although a randomized trial showed a decrease in hospital stay and transfusion, a subsequent retrospective review revealed a higher readmission rate, independent of a history of asthma.56,57
Transfusion has been shown to significantly improve oxygenation and clinical status in ACS.52 Although transfusion is usually effective at reversing ACS, because of the risks, including alloimmunization, transfusion not part of initial management unless severely ill. For most patients serial observations, with a low threshold for repeat chest radiographs is appropriate; reserving transfusion for progression despite conservative management. The decision of when to transfuse is contentious and data have been lacking. Promising studies suggest elevated secretory phospholipase A2 (sPLA2) levels predict the development of ACS in SCD.58 sPLA2 cleaves phospholipids, releasing free fatty acid, and is increasingly being associated with cardiovascular and pulmonary damage, including meconium aspiration. A randomized trial suggests early transfusion guided by sPLA2 levels, can prevent ACS.58 Though promising, sPLA2 assays are not routinely available and their role in management remains to be determined.
Although 20% to 70% of patients with ACS are transfused, views differ on performing a simple transfusion targeting a Hgb of approximately 10, or an exchange transfusion targeting the same Hgb while lowering the HbS to under 30%.51,52 Although both approaches improve oxygenation and are safe and effective, exchange transfusion requires exposure to more donors, increasing allo-immunization, is more time consuming, and may require central access in this population with increased thrombotic risk. Thus a simple transfusion is recommended for most situations.59,60 The decision between direct and exchange transfusion in worsening ACS patients with a high Hgb (>9 g/dL) remains unclear as a minimal amount of red cells can be transfused.
Although lacking formal studies, the use of BiPAP or CPAP in an attempt to stave off intubation makes sense pathophysiologically. There is no consensus as to when to intubate or the optimal ventilation strategy in ACS, thus patients are often treated as children with acute lung injury or ARDS. For those patients with refractory hypoxemia, both high-frequency oscillatory ventilation and venovenous extracorporeal membrane oxygenation have been successful.61–63 Several promising approaches to ACS including improving NO metabolism with inhaled NO or oral arginine, a precursor of NO; and inhibitors of sPLA2.64
Patients with ACS have developed plastic bronchitis. A review of 29 children hospitalized with ACS found 72% had plastic bronchitis, one requiring mechanical ventilation, suggesting there may be a role for intratracheal DNase.65,66
Stroke
See Appendix Table 84-C for a detailed care plan and Appendix Figure 84-H for a diagnostic approach.22 One of the most debilitating and under-appreciated complications of SCD is a cerebrovascular accident (CVA), or the acute disruption of cerebral blood supply resulting in loss of brain tissue function lasting longer than 24 hours.67 In contrast, transient ischemia attacks resolve within 24 hours, and usually less than 1 hour. This section will focus on CVA related to SCD; a detailed discussion of stroke can be found in Chapter 63. Before screening and prevention programs, 11% of children with HbS/S or HbS/β-thalassemia developed overt strokes (peak, 2 to 9 years), with another 17% having silent infarcts.68,69 Risk factors include anemia, moya moya, history of CVA, and high velocities upon transcranial Doppler (TCD) ultrasound.70 Additional risks for ischemic stroke include ACS and hypertension, whereas leukocytosis is a risk for hemorrhagic stroke. Patients with HbS/C disease and other compound heterozygotic states do not have such a significantly increased risk. Strokes are heterogeneous, being due to ischemia, hemorrhage, or thromboembolic events and involving large, medium, and small vessels. The type of CVA varies by age, with ischemic events more common in the first decade and hemorrhagic events more common after age 20 years. Pathophysiology varies tremendously depending on the type and site of stroke, for example anemia is associated with watershed infarcts, whereas thrombus formation and emboli affect more midsize to small vessels, and hemorrhagic strokes likely result from intimal hyperplasia and dilation at the site of previous insults.
Natural History
The natural history of overt ischemic infarcts has changed dramatically with screening TCDs and chronic transfusions. If untreated, 67% of patients have a recurrent infarct within 9 years, and this is reduced to 10% with chronic transfusions targeting a HbS of under 30%.71–73 Screening TCDs using a standardized approach identifies patients with high cerebral vessel flow, associated with an excessively high stroke risk within 3 years.74 This can now be decreased to 10% with chronic transfusions.75 Although physical manifestations often show significant improvement after a stroke, persistent neurocognitive abnormalities are common. A subgroup of patients have moya moya, multiple constrictions of large arteries leading to collaterals at high risk of hemorrhage, aneurysm and clot (see Appendix Video 84-1). This group may have particularly severe disease and may represent some of the 10% resistant to chronic transfusions. The natural history of hemorrhagic infarcts varies in large part with their severity at presentation and location, because these dictate possible neurosurgical or radiology-guided interventions. Silent infarcts can result in difficulties with academic attainment and achievement and cognitive dysfunction similar to that in overt strokes.76 Executive function deficits are most common, having a tremendous effect on a patient’s function in society, and ability to navigate the health care system.
Diagnosis
See Appendix Figure 84-H for a diagnostic approach.22 While diagnosis is suggested by a history of acute neurologic changes and abnormalities on neurologic physical exam, magnetic resonance imaging (MRI) and magnetic resonance angiogram confirm it. Notably, headache and signs of increased cranial pressure are more common in hemorrhagic stroke (see Chapter 59). Neurosurgical intervention for a hemorrhagic lesion should never be delayed; thus an emergent noncontrast CT is obtained to immediately rule out any surgically amenable lesions and MRI may be deferred, potentially until after therapy has been initiated (see Appendix Figure 84-H).77 Other etiologies of childhood stroke must be considered and ruled out including infection, thrombosis (thrombosis panel and anticardiolipin antibodies), cardiac embolic disease, masses and trauma with vascular damage.
Management
See Appendix Table 84-C for a detailed care plan.22 Acute care and monitoring is similar to that for other children with CVAs except exchange transfusion is suggested. Careful attention to signs of increased intracranial pressure and urgent neurosurgical consult in the setting of hemorrhagic stroke, assuring oxygenation, treating fevers to decrease metabolic demands, maintaining adequate cerebral perfusion pressure and monitoring blood sugars and electrolytes are discussed in detail in Chapter 63). Seizures should be treated, but there is no role for prophylaxis. Although tissue plasminogen activator (t-PA) has a role in non-SCD stroke, t-PA or anticoagulant therapy is not recommended in children, in part due to the different pathophysiology. While no studies have assessed the need for acute transfusion, the prevention of secondary events with a chronic transfusion program and the underlying pathophysiology has led to acute transfusion with a goal of maintaining a HbS under 30% being standard of care.77 This is most efficiently done with exchange transfusion with a final Hgb of 10g/dL. Care must be taken to avoid hypotension with blood withdrawal during the exchange. Once stabilized, extensive evaluation by physical, occupational, and speech therapy, as well as a neurocognitive evaluation are essential to define new postevent baselines and guide future needs.
Aplastic Crisis
See Appendix Figure 84-I for a detailed care plan.22 An acute worsening of baseline anemia (by 1 to 2 g/dL of Hgb) associated with decreased reticulocyte count (typically <1%), suggests aplastic crises and is caused by acute infection such as parvovirus B19. Sickle RBCs survive 10 to 14 days (vs. 60 to 100 days for normal cells); thus patients are dependent on a threefold to fivefold increased reticulocyte production and any decrease can lead to a transient red cell aplasia with rapid development of a profound anemia. Monitoring of Hgb (both absolute and compared with the individual’s baseline), reticulocyte count, and cardiovascular status are essential. Most parvovirus B19 will spontaneously resolve, however, intravenous gammaglobulin should be considered to hasten viral clearance if reticulocytopenia persists. Transfusion may be necessary if cardiovascularly unstable or if the hemoglobin acutely falls more than 2 g/dL. Profound anemia or cardiac compromise may require a slow transfusion (2 mL/kg/hr), diuretics, or even exchange transfusion to avoid congestive heart failure.
Splenic Sequestration
See Appendix Figure 84-I for a detailed care plan.22 Sequestration is characterized by an acutely enlarging spleen with a Hgb more than 2 g/dL below an individual’s baseline value. Mild to moderate thrombocytopenia may also be present. Splenic sequestration occurs in 10% to 30% of children with SCD, most commonly between the ages 6 months and 3 years, often following a febrile illness. Abdominal pain, nausea, and vomiting are common, and severe episodes of sequestration may progress rapidly to cardiovascular collapse and death. Transfusion is indicated when signs of cardiovascular instability are present and, as with aplastic crises, caution should be taken to avoid contributing to congestive heart failure. Though rare, emergent splenectomy may be required. Elective splenectomy is indicated for recurrent episodes of sequestration with cardiovascular compromise.
Pulmonary Hypertension
Despite being a major cause of death in SCD, pulmonary hypertension (PHTN) is underappreciated, in part due the uniquely high morbidity associated with mild elevations in pulmonary artery pressures (PAP).78 Adults with estimated PAPs greater than 30 mm Hg or a tricuspid regurgitation velocity jet velocity (TR jet) of greater than 2.5 m/sec have a strikingly higher mortality rate; thus it is essential to have a significantly lower threshold for aggressive intervention for PHTN in the hemoglobinopathy patient than in others.78 Pulmonary hypertension in children (defined as a PAP greater than 25 mm Hg at rest or greater than 30 mm Hg with activity) is present in approximately 30% of children, as well as adults, with SCD. With approximately 100,000 SCD patients in the United States, this translates to 30,000 cases, making SCD the leading cause of PHTN. Despite this, there is little awareness of this relationship in the hematology and cardiology communities.
Pathophysiology and Etiology
Assessment and management is complicated as multiple etiologies and risk factors contribute (see Chapter 48). Unlike most PHTN in childhood in which there is a single dominant cause (e.g., congenital malformation), multiple pathways contribute to the manifestation of PHTN in SCD and each must be addressed for optimal management. Pulmonary hypertension is more common in SCD patients with a “hemolytic phenotype” associated with priapism and leg ulcers, but independent of VOE. Thus patients often have a high LDH, bilirubin, and reticulocyte count, and low HCT which are associated with decreased NO production and increased NO destruction, that result in a NO resistant state. Frequently confounding factors in SCD include (1) hypoxic PHTN due to the high incidence of enlarged tonsils, obstructive sleep apnea, asthma, and chronic lung disease; (2) arterial obstructive PHTN secondary to increased coagulation and embolic disease; and (3) pulmonary venous hypertension due to cardiomyopathy. Contributing to the high morbidity in SCD are the protean manifestations of mild pulmonary hypertension (30 to 44 mm Hg). The majority of patients with SCD and PHTN will be asymptomatic or have mild decreases in exercise tolerance (e.g., 6-minute walk) yet have a 10-fold increased risk of death.78 Adults with PHTN may not experience significant effects until pressures become moderate to severe, which for SCD portends a high mortality not observed in other patient populations with comparable pressure. The dilemma is that while mild PHTN is a major risk for death in adults, the natural history in children is unknown, and a recent study suggested no increased risk of death within three years of diagnosis.79 Thus it is not clear how aggressively to treat children with SCD and PHTN.
