[level-membership-for-basic-science-category]
CHAPTER 10 Hemoglobin and the Hemoglobinopathies
At least a quarter of a million people are born in the world each year with one of the disorders of the structure or synthesis of hemoglobin (Hb), the hemoglobinopathies. The hemoglobinopathies therefore have the greatest impact on morbidity and mortality of any single group of disorders following mendelian inheritance. The mobility of modern society means that new communities with a high frequency of hemoglobinopathies have become established in countries whose indigenous populations have a low frequency. Awareness of this group of disorders is therefore important and many countries have introduced screening programs. In England and Wales, there are an estimated 600,000 healthy carriers of Hb variants. It is noteworthy that the hemoglobinopathies have served as a paradigm for our understanding of the pathology of inherited disease at the clinical, protein, and DNA levels.
Structure of Hb
Protein Analysis
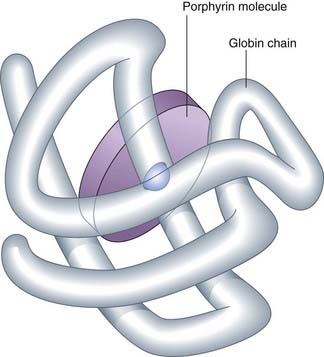
Analysis of the iron content of human Hb revealed that iron constituted 0.35% of its weight, from which it was calculated that human Hb should have a minimum molecular weight of 16,000 Da. In contrast, determination of the molecular weight of human Hb by physical methods gave values of the order of 64,000 Da, consistent with the suggested tetrameric structure, α2β2, with each of the globin chains having its own iron-containing group—heme (Figure 10.1).
Developmental Expression of Hemoglobin
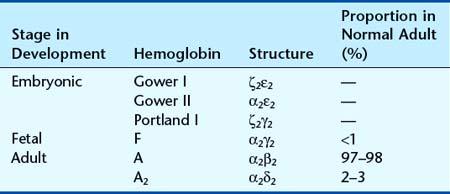
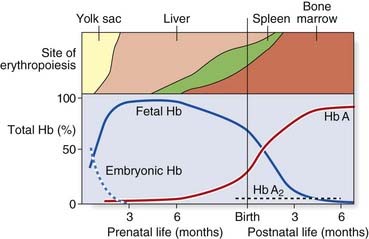
Analysis of Hb from embryos earlier in gestation revealed a developmental, or ontological, succession of different embryonic Hbs: Hb Gower I and II, and Hb Portland, which are produced transiently in varying amounts at different gestational ages. These are in tetramers of various combinations of α, or α-like, zeta (ζ) chains with β, or β-like, γ- and epsilon (ε) chains (Table 10.1). Although both the ζ chain and ε chain are expressed transiently in early embryonic life, the α chain and γ chain are expressed throughout development, with increasing levels of expression of the β chain toward the end of fetal life (Figure 10.2).
Globin Chain Structure
Globin Gene Mapping
The first evidence for the arrangement of the various globin structural genes on the human chromosomes was provided by analysis of the Hb electrophoretic variant, Hb Lepore. Comparison of trypsin digests of Hb Lepore with Hb from normal people revealed that the α chains were normal, whereas the non-α chains appeared to consist of an amino-terminal δ-like sequence and a carboxy-terminal β-like sequence. It was therefore proposed that Hb Lepore could represent a ‘fusion’ globin chain that had arisen as a result of a crossover coincidental with mispairing of the δ- and β-globin genes during meiosis as a result of the sequence similarity of the two genes and the close proximity of the δ- and β-globin genes on the same chromosome (Figure 10.3). If this hypothesis was correct, it was argued that there should also be an ‘anti-Lepore’ Hb (i.e., a β–δ-globin fusion product in which the non-α-globin chains contained β-chain residues at the amino-terminal end and δ-chain residues at the carboxy-terminal end). In the late 1960s, a new Hb electrophoretic variant, Hb Miyada, was identified in Japan, whereby analysis of trypsin digests showed it to contain β-globin sequence at the amino-terminal end and δ-globin sequence at the carboxy-terminal end, as predicted.
