15 Hematolymphoid Disorders
In the interval since the first edition of this textbook, there have been clinically significant revisions to the classification of lymphomas that affect the approach to diagnosing hematolymphoid proliferations. These include the identification of new “phenotypic” entities, such as the anaplastic large B cell lymphoma associated with ALK translocations,1–5 plasmablastic microlymphoma,6,7 cyclin D1+ large B cell lymphomas,8 and new “cytogenetic” entities, such as “double-hit” diffuse large B cell lymphomas.9 Other changes include the subdivision of old entities, such as diffuse large B cell lymphoma into genetic “activated” and “germinal center” subtypes.10–12 Diagnostic categories, such as atypical Burkitt lymphoma and Hodgkin-like anaplastic large T cell lymphoma, have been retired. This is evident in the revisions of the World Health Organization (WHO) publication on the pathology and genetics of hematopoietic and lymphoid tissues,5 which increasingly represents a clinicopathologic touchstone for oncology.
Twenty-first-century care requires both the generalist and the specialist to be familiar with the indications for the full range of molecular tests available, the specimen requirements for each, and their suitability and relevance to specific differential diagnostic setting. When presented with hematologic proliferations in the lung, pathologists must use an expanding array of special studies to formulate and resolve the differential diagnosis and provide the results of tests that aid in risk stratification.11 Microarray technology is an established method for both class discovery and class definition13,14 and in the not-too-distant future may become a part of the diagnostic and prognostic workup of patients with lymphoma.15 This chapter describes the tests and techniques of hematopathology and the diagnoses that these tests enable.
Special Studies
Immunohistochemistry
Specimen Requirements
Immunophenotypic analysis of tissue sections is an essential part of the evaluation of hematolymphoid proliferations in both nodal and extranodal sites. The range of antibodies and the increase in the number of rabbit monoclonal antibodies available for paraffin-embedded material continues to expand,16–19 but most differential challenges can be resolved with a handful of markers. Table 15-1 lists markers that cover all of the entities described in this chapter. Boxes 15-1 and 15-2 describe panels and the manner in which they might be used.
Table 15-1 Antibodies Useful in the Paraffin Section Evaluation of Hematolymphoid Proliferations
| Marker | Description |
|---|---|
| B Cell Lineage Markers | |
| CD10 | Positive in follicle center cell non-Hodgkin lymphoma (not lineage-specific; also present on some epithelial and stromal tumors) |
| CD19 | Early B cell marker (also present on B-LBL; not present on plasma cells) |
| CD20 | Mature B cell marker (not present on B-LBL; most plasma cells negative) |
| CD23 | Activated B cells |
| CD79a | Immature and mature B cells (present on B-LBL as well as plasma cells) |
| PAX5 | Immature B cells, including lymphoblasts, mature B cells; negative in plasma cells; also positive in some neuroendocrine tumors, including small cell carcinoma |
| CD138 | Plasma cells, some cases of classic Hodgkin lymphoma |
| MUM1 | In the proper context, postfollicular B cells and plasma cells |
| IgD | Immunoglobulin heavy-chain delta, present in benign mantle cells, some lymphomas |
| κ, λ | Immunoglobulin light chains (cell surface expression assessed by flow; cytoplasmic expression by immunohistochemistry) |
| T Cell Lineage Markers | |
| CD1a | Some immature T cells (thymocytes), Langerhans cells |
| CD2 | Pan T cell marker; may also be present on natural killer cells by flow cytometry |
| CD3 | T cells |
| CD4 | Helper/suppressor T cells |
| CD5 | Preferential T cell marker (also positive in some B cell neoplasms) |
| CD7 | T cells, some natural killer cells |
| CD8 | Cytotoxic T cells |
| CD43 | Preferential T cell marker (also positive in some B cell neoplasms and granulocytic proliferations) |
| CD56 | Natural killer cells and some T cells; also positive in some neuroendocrine tumors |
| CD57 | Natural killer cells and some T cells; also expressed on some neuroendocrine tumors |
| Monocyte/Macrophage/Accessory Cell Markers | |
| CD1a | Langerhans cells, some T cells (thymocytes) |
| CD14 | Monocytes (paraffin markers available, not widely used) |
| CD15 | Granulocytes, also positive in Hodgkin lymphoma and adenocarcinoma |
| CD21 | Follicular dendritic cells, some B cells |
| CD31 | PECAM-1; marks vascular endothelium and monocytes, macrophages, and histiocytes |
| CD33 | Granulocytes (paraffin marker available, not yet widely used) |
| CD68 | Macrophages, monocytes (two clones; KP1 and PGM1 have slightly different specificities) |
| CD163 | Hemoglobin scavenger receptor; expressed on macrophages and histiocytes, including histiocytic malignancies |
| Langerin | Langerhans cells, both in Langerhans cell histiocytosis and Langerhans cell sarcoma |
| Miscellaneous Markers | |
| ALK1 | Positive in some peripheral T cell lymphomas, also in some inflammatory myofibroblastic tumors |
| bcl6 | Transcriptional regulator positive in germinal center B cells as well as some lymphoblasts; may be positive in the lesional cells of some T cell neoplasms |
| bcl2 | Anti-apoptosis protein positive in virtually all lymphoid proliferations except benign germinal center B cells and Burkitt lymphoma |
| Cyclin D1 | Cell cycle regulator positive in mantle cell lymphoma, myeloma, and rare cases of large B cell lymphoma |
| CD45 | Leukocyte common antigen present on lymphocytes, blasts, monocytes, and L&H type Reed-Sternberg cells |
| Oct2 | Transcription factor in some B and T cells; also present in L&H type Reed-Sternberg cells |
| TdT | Terminal deoxynucleotidyl transferase, a marker of the blastic stage |
| EMA | Epithelial membrane antigen (positive in some large cell lymphomas and some plasmacytomas) |
| Ki67 | Proliferation marker that helps to identify proliferation centers in chronic lymphocytic lymphoma/small lymphocytic leukemia and is also useful in the multiparameter distinction of high-grade large B cell lymphoma and Burkitt lymphoma |
B-LBL, B cell lymphoblastic lymphoma: L&H, lymphocytic and histiocytic; PECAM, platelet-endothelial cell adhesion molecule.
Box 15-1 Immunophenotypic Profiles Associated with Lymphoid Neoplasia
Phenotypic Findings Indicative of Lymphoid Neoplasia
Aberrant Gain of a Preferential T Cell Marker on a Proliferation of B Cells
Phenotypic Findings Helpful in Classifying Neoplastic Small Lymphoid Proliferations
MUM1 Expression
Box 15-2 A Panel Approach to Immunophenotypic Analysis of Hematolymphoid Proliferations in the Lung
The judicious and cost-effective use of immunostains is maximized if the pathologist puts each marker ordered to specific purpose.20–23 In the workup of hematolymphoid lesions of the lung, there are four principal goals:
In working up hematologic tumors in the lung, the pathologist needs to be aware of certain pitfalls.20 It has long been recognized that CD138 (syndecan 1) marks both plasma cells and a range of epithelial tumors. More recently, PAX-5 has been shown in a broad range of neuroendocrine tumors,31 including small cell carcinoma and Merkel cell carcinoma.32 These studies show that, especially in the evaluation of lung tumors, no one marker should be the sole basis on which the B cell nature of a lung process is defined.33 Other pitfalls were reviewed recently by Yaziji and Barry.20
Flow Cytometry
Specimen Requirements
An advantage of flow cytometry over tissue section immunophenotyping is that three or four markers can be evaluated simultaneously on specific small- and large-cell populations. Through this detailed multiparameter profile, many lymphoproliferative disorders can be classified more accurately.34–37 However, flow cytometric phenotyping does not allow for visualization of the immunoarchitecture of the proliferation, which can be an important element of accurate classification of lymphomas, particularly in the lung, where marginal zone lymphoma (MaZL) is so common. In tissue section-based immunophenotyping, the pathologist can readily identify situations in which the lesional foci have disappeared from the sections used for the stains. In flow cytometric immunophenotyping, however, the pathologist must ensure that the lesional cells are present on a cytospin made from the disaggregated cells.
Most reference laboratories performing flow cytometry have established panels for specific clinical scenarios (lymphoma panel, adult leukemia panel, pediatric leukemia panel, expanded T cell panel) that are tailored to efficiently identify the classic immunophenotypic profiles characteristic of common disease entities.38–41 The appropriate panel should be used to address the diagnostic question. Perhaps 10% to 15% of the time, however, a particular lymphoma or leukemia will have a variant phenotype.41,42 Examples include a lack of CD10 in a lymphoma of follicular origin, acquisition of CD10 in hairy cell leukemia, CD5 expression in large cell lymphoma, CD19 expression in acute myeloid leukemia, apparent dim CD23 expression in mantle cell lymphoma, and a lack of CD23 expression in chronic lymphocytic leukemia. For this reason and because the immunoarchitecture is an important part of the disease definition for some lymphomas, both flow cytometry and immunohistochemistry may need to be performed in some cases, and the diagnostician should not be shy about doing both.
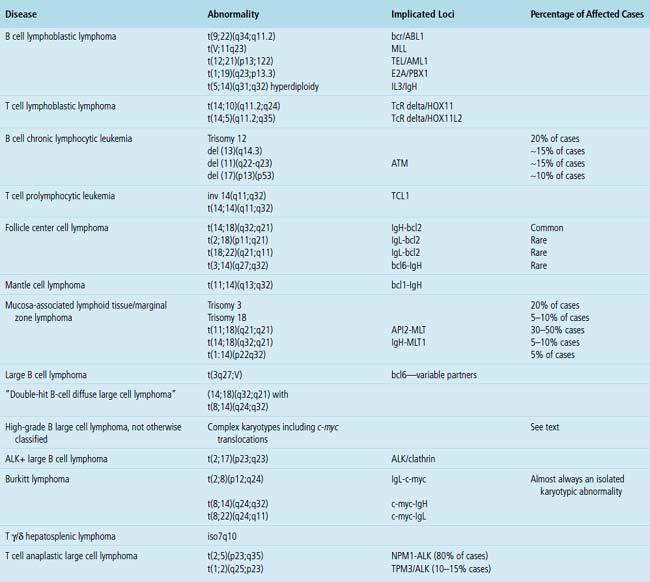
Cytogenetics
Specimen Requirements
Many leukemias and lymphomas have recurring chromosomal translocations that can be identified with FISH and classic cytogenetic testing.43–53 These tests are widely available at many referral laboratories, but they are expensive and time-consuming. Therefore, the pathologist must be aware of their indications, applications, and limitations and also must make the clinician aware of the time frame between biopsy and final results.
One of the most important applications of cytogenetics is in distinguishing between morphologically similar tumors with widely different aggressiveness or treatment protocols. The distinction between small lymphocytic lymphoma and mantle cell lymphoma is an example of the first type, and the distinction between Burkitt lymphoma and high-grade B cell lymphoma is an example of the second type. Although some translocations are characteristic of certain diseases, not all are as specific for a particular entity as once believed. For instance, c-myc–related translocations were once believed to be specific for Burkitt lymphoma, but are now known to be present in some large B cell lymphomas and in some lymphomas that populate the new WHO category of “B cell lymphoma unclassifiable with features intermediate between high-grade large B cell lymphoma and Burkitt lymphoma”5,48 (discussed later). The most common and clinically relevant karyotypic changes related to leukemias and lymphomas are enumerated in Table 15-2.
Molecular Genetics
Specimen Requirements
Over the last decade, molecular genetic analysis has gone from being an expensive test seldom ordered by non-hematopathologists to one routinely ordered when an atypical lymphoid infiltrate is encountered and flow cytometry was not performed or the results were not informative.36 General pathologists must be familiar with the range of molecular tests available, their applications and limits, and the small but real risk of misleading results. Although B5, Bouin, and Hollande fixatives yield excellent cytologic detail, they also denature DNA to the point that tissues fixed in these solutions are not usable for molecular analysis.54–57 Therefore, care should be taken during prosection to include sufficient tissue in formalin to allow for molecular studies if preliminary findings on touch preparations or frozen section suggest an hematolymphoid process.58
Polymerase chain reaction analysis may be used on formalin-fixed, paraffin-embedded material or archived snap-frozen tissue to document clonality,59–62 define lineage, evaluate for translocation events, and assess for the presence of infectious agents. PCR may also be used to speciate mycobacterial organisms. Clonality is assessed by examination of areas of the immunoglobulin heavy- and light-chain loci or the T cell receptor α/β or γ loci that are rearranged during normal lymphoid development.54 Specially designed primer sets that flank certain gene loci are used for PCR-based evaluations for certain translocations, a helpful solution when there are no commercially available FISH probes or if tissue suitable for FISH is not available.
As “objective” as they are, molecular genetic tests do not replace the thought process of diagnosis. These test results are adjunctive data that point in one direction or another, but the final diagnosis must be made by the pathologist.63 The results of molecular studies must be integrated into the “big picture” painted by the clinical setting, the morphologic findings, and the immunophenotypic results. Although clonality is used to define neoplasia, lack of clonality does not prove that a lesion is reactive. The finding of an oligoclonal band is meaningless to the treating physician unless the pathologist puts the result into the context of the morphologic and clinical data. Low tumor cell numbers in Hodgkin lymphoma, lymphomatoid granulomatosis, and T cell–rich B cell lymphoma may yield nonclonal results because of the dilutional effect of reactive cells. Similarly, non–B, non–T cell malignancies, such as natural killer cell lymphomas and myeloid leukemias, yield a polyclonal smear because the lesional cells are of neither B cell nor T cell lineage.