Management
Though no consensus has been reached, a three-part approach to treatment is reasonable. (1) Optimization of SCD care, with the goal of decreasing the hemolysis fueling NO imbalance. Hydroxyurea and chronic transfusions are typically used though these are not acute interventions. (2) Aggressive diagnosis and treatment of confounding factors (e.g., tonsillectomy and adenoidectomy, BiPAP or CPAP, and nighttime O2. (3) PHTN specific interventions, discussed in detail in Chapter 48. The mainstay of treatment of PHTN in the ICU is inhaled NO (iNO), which provides both direct vasodilation of the pulmonary vasculature and simultaneous reversal of the underlying disruption in NO metabolism. The major challenge with iNO is the development of rebound PHTN on discontinuation of therapy and difficulty in administration, thus driving a focus on alternative therapies. The phosphodiesterase (PDE)-5 inhibitor sildenafil is effective in facilitating weaning from iNO and in decreasing PAP over time in non-SCD PHTN80,81; however, a recent trial of sildenafil for PHTN in SCD was discontinued because of increased rates of VOE. Additional therapies that deserve consideration include (1) intravenous prostacyclin (epoprostenol), which reduced PAP in a small number of patients with SCD undergoing cardiac catheterization82; (2) endothelin receptor antagonists (e.g., bosentan and ambrisentan), which reduced PAP and improved 6-minute walk in adults with SCD and PHTN83; (3) inhaled iloprost, a prostacyclin analogue used in Europe, that has shown promising results in idiopathic PHTN, but its safety and efficacy in SCD has not been determined; and (4) oral arginine therapy, which reduced PAP within 5 days of therapy in a small number of patients with SCD and PHTN.84
Multiorgan Failure
Multiorgan failure syndrome is defined as severe pain associated with failure of at least two of the following organs: liver, lung, and kidney. It is often associated with severe pain in patients with previously mild disease and a relatively high Hgb.85 Widespread vaso-occlusion is thought to be responsible, though data is lacking. Patients present with an atypically severe VOE, fever, and sudden deterioration including a drop in Hgb and platelets, diffuse encephalopathy, and rhabdomyolysis. Death has been reported in up to 25% of patients.86 Exchange transfusion should be considered early, and can result in rapid recovery of organ function as well as survival. Antibiotics are often used, though many patients are culture negative. There are isolated reports of success with NO or plasma exchange in those with transfusion resistant disease.
Priapism
See Appendix Table 84-D for a detailed care plan.22 Priapism is common in males with SCD, often starting in childhood and frequently occurring during the early morning hours. Stuttering priapism is intermittent episodes lasting fewer than 2 to 4 hours, and is often recurrent and may precede a more severe episode. Severe episodes, lasting over 2 to 4 hours, need rapid intervention as they may result in permanent tissue damage and impotence.87 Initial interventions include hydration, warmth, micturition, activity, and analgesia. Pseudoephedrine can be used for prevention and treatment though no efficacy data is available. If these measures fail to lead to detumescence in 4 to 6 hours aspiration and irrigation with a dilute α-agonist (e.g., epinephrine, phenylephrine) by a urologist has been shown to be effective if done within 24 hours.88 Prevention with PDE-5 inhibitors is being investigated.
Renal Conditions
Passage through the vasa recta exposes red cells to hypoxia, acidosis, and hypertonicity, which are extreme enough conditions to make some trait cells sickle, and causing damage to the renal medulla. This can lead to multiple complications that can affect fluid balance and renal function including (1) Hyposthenuria, an inability to concentrate urine. It is critical to remain aware of this in the ICU as the production of dilute urine cannot be used as a marker for being euvolemic, and, patients are prone to dehydration, leading to pain episodes. This results in enuresis being common and worsened in the ICU by continuous intravenous fluids and opiates. (2) Hematuria: This can range from micro-, to gross hematuria associated with papillary necrosis. Conservative management with bed rest and maintenance of high urine output to avoid the development of clots usually suffices. Vasopressin has been used with some success, as has ε-amino caproic acid, though the latter should be used with caution as it can lead to clot. If recurrent transfusions are required or bleeding becomes life-threatening, resection of the involved region may be indicated. (3) Tubule dysfunction including an incomplete renal tubular acidosis worsened by the hyposthenuria, and hyperkalemia associated with the use of potassium-sparing diuretics, angiotensin-converting enzyme inhibitors, or β-blockers. (4) Chronic renal failure associated with glomerular injury and proteinuria and can progress to nephrotic syndrome. End-stage renal disease develops in 40% of patients with nephrotic syndrome, thus persistent proteinuria is an indication for an in-depth evaluation. Risk factors include hypertension, hematuria, proteinuria, and worsening anemia. Although there is no clear cure, angiotensin-converting enzyme inhibitors can reduce the proteinuria, and NSAIDs should be avoided. Worsening anemia can be treated with erythropoietin, though patients often need higher doses than others. Alternatively, transfusions can be done, keeping in mind the increased risk of fluid overload. Kidney transplantation is appropriate, and outcomes are similar to that of African Americans, albeit less than other Americans. Current success using combined hematopoietic stem cell and kidney transplants in a canine model raise the exciting possibility of curing both the hemoglobinopathy and renal failure simultaneously.89
Avascular Necrosis
Occlusion of endarterial vessels can lead to necrosis of juxta-articular bone. This occurs disproportionately in patients with concomitant α-thalassemia or HbS/C disease. Avascular necrosis can lead to significant pain and decrease in function. Conservative management with NSAIDs, opiates, and reduced weight bearing can allow for limited function while boney remodeling occurs, though with age this is less successful and progressive destruction leads to degenerative arthritis. Targets relate to size and weight bearing, with the hips, knees, and shoulders being most affected. In an attempt to avoid hip replacement, surgical core decompression had been promoted, but a specific, aggressive physical therapy program has been shown to be as effective.90 For severe cases, hip replacement is indicated.
Sleep Conditions
SCD patients are at increased risk for sleep-disordered breathing and obstructive sleep apnea, though the importance of this remains under debate.91,92 It has been suggested sleep disturbances increase the risk of VOE and pulmonary hypertension, though data is inconsistent. The etiology is not clear, but notably, SCD patients tend to have enlarged tonsils and adenoids and children with SCD have more severe nocturnal desaturations and hypercapnia than others with obstructive sleep apnea. Tonsillectomy and adenoidectomy often leads to resolution. Alternatively, auto-CPAP has been well tolerated in SCD and shown to improve sleep and cognitive function.93
Depression and Suicide
With recurrent pain and complications that can occur without warning, it is not surprising that anxiety and depression are increased in SCD.94,95 Interestingly, rates are higher than in cystic fibrosis, spina bifida, or diabetes, with 49% having anxiety symptoms in one study, and the risk of suicide in adults being increased.33,95–97
Surgery and Anesthesia
See Appendix Table 84-E for a detailed care plan.22 Major surgery in sickle cell is associated with increased perioperative risks including VOE pain, acute chest syndrome, and death, but these can be minimized with specific perioperative care. Preoperative transfusion was thought to provide benefit, leading to a randomized trial in which perioperative care was standardized and direct transfusion targeting a Hgb of 10 was compared with exchange transfusion targeting a Hgb of 10 with a HbS less than 30%.98 Outcomes for both arms appeared better than observed historically, and the two groups were equivocal except the exchange arm was exposed to more units of blood and had more alloimmunization. Thus a direct transfusion is preferred as it minimizes exposure to blood. Communication and coordination between anesthesia, surgery, and hematology is essential, and institutions are encouraged to have a standard perioperative care plan for SCD. Most patients are admitted overnight for O2, aggressive IS and observation, even if cleared to return home from a surgical standpoint. Because of poor compliance with incentive spirometry the use of BiPAP in the immediate post-operative period may be of use.99 Specific recommendations should be followed (Appendix Table 84-E), though some feel that with modern improvements in perioperative care, that routine transfusion is no longer needed.99,100
Therapies and Interventions
Hydroxyurea
Although hydroxyurea is not an acute intervention, the intensivist should be aware that many SCD patients are (or should) be prescribed hydroxyurea, as it can decrease pain, ACS episodes, and transfusion need while increasing hemoglobin and lifespan.10,101 Originally used in SCD to stimulate HbF production, the primary clinical benefits may stem from the relative leukopenia induced by this oral chemotherapeutic (see “Pathophysiology” above). Although good data exist for its use in adults, its role in children, especially the very young is an ongoing research question, but looks promising.102 Clinicians should remain aware that in addition to cytopenias, hydroxyurea can lead to a red cell macrocytosis and should not be alarmed by MCVs greater than 115.
Transfusion
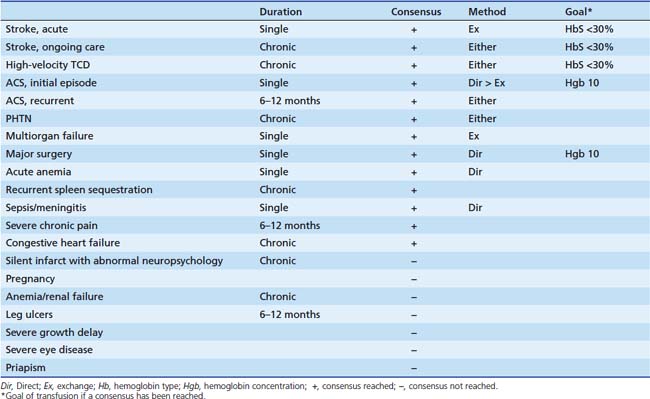
Though a mainstay of therapy, guidelines for transfusions in SCD are complex and vary by indication (Table 84-2).103–106 See Chapter 82 for general information regarding blood transfusion. SCD specific issues follow.