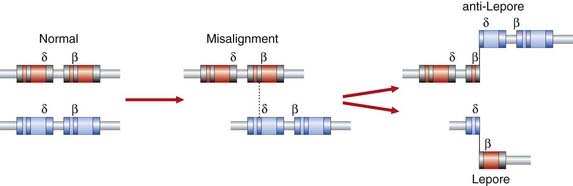
FIGURE 10.3 Mechanism of unequal crossing over which generates Hb Lepore and anti-Lepore.
(Adapted from Weatherall DJ, Clegg JB 1981 The thalassaemia syndromes. Blackwell, Oxford.)
Further evidence at the protein level for the physical mapping of the human globin genes was provided by the report of another Hb electrophoretic variant, Hb Kenya. Amino acid sequence analysis suggested it was a γ–β fusion product with a crossover having occurred somewhere between amino acids 81 and 86 in the two globin chains. For this fusion polypeptide to have occurred, it was argued that the γ-globin structural gene must also be in close physical proximity to the β-globin gene.
Little evidence was forthcoming from protein studies about the mapping of the α-globin genes. The presence of normal Hb A in individuals who, from family studies, should have been homozygous for a particular α chain variant, or obligate compound (double) heterozygotes (p. 114), suggested there could be more than one α-globin gene. In addition, the proportion of the total Hb made up by the α chain variant, in those heterozygous for those variants, was consistently lower (less than 20%) than that seen with the β chain variants (usually more than 30%), suggesting there could be more than one α-globin structural gene.
Globin Gene Structure
The detailed structure of globin genes has been made possible by DNA analysis. Immature red blood cells, reticulocytes, provide a rich source of globin messenger RNA (mRNA) for the synthesis of complementary DNA (cDNA)—reticulocytes synthesize little else! Use of β-globin cDNA for restriction mapping studies of DNA from normal individuals revealed that the non-α, or β-like, globin genes are located in a 50-kilobase (kb) stretch on the short arm of chromosome 11 (Figure 10.4). The entire sequence of this 50-kb stretch containing the various globin structural genes is known. Of interest are non-functional regions with sequences similar to those of the globin structural genes—i.e., they produce no identifiable message or protein product and are pseudogenes.
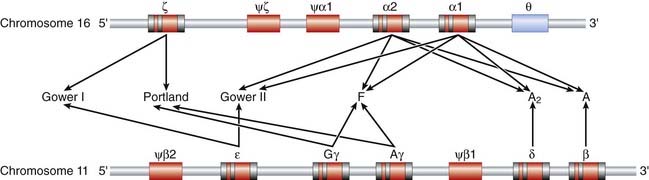
Studies of the α-globin structural genes have shown that there are two α-globin structural genes—α1 and α2—located on chromosome 16p (see Figure 10.4). DNA sequencing has revealed nucleotide differences between these two structural genes even though the transcribed α-globin chains have an identical amino acid sequence—evidence for ‘degeneracy’ of the genetic code. In addition, there are pseudo-α, pseudo-ζ, and ζ genes to the 5′ side of the α-globin genes, as well as an additional theta (θ)-globin gene to the 3′ side of the α1-globin gene. The θ-globin gene, whose function is unknown, is interesting because, unlike the globin pseudogenes, which are not expressed, its structure is compatible with expression. It has been suggested that it could be expressed in very early erythroid tissue such as the fetal liver and yolk sac.
Synthesis and Control of Hemoglobin Expression
Translation studies with reticulocyte mRNA have shown that α- and β-globin chains are synthesized in roughly equal proportions. In vitro studies have shown, however, that β-globin mRNA is slightly more efficient in protein synthesis than α-globin mRNA, and this difference is compensated for in red blood cell precursors by a relative excess of α-globin mRNA. The most important level of regulation of expression of the globin genes, as with other eukaryotic genes, appears to occur at the level of transcription (p. 18).