Normal Lymphoid Tissue in the Lung and the Concept of MALT
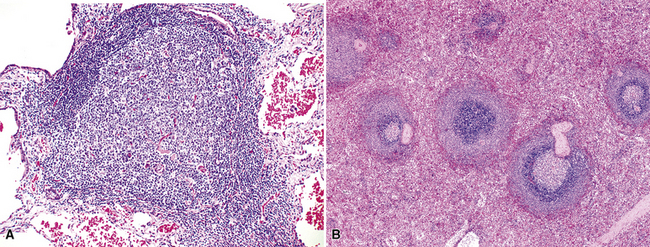
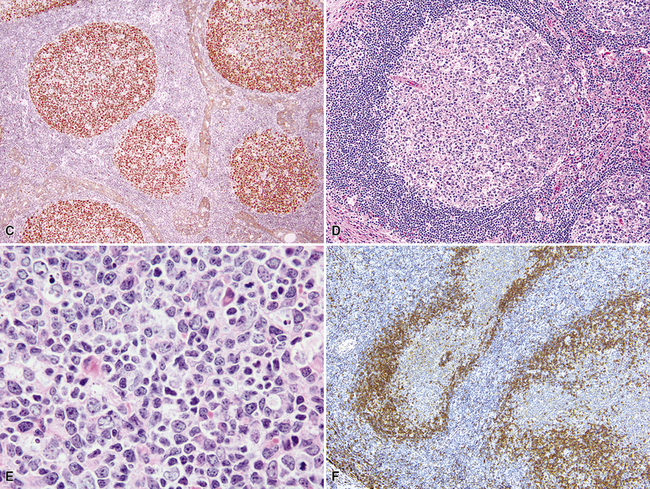
The lung contains an extensive lymphatic network that channels antigen-rich lymph centripetally toward the parenchymal, septal, hilar, and mediastinal lymph nodes. Organized lymphoid tissue in the periphery of the normal lung is limited to sparse submucosal aggregates of lymphocytes and intrapulmonary lymph nodes, but can be more substantial centrally and along bronchiolar branch points.64 Inhaled particulate matter is trapped in the mucus layer of the proximal airways, and some passes across patches of specialized epithelium, where it initiates primary and secondary immune responses. Inhaled irritants stimulate a principally monocyte/macrophage response, whereas inhaled immunogens promote a lymphocytic (or lymphoplasmacytic) response. In practice, inhalational exposures are seldom purely one or the other, and so the tissue response tends to be mixed and is not infrequently masked by fibrinous exudates and actively phagocytic macrophages. The lymphoid tissue proliferates as a result of nonexogenous stimuli as well. In autoimmune conditions and immunodeficiency states, there is an intrinsic dysregulation of lymphoid proliferation, and the lymphoid hyperplasia—“acquired mucosa-associated lymphoid tissue” (MALT) or “bronchus-associated lymphoid tissue”36,65–67 is less masked by acute-phase mucosal changes. Intrapulmonary lymph nodes, also a part of the pulmonary immune surveillance system, are uncommon, but when found, they are more often solitary, peripherally located, and located in the lower lung field. Their structure and immunoarchitectural compartments—and the diseases that affect them—are no different from those of extrapulmonary lymph nodes.
In addition, MALT has a close and specific relationship to the adjacent alveolar and bronchiolar epithelium, the “lymphoepithelium,” where antigen processing and presentation occur. Benign MALT has a distinctive immunoarchitecture, with discrete compartments: the B cell–rich follicles, the mantle, and the T cell–rich interfollicular regions. In some cases and in some areas, a marginal zone of intermediate-sized cells with moderate to abundant amounts of cytoplasm may be interposed between the mantle and the interfollicular regions, although this marginal zone is never as well developed as it is where MALT was originally recognized, in the spleen and Peyer patches (Fig. 15-1). The lymphocytes in these structures shuttle between the mucosa and the circulation to provide ongoing immune surveillance and response to antigens that diffuse through the airways. Between follicles, lymphocytes range from small and resting to intermediate in size and somewhat activated. Plasma cells may be commingled, none with Dutcher bodies. Where immunoblasts are present, their regular size, round nuclear shape, and smooth nuclear contour support a benign interpretation.
Very limited foci of lymphocytes likely have no clinical or radiologic correlate and may not require recognition with a diagnostic line in the report. However, lymphoid accumulations that either have a radiologic correlate or clearly associate with distortions of airways or air spaces should be mentioned (Box 15-3 and Table 15-3).
Reactive Lymphoid Proliferations
Clinicopathologic Patterns of Pulmonary Lymphoid Hyperplasia
The three main patterns of lymphoid hyperplasia—follicular bronchiolitis (FB), nodular lymphoid hyperplasia (NLH), and diffuse lymphoid hyperplasia (lymphoid interstitial pneumonia [LIP])—may be seen in isolation or may coexist in the same specimen.67,68 Much of the work in identifying specific benign and malignant lymphoid proliferations requires the diagnostician to recognize the intactness or the patterned disruption of the morphologic and immunologic landmarks of normal MALT (Fig. 15-2; see also Box 15-3 and Table 15-3).
Follicular Bronchiolitis
Follicular bronchiolitis is slightly more common in males than in females, involves the lungs bilaterally, and produces a centrilobular reticulonodular pattern of involvement on radiologic studies.69,70 In exceptional cases, opacities up to 1 cm in size may be seen. FB is most commonly seen in patients with congenital or acquired immunodeficiency (HIV, common variable immunodeficiency, or immunoglobulin A deficiency), collagen vascular disease (especially rheumatoid arthritis), or chronic obstructive pulmonary disease,70–73 and it may also be seen at the periphery of localized infectious processes of the lung.
Microscopically, the key feature is multiple foci of eccentric peribronchiolar accumulations of lymphoid tissue that distort and may narrow the bronchiolar lumen71,74–76 (Box 15-4 and Fig. 15-3). Confluent nodule-forming infiltrates larger than 1 cm should raise concern about lymphoma. The structure of benign MALT is preserved, with bcl2– germinal centers that are crisply demarcated by an immunoglobulin D–positive mantle zone and a polymorphous lymphocytic and histiocytic component at the interface with normal lung parenchyma. The proliferation may compress the airways, leading to postobstructive bronchiectasis in distal parenchyma. There is no interstitial involvement in the alveolar walls away from the bronchioles, and the air spaces are uninvolved (Fig. 15-4), a feature that distinguishes FB from LIP.69,70,75–77
Box 15-4 Features of Follicular Bronchiolitis
What Should Be Present
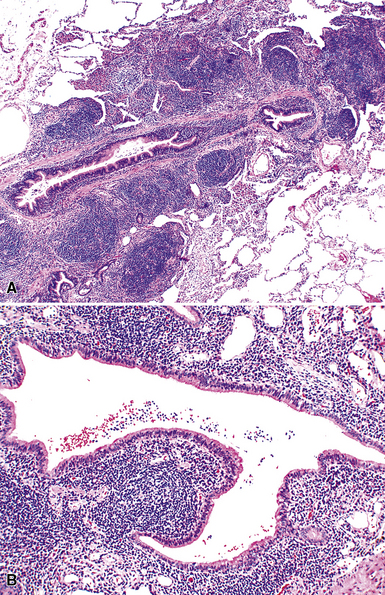
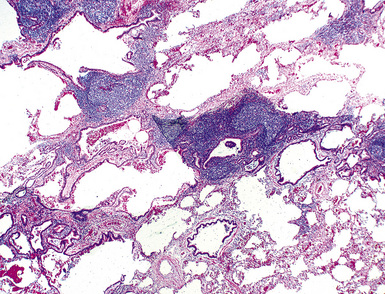
Differential diagnostic considerations include nonspecific chronic inflammation,78,79 which is not organized or airway-centered, and which usually extends into alveolar walls. NLH may enter into the differential diagnosis, and the distinction is as much quantitative as it is a perception of a mass-forming process that compresses adjacent normal lung parenchyma.80Constrictive bronchiolitis may be associated with lymphocytic accumulations around the bronchioles, but the cue to the correct diagnosis is concomitant peribronchiolar fibrosis with reduction in lumen size such that the bronchiole is significantly smaller in diameter than its accompanying arteriole. An elastin stain may be helpful in defining the architecture in such circumstances.
Nodular Lymphoid Hyperplasia
Nodular lymphoid hyperplasia is an extremely rare condition, with only one large series published, representing the Armed Forces Institute of Pathology experience over a decade. In older literature, “NLH” has been used synonymously with “pseudolymphoma,” an outdated and confusing term that should be discarded.80 It is a proliferation that is most commonly seen in adults, reported in the second to ninth decade. Whereas there are reports of NLH in patients with an altered immune state, such as autoimmune disorders, collagen vascular disease, or acquired immunodeficiency, in the Armed Forces Institute of Pathology series there was no special relationship with those conditions.80–82
Patients come to clinical attention because of cough or reasons unrelated to respiratory symptoms. One or several discrete subpleural or peripheral nodules are detected on radiography.81 If there is a reticulonodular pattern in the remaining lung, clinical concern for lymphoma as well as infectious etiologies is greater. In contrast to the lymphoid lesions of FB, the nodules of NLH are typically larger than 0.5 cm, but seldom larger than 5 cm.
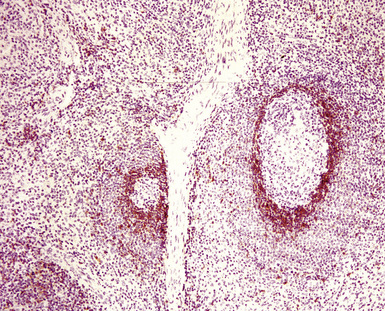
The lymphoid infiltrate of NLH forms a circumscribed nodule of lymphoid tissue (Box 15-5 and Fig. 15-5), with intact architecture, including germinal centers, a discrete mantle, and a preserved “paracortical” interfollicular zone. The process is not pleurally based, and there should not be a bronchiolar distribution.
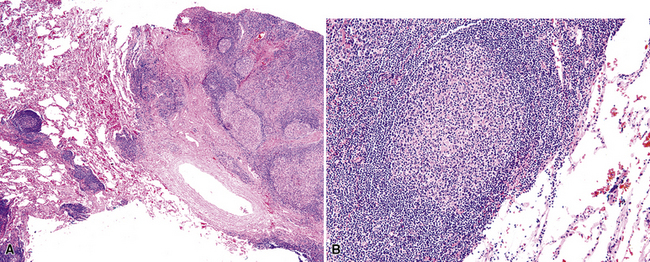
Figure 15-5 Nodular lymphoid hyperplasia is a discrete mass-forming lymphoid proliferation that is spherical or ovoid, in contrast to the linear parabronchial distribution of follicular bronchiolitis. A, Germinal centers are widely spaced, vary in size, and maintain the benign immunoarchitectural landmarks seen in Figure 15-2. B, Pale-staining monocytoid cells seen in marginal zone lymphoma are not present.
Within the mass, follicular architecture predominates.80,83 Cells within nodules retain centrocytic and centroblastic morphologic features, and the mantle zone retains its population of small cells with deeply basophilic round nuclei and scant cytoplasm. Small resting lymphocytes, stromal elements, and plasma cells fill the interfollicular zone, which may also contain patchy accumulations of histiocytes. Lesional foci of NLH remain sharply circumscribed from the surrounding lung parenchyma, with at least a partial immunoglobulin D–positive mantle (Fig. 15-6), without significant extension along the lobar septa or into the alveolar walls, in contrast to LIP. Plasma cells with Russell bodies and Mott cells may be present, but destructive lymphoepithelial lesions, Dutcher bodies, and follicular colonization84 should not be identified.