Choice of Product
Packed RBCs should be leukodepleted to minimize febrile reactions and alloimmunization. Although transfusion from sickle trait donors is safe and effective, HbS negative blood should be requested to allow accurate determination of posttransfusion HbS levels if needed. Due to racial, and therefore antigenic, differences between patients and the primarily white U.S. donor pool, alloimmunization is common, even after a single transfusion. This can significantly hinder the ability to provide future transfusion support. An extended cross-match for antigens of clinical significance that vary between racial groups should always be requested and should include Rh (Cc, D, Ee), and Kell in addition to ABO.105 The intensivist must be aware that this matching, or finding compatible units for highly immunized patients, may take considerable time, thus a type and cross should be sent well before blood is needed, and in some emergencies extended matching may need to be forgone. Unlike patients with hematologic malignancies there is no need for blood product irradiation or CMV negative selection.
Type and Goals of Transfusion
Transfusion increases O2 carrying capacity while decreasing the percent of HbS and different modes of transfusion affect these differentially. Few clinical trials are available but for several acute indications direct and exchange transfusions provided similar benefit, thus in these instances direct transfusion is preferred to minimize alloimmunization. The decision in chronic transfusion is more difficult as exchange transfusion results in less iron overload at the cost of increased exposure to blood. Manual exchange transfusions can be initiated rapidly with a single intravenous infusion, but are laborious and time consuming. Automated exchange (erythrocytapheresis) is rapid but not universally available, requires two large-bore intravenous infusions or a central pheresis-specific catheter and cannot be done in small children because of the significant volume and pressure shifts (size limits vary with the machine used). The target hemoglobin concentration or HbS percentage varies with indication (see Table 84-2) but in most cases the final Hb should never be higher than 11. Studies suggest higher Hb increase oxygen carrying capacity, but O2 delivery falls. Formulas for calculating blood volumes for routine and exchange transfusions are available (Appendix Figure 84-K).
Indications
The reflective tendency to transfuse SCD patients should be avoided. Because of alloimmunization and other risks, attempts should be made to avoid transfusion unless there is a clear clinical indication. Transfusions may be one-time, or chronic, the latter not being an ICU issue. Table 84-2 summarizes guidelines and additional information is in specific sections.103,104,106
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) remains the one cure for SCD and is recommended for young patients with severe disease and a suitable donor. This is not appropriate in the acute setting, and there is debate over what defines a young patient and suitable donor, as well as what the optimal conditioning regime is. The major role of the ICU is supportive care post-transplant (see Chapter 83). Pediatric patients receiving a matched sibling transplant after a myeloablative conditioning regime had an 85% event-free survival and an approximately 94% overall survival with approximately 10% graft failure (reverting to their endogenous marrow) and 12% to 20% significant GVHD.107,108 Attempts to adapt a less toxic, nonablative “mini transplant” regimen have been hampered by a high incidence of graft failure, potentially contributed to by a long history of exposure to donor antigens in transfusions. Initial attempts have found current regimes overly toxic to adults, presumably due to the accrued end-organ damage from SCD before transplant. Currently multiple SCD research transplant studies are available and address optimizing the conditioning regime and extending transplants to older patients, and to using alternate donors (unrelated, cord blood, haplo-identical).
Nitric Oxide
SCD often results in a NO resistant state. Both hemolysis related and vaso-occlusion related complications may directly benefit from NO-related therapies. Because NO is incredibly labile, multiple modes of delivery are being investigated, including providing substrate (e.g., arginine), providing co-factors for its generation (e.g., 6R-BH4), providing NO or direct metabolites intravenously or by inhalation, or modulating the effects of NO (e.g., sildenafil).109
Thalassemia
Molecular Description and Epidemiology
In contrast to a single mutation being responsible for sickle cell, and the mutant peptide directly leading to the pathophysiology, a broad spectrum of mutations lead to thalassemia and it is the residual unpaired phenotypically normal globin chains that incite damage. The hallmark of thalassemia is an imbalance in the ratio of α- and β-chains, not a deficiency in chains. The distinction becomes important when one considers α-/β-thalassemia compound heterozygotes, or duplicated (excess) globin genes. The broad spectrum of mutations responsible for thalassemia truly represents a textbook compendium of mechanisms of mutation, but common to all is an altered level of chain production. Gene deletions account for the majority of α-thalassemias, leading to quantum incremental changes in phenotype depending on whether 1, 2, 3, or all 4 adult α-globin genes are missing. In contrast to α-thalassemia, a broad range of mutations lead to β-thalassemia, resulting in a continuum of clinical phenotypes. As with sickle cell trait, thalassemia trait is thought to provide increased fitness in malaria zones. It is estimated that 1.5% of humans are heterozygous for a thalassemia, with about 0.05% having clinically significant disease. Because of the coexistence of α-/β-thalassemia in the same regions compound heterozygotes are common, leading to a huge spectrum of disease and complicating diagnostics.
Laboratory and Diagnostics
Although the sine quin non of diagnostics is the demonstration of an α- to β-globin chain imbalance, this is not practical for routine testing, thus most clinical laboratories rely on a variation of a thalassemia screen. The screen distinguishes thalassemia from iron deficiency, and α- from β-thalassemia. Due to the wide range of defects, and frequent mixed α- and β-thalassemia patients, it is impossible to state absolutes for diagnostic interpretation, and there should be a low threshold to consult with hematology. Components of the screen include (1) a complete blood count where emphasis is placed on the MCV which is decreased in proportion to hemoglobin production, the RBC to assess the degree of compensation for the decreased cell size, the hemoglobin and the smear; (2) a hemoglobin electrophoresis or HPLC to quantitate normal, and variant hemoglobins; (3) an inclusion body or Brilliant Cresyl Blue preparation to semi-quantitatively assess excess β-like chains and distinguish α- and β-thalassemia, and a (4) zinc-protoporphyrin or similar screen to exclude iron deficiency. α-Thalassemia results in excess β-like chains that form tetramers (β4: Hb H, γ4: Hb Barts) that precipitate in proportion to the number of missing α-genes, resulting in a positive inclusion body prep (Table 84-3). A negative study in the face of adequate iron is suggestive of β-thalassemia. The diagnosis of β-thalassemia is less direct. Excess α-chains are less stable and degrade, thus the inclusion body prep is normal and no α-tetramers are detectable on electrophoresis or HPLC. Frequently β-thalassemia results in up regulation of HbA2 or HbF; thus the presence of either is suggestive of the diagnosis. Many mistakenly exclude the diagnosis if HbA2 or HbF is not elevated, but this is not appropriate (e.g., deletion of the δ and β genes results in thalassemia but HbA2 is not elevated).

Forms and Variations
β-Thalassemia
In contrast to α-thalassemia, the plethora of β-gene mutations leads to a continuous spectrum of disease. β0-thalassemia denotes non-expressing alleles, whereas β+-thalassemia is used for genes that express a reduced amount of normal protein. Most mutations do not eliminate ε- or γ-gene expression, thus it is unusual for clinical complications to occur until several months of life, when infants become dependent on β-globin chains. Nomenclature is based on this continuum of phenotypic severity rather than genotype. β-thalassemia trait, also referred to as thalassemia minor, is due to a single silent allele and is benign and clinically similar to α-thalassemia trait. Although the term thalassemia major (aka Cooley anemia) is reserved for transfusion dependent phenotypes, those patients with clinical sequelae, but who are not dependent on transfusions for survival are referred to as having thalassemia intermedia. The latter require the most clinical decision-making as there is increasing awareness of the high morbidity in many of these patients.
Assessment of Iron Overload
Serum ferritin is a poor indicator of iron stores.110 Though inexpensive and easily obtained, it is elevated with inflammation and does not correlate well with tissue iron content, especially in patients receiving chelation therapy. Serial observations may help track trends but this is not a substitute for direct tissue measurements. While previously liver biopsy was the gold standard for assessment of liver iron content (LIC) it is limited by (1) its invasiveness and risk, (2) it not correlating with accumulation in the heart and other target organs, and (3) sampling errors, as only a small region of liver is assessed while iron accumulation is heterogeneous throughout the liver. MRI-based assessment of iron overload is based upon relaxation times correlating with iron content. These techniques are evolving rapidly, making it essential to discuss options with local radiologists, as standards and practices vary greatly. Ferriscan was the first Food and Drug Administration-approved MRI assessment of iron, obviating biopsies, but it is limited to the assessment of hepatic iron.111 In contrast, T2∗ imaging is increasingly available and allows simultaneous assessment of hepatic and cardiac iron, and potentially, other target organs.112 A lower T2∗ correlates with higher degrees of iron overload (see “Cardiac Failure” below).
Spectrum of Disease
Although a summary of clinical problems and management are listed below and in Appendix Figure 84-N, please refer to the Northern California Comprehensive Thalassemia Center standard of care guidelines113 for more details: http://thalassemia.com/documents/thalhandbook2008.final.pdf.
Anemia
Although there is much debate as to the optimal Hgb that should be targeted, in the ICU one must factor in the acuteness of the anemia, the degree of cardiovascular compromise, and the patient’s baseline Hgb. A baseline level of under 7 g/dL is often the threshold for initiating chronic transfusions, though a higher threshold is used if growth, skeletal malformations or extramedullary erythropoiesis become problematic. The goal should be to maintain a Hgb of 9 to 10 g/dL pretransfusion (10 to 12 g/dL if cardiac disease is present), but no higher than 14 g/dL posttransfusion. As with SCD, profound anemia or cardiac compromise may require a slow transfusion, diuretics, or even exchange transfusion to avoid congestive heart failure. Blood products should be leuko-reduced to minimize alloimmunization, febrile nonhemolytic transfusion reactions, and CMV transmission. As with SCD, blood should be matched for ABO, Rh (Cc, D, Ee), and Kell. Family and related donor transfusion should be avoided if a hematopoietic stem cell transplant is to be considered as alloimmunization to donor antigens increases graft rejection.
Transfusion-Related Complications
Chronic or frequent transfusions increase infectious risk (described in Chapter 82) as well the risk of alloimmunization. Because of inappropriately low hepcidin levels, the ability of the body to absorb iron is unopposed, leaving blood loss and sloughing of endothelial and skin cells as the only mechanism to decrease iron in the face of iron overload. This is estimated to lead to a loss of 1 mg/day in an adult that pales in comparison to the approximately 200 mg of iron in each unit of blood. Although previous focus was on hepatic iron overload and cirrhosis, there is increasing awareness of iron overload in the heart, pituitary and endocrine organs leading to dysfunction (the the following section), and excessive melanin production leading to “bronze” pigmentation. Iron overload requires chelation, which can result in additional complications (Appendix Table 84-G).113 Because of its short half-life, deferoxamine therapy is given continuously (subcutaneously or intravenously), and complications may include oto- or ophthalmologic toxicity, allergic reactions with life threatening hypotension (primarily when given intravenously), growth failure, metaphyseal cartilaginous dysplasia, bacterial infections (e.g., Yersinia), and inadvertent chelation of other cations including zinc, copper, selenium, and calcium. The need for continuous subcutaneous infusions often leads to problems with compliance, in part overcome by the use of oral deferasirox. Though quality of life may be improved, compliance issues remain, and more than 30% of patients on deferasirox have a reversible increase in creatinine; renal and hepatic failure has been reported.110 Optimal dosing of either drug is determined over time, taking into account the degree of overload, transfusion regime, and whether a neutral, or negative iron balance is desired. Please see the cardiac failure section for emergent dosing and new approaches.