Disorders of Hemoglobin
Structural Variants/Disorders
In 1975, Ingram demonstrated that the difference between Hb A and Hb S lay in the substitution of valine for glutamic acid in the β chain. Since then, more than 300 Hb electrophoretic variants have been described due to a variety of types of mutation (Table 10.2). Some 200 of these electrophoretic variants are single amino acid substitutions resulting from a point mutation. The majority are rare and not associated with clinical disease. A few are associated with disease and relatively prevalent in certain populations.
| Type of Mutation | Examples | Chain/Residue(s)/Alteration |
|---|---|---|
| Point (>200 variants) | Hb S | β, 6 glu to val |
| Hb C | β, 6 glu to lys | |
| Hb E | β, 26 glu to lys | |
| Deletion (shortened chain) | Hb Freiburg | β, 23 to 0 |
| Hb Lyon | β, 17–18 to 0 | |
| Hb Leiden | β, 6 or 7 to 0 | |
| Hb Gun Hill | β, 92–96 or 93–97 to 0 | |
| Insertion (elongated chain) | Hb Grady | α, 116–118 (glu, phe, thr) duplicated |
| Frameshift (insertion or deletion of multiples other than 3 base pairs) | Hb Tak, Hb Cranston | β*, +11 residues, loss of termination codon, insertion of 2 base pairs in codon 146/147 |
| Hb Wayne | α*, +5 residues, due to loss of termination codon by single base-pair deletion in codon 138/139 | |
| Hb McKees Rock | β*, –2 residues, point mutation in 145, generating premature termination codon | |
| Chain termination | Hb Constant Spring | α*, +31 residues, point mutation in termination codon |
| Fusion chain (unequal crossing over) | Hb Lepore/anti-Lepore | Non-α, δ-like residues at N-terminal end and β-like residues at C-terminal end, and vice versa, respectively |
| Hb Kenya/anti-Kenya | Non-α, γ-like residues at N-terminal end and β-like residues at C-terminal end, and vice versa, respectively |
* Residues are either added (+) or lost (–).
Types of Mutation
Point mutation
A point mutation that results in substitution of one amino acid for another can lead to an altered hemoglobin, such as Hb S, C, or E, which are missense mutations (p. 23).
Deletion
There are a number of Hb variants in which one or more amino acids of one of the globin chains is missing or deleted (p. 24) (e.g., Hb Freiburg).
Insertion
Conversely, there are variants in which the globin chains are longer than normal because of insertions (p. 24), such as Hb Grady.
Frameshift mutation
Frameshift mutations involve disruption of the normal triplet reading frame—i.e., the addition or removal of a number of bases that are not a multiple of three (p. 25). In this instance, translation of the mRNA continues until a termination codon is read ‘in frame’. These variants can result in either an elongated or a shortened globin chain.
Clinical Aspects
Some Hb variants are associated with disease, but many are harmless and do not interfere with normal function, having been identified coincidentally in the course of population surveys. The more common that interfere with normal Hb function are shown in Table 10.3.
Table 10.3 Functional Abnormalities of Structural Variants of Hemoglobin
| Clinical Features | Examples |
|---|---|
| Hemolytic Anemia | |
| Sickling disorders | HbS/S, HbS/C disease, or HbS/O (Arab), HbS/D (Punjab), HBS/β-thalassemia, HbS/Lepore |
| Other rare homozygous sickling mutations—HbS-Antilles, Hb S-Oman) | |
| Unstable hemoglobin | Hb Köln |
| Hb Gun Hill | |
| Hb Bristol | |
| Cyanosis | |
| Hemoglobin M (methemoglobinemia) | Hb M (Boston) |
| Hb M (Hyde Park) | |
| Low oxygen affinity | Hb Kansas |
| Polycythemia | |
| High oxygen affinity | Hb Chesapeake |
| Hb Heathrow | |
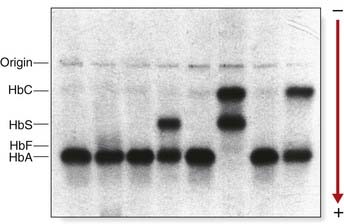
The structural variants of Hb identified by electrophoretic techniques probably represent a minority of the total number of variants that exist, as it is predicted that only one-third of the possible Hb mutations that could occur will produce an altered charge in the Hb molecule, and thereby be detectable by electrophoresis (Figure 10.5).