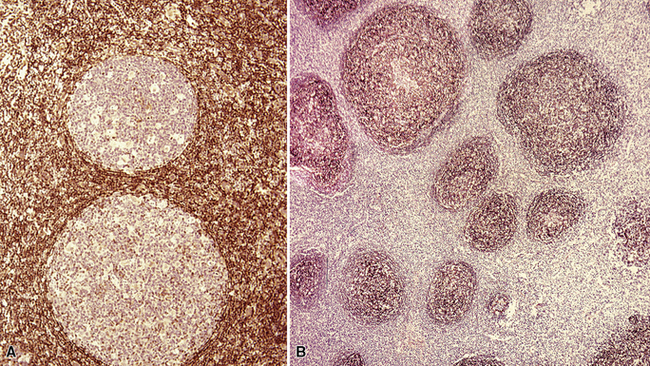
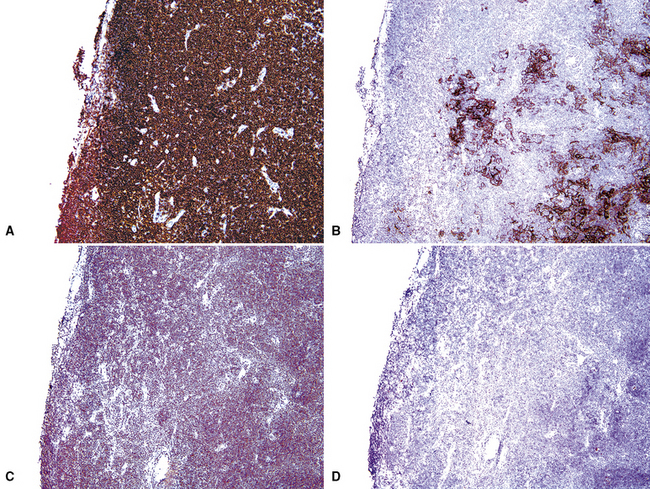
If the nodule was not sampled for flow cytometry, a useful immunohistochemical panel includes CD20, CD21, CD3, bcl2, bcl6, and immunoglobulin D, as well as kappa, lambda, MUM1, and cytokeratin. The immunoglobulin D–positive mantle should be present and fairly crisply demarcated from the immunoglobulin D–negative germinal center inside and the immunoglobulin D–negative paracortex beyond. The germinal center cells should have a bcl2–, bcl6+ phenotype. The bcl6+, CD20+ B lymphocytes within the follicles are definitionally polytypic, as are the plasma cells and immunoglobulin D–positive mantle cells. The interfollicular areas are rich in CD3+ T cells. CD21 staining in NLH highlights the compact nature of the follicular dendritic cell network (Fig. 15-7). A disrupted appearance, particularly if associated with a significant bcl2+, bcl6–, or MUM1+ population of B cells within the follicles or an increase in the number of interfollicular B cells, suggests the diagnosis of MaZL (see Table 15-3).82
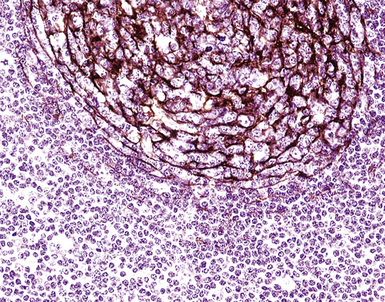
Figure 15-7 The CD21+ follicular dendritic meshwork of nodular lymphoid hyperplasia is sharply circumbscribed, in contrast to the ragged appearance of the network in marginal zone lymphoma, which is seen in Figure 15-17B.
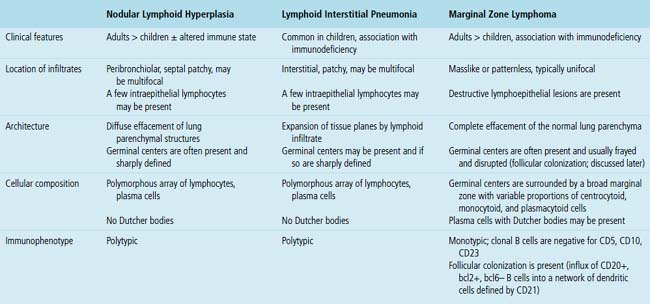
Because of its rarity, NLH is a diagnosis that should be approached with caution and made only after all necessary studies to exclude lymphoma are performed. In practice, if neither flow cytometry nor immunohistochemistry yields a secure diagnosis, molecular studies are a reasonable final step. Principal microscopic differential considerations in transbronchial or transthoracic biopsy specimens may include a particularly robust FB, LIP, and various low-grade lymphomas. In contrast to FB, NLH is mass-forming and displaces significant amounts of lung parenchyma.80LIP is readily excluded by radiologic correlation because it is generally diffuse and bilateral and is not mass-forming, and it is primarily interstitial, with extensive infiltration of the alveolar walls, whereas NLH displaces normal lung tissue as a mass.
In contrast to follicular lymphoma, the germinal centers of NLH are widely spaced, vary in size, exhibit light zone/dark zone polarity, and are demarcated by a distinct immunoglobulin D–positive mantle zone composed of cytologically bland small lymphocytes.5 The finding of bcl2 positivity within nodules in excess of what CD3+ intrafollicular T cells would yield, or the finding of significant numbers of bcl6+, CD20+ B cells outside of the germinal centers should raise concern about follicular lymphoma. If flow cytometry is not available, molecular assessment for clonality and disease-defining translocations should be pursued (PCR, FISH). MUM1 immunostaining may be helpful in distinguishing follicular lymphoma from MaZL.
Marginal zone lymphoma is the most challenging element of the differential diagnosis. Because MaZL is much more common than NLH, it can be argued that molecular studies should be pursued in all cases before a benign diagnosis is rendered. The architecture may be identical to that of NLH, or there may be some blurring of the mantle zone separating the germinal centers from the intervening cellularity. A significant population of monocytoid cells (intermediate size, round or reniform nucleus, sufficient quantities of pale cytoplasm that nuclei are widely spaced on routine sections), either within or between nodules, favors MaZL. The nodules may exhibit “follicular colonization” by MaZL,84 which is seen as displacement of the CD20+, bcl6+, bcl2–, MUM1– centrocytes and centroblasts of the normal follicle by the MaZL cells (CD20+, bcl6–, bcl2+, MUM1+; discussed later). The mantle zone in MaZL is eroded or distorted on immunoglobulin D stain, and the follicular dendritic cell meshwork is diffused through the dilutional effects of the infiltrating MaZL cells. Although they are not diagnostic of MaZL, destructive lymphoepithelial lesions should be sought on cytokeratin stain.
Lymphoid Interstitial Pneumonia and Diffuse Lymphoid Hyperplasia
The pattern of LIP may be seen in both children and adults, and up to 40% of cases are eventually attributable to a specific underlying condition. It is more common in the setting of altered immune states, such as autoimmune conditions and connective tissue disorders (especially Sjögren syndrome85), AIDS,86,87 and congenital immunodeficiency states,88 and after bone marrow transplant. It may also be a tissue response pattern to infections such as mycoplasma, chlamydia, Epstein-Barr virus (EBV), and legionella.89 In some series, females are affected disproportionately,69 perhaps because of the common collagen vascular disease association. Older literature doubtless includes cases of MALT lymphoma, which may skew both outcome and the clinicopathologic parameters that have been associated with LIP.
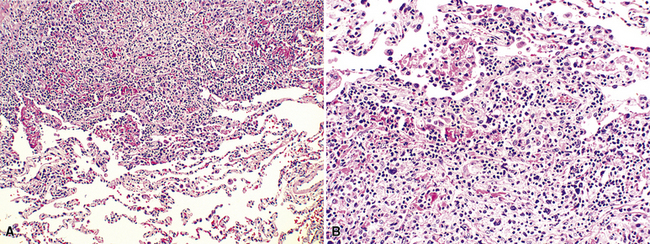
Patients with LIP present with a cough and slow but progressive shortness of breath, and radiologic studies usually show bilateral basilar patchy opacities or reticulonodular infiltrates.69,87,89–91 Cysts have been reported in LIP,92 but correlation with clinical findings suggests that the cysts are more likely a manifestation of the underlying condition (Sjögren syndrome) than of LIP. Some patients have systemic symptoms, such as fever and weight loss, and many (70%) have polyclonal hypergammaglobulinemia; both the clinical and laboratory abnormalities likely reflect the underlying altered immune state.
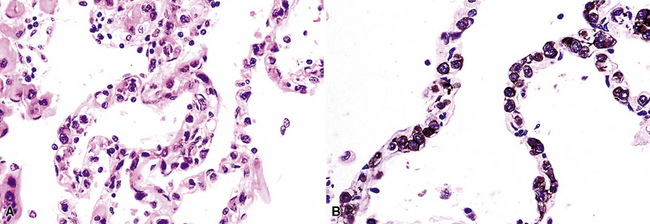
In contrast to FB and NLH, the infiltrate of LIP has a dominant interstitial pattern of distribution, although the constituent cellular components are otherwise similar. Small, cytologically bland lymphocytes and intermingled plasma cells distend the alveolar walls, with accentuation along the bronchovascular bundles and lobular septae (Box 15-6 and Fig. 15-8). Aggregates of histiocytes or poorly formed granulomas may be present, but neutrophils and eosinophils are scarce. A few germinal centers may be present.69,87,90 An intraepithelial component mimicking the lymphoepithelial lesions of MALT lymphoma and a coalescence in and around the microvasculature have been described, but truly destructive changes are not part of LIP (Fig. 15-9).
Box 15-6 Features of Diffuse Lymphoid Hyperplasia/Lymphoid Interstitial Pneumonia
What Should Be Present
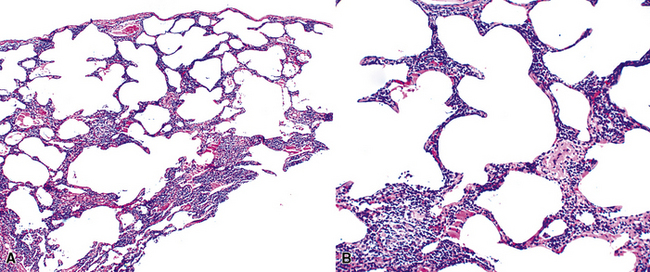
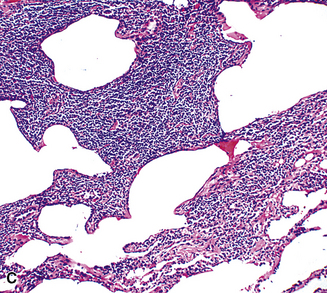
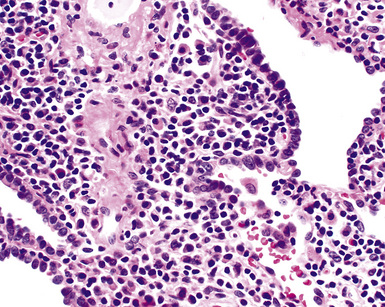
Figure 15-9 Lymphoid interstitial pneumonia shows composition by morphologically mature lymphocytes. Most are CD3+ T cells, a helpful feature in distinguishing this condition from marginal zone lymphoma. Lymphocytes may abut the epithelium of the distal airways, but they do not form destructive aggregations, as seen in marginal zone lymphoma and in Figure 15-15.
An LIP-like pattern has been described in the lung in the setting of atypical infectious mononucleosis.53,93,94 In these few reports, the alveolar walls and interstitium were expanded by a mixture of lymphocytes, plasma cells, and transformed lymphocytes (immunoblasts), and there was a patchy alveolar exudate. In situ hybridization for EBV-encoded ribonucleotides (EBERs) is the most sensitive and specific means of identifying the virally mediated nature of the process. Immunosuppressed patients are at increased risk for EBV-related lymphoid proliferations, and biopsy is usually undertaken to assess for a specific infectious process. In the transplant setting, the terminology of post-transplant lymphoproliferative disorders (PTLDs) should be used (discussed later).
Occasional germinal centers and focal intraepithelial accumulations of lymphocytes may prompt consideration of MaZL/MALT lymphoma, but the bilateral and interstitial (rather than unifocal and mass-forming) nature of LIP, as well as the lack of a dominant B cell population in the interfollicular areas, provides a strong and objective means of excluding this possibility. Because of the interstitial distribution, dominant T cell population, and cytologic heterogeneity of LIP, distinction from pulmonary presentation of systemic lymphomas, such as small lymphocytic lymphoma, mantle cell lymphoma, or follicle center cell lymphoma is seldom an issue. In difficult cases, or when diagnostic material is limited, immunohistochemistry (CD20, CD3) usually permits a definite diagnosis (discussed later; see Box 15-3 and Table 15-3).
Castleman Disease
Castleman disease includes two distinct conditions. One, the “hyaline vascular variant,” is almost invariably presents via mass effect of a solitary mediastinal mass or central lymphadenopathy in otherwise asymptomatic individuals and is cured by complete resection. The other, the “plasma cell variant,” often manifests with systemic symptoms and multifocal disease, and has significant associated morbidity and mortality. What brings them together95 is that, first, the two histologies may coexist in the same lymph node, and second, they were characterized in separate publications by the same individual, Benjamin Castleman.
Hyaline Vascular Variant
The hyaline vascular variant of Castleman disease (HVCD) arises in axial node groups of the mediastinum and abdomen, although it may extend along the hilum to involve peribronchial lymph nodes. Most patients have no systemic symptoms and present because of mass effect (e.g., airway compression or superior vena cava syndrome) or have mediastinal adenopathy detected during radiologic studies performed for other reasons.96–98 A patient who has “B” symptoms and diffuse adenopathy likely has a mixed type of Castleman disease, in which the histologic features of the plasma cell variant are present elsewhere (discussed later). In contrast to the plasma cell variant of Castleman disease (PCCD), there is no consistent abnormality in laboratory findings.