Cardiac Failure
Cardiac failure, defined as a low ejection fraction with a component of cardiomyopathy, is the major cause of death in thalassemic patients with iron overload. A thalassemic patient in failure should be assumed to have cardiac iron overload until proven otherwise. Risk factors include transfusion history and underchelation. Excess unbound iron from transfusion and inappropriate gastrointestinal absorption freely penetrates cardiac myocytes leading to progressive tissue damage. Iron accumulates predominantly in the epicardial portion of the ventricular septum and ventricular free walls, stimulating the production of free radicals, resulting in peroxidative tissue damage, decreased cardiac contractility and dysrhythmias. This, in combination with the high output state from chronic anemia, results in the early development of cardiac failure. Cardiac iron overload occurs years after the liver becomes overloaded. Initial cardiac iron deposition increases the influx of additional iron resulting in a rapid accumulation and the potential for rapid decline, a wide range of dysrhythmias and failure after years of normal function. The clinical presentation is that of congestive heart failure; however, some present solely with abdominal pain due to liver distension. Clinicians should always have a low threshold to evaluate for cardiac dysfunction. Before the introduction of iron chelation therapy in the 1960s, the majority of patients with thalassemia died from congestive heart failure by age 16 years. With advances in both transfusion and chelation therapy survival has increased, with 80% of patients living to age 40 years.114 Lifespan is highly dependent on the underlying thalassemia as well as the transfusion and chelation history.
Assessments
Cardiac iron accumulation and function must both be assessed. Ferritin and assessments of LIC cannot be used, because kinetics of iron loading and unloading differ between the organs. Iron assessment by T2∗ is invaluable as one can assess cardiac iron, as well as LIC.112 A T2∗ of over 20 ms is not associated with increased cardiac risk. In contrast, a value of 10 to 20 ms denotes overload and increased cardiac risk, and a T2∗ of under 10 ms portends a high risk of cardiac dysfunction and an emergent situation requiring aggressive chelation (Appendix Table 84-H).110,113,115 Echocardiography is essential, and left ventricular dysfunction is highly suggestive of iron overload.116 Given the high output state, some suggest the cutoff for a normal ejection fraction in thalassemics should be 60%.117 Electrocardiogram often revels biventricular dysfunction with left ventricular hypertrophy, a prolonged PR interval, bradycardia, ST-T wave change, and T-wave inversions. Iron-related parathyroid, and thyroid and adrenal dysfunction, as well as vitamin D and thiamine status should be investigated for as these impact cardiac function.
Management
Optimal chelation strategies in emergent situations are evolving rapidly, thus a hematologist or local expert on chelation should be consulted. While deferoxamine is proven to chelate cardiac as well as liver iron, increasing data suggests that deferasirox may be effective as well. Deferiprone (L1) is small molecular weight oral chelator awaiting Food and Drug Administration approval that has been used extensively in Europe and studies suggest it may be superior for removal of cardiac iron, but its major limitation is the risk of agranulocytosis. The availability of additional agents is leading to new regimes for cardiac iron removal making use of dual therapy with agents with nonoverlapping toxicity profiles.
Therapies and Interventions
Hematopoietic Stem Cell Transplantation
Currently HSCT remains the only proven cure for thalassemic patients. Long transfusion histories increase the chance of graft failure, necessitating an aggressive myeloablative conditioning regimen, which in turn can lead to a high treatment-related toxicity if significant end-organ damage is present. Success is dependent on having an adequate donor, and the degree of end-organ damage. The latter is assessed using the Lucarelli staging system that factors in the degree of hepatomegaly, the degree of portal fibrosis, and the quality of the chelation treatment given before the transplant.118 With improved pretransplant care and increased myeloablation and immunosuppression, even the highest risk group has done well with matched-sibling donors with a 90% event-free survival, 96% overall survival, and 7% rejection rate.119
Inducers of Other β-like Genes
As expression of other β-like globin genes will ameliorate disease severity, numerous agents have been tried. Results from clinical trials vary in part by genotype, but the discussion in this chapter will be limited to a generic approach. Histone de-acetylases such as the short chain fatty acid butyrate initially showed promise in increasing expression from the δ-genes, but have showed variable efficacy.120–122 Current efforts are focused on developing related compounds such as additional histone acetylases (e.g., suberoylanilide hydroxamic acid), and additional short chain fatty acids that may induce embryonic globin genes. Hydroxyurea is less efficacious than in sickle cell, but some success has been observed in combination with erythropoietin.123 Previously, promising results with the demethylating agent 5-aza-cytidine led to the development of the DNA methyltransferase inhibitor decitabine that results in demethylation and activation of fetal globin genes.123,124 Early studies, and the potential to develop an oral form make decitabine an exciting future prospect for the treatment of SCD and β-thalassemia.
Acknowledgment
We to thank Gabrielle Seibel for critical reading and review.
References are available online at http://www.expertconsult.com.
1. World Health Organization: Control of hereditary diseases: report of a who scientific group, Geneva, 1996, WHO
2. Stamatoyannopoulos G. Molecular and cellular basis of hemoglobin switching. In: Steinberg M.H., Forget B.G., Higgs H.R., Nagel R.L., editors. Disorders of hemoglobin. Cambridge, UK: Cambridge University Press; 2001:131-145.
3. Austin H., Key N.S., Benson J.M., et al. Sickle cell trait and the risk of venous thromboembolism among blacks. Blood. 2007;110:908-912.
4. Mitchell B.L. Sickle cell trait and sudden death–bringing it home. J Natl Med Assoc. 2007;99:300-305.
5. Gill F.M., Sleeper L.A., Weiner S.J., et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776-783.
6. Platt O.S., Brambilla D.J., Rosse W.F., et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639-1644.
7. Serjeant G.R., Higgs D.R., Hambleton I.R. Elderly survivors with homozygous sickle cell disease. N Engl J Med. 2007;356:642-643.
8. Wierenga K.J., Hambleton I.R., Lewis N.A. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. Lancet. 2001;357:680-683.
9. Bakanay S.M., Dainer E., Clair B., et al. Mortality in sickle cell patients on hydroxyurea therapy. Blood. 2005;105:545-547.
10. Steinberg M.H., Barton F., Castro O., et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645-1651.
11. Bunn H.F. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762-769.
12. Kato G.J., Gladwin M.T., Steinberg M.H. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37-47.
13. Morris C.R. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology. 2008:177-185.
14. Steinberg M.H. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. Sci World J. 2008;8:1295-1324.
15. Miller S.T., Sleeper L.A., Pegelow C.H., et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342:83-89.
16. Fitzhugh C.D., Hsieh M.M., Bolan C.D., et al. Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy. 2009;11:464-471.
17. Steinberg M.H., Lu Z.H., Barton F.B., et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter study of hydroxyurea. Blood. 1997;89:1078-1088.
18. Ware R.E., Eggleston B., Redding-Lallinger R., et al. Predictors of fetal hemoglobin response in children with sickle cell anemia receiving hydroxyurea therapy. Blood. 2002;99:10-14.
19. Machado RF, Barst RJ, et al: safety and efficacy of sildenafil therapy for doppler-defined pulmonary hypertension in patients with sickle cell disease: preliminary results of the Walk-PHaSST clinical trial. Presented at the American Society of Hematology. Annual Meeting and Exposition, New Orleans, LA, 2009.
20. Schmidtko A., Tegeder I., Geisslinger G., No N.O. no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32:339-346.
21. The management of sickle cell disease. Bethesda, 2002, National Heart, Lung, and Blood Institute.
22. Bender M.A., White R., Bloomquist T.E.Sickle cell disease: critical elements of care 2006:ed 4 Seattle Children’s Hospital Seattle
23. Shapiro B.S., BB K. The acute painful episode. In: Embury S.H., Hebbel R.P., Mohandas N., Steinberg M.H., editors. Sickle cell disease: basic principles and clinical practice. Philadelphia: Lippincott-Raven; 1994:531-543.
24. Benjamin LJ, Dampier CD, Jacox AK, et al. Guideline for the management of acute and chronic pain in sickle-cell disease. Clinical practice guideline series, vol 1, Glenville, IL, 1999, American Pain Association.
25. Rees D.C., Olujohungbe A.D., Parker N.E., et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematol. 2003;120:744-752.
26. Todd K.H., Green C., Bonham V.L.Jr., et al. Sickle cell disease related pain: crisis and conflict. J Pain. 2006;7:453-458.
27. Platt O.S., Thorington B.D., Brambilla D.J., et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11-16.
28. Smith W.R., Bovbjerg V.E., Penberthy L.T., et al. Understanding pain and improving management of sickle cell disease: the PiSCES study. J Natl Med Assoc. 2005;97:183-193.
29. Rogovik A.L., Med Y.L., Kirby M.A., et al. Admission and length of stay due to painful vasoocclusive crisis in children. Am J Emerg Med. 2009;27:797-801.
30. Vasavda N., Ulug P., Kondaveeti S., et al. Circulating DNA: a potential marker of sickle cell crisis. Br J Hematol. 2007;139:331-336.
31. Ballas S.K. Sickle cell disease: clinical management. Baillieres Clin Hematol. 1998;11:185-214.
32. Edwards C.L., Wellington C.C. Adoption status and psychological distress in black patients with sickle cell disease. J Natl Med Assoc. 2009;101:1070-1071.
33. Simon K., Barakat L.P., Patterson C.A., Dampier C. Symptoms of depression and anxiety in adolescents with sickle cell disease: the role of intrapersonal characteristics and stress processing variables. Child Psychiatry Hum Dev. 2009;40:317-330.
34. Beiter J.L.J., Simon H.K., Chambliss C.R., et al. Intravenous ketorolac in the emergency department management of sickle cell pain and predictors of its effectiveness. Arch Pediat Adolesc Med. 2001;155:496-500.
35. van Beers E.J., van Tuijn C.F.J., Nieuwkerk P.T. Patient-controlled analgesia versus continuous infusion of morphine during vaso-occlusive crisis in sickle cell disease, a randomized controlled trial. Am J Hematol. 2007;82:955-960.