Sickle Cell Disease
This severe hereditary hemolytic anemia was first recognized clinically early in the twentieth century, but in 1940 red blood cells from affected individuals with sickle cell (SC) disease were noted to appear birefringent when viewed in polarized light under the microscope, reflecting polymerization of the sickle hemoglobin. This distorts the shape of red blood corpuscles under deoxygenated conditions—so-called sickling (Figure 10.6). Pauling, in 1949, using electrophoresis, showed that it had different mobility to HbA and called it HbS, for sickle.
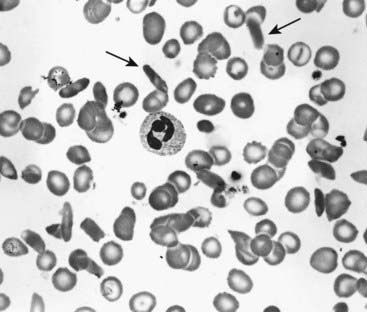
FIGURE 10.6 Blood film showing sickling of red cells in sickle-cell disease. Sickled cells are arrowed.
(Courtesy Dr. D. Norfolk, General Infirmary, Leeds, UK.)
Clinical Aspects of SC Disease
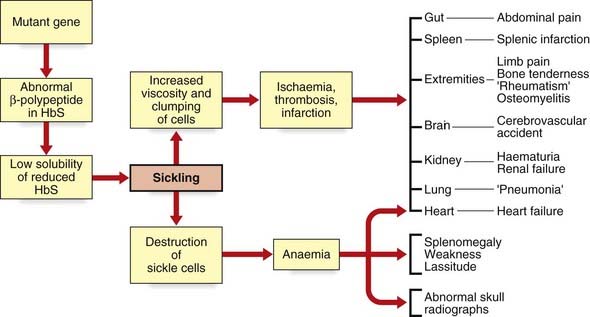
SC disease, following autosomal recessive inheritance, is the most common hemoglobinopathy; in the United Kingdom, about 310,000 individuals are carriers, and approximately 400 pregnancies are affected annually. The disease is especially prevalent in those areas of the world where malaria is endemic. The parasite Plasmodium falciparum is disadvantaged because the red cells of SC heterozygotes are believed to express malarial or altered self-antigens more effectively, resulting in more rapid removal of parasitized cells from the circulation. SC heterozygotes are therefore relatively protected from malarial attacks. They are therefore biologically fitter, the SC gene can be passed on to the next generation, and over time this has resulted in relatively high gene frequency in malarial-infested regions (see Chapter 8). The clinical manifestations are manifold and include painful sickle cell crisis, chest crisis, aplastic crisis, splenic sequestration crisis, priapism, retinal disease, and cerebrovascular accident. Pulmonary hypertension may occur and heart failure can accompany severe anemia during aplastic or splenic sequestration crises. All of these are the result of deformed, sickle-shaped red cells, which are less able to change shape and tend to obstruct small arteries, thus reducing oxygen supply to the tissues (Figure 10.7). Sickled cells, with damaged cell membranes, are taken up by the reticuloendothelial system. The shorter red cell survival time leads to a more rapid red cell turnover and, consequently, anemia.
Disorders of Hemoglobin Synthesis
There are similarities in the pathophysiology of all forms of thalassemia, though excessive α chains are more haemolytic than excessive β chains. An imbalance of globin-chain production results in the accumulation of free globin chains in the red blood cell precursors which, being insoluble, precipitate, resulting in hemolysis of the red blood cells (i.e., a hemolytic anemia). The consequence is compensatory hyperplasia of the bone marrow.
α-Thalassemia
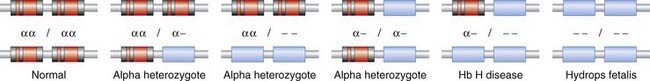
This results from underproduction of the α-globin chains and occurs most commonly in Southeast Asia but is also prevalent in the Mediterranean, Middle East, India, and sub-Saharan Africa, with carrier frequencies ranging from 15% to 30%. There are two main types of α-thalassemia, with different severity: the severe form, in which no α chains are produced, is associated with fetal death due to massive edema secondary to heart failure from severe anemia—hydrops fetalis (Figure 10.8). Analysis of Hb from such fetuses reveals a tetramer of γ chains, originally called Hb Barts.