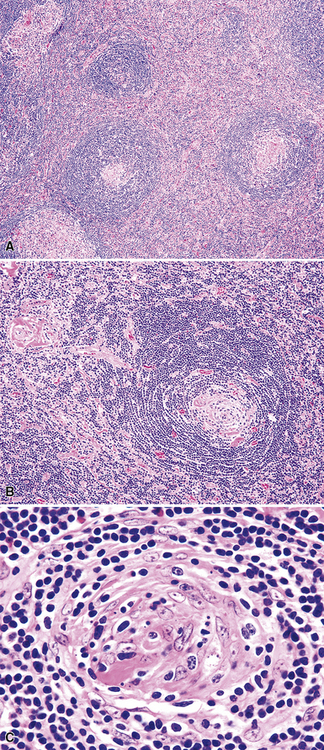
At low power, the architecture is nodular, composed of small and involuted germinal centers with expansive immunoglobulin D–positive mantles formed of small lymphocytes, often in a laminated or “onion skinning” array. Multiple germinal centers may be found in the boundaries of a single mantle zone, and in opportune sections, the lymphoid depletion in the germinal centers unmasks the radially penetrating high endothelium—the “lollipop” motif (Box 15-7 and Fig. 15-10). Sinuses between the regressed follicles are imperceptible usually because they are absent. At high power, the germinal centers are depleted of lymphocytes and contain both extracellular matrix and abundant follicle dendritic cells. The interfollicular zone includes plasmacytoid monocytes, stromal myoid cells, histiocytes, dendritic cells, and lymphocytes.99–101
Box 15-7 Features of Castleman Disease, Hyaline Vascular Type
What Should Be Present
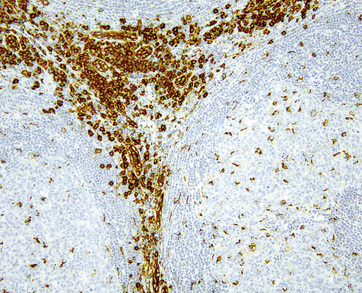
Follicle dendritic cells within the germinal centers are CD21+ and S-100–, and are distributed as dense aggregates rather than as circumscribed meshworks with intermingled centrocytes and centroblasts. Their processes extend in a laminar array beyond the germinal center border such that the mantle cells align along them. The lymphocytes of the mantle zone represent a mixture of CD20+ B cells, and the plasmacytoid monocytes are both CD4+ (but CD3–) and CD68+.99,100,102 Plasmacytoid monocytes aggregate in clusters between regressed follicles, and are demonstrable on HECA-452 (Fig. 15-11).
Differential diagnostic considerations may include a Castleman-like reaction to tumor103,104 and a variety of non-Hodgkin lymphomas. Mantle cell lymphoma is clonal and positive for cyclin D1, and in partially involved nodes, the sinuses may be compressed but still present. Follicular lymphoma rarely (if ever) has an immunoglobulin D–positive mantle zone that is broader than the follicle within, and the nodules are cellular and rich in bcl6+ B cells, rather than depleted. HIV-related lymph node changes, particularly the depleted form, which has regressed germinal centers, may enter into the differential diagnosis and may be difficult to exclude, but the laminated array of mantle cells is generally undeveloped, plasmacytoid monocytes are few or absent, and sinuses are present and generally congested with histiocytes. The angioimmunoblastic lymphadenopathy (AILD) type of peripheral T cell lymphoma is discussed in the differential diagnosis with HVCD because both have abnormal follicular structures, but in AILD-PTCL the follicles are generally enlarged and fragmented, not regressed, and flow cytometry may show the loss of a pan T cell antigen on T cells. Immunohistochemistry shows bcl6 expression in CD3+ T cells. Molecular studies often document at least a clonal T cell population and sometimes also a clonal B cell population. Double antibody immunostains highlight a special population of bcl6+ T cells in perivascular areas. If there is cytoatypia or a mass-forming coalescence of follicular dendritic cells, consideration should be given to an evolving follicular dendritic cell tumor (discussed later).
Outcome is excellent in patients with fully resected localized HVCD.105
Plasma Cell Variant
The plasma cell variant of Castleman disease is most frequently encountered in HIV-positive patients and the elderly, and presenting pulmonary symptoms include shortness of breath, productive cough, and fevers. When it involves the chest structures, PCCD is more often found in the central lymph nodes, with secondary extension into the centrilobular regions of lung tissue. Radiologic studies may show adenopathy alone or concurrent with bilateral interstitial infiltrates.106 Laboratory studies may show cytopenia, an elevated erythrocyte sedimentation rate, and hypergammaglobulinemia—“hyper-IL-6 syndrome.”107,108
Both localized and multicentric expressions of PCCD occur. Taken together, they are far less common than HVCD. When localized, the adenopathy is typically axial (mediastinum or abdomen and, less commonly, in the neck) and node-based. When the disease is multicentric, patients typically have generalized lymphadenopathy and hepatosplenomegaly, and may have symptoms fitting POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, “M” spike, skin disorder109).
The low-power appearance is that of follicular hyperplasia with marked interfollicular plasmacytosis (Box 15-8 and Fig. 15-12). In contrast to HVCD, the subcapsular and medullary sinuses remain patent; the germinal centers are hyperplastic and large and contain amorphous eosinophilic material and have a discrete, if thinned, mantle zone.99 Because the potential for lymph nodes to be involved by more than one process (e.g., Castleman disease and Hodgkin lymphoma or plasmablastic non-Hodgkin lymphoma), a careful search for a second diagnosis should be made before the solo diagnosis of PCCD is rendered.
Box 15-8 Features of Castleman Disease, Plasma Cell and Multicentric Types
What Might Be Present
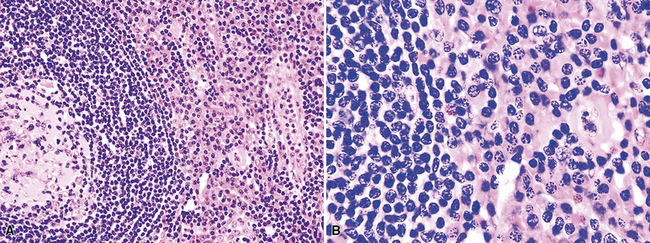
Flow cytometric analysis identifies only polytypic B lymphocytes and phenotypically normal T cells. Routine immunostaining shows a polytypic population of plasma cells. Careful scrutiny of the lambda light-chain stain may show a population of immunoblasts in the perifollicular region. These, all immunoglobulin M-lambda, may be monoclonal on PCR and part of a “microlymphoma” or polyclonal at the genetic level.99,110,111
Clinically, PCCD may be smouldering or aggressive, but it is inexorable and is associated with a high rate of morbidity and mortality as a result of infection and progressive renal, hepatic, or immune system dysfunction.112–114 Median survival time is 2 to 3 years for patients with multicentric disease. A subset of patients has Kaposi sarcoma (approximately 10%), large cell B cell lymphoma, Hodgkin disease, plasmacytoma, myeloma, or POEMS syndrome (up to 20% in HIV-positive patients).
Neoplastic and Malignant Lymphoid Proliferations
Primary Lung Lymphomas
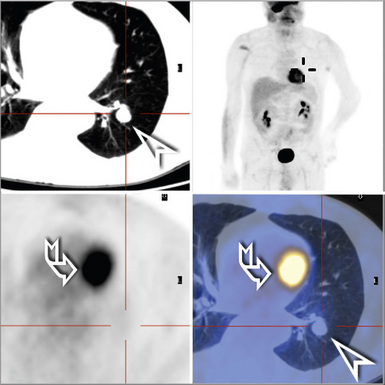
Regardless of the histologic type, adults are most commonly affected, and primary pulmonary lymphoma is quite rare in the pediatric population.115,116 The designation of primary pulmonary lymphoma is restricted to de novo lymphomas that present with lung-limited disease. Other than hilar node involvement, no evidence of extrapulmonary disease is evident on staging at presentation or on restaging studies repeated after a period of observation. The historical definition of disease that remains localized to the lung over the course of a several-month period of observation117 was likely overinclusive: Up to 40% of patients whose disease was defined in this way progress to stage IV systemic disease or even die of disease within 6 months of initial diagnosis.118 With the routine use of positron emission tomography scans, initial radiologic staging may replace this approach. Strictly defined, patients with primary pulmonary lymphoma should have little disease-related morbidity and mortality119,120 unless the disease acquires systemic manifestations or progresses to involve extrathoracic sites.
Mucosa-Associated Lymphoid Tissue Lymphoma
Although it was not recognized as a distinct and separate type of lymphoma until the early 1990s, MaZL of MALT type is the most common type of primary pulmonary lymphoma. Unlike MaZL of the stomach, thyroid, and salivary gland, however, it has an inconstant association with infectious agents or specific autoimmune conditions.121–123 Recent studies have shown that 40% of cases contain a t(11;18) involving API2 and MALT1.124 In contrast, other nongastric MALT-type lymphomas rarely harbor this translocation and arise much more often in the setting of an autoimmune condition. Although some patients are entirely asymptomatic, many present with cough, fever, or unexplained weight loss,121–123 and radiologic studies most commonly show solitary or multiple discrete nodules.125,126 Findings of serum protein electrophoresis are positive in up to 30% of patients, and staging shows extrathoracic disease in one third of patients. The disease is almost entirely restricted to adults. Occasionally, the tumor starts in the thymus.127
Following the paradigm of benign MALT, MaZL is composed of cells that morphologically and phenotypically resemble the mature B cells that form the outer rim of the malpighian corpuscles of the spleen and their counterpart in the organized lymphoid tissue of Peyer patches in the terminal ileum.121,128–130 At low power, MaZL may have a nodular or diffuse pattern, and at the periphery, the lesion may extend along intact alveolar walls in discontinuous fashion (Fig. 15-13), occasionally with a low-power beading motif. Nodularity may be inconspicuous, but where it is present, it corresponds to residual benign germinal centers that have been infiltrated (“colonized”) to a greater or lesser degree by tumor cells.84 Much of the neoplastic proliferation is present between the nodules and is composed of a mixture of small, resting lymphocytes, monocytoid cells (oval or reniform nuclei, condensed chromatin, and moderate amounts of pale-staining cytoplasm), and plasmacytoid forms (Box 15-9 and Fig. 15-14).
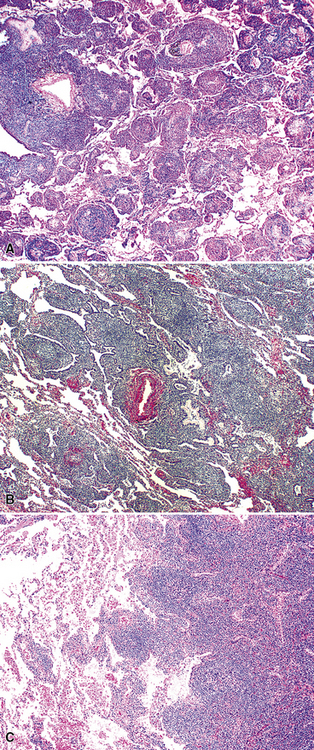
Box 15-9 Features of Low-Grade Mucosa-Associated Lymphoid Tissue Lymphoma
What Should Be Communicated in the Report
IHC, immunohistochemistry; SPEP, serum protein electrophoresis; UPEP, urine protein electrophoresis.
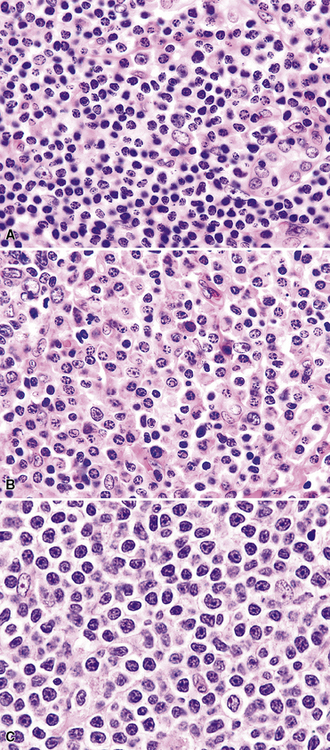
Figure 15-14 Although the histologic features of marginal zone lymphoma are often described as “heterogenous,” in any given high-power field, a fairly uniform population of cells is present. In some areas, this is “centrocytoid” (A); in others it is plasmacytoid, with Dutcher bodies (B); and in yet others it is monocytoid (C). In all of these patterns, even the “centrocytoid” pattern, the heterogenous mix of centrocytes, centroblasts, dendritic cells, and tingible body macrophages of normal germinal centers (as seen in Fig. 15-2E) is not present.