36. Buchanan I.D., Woodward M., Reed G.W. Opioid selection during sickle cell pain crisis and its impact on the development of acute chest syndrome. Pediatr Blood Cancer. 2005;45:716-724.
37. Finkelstein Y., Schechter T., Garcia-Bournissen F., et al. Is morphine exposure associated with acute chest syndrome in children with vaso-occlusive crisis of sickle cell disease? A 6-year case-crossover study. Clin Ther. 2007;29:2738-2743.
38. Howland M.A., Goldfrank L.R. Why meperidine should not make a comeback in treating patients with sickle cell disease. Ann Emerg Med. 2008;51:203-205.
39. Yaster M., Tobin J.R., Billett C., et al. Epidural analgesia in the management of severe vaso-occlusive sickle cell crisis. Pediatrics. 1994;93:310-315.
40. Phillips W.J., Gadiraju S., Dickey S., et al. Dexmedetomidine relieves pain associated with acute sickle cell crisis. J Pain Symp Manag. 2007;34:346-349.
41. Telfer P.T., Lahoz C., Ali K., et al. Intranasal diamorphine for acute sickle cell pain. Arch Dis Child. 2009;94:979-980.
42. Qari M.H., Aljaouni S.K., Alardawi M.S., et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost. 2007;98:392-396.
43. Powars D., Overturf G., Weiss J., et al. Pneumococcal septicemia in children with sickle cell anemia. Changing trend of survival. JAMA. 1981;245:1839-1842.
44. Vichinsky E., Hurst D., Earles A., et al. Newborn screening for sickle cell disease: effect on mortality. Pediatrics. 1988;81:749-755.
45. Gaston M.H., Verter J.I., Woods G., et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314:1593-1599.
46. Halasa N.B., Shankar S.M., Talbot T.R., et al. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis. 2007;44:1428-1433.
47. Hord J., Byrd R., Stowe L., et al. Streptococcus pneumoniae sepsis and meningitis during the penicillin prophylaxis era in children with sickle cell disease. J Pediatr Hematol Oncol. 2002;24:470-472.
48. Adamkiewicz T.V., Silk B.J., Howgate J., et al. Effectiveness of the 7-valent pneumococcal conjugate vaccine in children with sickle cell disease in the first decade of life. Pediatrics. 2008;121:562-569.
49. Norris C.F., Smith-Whitley K., McGowan K.L. Positive blood cultures in sickle cell disease: time to positivity and clinical outcome. J Pediatr Hematol Oncol. 2003;25:390-395.
50. Bjornson A.B., Falletta J.M., Verter J.I., et al. Serotype-specific immunoglobulin G antibody responses to pneumococcal polysaccharide vaccine in children with sickle cell anemia: effects of continued penicillin prophylaxis. J Pediatr. 1996;129:828-835.
51. Vichinsky E.P., Styles L.A., Colangelo L.H., et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787-1792.
52. Vichinsky E.P., Neumayr L.D., Earles A.N., et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855-1865.
53. Schindler M.B. Treatment of atelectasis: where is the evidence? Crit Care. 2005;9:R351-356.
54. Hsu L.L., Batts B.K., Rau J.L. Positive expiratory pressure device acceptance by hospitalized children with sickle cell disease is comparable to incentive spirometry. Respir Care. 2005;50:624-627.
55. Morris C.R. Asthma management: reinventing the wheel in sickle cell disease. Am J Hematol. 2009;84:234-241.
56. Bernini J.C., Rogers Z.R., Sandler E.S. Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood. 1998;92:3082-3089.
57. Strouse J.J., Takemoto C.M., Keefer J.R. Corticosteroids and increased risk of readmission after acute chest syndrome in children with sickle cell disease. Pediatr Blood Cancer. 2008;50:1006-1012.
58. Styles L.A., Schalkwijk C.G., Aarsman A.J., et al. Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood. 1996;87:2573-2578.
59. Turner J.M., Kaplan J.B., Cohen H.W., Billett H.H. Exchange versus simple transfusion for acute chest syndrome in sickle cell anemia adults. Transfusion. 2009;49:863-868.
60. Velasquez M.P., Mariscalco M.M., Goldstein S.L., Airewele G.E. Erythrocytapheresis in children with sickle cell disease and acute chest syndrome. Pediatr Blood Cancer. 2009;53:1060-1063.
61. Pelidis M.A., Kato G.J., Resar L.M.S., et al. Successful treatment of life-threatening acute chest syndrome of sickle cell disease with venovenous extracorporeal membrane oxygenation. J Pediatr Hematol Oncol. 1997;19:459-461.
62. Trant C.A.Jr., Casey J.R., Hansell D., et al. Successful use of extracorporeal membrane oxygenation in the treatment of acute chest syndrome in a child with severe sickle cell anemia. ASAIO J. 1996;42:236-239.
63. Wratney A.T., Gentile M.A., Hamel D.S., Cheifetz I.M. Successful treatment of acute chest syndrome with high-frequency oscillatory ventilation in pediatric patients. Respir Care. 2004;49:263-269.
64. Hajeri A.A., Serjeant G.R., Fedorowicz Z. Inhaled nitric oxide for acute chest syndrome in people with sickle cell disease. Cochrane Database Sys Rev. 23, 2008. CD006957
65. Manna S.S., Shaw J., Tibby S.M., Durward A. Treatment of plastic bronchitis in acute chest syndrome of sickle cell disease with intratracheal rhDNase. Arch Dis Child. 2003;88:626-627.
66. Moser C., Nussbaum E., Cooper D.M. Plastic bronchitis and the role of bronchoscopy in the acute chest syndrome of sickle cell disease. Chest. 2001;120:608-613.
67. Wong W.Y., Powars D.R. Overt and incomplete (silent) cerebral infarction in sickle cell anemia: diagnosis and management. Hematol Oncol Clin North Am. 2005;19:839-855.
68. Ohene-Frempong K., Weiner S.J., Sleeper L.A., et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288-294.
69. Pegelow C.H., Macklin E.A., Moser F.G., et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. 2002;99:3014-3018.
70. Hoppe C. Defining stroke risk in children with sickle cell anaemia. Br J Haematol. 2005;128:751-766.
71. Pegelow C.H., Adams R.J., McKie V., et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126:896-899.
72. Powars D., Wilson B., Imbus C. The natural history of stroke in sickle cell disease. Am J Med. 1978;65:461-471.
73. Russell M.O., Goldberg H.I., Hodson A., et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. 1984;63:162-169.
74. Adams R., McKie V., Nichols F., et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992;326:605-610.
75. Adams R.J., McKie V.C., Hsu L., et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11.
76. Schatz J., Brown R.T., Pascual J.M., et al. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology. 2001;56:1109-1111.
77. The management of sickle cell disease, ed 4, Bethesda MD, 2002, National Heart, Lung, and Blood Institute, p 83–99.
78. Gladwin M.T., Sachdev V., Jison M.L., et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Eng J Med. 2004;350:886-895.
79. Lee M.T., Small T., Khan M.A., et al. Doppler-defined pulmonary hypertension and the risk of death in children with sickle cell disease followed for a mean of three years. Br J Haematol. 2009;146:437-441.
80. Peiravian F., Amirghofran A.A., Borzouee M., et al. Oral sildenafil to control pulmonary hypertension after congenital heart surgery. Asian Cardiovasc Thorac Ann. 2007;15:113-117.
81. Raja S.G., Danton M.D., MacArthur K.J., Pollock J.C. Effects of escalating doses of sildenafil on hemodynamics and gas exchange in children with pulmonary hypertension and congenital cardiac defects. J Cardiothorac Vasc Anesth. 2007;21:203-207.
82. Castro O., Hoque M., Brown B.D. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257-1261.
83. Minniti C.P., Machado R.F., Coles W.A., et al. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol. 2009;147:737-743.
84. Morris C.R., Sidney M., Morris J., et al. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Res Crit Care Med. 2003;168:63-69.
85. Hassell K.L., Eckman J.R., Lane P.A. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med. 1994;96:155-162.
86. Perronne V., Roberts-Harewood M., Bachir D., et al. Patterns of mortality in sickle cell disease in adults in France and England. Hematol J. 2002;3:56-60.
87. Rogers Z.R. Priapism in sickle cell disease. Hematol Oncol Clin North Am. 2005;19:917-928. viii
88. Mantadakis E., Ewalt D.H., Cavender J.D. Outpatient penile aspiration and epinephrine irrigation for young patients with sickle cell anemia and prolonged priapism. Blood. 2000;95:78-82.
89. Kuhr C.S., Yunusov M., Sale G. Long-term tolerance to kidney allografts in a preclinical canine model. Transplantation. 2007;84:545-547.
90. Neumayr L.D., Aguilar C., Earles A.N., et al. Physical therapy alone compared with core decompression and physical therapy for femoral head osteonecrosis in sickle cell disease. Results of a multicenter study at a mean of three years after treatment. J Bone Joint Surg Am. 2006;88:2573-2582.
91. Kaleyias J., Mostofi N., Grant M., et al. Severity of obstructive sleep apnea in children with sickle cell disease. J Pediatr Hematol Oncol. 2008;30:659-665.
92. Sedrak A., Rao S.P., Miller S.T., et al. A prospective appraisal of pulmonary hypertension in children with sickle cell disease. J Pediatr Hematol Oncol. 2009;31:97-100.
93. Marshall M.J., Bucks R.S., Hogan A.M., et al. Auto-adjusting positive airway pressure in children with sickle cell anemia: results of a phase I randomized controlled trial. Haematologica. 2009;94:1006-1010.
94. Gil K.M., Anthony K.K., Carson J.W., et al. Daily coping practice predicts treatment effects in children with sickle cell disease. J Pediatr Psychol. 2001;26:163-173.
95. Hasan S.P., Hashmi S., Alhassen M., et al. Depression in sickle cell disease. J Natl Med Assoc. 2003;95:533-537.
96. Edwards C.L., Green M., et al. Depression, suicidal ideation, and attempts in black patients with sickle cell disease, J Natl Med Assoc. 2009;101:1090-1095.
97. Levenson J.L., McClish D.K., Dahman B.A., et al. Depression and anxiety in adults with sickle cell disease: the PiSCES project. Psychosom Med. 2008;70:192-196.
98. Vichinsky E.P., Haberkern C.M., Neumayr L., et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med. 1995;333:206-213.
99. Leff D.R., Kaura T., Agarwal T. A nontransfusional perioperative management regimen for patients with sickle cell disease undergoing laparoscopic cholecystectomy. Surg Endosc. 2007;21:1117-1121.