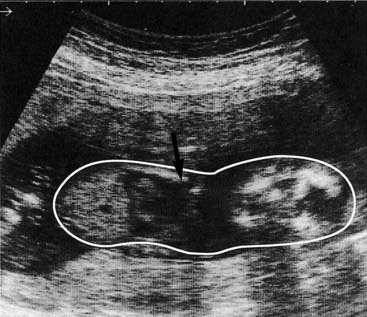
Mutational basis of α-thalassemia
The absence of α chain synthesis in hydropic fetuses, and partial absence in Hb H disease, was confirmed using quantitative mRNA studies from reticulocytes. Studies comparing the quantitative hybridization of radioactively labeled α-globin cDNA to DNA from hydropic fetuses, and in Hb H disease, were consistent with the α-globin genes being deleted. Restriction mapping studies of the α-globin region revealed two α-globin structural genes on chromosome 16p. The various forms of α-thalassemia are to be mostly the result of deletions of one or more of these structural genes (Figure 10.9). These deletions are thought to have arisen as a result of unequal crossover events in meiosis, more likely to occur where genes with homologous sequences are in close proximity. Support for this hypothesis comes from the finding of the other product of such an event (i.e., individuals with three α-globin structural genes located on one chromosome).
β-Thalassemia
By now the reader will deduce that this is caused by underproduction of the β-globin chain of Hb. Production of β-globin chains may be either reduced (β+) or absent (β0). Individuals homozygous for β0-thalassemia mutations have severe, transfusion-dependent anemia. Approximately 1:1000 Northern Europeans are β-thalassemia carriers and in United Kingdom, on average, 22 babies with β0 thalassemia are born annually and roughly 860 people live with the condition; there are an estimated 327,000 carriers.
Mutational basis of β-Thalassemia
β-Thalassemia is rarely the result of gene deletion and DNA sequencing is often necessary to determine the molecular pathology. In excess of 100 different mutations have been shown to cause β-thalassemia, including point mutations, insertions, and base-pair deletions. These occur within both the coding and non-coding portions of the β-globin genes as well as the 5′ flanking promoter region, the 5′ capping sequences (p. 19) and the 3′ polyadenylation sequences (p. 19) (Figure 10.10). The various mutations are often unique to certain population groups and can be considered to fall into six main functional types.
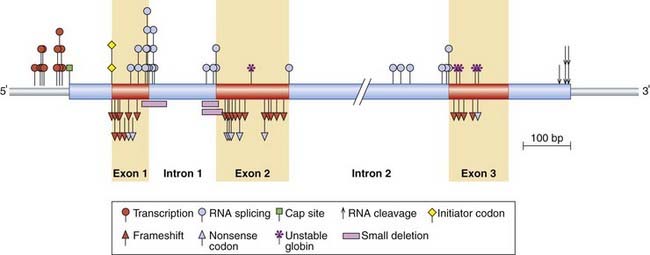
FIGURE 10.10 Location and some of the types of mutation in the β-globin gene and flanking region that result in β-thalassemia.
(Adapted from Orkin SH, Kazazian HH 1984 The mutation and polymorphism of the human β-globin gene and its surrounding DNA. Annu Rev Genet 18:131–171.)
mRNA splicing mutations. Mutations involving the invariant 5′ GT or 3′ AG dinucleotides of the introns in the β-globin gene or the consensus donor or acceptor sequences (p. 19) result in abnormal splicing with consequent reduced levels of β-globin mRNA. The most common Mediterranean β-thalassemia mutation leads to the creation of a new acceptor AG dinucleotide splice site sequence in the first intron of the β-globin gene, creating a ‘cryptic’ splice site (p. 25). The cryptic splice site competes with the normal splice site, leading to reduced levels of the normal β-globin mRNA. Mutations in the coding regions of the β-globin region can also lead to cryptic splice sites.
RNA modification mutations. Mutations in the 5′ and 3′ DNA sequences, involved respectively in the capping (p. 19) and polyadenylation (p. 19) of the mRNA, can result in abnormal processing and transportation of the β-globin mRNA to the cytoplasm, and therefore reduced levels of translation.