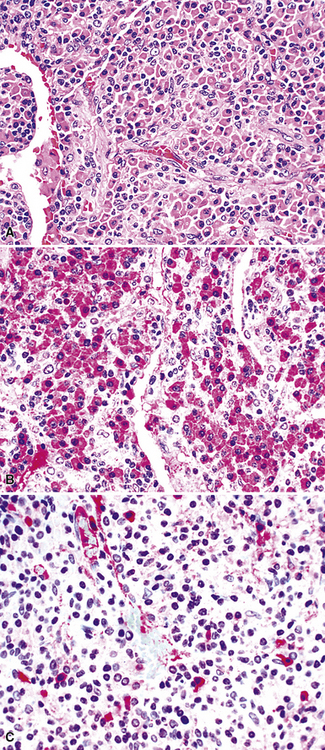
Because of their ontogenic relationship to lymphocytes that home to mucosal surfaces, tumor cells in MaZL have a tendency to form destructive lymphoepithelial lesions (Fig. 15-15), a characteristic feature of this disease, although one that is not independently diagnostic of neoplasia in general or MALToma in particular.68 In some cases, monocytoid morphologic features dominate, whereas in others, the cells more closely resemble centrocytes within germinal centers. Transbronchial biopsy should be approached cautiously because plasmacytoid differentiation may be so striking superficially that the differential diagnosis includes plasmacytoma131 (Fig. 15-16). With fuller representation (e.g., on wedge biopsy), a concomitant lymphoid component is often identified, however, permitting accurate classification. Occasional cases may have a few cells with intracytoplasmic crystalline immunoglobulin132–135 or amyloid deposits.136–140
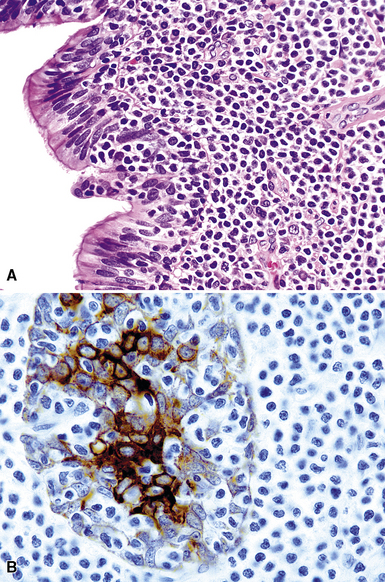
The lesional cells of MaZL are CD19+ and CD20+ and do not co-express CD5, CD10, or CD23, Tdt, cyclin D1, or bcl6. In small biopsy specimens, a cytokeratin stain may help to identify lymphoepithelial lesions, and the combination of CD21, bcl2, and bcl6 stains highlights germinal centers colonized and overrun by tumor cells68,121,130 that are bcl2+, bcl6– and have a disrupted network of follicular dendritic cells84 (Fig. 15-17). Bcl10 protein expression has been shown to correlate with the presence of the t(11;18)(API2/MALT1) translocation, and a positive result by paraffin section immunohistochemistry can be a helpful adjunct to classification.124
Although MaZL resembles NLH, the latter has a polymorphous population of lymphocytes and histiocytes in the interfollicular areas. In addition, it contains benign, “uncolonized” germinal centers with a CD20+, bcl2-, bcl6+ phenotype and has interfollicular areas rich in T cells, not B cells68 (see Box 15-3 and Table 15-3). Importantly, there is no evidence of a monotypic population of B cells on flow cytometry, and NLH is nonclonal on PCR-based molecular studies. Small biopsy specimens of MaZL may have some degree of morphologic overlap with LIP, but MaZL tends to overrun normal structures and the process does not have the predominant interstitial localization associated with LIP. Distinction from other lymphomas rests on the findings of immunophenotyping studies5 (see Boxes 15-2 and 15-3 and Table 15-3), although colonization of the germinal centers and heterogeneity of the lesional infiltrate are two features that favor MaZL. Distinction from follicular lymphoma with florid marginal zone differentiation141 requires careful attention to immunohistochemical assessment for follicular colonization and may also require FISH for the t(14;18) and t(11;18) translocations. It is important to be attentive to the possibility that the biopsy specimen contains more than one lymphoma, because “collisions” occur.142
Diffuse Large B Cell Lymphoma, Variants, and Subtypes
Large cell lymphoma of B cell type (B-LCL) is the second most common type of primary pulmonary lymphoma5,143,144 and most commonly affects older adults in the sixth and seventh decades. Patients present with cough or dyspnea; hemoptysis is rare. Most lesions are solitary, solid, and off-white, and have a discrete border with adjacent normal lung parenchyma. A subset of B-LCL arises in patients with a preexisting or concurrent low-grade lymphoma, such as MaZL, small lymphocytic lymphoma, or follicular lymphoma. Rapidly proliferating tumors may have central cavitation on computed tomography, as a result of tumoral necrosis.
The neoplastic nature of B-LCL is readily evident from the dominant population of large cells as well as the destructive manner in which it obliterates the lung parenchyma. The lesional cells are large (20–30 microns) and form confluent, discohesive sheets of cells. Cytologic features vary from case to case, although usually the tumor cells have coarse chromatin, distinct nucleoli, and abundant cytoplasm (Fig. 15-18). Essentially all are CD19+, CD20+, and CD79a, and those with a follicle center cell origin also express CD10 and bcl6.5
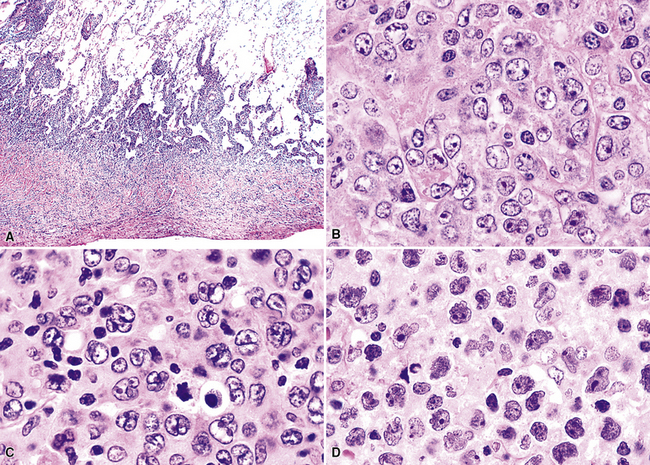
Differential diagnostic considerations can include primary or metastatic carcinoma, metastatic melanoma, and other epithelioid malignancies (especially large cell neuroendocrine carcinoma), all resolvable on phenotypic analysis. Some cases of B-LCL may have a high content of reactive T cells or histiocytes (“T cell–rich large B cell lymphoma” [TcRBCL])145 (Fig. 15-19) and may be impossible to distinguish from metastatic lymphoepithelioma-type nasopharyngeal carcinoma or even Hodgkin lymphoma without immunohistochemical studies.
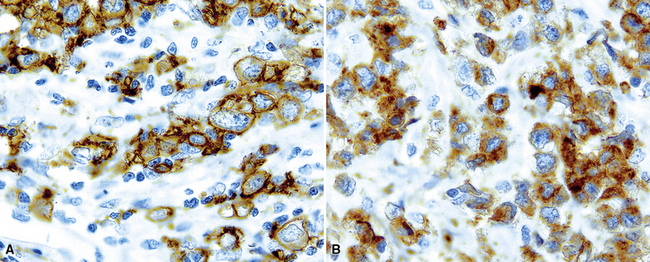
Several histologic and immunophenotypic variants bear mentioning because of specific differential considerations. Although most have been reported first in lymph nodes, there is no reason why the same tumor could not involve the lung. The immunoblastic variant of B-LCL has vesicular chromatin, thick nuclear membrane, a prominent and centrally placed eosinophilic nucleolus, and abundant amphophilic cytoplasm. A morphologic variant may confer a worse prognosis5 (Fig. 15-20) and may mimic anaplastic myeloma, plasmablastic lymphoma,146 carcinoma, and melanoma as well as some dendritic cell tumors. A panel including CD138, cytoplasmic immunoglobulin (cIg), pancytokeratin, CD45, MART1, S-100, and CD21 may be helpful in such cases. The presence of CD5+ B-cell diffuse large cell lymphoma (B-DLCL)147 is rare but recognized, and where this phenotype occurs, cyclin D1 staining and well-prepared slides of well-fixed tissue can be helpful in excluding mantle cell lymphoma. ALK+ B-DLCL, although rare, is now well characterized3 (Fig. 15-21), and like the T cell counterpart, may mimic carcinoma, histiocytic malignancy, and melanoma. These rare variants are usually negative for CD20, CD30 (unlike their T cell counterpart!), CD45, and PAX5, and positive for epithelial membrane antigen. Negative pancytokeratin results help to exclude carcinoma, and positive results for CD138, MUM1, cytoplasmic ALK1, and cytoplasmic immunoglobulin light chains point toward the correct diagnosis. FISH or PCR for the t(2;17)(ALK/clathrin) translocation is confirmatory.
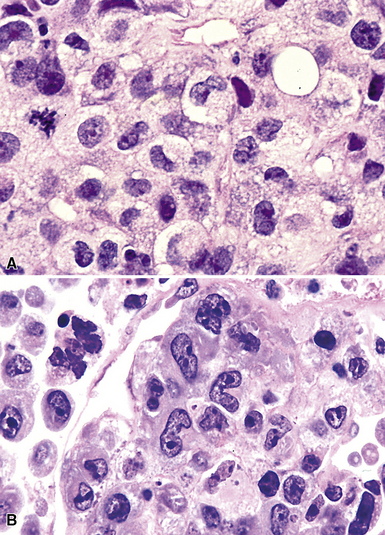
In some cases, a substantial reactive population of T cells or histiocytes may disperse the lesional large B cells such that flow cytometry and molecular studies do not identify a B cell clone. T cell/histiocyte-rich large B cell lymphoma is the prototype in this category,145 and consideration can be given to lymphoepithelioma, metastatic nasopharyngeal carcinoma, and Hodgkin lymphoma of either the classic or the nodular lymphocyte-predominance type. Pancytokeratin, CD30, CD21, CD57, PAX5, and Oct2 can be used to identify such cases. Believed to reflect clonal escape of virally infected cells in older patients with age-related deterioration of immunity, EBV+ DLCL of the elderly148,149 also includes a mix of small and large cells. A cue to this diagnosis is the finding of a polymorphous spectrum of large immunoblasts, Reed-Sternberg (RS)-like cells, and medium-sized transitional lymphoplasmacytoid forms, all diluted by small, reactive lymphocytes in a patient older than 70 years. CD30 findings may be positive, making the exclusion of classic Hodgkin lymphoma difficult, although EBER/EBV-ISH positivity and strong expression of CD20 or CD79a in the large cells help to secure the diagnosis. Lymphomatoid granulomatosis is another special subtype of EBV-related B-LCL, described later.
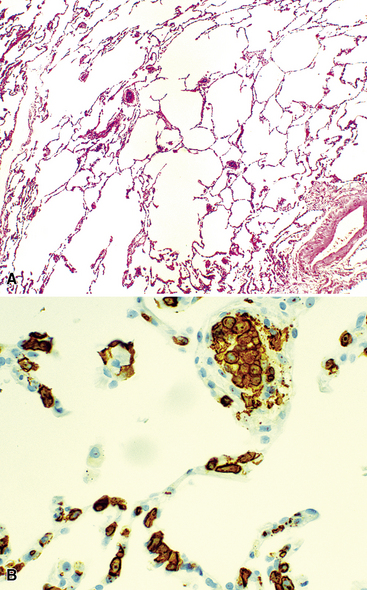
There are also several subtypes of B-DLCL with distinctive clinical aspects that should be addressed. An intravascular (“angiotrophic”) variant of B-LCL150 presents without adenopathy, organomegaly, or mass-forming lesions in solid organs, and may only be recognized when the patient undergoes biopsy of sites without clinical evidence of lymphomatous involvement, including the lung (Figs. 15-22 and 15-23). The vessels are filled with aggregations of large cells with coarse chromatin and a high nucleus-to-cytoplasm ratio, clearly different from resting lymphocytes or monocytes. Although it may have a prominent pulmonary component, the disease is never restricted to the lung and should be regarded as an aggressive, systemic lymphoma from the outset.
Primary thymic/mediastinal large B cell lymphoma usually presents with mass-related symptoms (e.g., superior vena cava syndrome) in young patients, and when it disseminates, it tends to involve extranodal sites (kidney, adrenal, liver) as much as or more than the development of generalized adenopathy with marrow involvement. The tumor can closely mimic classic Hodgkin lymphoma.150,151 Because of tumor-related fibrosis (Fig. 15-24), the inherent fragility of cells, and the known lack of surface immunoglobulin display in these tumor cells, flow cytometry findings are often negative. Because many patients with primary thymic/mediastinal large B cell lymphoma express CD30, exclusion of classic Hodgkin lymphoma relies on assessment of the expression of Oct2, PAX5, CD20, CD79a, and CD23, and on identifying the packeting of tumor cells within long, thin slips of collagenous fibrosis (Fig. 15-25). Treatment for DLCL and treatment for classic Hodgkin lymphoma are fundamentally different, so when there is any degree of ambiguity, gene rearrangements can be pursued. This adjunctive study may not allow for complete clarity in all cases, leaving some in a “gray zone” that the WHO Classification refers to as “B cell lymphoma unclassifiable with features intermediate between B-DLCL and classic Hodgkin lymphoma.” 5
Lymphomatoid Granulomatosis
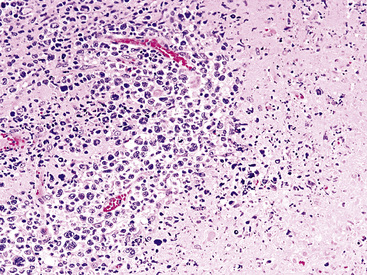
Although the vast majority of the cellularity of lymphomatoid granulomatosis (LyG, also known as “angiocentric immunoproliferative lesions,” “polymorphic reticulosis,” or “midline lethal granuloma”) is of T lineage, studies have shown that the neoplastic cells in this process are a clonal population of EBV-infected B cells.152–154 Consequently, LyG belongs in the category of B lineage lymphoproliferative disorders. It affects primarily adults, and many cases arise in the setting of immunodeficiency.155 Most patients have a prodromal phase of fever and nonspecific symptoms that may relate to pulmonary or sinonasal disease (cough, epistaxis). Skin, subcutaneous tissues, and the central nervous system may also be involved,156 yielding nonpulmonic signs and symptoms that may suggest the correct diagnosis. Radiologic studies usually show multiple opacities and nodules, with or without cavitation,157 and mediastinal adenopathy is rare. In resection specimens, the tumoral masses are centrally located within the lung and have an homogenous off-white appearance on cut section.