100. Firth P.G., Head C.A. Sickle cell disease and anesthesia. Anesthesiology. 2004;101:766-785.
101. Charache S., Terrin M.L., Moore R.D., et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317-1322.
102. Thornburg C.D., Dixon N., Burgett S., et al. A pilot study of hydroxyurea to prevent chronic organ damage in young children with sickle cell anemia. Pediatr Blood Cancer. 2009;52:609-615.
103. Josephson C.D., Su L.L., Hillyer K.L., Hillyer C.D. Transfusion in the patient with sickle cell disease: a critical review of the literature and transfusion guidelines. Transfus Med Rev. 2007;21:118-133.
104. Ohene-Frempong K. Indications for red cell transfusion in sickle cell disease. Semin Hematol. 2001;38:5-13.
105. Vichinsky E. Consensus document for transfusion-related iron overload. Semin Hematol. 2001;38:2-4.
106. Wanko S.O., Telen M.J. Transfusion management in sickle cell disease. Hematol Oncol Clin North Am. 2005;19:803-826.
107. Bhatia M., Walters M.C. Hematopoietic cell transplantation for thalassemia and sickle cell disease: past, present and future. Bone Marrow Transplant. 2008;41:109-117.
108. Michlitsch J.G., Walters M.C. Recent advances in bone marrow transplantation in hemoglobinopathies. Curr Mol Med. 2008;8:675-689.
109. Kato G.J., Gladwin M.T. Evolution of novel small-molecule therapeutics targeting sickle cell vasculopathy. JAMA. 2008;300:2638-2646.
110. Kwiatkowski J.L. Oral iron chelators. Pediatr Clin North Am. 2008;55:461-482.
111. St Pierre T.G., Clark P.R., Chua-anusorn W., et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105:855-861.
112. Wood J.C. Magnetic resonance imaging measurement of iron overload. Curr Opin Hematol. 2007;14:183-190.
113. Vichinsky E., Levine L., et al. Standards of care guidelines for thalassemia. Oakland, CA: Children’s Hospital & Research Center Oakland; 2008.
114. Borgna-Pignatti C., Rugolotto S., De Stefano P., et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89:1187-1193.
115. Patton N., Brown G., Leung M., et al. Observational study of iron overload as assessed by magnetic resonance imaging (mri) in an adult population of transfusion dependent patients with beta thalassaemia: significant association between low cardiac T2∗ < 10 ms and the occurrence of cardiac events. Intern Med J. 2010. 40(6):419–426,
116. Anderson L.J., Holden S., Davis B., et al. Cardiovascular T2-star (T2∗) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171-2179.
117. Pepe A., Positano V., Santarelli M.F., et al. Multislice multiecho T2∗ cardiovascular magnetic resonance for detection of the heterogeneous distribution of myocardial iron overload. J Magn Reson Imaging. 2006;23:662-668.
118. Lucarelli G., Andreani M., Angelucci E. The cure of thalassemia by bone marrow transplantation. Blood Rev. 2002;16:81-85.
119. Sodani P., Gaziev D., Polchi P., et al. New approach for bone marrow transplantation in patients with class 3 thalassemia aged younger than 17 years. Blood. 2004;104:1201-1203.
120. Fathallah H., Atweh G.F. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology. 2006:58-62.
121. Perrine S.P., Castaneda S.A., Boosalis M.S. Induction of fetal globin in beta-thalassemia: cellular obstacles and molecular progress. Ann N Y Acad Sci. 2005;1054:257-265.
122. Testa U. Fetal hemoglobin chemical inducers for treatment of hemoglobinopathies. Ann Hematol. 2009;88:505-528.
123. Perrine S.P. Fetal globin stimulant therapies in the beta-hemoglobinopathies: principles and current potential. Pediatr Ann. 2008;37:339-346.
124. Saunthararajah Y. Decitabine and sickle cell disease: molecular therapy for a molecular disease. Pediatr Hematol Oncol. 2007;24:465-468.
125. Bender M.A., Hobbs W. Sickle cell disease genereviews at genetests: medical genetics information resource. Seattle: University of Washington; 2009.
126. Higgs D.R., Bowden D.K. Clinical and laboratory features of alpha-thalassemia syndromes. In: Steinberg M.H., Forget B.G., Higgs D.R., Nagel R.L., editors. Disorders of hemoglobin. Cambridge, UK: Cambridge University Press; 2001:431-469.
127. Galanello R., Cao A. Alpha-thalassemia genereviews at genetests: medical genetics information resource. Seattle: University of Washington; 2008.
128. Wayne A.S., Kevy S.V., Nathan D.G. Transfusion management of sickle cell disease. Blood. 1993;81:1109-1123.
129. Rund D., Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353:1135-1146.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 84 Hemoglobinopathies
Appendix materials for this chapter are available online at http://www.expertconsult.com.
Perspective
Hemoglobinopathies are plentiful as many provide a protective effect to malaria. Although 5% of humans have hemoglobin variants, an estimated 300,000 affected homozygotes are born annually.1 Contrary to popular beliefs, hemoglobinopathies do not provide resistance to infection, but rather diminish the chance of death once infected, allowing the development of partial immunity. Although sickle cell and thalassemia trait increase fitness, homozygotes in malaria endemic areas often do not survive into their teens. Although this strong protective effect resulted in a high incidence of hemoglobinopathies in the equatorial malaria zones, the slave trade and more recent voluntary migrations has led to the worldwide distribution of these conditions.
The Globin Gene Loci
The α- and β-like genes reside within multigene loci and are transcribed at unparalleled levels in both tightly tissue specific, and developmentally specific patterns.2 This, and their involvement in human disease, has made these loci paradigms for gene regulation and pathophysiology. The β-globin gene was the first sequenced, and sickle cell was the first molecularly defined and diagnosed disease. Most forms of mutation (e.g., point, splicing, deletion, gene rearrangement) are observed in thalassemia, making these the quintessential teaching loci. This high variety and incidence of mutations result in frequent compound heterozygotes with a wide spectrum of clinical manifestations.
The five genes of the β-globin locus reside in a cluster on chromosome 11. The genes are expressed in an erythroid, and developmentally stage-specific manor; the ε, Aγ and Gγ, and δ and β genes being expressed primarily during the embryonic, fetal, and postnatal periods, respectively. At birth 95% of β-like chains are γ, with the rest being β. This ratio gradually inverts during the first year of life, explaining why phenotypes limited to the β-globin gene such as sickle cell and most β-thalassemias do not manifest until several months of age. Expression of the chromosome 16 based α-like genes differs; the embryonic ζ-gene parallels the expression of ε, but the twin α-genes are expressed from the fetal period onward. Thus α abnormalities manifest in utero, potentially with devastating consequences (e.g., hydrops fetalis). The resultant hemoglobin α-, β-heterotetramers (HbS) are developmentally expressed; embryonic: Hb Gower1 (ζ2,ε2), Hb Gower2 (α2,ε2), and Hb Portland (ζ2,γ2); fetal: HbF (fetal) (α2,γ2) and adult: HbA2 (α2,δ2) and HbA (Adult) (α2, β2) (Appendix Figure 84-A). Although many suggest HbF, with its higher oxygen affinity, evolved to aid the fetus in “stealing” oxygen from the mother, this is not supported experimentally, the advantage of HbF may in fact lie in differential transport of NO or carbon dioxide. Unfortunately, despite the intensive study, multiple Nobel prizes and the clinical importance of the globin loci, little of the molecular understanding of the loci has been translated to clinical care. The holy grail of manipulating expression from the β-locus remains elusive. Inhibiting adult sickle or thalassemic genes, while concomitantly activating expression from the normal fetal genes would alleviate most disease, obviating the need for this chapter.
Sickle Cell Disease
Molecular Description and Epidemiology
Sickle cell disease (SCD) is a group of single gene, autosomal recessive disorders common in people originating from specific regions of Africa or India and in those of Hispanic descent. SCD encompasses a group of disorders characterized by the presence of a single nucleotide change in the adult β-globin gene, and a second abnormal allele allowing the sickle hemoglobin to polymerize. Substitution of an adenine (A) for a thymidine (T) in codon 6 results in replacement of a hydrophilic glutamic acid residue with a hydrophobic valine residue. The allosteric changes in hemoglobin structure occurring with deoxygenation leads to exposure of a destabilizing valine-containing pocket. The high concentration of hemoglobin in erythrocytes (MCHC) allows for the alignment of these pockets, leading to polymerization and transition to the classic sickle cell morphology (Figure 84-1) and the initiation of downstream events leading to pain and chronic end-organ damage. The presence of HbA in sickle trait, or HbF in the newborn, attenuate polymerization, diminishing the clinical consequences of HbS. In the United States, approximately 1 in 14 and 1 in 400 African Americans have sickle trait and SCD, respectively, with approximately 100,000 cases and approximately 1000 new births per year (in contrast to approximately 1 million births per year in Africa). With increased ethnic mixing, compound heterozygous forms of SCD are increasingly being observed (e.g., S/β-thalassemia or HbS/E). Sadly, though simple public health measures have dramatically decreased mortality rates in the United States, the countries with the highest incidence of SCD often lack such resources and children with SCD often do not reach their teens.
Sickle Cell Trait
Once the presence of HbS has been confirmed, the differential diagnosis consists of severe forms, less severe forms, and carrier states. Sickle cell trait (SCT) is distinguished from SCD by the presence of a normal β-globin allele and more HbA than HbS (typically 40% HbS) (Table 84-1). SCT does not result in the classic spectrum of sickle-related complications or alter red cell indices, but can lead to clinical abnormalities. Impaired renal concentrating abilities and intermittent microhematuria is common. Although of unclear clinical significance, this damage may interact with other pathophysiology in the African-American population (e.g., diabetes), explaining the high risk of chronic renal disease. In addition, there may be an increased risk of venous thromboembolism.3 Most controversial is the concern that extremes of physical exertion, dehydration, and/or altitude may induce sickle cell vaso-occlusive events in some individuals with sickle cell trait.4 This is particularly relevant to student athletes for whom no formal activity restrictions are recommended, but empirically, aggressive hydration during extremes of physical exertion is generally recommended.
Spectrum of Sickle Cell Disease Genotypes
Although homozygous sickle cell (Hb S/S, often called sickle cell anemia) is most common, it is essential to be aware of additional genotypes that result in a similar spectrum of disease (see Table 84-1). That said, once in the intensive care unit (ICU), the form of SCD plays little role in decision-making. In HbS/β+-thalassemia and HbSβ0-thalassemia, the lower the expression of β-like chains from the thalassemic allele, the lower the hemoglobin concentration (Hgb) and MCV, and the higher the percentage of HbS and polymerization, resulting in a more severe clinical phenotype. In contrast, when HbS is expressed in conjunction with HbC, HbD, or Hb O-Arab or several other variants, near normal amounts of hemoglobin are made, but these forms are less able to attenuate HbS polymerization than HbA and result in milder sickle syndromes.