Clinical aspects of β-thalassemia
Children with thalassemia major, or Cooley’s anemia as it was originally known, usually present in infancy with a severe transfusion-dependent anemia. Unless adequately transfused, compensatory expansion of the bone marrow results in an unusually shaped face and skull (Figure 10.11). Affected individuals used to die in their teens or early adulthood from complications resulting from iron overload from repeated transfusions. However, daily use of iron-chelating drugs, such as desferrioxamine, has greatly improved their long-term survival.

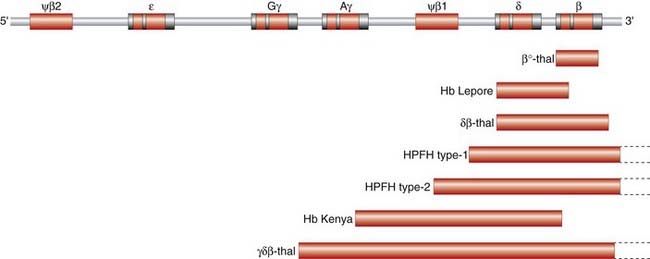
Hereditary Persistence of Fetal Hemoglobin
Mutational basis of HPFH
Some forms of HPFH are due to deletions of the δ- and β-globin genes, whereas non-deletion forms may have point mutations in the 5′ flanking promoter region of either the Gγ or Aγ globin genes near the CAAT box sequences (pp. 21–22), which are involved in the control of Hb gene expression.
Clinical Variation of the Hemoglobinopathies
The marked mutational heterogeneity of β-thalassemia means that affected individuals are often compound heterozygotes (p. 114), i.e., they have different mutations in their β-globin genes, leading to a broad spectrum of severity, including intermediate forms—thalassemia intermedia—which require less frequent transfusions.
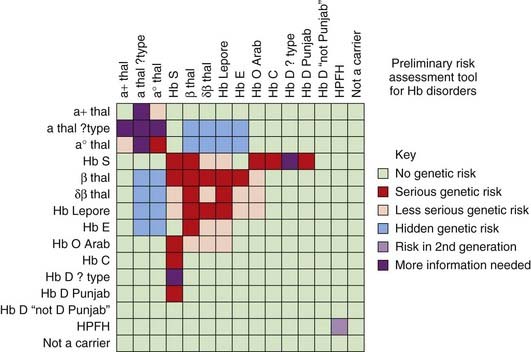
Certain areas of the world show a high prevalence of all the hemoglobinopathies and, not unexpectedly, individuals may have two different disorders of Hb. In the past, precise diagnoses were difficult but the arrival of DNA sequencing has greatly helped to solve conundrums—e.g., individuals heterozygous for both Hb S and β-thalassemia (compound heterozygotes; see p. 114). Certain combinations can result in a previously unexplained mild form of what would reasonably be anticipated to be a severe hemoglobinopathy. For example, deletion of one or two of the α-globin genes in a person homozygous for β-thalassemia results in a milder illness because there is less of an imbalance in globin chain production. Similarly, the presence of one form of HPFH in a person homozygous for β-thalassemia or sickle cell can contribute to amelioration of the disease as the increased production of γ-globin chains compensates for the deficient β-globin chain production. The relative severity of different homozygous or compound heterozygous hemoglobinopathy states is helpfully summarized in a risk assessment tool produced by the NHS Sickle Cell and Thalassaemia Screening Programme (Figure 10.13).
Antenatal and Newborn Hemoglobinopathy Screening
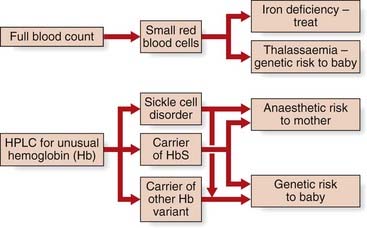
The decision to perform antenatal screening is in some regions guided by the findings of an ethnicity and family history questionnaire administered to the pregnant woman. Initial screening is undertaken on a simple full blood count, looking for anemia (Hb <11 g/dl) and microcytosis MCH (mean corpuscular hemoglobin/0 <27 pg). These findings prompt electrophoresis by high performance liquid chromatography, summarized in Figure 10.14. As with all screening, this program aims to reduce the burden of health care in the long term, which is entirely appropriate for the most common single-gene conditions in humans.