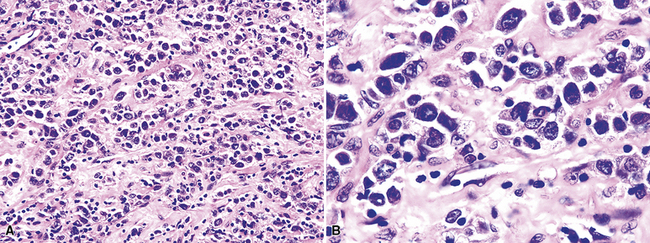
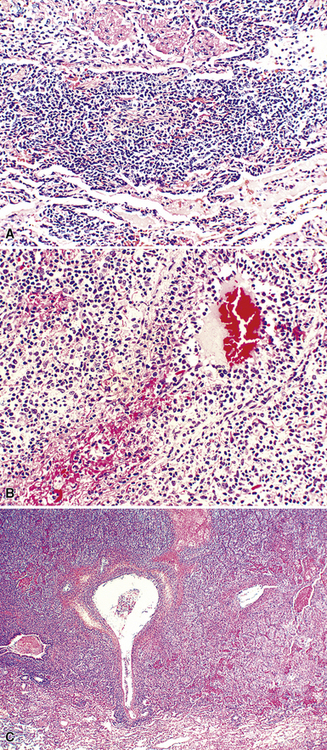
Transbronchial and transthoracic biopsy specimens usually yield insufficient diagnostic material to secure the diagnosis, and a wedge biopsy is usually required. Extensive necrosis, angio-occlusion, and vascular damage (Box 15-10 and Fig. 15-26) in the context of a mixed lymphohistiocytic infiltrate are the histologic hallmarks of LyG.152,156 The lymphoid component includes small cytotoxic T lymphocytes,158 intermediate-size activated forms, and large neoplastic B cells that closely resemble centroblasts or immunoblasts. The large lesional B cells are diffusely dispersed and have coarse chromatin, distinct or prominent nucleoli, and moderate amounts of cytoplasm. The process is both angiocentric (accumulations of viable cells around the arterioles and venules) and angiodestructive (mural invasion, lumenal occlusions, and disruption of vessel walls), and the endothelial cells are plump and activated. Necrosis may be present and is usually focal. Diagnostic RS cells are not present, although some cells may have features reminiscent of mononuclear variants.
Box 15-10 Features of Lymphomatoid Granulomatosis
What Should Be Communicated in the Report
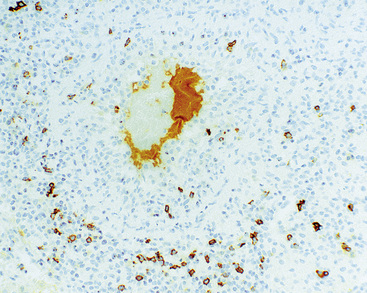
Three categories, or “grades” have been described according to the density of large lesional B cells. At the low end of the spectrum, in grade I LyG, the proliferation is polymorphous and is composed of cytologically bland small lymphocytes, plasma cells, histiocytes, and very rare large lesional cells that may be evident only on CD20 stains (Fig. 15-27) and EBER-ISH. Koss119 proposed objective boundaries of fewer than five EBV+ cells per high-power field for grade I LyG, although the WHO Classification of hematologic malignancies does not include this recommendation.
Greater numbers of large lesional cells (in the range of 5–20 cells) and more abundant levels of necrosis (Fig. 15-28) are the primaryfindings that distinguish grade II LyG from grade I LyG, although the large centroblastic/immunoblastic cells remain widely dispersed and may be difficult to find on routine stains. According to Koss’s119 proposal, EBV+ B cells are present at a density of 5 to 20 per high-power field, on average. Grade III LyG has all of the hallmark features of a high-grade lymphoma—high content of mitotically active large atypical cells and necrosis (Fig. 15-29), often in sheets and syncitia—although the small lymphoid component persists. When the angiocentric component takes on a monomorphic quality and moderate or marked atypia is noted in the small lymphocytes, the process is best classified as grade III.70,93,94
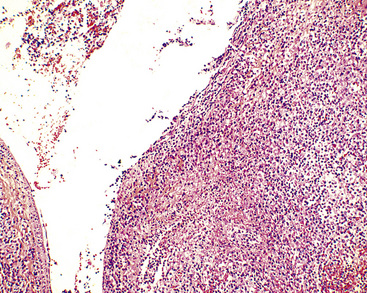
Figure 15-28 Even at the point at which lymphomatoid granulomatosis is mass-forming, there are no “granulomatous” foci.
The majority of the small lymphoid component is composed of CD2+, CD3+, and CD4+ T helper cells, with lesser numbers of CD8+ T killer cells and CD16/CD56+ natural killer cells. CD79a and CD20 staining is present in the large-cell component only, and it helps to identify lesional cells that may not be readily evident on hematoxylin and eosin stains. Latent membrane expression and EBERs are present in the lesional large B cells but not the T cells of LyG (Fig. 15-30).157–159 CD30 may be expressed, and in difficult cases, CD15, PAX5, Oct2 and other markers may be helpful in excluding classic Hodgkin lymphoma. Because lung involvement by classic Hodgkin lymphoma is exceptionally rare without mediastinal involvement, radiologic correlation may also be helpful.
In contrast to LyG, peripheral T cell lymphoma lacks the atypical large CD20+ cells and instead has significant cytologic atypia of small, intermediate, and large cells; patches of cells with small nuclei and abundant pale-staining cytoplasm; tissue eosinophilia; aberrant loss of a stage-specific pan T cell marker (CD2, CD3, CD5, or CD7) (Box 15-11; see also Box 15-2); and usually clonal T cell gene rearrangements by PCR analysis. If there is sinonasal disease, T/natural killer (NK) cell lymphomas of the “nasal type” enter into the differential diagnosis. Because these T/NK lymphomas can be angiocentric and angiodestructive as well as EBV+, the presence of CD20+ lesional large cells is the distinguishing feature. An abundance of CD56+ natural killer cells and an absence of CD20+ large cells would favor natural killer cell lymphomas of the non-nasal type.
Box 15-11 Interpretation of Flow Cytometry on Hematolymphoid Proliferations in the Lung
Hodgkin Lymphoma
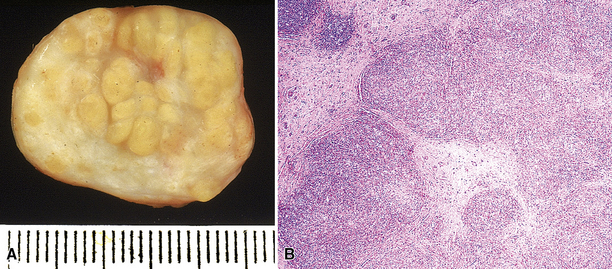
Hodgkin lymphoma is a tumor of lymphoid lineage160,161 in which the neoplastic cells are in the vast numeric minority. The 2008 edition of the WHO Classification of hematologic malignancy includes two diagnostic categories—classic Hodgkin disease and lymphocyte-predominance Hodgkin lymphoma (LPHL). The distinction is based on the phenotype of the lesional cells as well as the nature of the background infiltrate. Most patients with pulmonary Hodgkin lymphoma present with concomitant node-based or mediastinal disease, and the diagnosis is established by lymph node biopsy. In rare cases, however, clinical, pathologic, and radiologic staging shows only pulmonary involvement, invariably mass-forming rather than interstitial (Box 15-12 and Fig. 15-31).161
Box 15-12 Features of Classic Hodgkin Lymphoma
What Should Be Absent
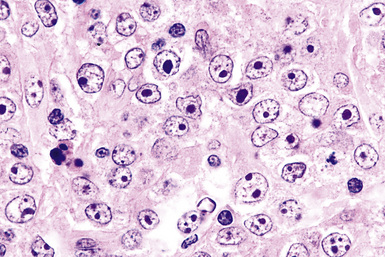
Distinguishing RS cells from mimics can be difficult, and when the cells are few in number, serial sections to find diagnostic forms may be necessary. The cell itself should be six- to eightfold larger than a resting lymphocyte, and the nucleus should be four- to fivefold larger than a small resting lymphocyte. In addition, it should have a thick nuclear membrane and a bilobed, multilobated, or wreath-shaped configuration. The nucleolus approaches or exceeds the size of the whole nucleus of resting lymphocytes and is often rimmed by a halo of pale nonstaining chromatin162,163 (Fig. 15-32). Cytoplasm is abundant and may be homogenously eosinophilic or feathered and retracted (the latter being the “lacunar variant”) (Fig. 15-33). RS-like cells may be seen in other clinical contexts, including primary mediastinal large B cell lymphoma, small lymphocytic lymphoma/chronic lymphocytic leukemia, and acute infectious mononucleosis, and so their presence cannot be the only criterion for diagnosis.
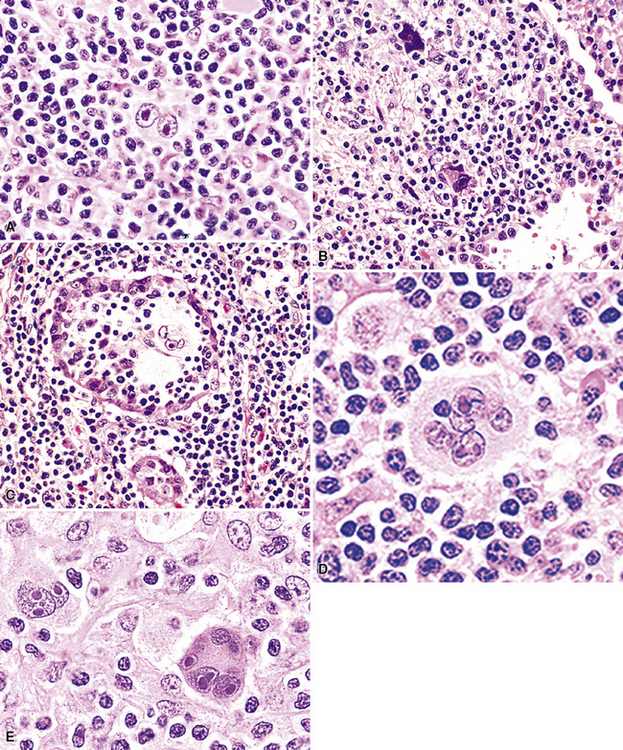
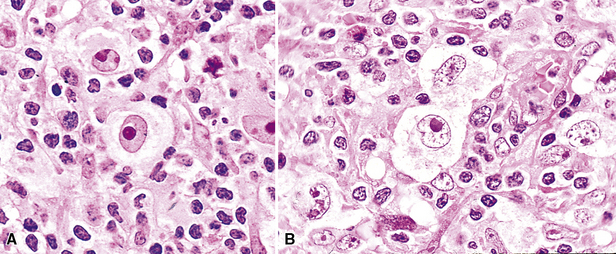
The milieu of Hodgkin lymphoma varies from area to area and from case to case, and in classic Hodgkin lymphoma, the background infiltrate ranges from a monotony of cytologically bland small T lymphocytes to a polymorphous mixture of lymphocytes, histiocytes, plasma cells, eosinophils, and neutrophils. When sclerosis is present, it consists of broad bands of collagenized fibrosis that encases cellular nodules and is evident grossly and microscopically (Fig. 15-34) rather that the delicate slips of collagen that encircle packets of cells in primary mediastinal large B cell lymphoma. In the lung, the mixed inflammatory milieu of Hodgkin lymphoma may overlap substantially with reactive conditions, such as hypersensitivity pneumonitis, collagen vascular disease, and infection. Apart from infection, however, these conditions are not mass-forming, and none of them contain diagnostic RS cells.
Immunophenotypic staining of RS cells typically yields a CD30+, CD20–, LCA– Oct2–, BOB.1– phenotype (Fig. 15-35). CD15 expression is reported in 60% to 70% of cases, depending on the series, and so is not an essential finding to establish the diagnosis. Weak and granular CD20 reactivity may be present in some cases162–165 (Table 15-4), as is weak nuclear staining for PAX5. CD79a expression, however, should be lacking. In the lung, differential diagnostic considerations for classic Hodgkin lymphoma with abundant sclerotic stroma include inflamed solitary fibrous tumors of the pleura, inflammatory myofibroblastic tumor, sclerosing mediastinitis, and infection. Without the sclerosis (particularly if the patient has a known cause of immunity), consideration should be given to senile EBV+ B-LCL of the elderly, LyG, and potentially reversible lymphoproliferative disorders arising in the setting of methotrexate therapy and transplantation.