Natural History
The clinical manifestations of SCD result from intermittent episodes of vascular occlusion leading to tissue ischemia/reperfusion injury and variable degrees of hemolysis, both of which contribute to multiorgan dysfunction. The severity of disease manifestations varies widely in individuals with the same genotype and is affected by (1) known genetic modifiers, e.g., α-thalassemia that lowers the MCHC; (2) genetic modifiers of additional pathways contributing to the pathophysiology of SCD (below); (3) behavioral and environmental factors, (e.g., family and community support, racial disparities and access to health care); and (4) psychological factors, (e.g., coping with chronic disease, recurrent pain, and a high rate of depression). The spectrum of manifestations changes with age, and is beautifully described in Gill et al.5 Previously, median survival in the United States for those with SCD was 42 years for men and age 48 years for women6; however, considerable improvements in longevity have been suggested.7,8 The main causes of death are infection, acute chest syndrome, pulmonary artery hypertension, and cerebrovascular events.9,10 What is not fully appreciated by pediatricians is that this increased lifespan is often associated with significant morbidity related to chronic organ damage.
Pathophysiology
Although unable to fully provide the elegant details of the rapidly evolving insights into the multifaceted pathophysiology of SCD, this chapter outlines key pathways with direct relevance to management basics. While the initial era of understanding pathophysiology focused on HbS polymerization and red cell “sickling,” this was followed by an awareness of the critical role of endothelial interactions and inflammation and later, of hemolysis and perturbations of NO metabolism (Figure 84-2).11–14
Red Cells, Inflammation, and the Endothelium
Crystallization leads to deformation of erythrocytes and oxidative damage to cell membrane proteins, leading to the activation of pathways contributing to the stimulation of white cells and inflammation, endothelial damage and the initiation of the clotting cascade and platelet aggregation. Membrane damage triggers movement of phosphatidylserine to the outer leaf of the red cell membrane where it acts as a substrate for factor V and VIII binding, promoting the clotting cascade. Reticulocytes and sickled cells expose increased adherence proteins, slowing flow and increasing CTT and enhancing binding to other cell types. Binding to platelets leads to platelet activation and aggregation, whereas binding to neutrophils stimulates the oxidative burst, damaging endothelium and exposing tissue factor, further promoting coagulation, platelet aggregation, and clot formation. In addition, bound white cells release cytokines, increasing inflammatory cell recruitment and division, perpetuating the process. Thus, sickle cell represents an activated, inflammatory state in which the CTT is easily compromised. This increases the likelihood of additional cells sickling and blood flow becoming further compromised, resulting in ischemic damage, inflammation, and propagation of the cycle. This activation and integration of multiple pathways explains the tremendous clinical heterogeneity observed in people with the same hemoglobin genotype, as well as why patients can show rapid clinical decline. The clinical importance of these paths in SCD is re-enforced by an elevated white blood cell (WBC) count being a risk factor for pain episodes, acute chest syndrome, and early death.6,15 This also explains why infection and trauma are frequent triggers for sickle related pain, and why granulocyte-colony stimulating factor (G-CSF) has led to death in SCD.16 Therapeutically, this points to the importance of nonsteroidal anti-inflammatory drugs (NSAIDs) in affecting the underlying pathophysiology as well as providing analgesia, and why clinical response to hydroxyurea is best correlated with a decrease in WBC.17,18 Finally the identification of these interacting pathways identifies potential targets for new therapies affecting inflammation and cell-endothelial interactions.
Hemolysis and Nitric Oxide Homeostasis
In contrast to the vaso-occlusive complications that correlate with a high WBC and diminished HbF, recently a subset of complications including pulmonary hypertension, skin ulcers, and priapism were found to correlate with increased lactate dehydrogenase and bilirubin and reticulocyte count.12–14 This defines a hemolytic phenotype arising from disturbances in NO hemostasis, and as described at the end of this section, this may be responsible for the inverse association of hemolysis and pain. Hemoglobin accelerates destruction of NO several thousand-fold. Normally laminar flow positions red cells centrally in vessels sequestering hemoglobin away from the endothelium where NO is synthesized and utilized. Hemolysis releases hemoglobin (Fe++) that is now free to diffuse and reduce NO to nitrate with resultant formation of methemoglobin (Fe+++). In addition, hemolysis releases erythrocyte arginase, resulting in arginine depletion, the precursor for NO synthesis. Thus chronic hemolysis leads to decreased NO synthesis and increased destruction, resulting in altered NO hemostasis and increased tone of small vessels, slowing flow and increasing sickling. Optimal management of hemolysis related pathophysiology remains uncertain. Hemolysis prevention with hydroxyurea is appropriate and well accepted, and agents to prevent cellular dehydration (e.g., Mg++ or ICA-17043) are being investigated, but neither is appropriate for acute management. NO metabolism is directly modified with class 5 phosphodiesterase inhibitors (e.g., sildenafil) or by administering NO or related metabolites, and both approaches are being actively investigated for both acute and chronic use. Endogenous sulfur compounds may be tightly integrated with NO pathways, thus a therapeutic role of sulfur compounds may emerge.
In an exciting new paradigm for understanding SCD pain suggested by Gladwin and colleagues,19 NO has been found to affect the processing of nociceptive signals, that may explain why patients with more severe hemolytic disease have fewer vaso-occlusive events (VOE). NO and resultant cGMP signaling increase the hyperexcitability of nociceptive neurons.20 Thus as hemolysis depletes NO, it potentially attenuates pain signaling. Conversely, this may explain why SCD patients with fewer markers of hemolysis have more VOE. The implications of this biochemical pathway on pain management in SCD and other states of high hemolysis such as cardiac bypass are not known (see “Pain” below).
Clinical Manifestations
Although a summary of clinical problems and management are listed in the following section, the reader is referred to the National Heart Lung and Blood Institute standard of care guidelines21 for more details: www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf.
Pain
See Appendix Table 84-A for a detailed care plan, and Appendix Figures 84-B, C, and D for overviews of pain management.22 Vaso-occlusive events, defined as the acute onset of severe pain secondary to ischemic tissue injury, are a hallmark of SCD. The complexities of SCD pain are best summarized by Shapiro and Ballas: “Vaso-occlusion is a physiological process, but the resultant pain is a biopsychosocial phenomenon. Psychosocial issues such as coping skills, social context, personality, mood, and interactions with the health care system mingle with the biologic factors and contribute to the expression of the illness.”23 SCD pain can be acute, recurrent and/or chronic, and is complicated by co-existing chronic disease as well as social and racial overlays. The need for aggressive and rapid treatment is critical and well documented in guidelines of the American Pain Society and British Society of Hematology and should be consulted.24,25 Although aggressive intervention is essential for humane care and physiologic improvements (e.g., improving respiratory mechanics when having rib infarct pain), too often racial attitudes and concerns of drug seeking prevent sufficient delivery to patients in excruciating pain.26 Pain is the most common reason for hospitalization in SCD. The impact of pain is extremely heterogenous in SCD, likely because of differences in genetic background affecting the pathways described above, environment and psychosocial and behavioral variables. In one cooperative study, 80% of patients sought care once or less per year, whereas 5% of patients accounted for one third of hospitalizations.27 Recent daily pain diary assessments suggest these prior studies underestimate the routine pain many experience.28 Risk factors for pain include a high Hgb, high WBC, coexisting alpha thalassemia, and increased levels of cell free DNA, whereas HbF is protective.27,29,30
Pathophysiology, Diagnosis, and Presentation
Pain can start spontaneously but common precipitating factors relate to the pathophysiology and include weather changes (especially cold), dehydration, inflammation, and infections, all of which increase CTT. Occlusion results in ischemic/reperfusion injury and the release of multiple inflammatory mediators that activate nociceptors, evoking a pain response. Pain may involve any part of the body though most commonly affecting the back and long bones. The unpredictability of the pain contributes to the psychological component of pain, potentially interfering with coping mechanisms.31 Recurrent and chronic pain contribute to a decreased quality of life, decreased function, depression and suicidal ideations.28,32,33 Diagnosis is based on a qualitative description, typically biting or gnawing, throbbing, and fatiguing; and quantitative description, that must be trusted (Figure 84-E). The description is essential to help rule out other etiologies of pain, because no physical exam, laboratory, or radiographic studies can confirm or negate this subjective complaint. Physical findings can include swelling, warmth, erythema and tenderness, but exams can be normal and critically, it must be re-enforced that patients can appear without distress. The role of laboratory and radiologic studies is to rule out other etiologies of pain; particularly important for sickle mesenteric pain that must be distinguished from other causes of abdominal pain (e.g., ultrasound to rule out appendicitis or cholecystitis, or tapping a joint to rule out infectious bone or joint processes). Serial quantitative assessments with a developmentally stage specific pain scale is standard of care, though fraught with problems.
Management
See Appendix Table 84-A for a detailed care plan, and Appendix Figures 84-B to 84-D for overviews of pain management.22 An effective management strategy addresses the underlying tissue damage, nociception, history of pain episodes, and doses of medications required to achieve acceptable analgesia, baseline pain medications, history of tolerance, and how people process pain and coexisting depression. Although many are tempted to begin with analgesic medications, there are several other key components to management, such as the following: (1) environmental manipulation. A dark, calm, quiet environment with supportive interactions supports pain reduction. (2) Complementary methods. Many benefit from distraction, prayer, biofeedback, self-hypnosis and similar modalities. (3) Addressing pathophysiology and triggers. Fluids to maintain euvolemia, warmth and NSAIDs decrease CTT and promote conditions less likely to result in vaso-occlusion. (4) Adjuncts. Physical therapy, ambulation, and incentive spirometry should be encouraged to maintain blood flow and prevent atelectasis and acute chest syndrome (ACS). Additional adjuvants such as massage and acupuncture have been tried. After these have been addressed, providers may focus on analgesic medications.