Cay JC, Phillips JA, Kazazian HH. Haemoglobinopathies and thalassemias. In: Rimoin DL, Connor JM, Pyeritz RE, editors. Principles and practice of medical genetics. 3 ed. Edinburgh: Churchill Livingstone; 1996:1599-1626.
A useful, up-to-date, concise summary of the hemoglobinopathies.
Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Pediatr Soc. 1925;37:29-40.
The original description of β-thalassemia.
Pauling L, Itano HA, Singer SJ, Wells IC. Sickle-cell anaemia, a molecular disease. Science. 1949;110:543-548.
The first genetic disease in which a molecular basis was described, leading to a Nobel Prize.
Serjeant GR. Sickle cell disease, 2 ed. Oxford: Oxford University Press; 1992.
Excellent, comprehensive text covering all aspects of this important disorder.
Weatherall DJ, Clegg JB, Higgs DR, Woods WG. The hemoglobinopathies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. 7 ed. New York: McGraw Hill; 1995:3417-3484.
A very comprehensive, detailed account of hemoglobin and the hemoglobinopathies.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 10 Hemoglobin and the Hemoglobinopathies
At least a quarter of a million people are born in the world each year with one of the disorders of the structure or synthesis of hemoglobin (Hb), the hemoglobinopathies. The hemoglobinopathies therefore have the greatest impact on morbidity and mortality of any single group of disorders following mendelian inheritance. The mobility of modern society means that new communities with a high frequency of hemoglobinopathies have become established in countries whose indigenous populations have a low frequency. Awareness of this group of disorders is therefore important and many countries have introduced screening programs. In England and Wales, there are an estimated 600,000 healthy carriers of Hb variants. It is noteworthy that the hemoglobinopathies have served as a paradigm for our understanding of the pathology of inherited disease at the clinical, protein, and DNA levels.
Structure of Hb
Protein Analysis
Analysis of the iron content of human Hb revealed that iron constituted 0.35% of its weight, from which it was calculated that human Hb should have a minimum molecular weight of 16,000 Da. In contrast, determination of the molecular weight of human Hb by physical methods gave values of the order of 64,000 Da, consistent with the suggested tetrameric structure, α2β2, with each of the globin chains having its own iron-containing group—heme (Figure 10.1).
Developmental Expression of Hemoglobin
Analysis of Hb from embryos earlier in gestation revealed a developmental, or ontological, succession of different embryonic Hbs: Hb Gower I and II, and Hb Portland, which are produced transiently in varying amounts at different gestational ages. These are in tetramers of various combinations of α, or α-like, zeta (ζ) chains with β, or β-like, γ- and epsilon (ε) chains (Table 10.1). Although both the ζ chain and ε chain are expressed transiently in early embryonic life, the α chain and γ chain are expressed throughout development, with increasing levels of expression of the β chain toward the end of fetal life (Figure 10.2).
Globin Chain Structure
Globin Gene Mapping
The first evidence for the arrangement of the various globin structural genes on the human chromosomes was provided by analysis of the Hb electrophoretic variant, Hb Lepore. Comparison of trypsin digests of Hb Lepore with Hb from normal people revealed that the α chains were normal, whereas the non-α chains appeared to consist of an amino-terminal δ-like sequence and a carboxy-terminal β-like sequence. It was therefore proposed that Hb Lepore could represent a ‘fusion’ globin chain that had arisen as a result of a crossover coincidental with mispairing of the δ- and β-globin genes during meiosis as a result of the sequence similarity of the two genes and the close proximity of the δ- and β-globin genes on the same chromosome (Figure 10.3). If this hypothesis was correct, it was argued that there should also be an ‘anti-Lepore’ Hb (i.e., a β–δ-globin fusion product in which the non-α-globin chains contained β-chain residues at the amino-terminal end and δ-chain residues at the carboxy-terminal end). In the late 1960s, a new Hb electrophoretic variant, Hb Miyada, was identified in Japan, whereby analysis of trypsin digests showed it to contain β-globin sequence at the amino-terminal end and δ-globin sequence at the carboxy-terminal end, as predicted.