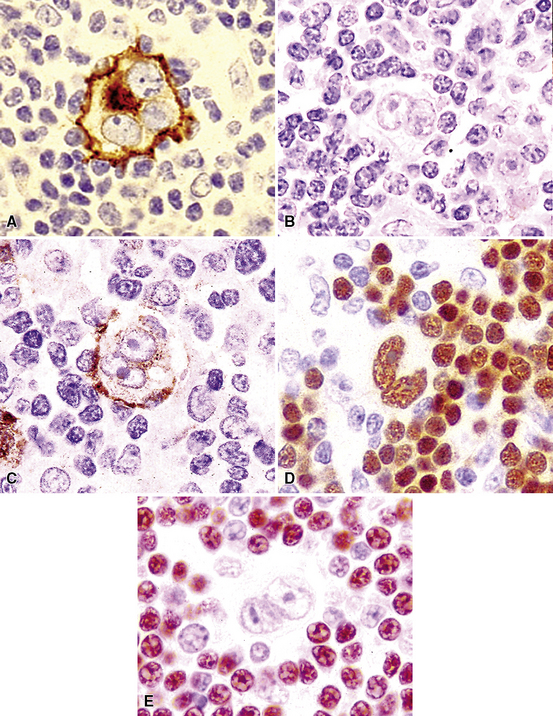
Table 15-4 Immunophenotypical Comparison of Classic Hodgkin Lymphoma and Differential Considerations
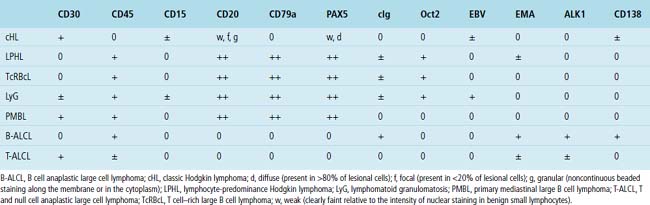
The “nonclassic” or lymphocyte-predominance subtype of Hodgkin lymphoma, LPHL, has not been reported as a primary lung tumor or as primary localized mediastinal disease. This disease is often restricted to a single lymph node at presentation, and it pursues an indolent clinical course. In the vast majority of patients, survival is similar to that of age-matched individuals without LPHL.128 Great care should be taken in evaluating lung biopsy specimens with features suggestive of LPHL: The phenotype of the lymphocytic-histiocytic (“L&H”) RS cell variants in LPHL (CD15–, CD30–, CD20+, PAX5+, CD45+ Oct2+, BOB.1+; see Table 15-4) is identical to that seen in the more aggressive TcRBCL.129,130,166,167 A large specimen is essential in resolving such a differential diagnosis because the B cell–rich milieu is as much a part of the disease definition as the nature of the lesional cells (Fig. 15-36).
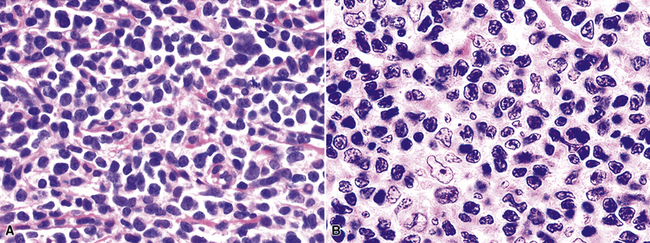
Systemic Lymphoproliferative Disorders That May Secondarily Involve the Lung, Pleura, or Mediastinum
Secondary pulmonary lymphoma is a lymphoma diagnosed in the lung of a patient who either has a previous or concurrent nodal diagnosis of lymphoma or has evidence of systemic lymphoma during a relatively short interval after presentation. The diagnostic criteria for lymph node biopsies should be applied.5,168
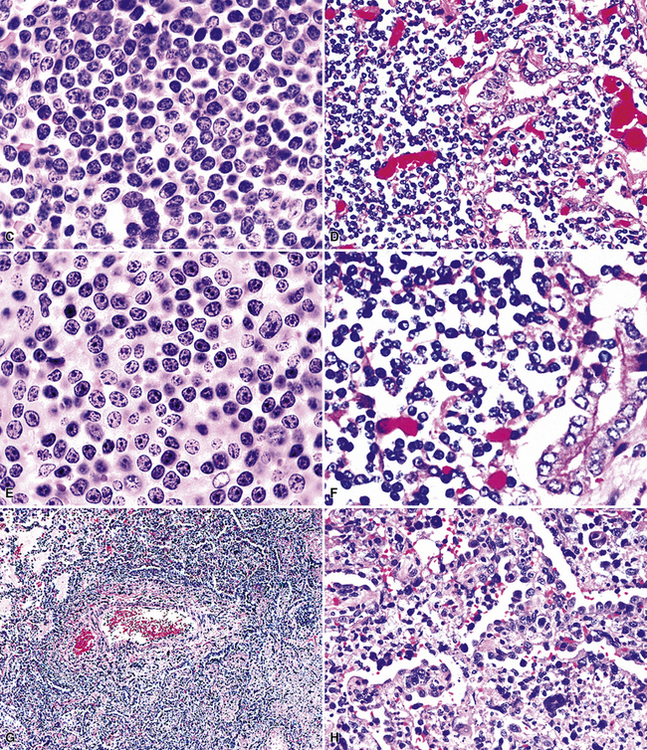
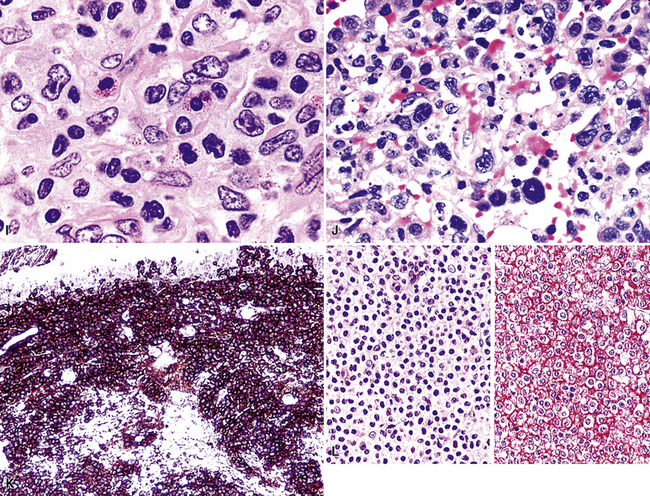
In the United States, follicular lymphoma, systemic large B cell lymphoma, small lymphocytic lymphoma, and mantle cell lymphoma (Fig. 15-37) are the most common systemic lymphomas to secondarily involve extranodal sites. Careful attention to cytologic detail, a broad immunohistochemistry panel, and above all, the clinical history, should permit complete diagnosis in most cases (Table 15-5). Although all of these lymphomas may remain localized for a period, all have a significant risk of disseminating to other sites. Cues to the diagnosis of follicular lymphoma include a mixture of small, intermediate, and large cleaved cells, including cells with irregular, cleaved, elongated “twisted towel,” and gourd-shaped nuclei. Mantle cell lymphoma, by contrast, is composed of uniform small lymphocytes with condensed chromatin and a minimally irregular nuclear profile. Pink histiocytes are often evenly commingled. Small lymphocytic lymphoma is usually composed of small cells with round nuclei with a smooth nuclear contour. Some patients have a greater degree of nuclear contour irregularity (“atypical” chronic lymphocytic leukemia) that is similar to mantle cell lymphoma cytologically. However, the cue to the correct classification is a second cell population of paraimmunoblasts and immunoblasts, which often accumulate together in pale-staining areas (“proliferation centers”).169–171MaZL (discussed in detail earlier) not infrequently involves the lung secondarily after presentation in other mucosal sites. As in secondary B-LCL, MaZL cannot be distinguished from a primary lung lymphoma on purely morphologic grounds and diagnosis requires correlation with clinical history and full radiologic staging. Immunohistochemical studies are central to the accurate diagnosis and classification of all of these conditions (see Table 15-5).
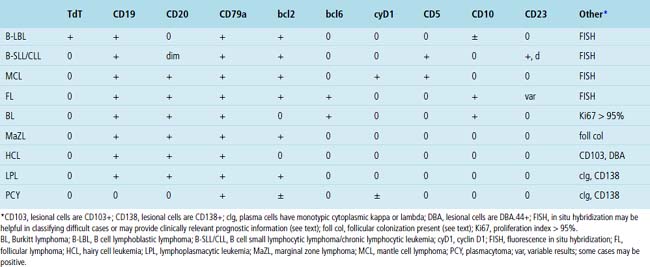
High-Grade Lymphomas
The histologic features characteristic of Burkitt lymphoma include uniform, intermediate cell size; uniformly round or ovoid nuclei, with coarse chromatin and dispersed, often peripheral, small nucleoli; and a rim of amphophilic cytoplasm with crisp separation from the adjacent cells (sometimes called “squaring off” of cytoplasm).172–174 The proliferation is uniform, with no commingled small cells (Fig. 15-38). Either apoptotic single-cell necrosis or geographic tumoral necrosis may be present, often accompanied by dispersed tingible body macrophages. Exceptional cases may show “tumoral pneumonia,” a consolidative picture radiologically because of alveolar filling by tumor cells, extravasated serum, and fibrin.67 This can also introduce confounding background staining into immunostains. A CD20+, CD10+, bcl6+, bcl2– profile with more than 95% of tumor cells exhibiting nuclear positivity for Ki68 is characteristic. In some cases, CD10 expression may be weak or absent, but bcl6 expression confirms the follicular stage of maturation. When an other-than-characteristic immunophenotype is obtained, cytogenetic testing should direct the final classification. The outdated term “atypical Burkitt lymphoma” should be avoided.
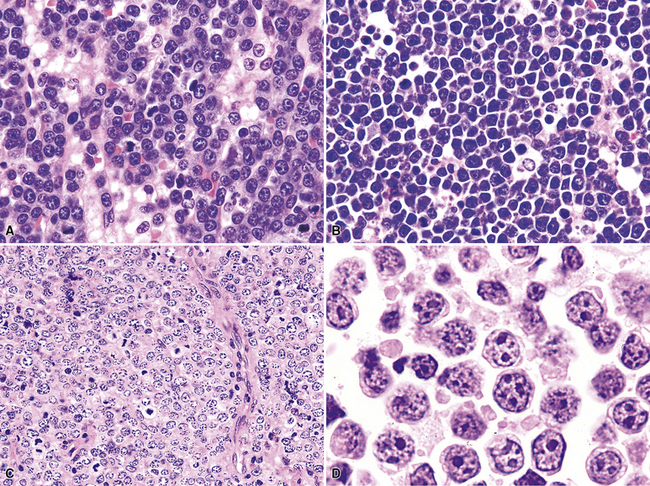
“B cell lymphoma, unclassifiable with features intermediate between DLBCL and BL” (BCL-U) is a heterogeneous category proposed by the WHO Classification.5 This category includes cases that have the characteristic morphologic features of Burkitt lymphoma and a phenotype that is not characteristic of BL. It also includes cases with morphologic features closer to those of large B cell lymphoma (greater degree of variation in cell size and nuclear contour irregularity; Fig. 15-39) and high-grade histologic features with a phenotype that is consistent with BL. Patients with either of these two patterns should be further evaluated with cytogenetics. An isolated c-myc translocation with either immunoglobulin H or immunoglobulin L loci supports a BL diagnosis, but the finding of a complex karyotype or a c-myc translocation with a gene other than immunoglobulin H or immunoglobulin L should prompt the use of BCL-U with a comment about why further classification is not possible or reconsideration of the diagnosis of B-DLCL with a clear delineation of the histologically high-grade nature of the process (Table 15-6).
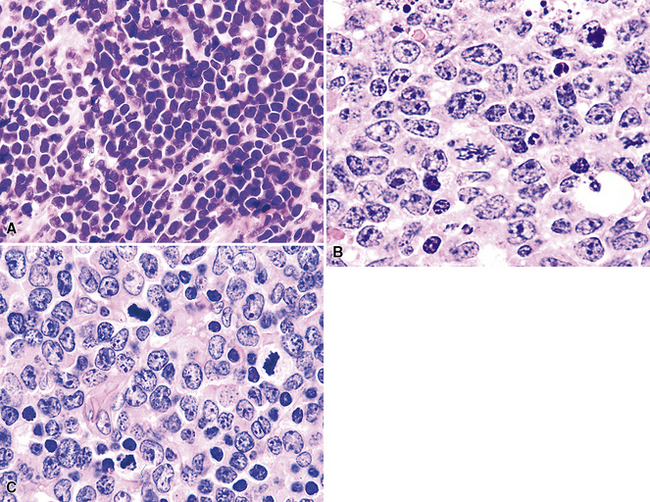
Table 15-6 Correlation of the Morphologic Features, Phenotype, and Cytogenetics in Burkitt and Burkitt-like High-Grade Lymphomas
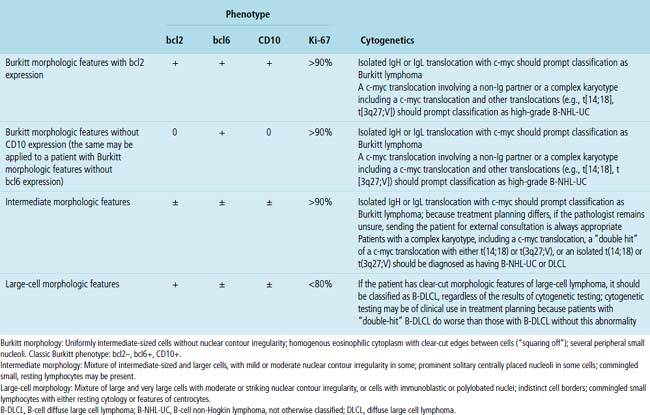
High-grade large B cell lymphoma, in contrast to BL, is more heterogeneous, with a minor population of commingled small cells. In the lesional cell population there is wide variation in cell size, including cells three to four times the size of small resting lymphocytes; cells with irregular, notched, bilobed, or polylobated nuclei; and usually a blurring of cell borders (no “squaring off”) (Fig. 15-40). An additional cue to the diagnosis is commingled small resting lymphocytes among the large lymphoma cells. More variable results for CD10 and bcl6 are reported in this setting, and at least 20% of the tumor cells are bcl2+. The majority of these cases have translocations involving either the bcl2 or bcl6 gene or a complex karyotype, and they only rarely have isolated c-myc translocation175 (see Table 15-6).