Patients with SCD are the experts in their own pain management. Ideally patients and staff have agreed on individualized, predefined guidelines for pain management taking into account the patient’s degree of opiate tolerance, and side effect profile, and these are documented online or on a patient identification card. Early initiation of NSAIDs is essential, although the choice of NSAID is controversial (e.g., ibuprofen is a better anti-inflammatory, whereas ketoralac is a better analgesic). Notably, half of children will have resolution of pain with intravenous ketorolac.34 For severe episodes, rapid and repeated doses of opiates (dosed every 20 minutes), followed by transition to patient controlled analgesia (if developmentally appropriate) is the accepted standard.35 Although no consensus on the opiate of choice exists, many controversies remain. There are conflicting reports regarding an increased incidence of acute chest syndrome with morphine, with some suggesting decreased rates when nalbuphine is used.36,37 Many avoid meperidine due to concerns of increased seizures and neuroexcitation.38 The goal of opiate use is to balance providing sufficient pain relief to allow one to rest and perform incentive spirometry, while avoiding oversedation, which contributes to the development of pulmonary processes. Certain subsets of patients may benefit from regional anesthesia including epidural,39 dexmedetomidine,40 clonidine, gabapentin, ketamine, and intranasal diamorphine41; although few data exist evaluating their efficacy. One additional promising agent is low molecular weight heparin, which targets the pro-thrombotic and inflammatory state in SCD, Notably a recent trial reported fewer days of severe pain and length of stay with this intervention.42
As mentioned in the discussion of the pathophysiology of SCD, hemolysis leads to depletion of NO and this may attenuate nociceptive signaling through modulation of cGMP signaling.19 This has substantial implications for SCD where, as described in the following section, modulators of NO metabolism are being increasingly employed therapeutically. Expected outcomes are not obvious as additional signaling pathways modulate cGMP activity, and NO and cGMP production can decrease pain under some conditions.20 Some of this complexity is explained by the differential effects of modulating distinct sets of NO synthase genes and proteins.20 Thus, ultimately agents that specifically modulate distinct arms of these pathways in specific regions of the body (e.g., periphery versus central nervous system) will be required to extract the full clinical benefits of NO metabolism manipulation for SCD while maintaining the benefit of NO deletion on chronic pain.
Sepsis
See Appendix Figure 84-F for a detailed care plan.22 Sepsis is a major cause of morbidity and mortality in SCD. Strikingly, in the 1970s, 20% of SCD patients in the United States died before age 6 years, primarily from sepsis with encapsulated organisms, particularly Streptococcus pneumoniae.5 Many patients were diagnosed with SCD when presenting with sepsis, and diagnosed families often did not appreciate that fever in a toddler was an emergency. This led to newborn screening programs and the development of comprehensive care programs focused on education and immunizations. As expected, this did not affect the incidence of sepsis, but deaths from sepsis plummeted,43,44 and further decreased with the initiation of prophylactic penicillin in those younger than 6.45 Widespread use of pneumococcal and Hib vaccines further decreased severe pneumococcal disease to 2.3 events per 100 patient-years, with ages 6 to 36 months being the highest risk group.46,47
Pathophysiology and Etiology
The developmental timing of sepsis risk correlates with decreased splenic function because of infarcts leading to functional asplenia, which can be observed as early as 3 months. The resultant defects in cellular immunity, the alternate complement pathway and decrease in memory cells and opsonizing antibodies reduces clearance of encapsulated organisms. Together these lead to a lessened response to the 23-valent polysaccharide vaccine requiring routine reimmunization and results in a 100 to 400 times increased risk of bacteremia with encapsulated pathogens. SCD patients have shown a strong response to the conjugated PCV-7 vaccine, resulting in a two-thirds reduction in invasive pneumococcal disease.48 Serotypes not covered by the PCV-7 are increasing in prevalence, prompting the development of the PCV-13 vaccine. Although S. pneumoniae accounts for the majority of invasive infections other major pathogens include Haemophilus influenzae type b, Escherichia coli urosepsis, and Salmonella and Staphylococcus osteomyelitis.49
Management
See Figure 84-F for a detailed care plan.22 Although the pathophysiology, diagnosis, and management of sepsis are presented in Chapter 103, several aspects are unique to SCD, including (1) a focus on prevention with aggressive vaccinations with PCV-13, the conjugated 23-valent vaccine (and recurrent boosters from falling titers), Hib, and meningococcus50; (2) prophylactic penicillin given through age 6 years; (3) the critical role of family and provider education in seeking medical care and parenteral antibiotics emergently for fevers higher than 38.3° C. Pneumococcus has doubling time of 20 minutes; thus a 2-hour delay can result in a 50-fold increase in bacterial burden; (4) antibiotic choice should be driven by clinical presentation, but for empiric therapy ceftriaxone is preferred over cefotaxime and cefuroxime due to increasing resistance of pneumococcus; and (5) possible transfusion if cardiovascularly unstable. Evolving care practices will need to account for (1) the influence of hydroxyurea, (2) increasing antibiotic resistance, and (3) the recent release of the PCV-13 vaccine. (Hydroxyurea can result in regrowth of splenic tissue, and early use in young children may prevent autoinfarction, but how this correlates with preservation of immune function and resistance to encapsulated organisms is unknown.)
Acute Chest Syndrome
See Appendix Table 84-B for a detailed care plan.22 ACS is clinically heterogeneous and its definitions vary in the literature, making comparisons of studies difficult. The most general definition is a new nonatelectatic infiltrate on chest radiograph in a patient with SCD, though others add requirements for fever or respiratory symptoms (Figure 84-3). ACS is the second leading cause of hospitalization in SCD. ACS can progress in hours resulting in a mortality rate of 1% in children (vs. 4% in adults), with most deaths occurring in children younger than age 3 years.51 ACS accounts for 25% of deaths.51 Risk factors include a history of asthma, high baseline Hgb and low HbF. Factors associated with mortality include a prior episode of ACS, development of respiratory failure within 48 hours of presentation, sepsis, and simultaneous presentation with pain.51 Etiologies vary by age and included bacterial infections with typical and atypical organisms, viral infections, fat emboli, and pulmonary hemorrhage and multiple etiologic factors are often present (see also Appendix Figure 84-G).52
Presentation
ACS frequently occurs 2 to 3 days into a VOE. Consistent with the variety of etiologies, presenting signs and symptoms are variable and vary with age (e.g., young children are more likely to present with fever while older adolescents are more likely to present with chest pain).51 Notably, no single or pattern of signs and symptoms predicts ACS, and up to 35% of patients will have a normal pulmonary exam.51 Therefore there should always be a low threshold to obtain a chest radiograph. Infiltrates are most common in the lower lobes, and pleural effusions are more common in adults.51,52 Leukocytosis and significant drops in hemoglobin and platelets are common.51 Significant morbidity is associated with ACS including pneumothorax and empyema and 14% of patients developed respiratory failure in one study.52 Risk factors for respiratory failure include (1) extensive lobar involvement, (2) platelet count less than 199,000/µL and (3) history of cardiac disease.52
Management
See Appendix Table 84-B for a detailed care plan.22 Management of ACS can be addressed in stages; initial conservative interventions are followed by transfusion for clinical decline or if severely ill and finally biphasic positive airway pressure (BiPAP) or ventilatory support if needed. Initial care should consist of (1) antibiotics: a cephalosporin to cover encapsulated organisms, particularly S. pneumoniae, and a macrolide because Mycoplasma and Chlamydia are the most common infectious pathogens; (2) oxygen to maintain saturations more than 94%; (3) judicious fluid resuscitation to maintain euvolemia while avoiding overly aggressive fluid resuscitation that may worsen cardiac and respiratory status due to a combination of severe anemia and decreased cardiac function; and (4) prevention of atelectasis. Incentive spirometry (IS) has been shown to prevent progression, and chest physiotherapy and ambulation may be of benefit.53 Although IS is often limited by chest pain and patient compliance, use of a positive expiratory pressure (PEP) device has been shown to be as effective and better tolerated.54 The use of BiPAP or continuous positive airway pressure (CPAP) as a preventive measure is untested but appropriate as early intervention may prevent the need for intubation. (5) Opiates should be carefully titrated to minimize splinting and allow IS, while minimizing respiratory depression (small opiate boluses before IS may be helpful). Pain relief can improve respiratory mechanics, improving clinical status. (6) Bronchodilators are indicated as asthma is common in SCD and its presence increases the risk of ACS, and a subset of patients respond independent of documented wheezing.55 (7) The use of corticosteroids remains controversial. Although a randomized trial showed a decrease in hospital stay and transfusion, a subsequent retrospective review revealed a higher readmission rate, independent of a history of asthma.56,57
Transfusion has been shown to significantly improve oxygenation and clinical status in ACS.52 Although transfusion is usually effective at reversing ACS, because of the risks, including alloimmunization, transfusion not part of initial management unless severely ill. For most patients serial observations, with a low threshold for repeat chest radiographs is appropriate; reserving transfusion for progression despite conservative management. The decision of when to transfuse is contentious and data have been lacking. Promising studies suggest elevated secretory phospholipase A2 (sPLA2) levels predict the development of ACS in SCD.58 sPLA2 cleaves phospholipids, releasing free fatty acid, and is increasingly being associated with cardiovascular and pulmonary damage, including meconium aspiration. A randomized trial suggests early transfusion guided by sPLA2 levels, can prevent ACS.58 Though promising, sPLA2 assays are not routinely available and their role in management remains to be determined.
Although 20% to 70% of patients with ACS are transfused, views differ on performing a simple transfusion targeting a Hgb of approximately 10, or an exchange transfusion targeting the same Hgb while lowering the HbS to under 30%.51,52 Although both approaches improve oxygenation and are safe and effective, exchange transfusion requires exposure to more donors, increasing allo-immunization, is more time consuming, and may require central access in this population with increased thrombotic risk. Thus a simple transfusion is recommended for most situations.59,60 The decision between direct and exchange transfusion in worsening ACS patients with a high Hgb (>9 g/dL) remains unclear as a minimal amount of red cells can be transfused.
Although lacking formal studies, the use of BiPAP or CPAP in an attempt to stave off intubation makes sense pathophysiologically. There is no consensus as to when to intubate or the optimal ventilation strategy in ACS, thus patients are often treated as children with acute lung injury or ARDS. For those patients with refractory hypoxemia, both high-frequency oscillatory ventilation and venovenous extracorporeal membrane oxygenation have been successful.61–63 Several promising approaches to ACS including improving NO metabolism with inhaled NO or oral arginine, a precursor of NO; and inhibitors of sPLA2.64
Patients with ACS have developed plastic bronchitis. A review of 29 children hospitalized with ACS found 72% had plastic bronchitis, one requiring mechanical ventilation, suggesting there may be a role for intratracheal DNase.65,66
Stroke
See Appendix Table 84-C for a detailed care plan and Appendix Figure 84-H for a diagnostic approach.22 One of the most debilitating and under-appreciated complications of SCD is a cerebrovascular accident (CVA), or the acute disruption of cerebral blood supply resulting in loss of brain tissue function lasting longer than 24 hours.67
[/not-level-membership-for-pediatrics-category]