FIGURE 10.3 Mechanism of unequal crossing over which generates Hb Lepore and anti-Lepore.
(Adapted from Weatherall DJ, Clegg JB 1981 The thalassaemia syndromes. Blackwell, Oxford.)
Further evidence at the protein level for the physical mapping of the human globin genes was provided by the report of another Hb electrophoretic variant, Hb Kenya. Amino acid sequence analysis suggested it was a γ–β fusion product with a crossover having occurred somewhere between amino acids 81 and 86 in the two globin chains. For this fusion polypeptide to have occurred, it was argued that the γ-globin structural gene must also be in close physical proximity to the β-globin gene.
Little evidence was forthcoming from protein studies about the mapping of the α-globin genes. The presence of normal Hb A in individuals who, from family studies, should have been homozygous for a particular α chain variant, or obligate compound (double) heterozygotes (p. 114), suggested there could be more than one α-globin gene. In addition, the proportion of the total Hb made up by the α chain variant, in those heterozygous for those variants, was consistently lower (less than 20%) than that seen with the β chain variants (usually more than 30%), suggesting there could be more than one α-globin structural gene.
Globin Gene Structure
The detailed structure of globin genes has been made possible by DNA analysis. Immature red blood cells, reticulocytes, provide a rich source of globin messenger RNA (mRNA) for the synthesis of complementary DNA (cDNA)—reticulocytes synthesize little else! Use of β-globin cDNA for restriction mapping studies of DNA from normal individuals revealed that the non-α, or β-like, globin genes are located in a 50-kilobase (kb) stretch on the short arm of chromosome 11 (Figure 10.4). The entire sequence of this 50-kb stretch containing the various globin structural genes is known. Of interest are non-functional regions with sequences similar to those of the globin structural genes—i.e., they produce no identifiable message or protein product and are pseudogenes.
Studies of the α-globin structural genes have shown that there are two α-globin structural genes—α1 and α2—located on chromosome 16p (see Figure 10.4). DNA sequencing has revealed nucleotide differences between these two structural genes even though the transcribed α-globin chains have an identical amino acid sequence—evidence for ‘degeneracy’ of the genetic code. In addition, there are pseudo-α, pseudo-ζ, and ζ genes to the 5′ side of the α-globin genes, as well as an additional theta (θ)-globin gene to the 3′ side of the α1-globin gene. The θ-globin gene, whose function is unknown, is interesting because, unlike the globin pseudogenes, which are not expressed, its structure is compatible with expression. It has been suggested that it could be expressed in very early erythroid tissue such as the fetal liver and yolk sac.
Synthesis and Control of Hemoglobin Expression
Translation studies with reticulocyte mRNA have shown that α- and β-globin chains are synthesized in roughly equal proportions. In vitro studies have shown, however, that β-globin mRNA is slightly more efficient in protein synthesis than α-globin mRNA, and this difference is compensated for in red blood cell precursors by a relative excess of α-globin mRNA. The most important level of regulation of expression of the globin genes, as with other eukaryotic genes, appears to occur at the level of transcription (p. 18).
Disorders of Hemoglobin
Structural Variants/Disorders
In 1975, Ingram demonstrated that the difference between Hb A and Hb S lay in the substitution of valine for glutamic acid in the β chain. Since then, more than 300 Hb electrophoretic variants have been described due to a variety of types of mutation (Table 10.2). Some 200 of these electrophoretic variants are single amino acid substitutions resulting from a point mutation. The majority are rare and not associated with clinical disease. A few are associated with disease and relatively prevalent in certain populations.
| Type of Mutation | Examples | Chain/Residue(s)/Alteration |
|---|---|---|
| Point (>200 variants) | Hb S | β, 6 glu to val |
| Hb C | β, 6 glu to lys | |
| Hb E | β, 26 glu to lys | |
| Deletion (shortened chain) | Hb Freiburg |