Immunoproliferative Disorders
Plasmacytoma
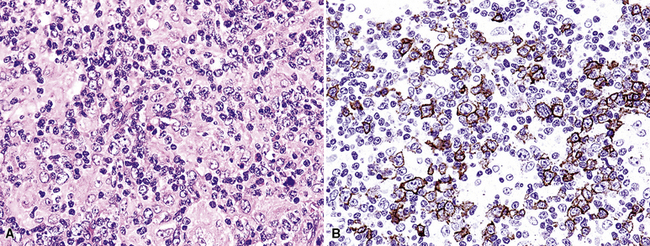
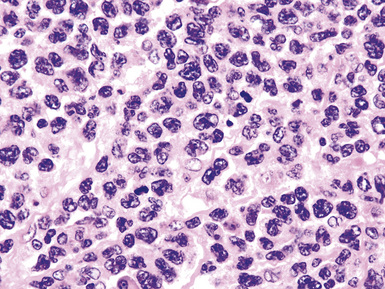
Extraosseous plasmacytoma is uncommon and usually involves the upper (rather than lower) aerodigestive tract.176 Primary pulmonary plasmacytomas, which are far less common than secondary pulmonary involvement by a disseminated plasma cell dyscrasia (i.e., multiple myeloma), have discretely demarcated edges and are brown or dark tan on cut section.177–180 Amyloid deposition may be evident grossly as white nodules or streaks in large tumors. Microscopically, the typical plasmacytoma is composed of syncitia of plasma cells with no residual germinal centers and with negligible numbers of intermingled small lymphocytes. Occasionally, lesional cells of plasmacytomas have a pleomorphic or anaplastic appearance (Fig. 15-41A) and may mimic bronchogenic carcinoma or metastatic melanoma. The amphophilic cytoplasm and paranuclear clear zone are preserved in sufficient numbers of cells to suggest the correct diagnosis in most cases.
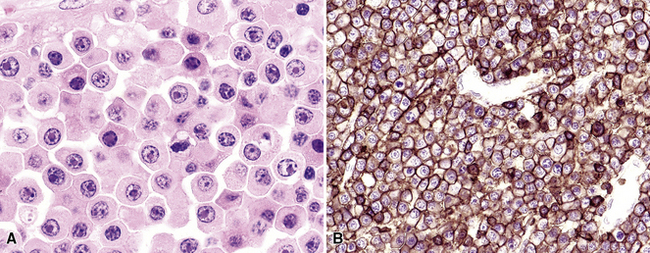
Flow cytometric analysis of plasmacytomas yields confusing “false negative” results. These terminally differentiated B cells are usually negative for CD45 (leukocyte common antigen) and the B cell markers PAX5, CD19, and CD20, and they do not have sufficient quantities of surface immunoglobulin expression to appear positive on flow cytometry (see Box 15-11). The diagnosis rests on the finding of CD138 expression (see Fig. 15-41B) and a restricted pattern of cytoplasmic immunoglobulin expression, which is easiest to show on paraffin sections. Other markers that may be positive include CD30 and epithelial membrane antigen, but neither is lineage-specific, and both should be interpreted in the context of cytokeratin, S-100 protein, and other marker results.
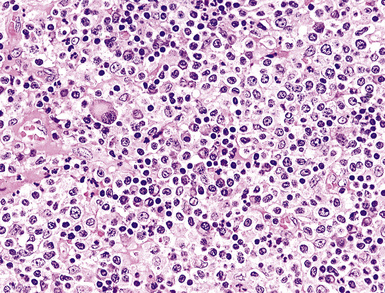
When more than 20% to 30% of a plasmacytic proliferation is composed of small lymphocytes, consideration should be given to small lymphocytic lymphoma, lymphoplasmacytic lymphoma (Fig. 15-42), and MaZL.181–183 All have a CD45+, CD20+, surface immunoglobulin (sIg+) profile for the lymphoid population, whereas plasmacytomas are negative for these markers (see Table 15-5). Findings that favor MaZL include colonized germinal centers and a polymorphous array of monocytoid, centrocytoid, and plasmacytoid cells. If residual germinal centers are detected in the setting of extreme plasmacytosis, consideration should be given not only to the possibility of MaZL but also to PCCD. Because it is impossible to distinguish between a solitary plasmacytoma and pulmonary involvement by a systemic plasma cell dyscrasia (multiple myeloma), all patients with biopsy-proven pulmonary plasmacytoma should be fully evaluated with serum and urine protein electrophoresis, skeletal survey, and bone marrow biopsy.
Patients with a previously diagnosed plasma cell dyscrasia (or MaZL) may have nodular deposits of amyloid within the lung. These may be solitary or multiple on chest radiograph, and the patient is usually asymptomatic. The material is homogenously eosinophilic and nonfibrillary, and it may contain intermingled lymphocytes, plasma cells, and transitional lymphoplasmacytoid forms. Congo red stain shows orangeophilia in regular light and green birefringence in polarized light in most cases, although noncongophilic amyloidosis occurs (Fig. 15-43). Although accumulations of amyloid are most closely associated with plasma cell dyscrasias, such as multiple myeloma, they also may be seen in association with MaZL and lymphoplasmacytic lymphoma as well as in benign settings.136–140
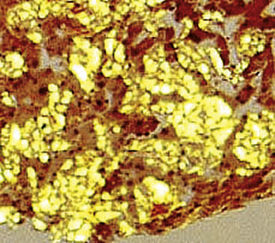
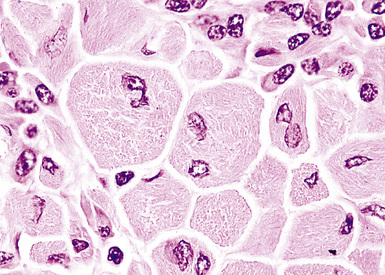
Crystal storing histiocytosis is a rare but distinctive manifestation of immunoglobulin deposition that mimics adult rhabdomyoma. Bone, spleen, lymph node, stomach, thymus, and sinonasal mucosa as well as lung have all been sites of infiltration by this mass-forming proliferation of large polygonal histiocytes that contain large periodic acid/Schiff–positive, phosphotungstic acid-hematoxylin–positive crystalloids (Figs. 15-44 and 15-45). The tumor can be distinguished from rhabdomyoma and plasmacytoma phenotypically by its CD68+, SMA–, CD138– phenotype; the cytoplasmic crystalloids are positive for immunoglobulin light-chain stains, usually of the kappa type (Fig. 15-46).132–135
Immunodeficiency-Related Lymphoproliferative Disorders
Post-transplant Lymphoproliferative Disorders
The immunosuppressed post-transplant state predisposes solid-organ transplant recipients to the development of lymphoid proliferations, often EBV-related and of B lineage184,185 (Table 15-7). For most patients, the disease is systemic at the time it is detected clinically, but for many patients who undergo lung transplant, PTLD arises in and remains localized to the graft. Radiographic findings vary from bilateral reticulonodular infiltrates to discrete single or multiple nodules or masses, with the latter most common in patients with high-grade histologic features.186
Table 15-7 Histologic, Phenotypic, and Genetic Characteristics of Post-transplant Lymphoproliferative Disorders
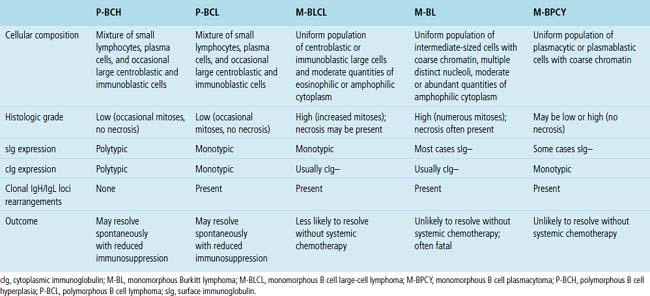
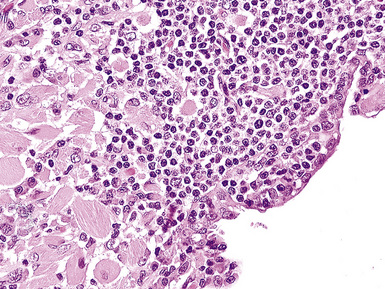
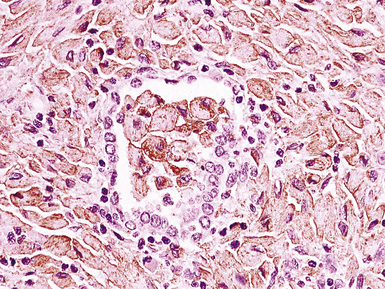
Classification of PTLDs integrates morphologic, phenotypic, and genetic data186 (see Table 15-7). Polymorphous PTLD is composed of a mixture of lymphocytes, plasma cells, and transitional lymphoplasmacytoid forms (Fig. 15-47). In this setting, a polytypic pattern of light-chain expression and the lack of clonality on Southern blot analysis are compatible with classification as “polymorphous B cell hyperplasia” or “nonclonal polymorphous PTLD.” With these early lesions, the lymph node architecture is preserved and the disease is usually more localized, so dissemination with lung involvement is uncommon. If there is a monotypic population of B cells seen on flow cytometry, or if gene rearrangement studies identify a clonal population, the proliferation is best diagnosed as “polymorphous B cell lymphoma” or “clonal polymorphous PTLD.” Clonal processes are more commonly associated with architectural distortion of the lymph node architecture and may be more likely to involve the lung. Monomorphous PTLDs have a uniform appearance and histologic features identical to those of large cell lymphoma, Burkitt lymphoma, or plasmacytoma occurring in an immunocompetent patient (Fig. 15-48). Most cases contain EBV, which can be detected with either immunohistochemistry or in situ hybridization. T lineage PTLDs with no relationship to EBV also occur, and a number have been shown to be of a special T γ/δ hepatosplenic type.
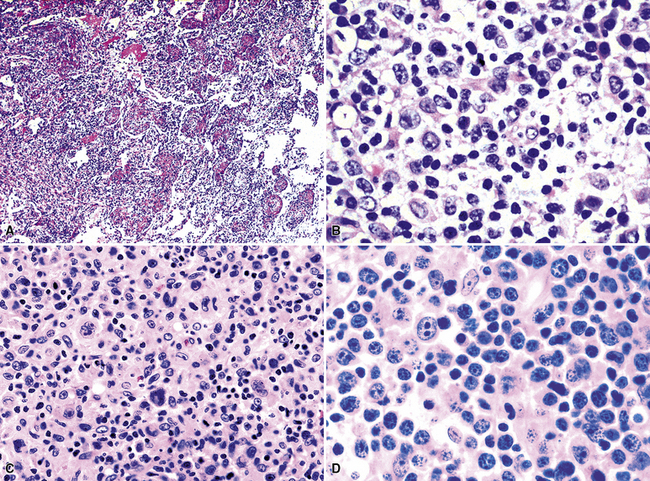
Polymorphic PTLDs are, in general, more likely than their monomorphic counterparts to regress if immunosuppression is reduced. The same can be said of polytypic or polyclonal proliferations relative to monotypic or monoclonal tumors. Therefore, classification plays an important role in initial treatment planning, and it is also an important indicator of prognosis.186 Transplant patients can have PTLDs in multiple sites, and whereas one might be polymorphous, there may be a monomorphous PTLD elsewhere. For this reason, therapeutic planning requires an integration of pathologic findings with clinical parameters of disease aggressiveness, which may not always be available to the pathologist in a timely manner.
T Lineage Lymphoid Malignancies
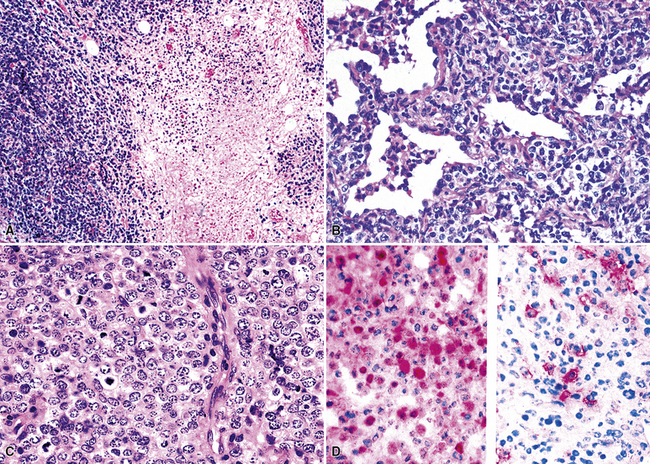
Most patients with peripheral T cell lymphoma (PTCL) are clinically ill with disseminated disease at presentation, and they may have features such as spiking fevers, rash, and lung infiltrates.187 Radiologic findings may show bilateral miliary nodules or reticulonodular infiltrates simulating interstitial disease, although the finding of one or more masses is most common. There is a systemic distribution of disease at presentation, and the initial diagnosis is usually based on lymph node biopsy.187 Lung biopsy may be performed at any point during presentation or treatment to distinguish among infection, treatment-resistant lymphomatous infiltrates, and medication-related interstitial lung disease. The histologic hallmark of most cases of PTCL is the following triad: (1) a spectrum of atypical small, intermediate, and large cells with irregular nuclear shapes and clear cytoplasm; (2) hypervascularity; and (3) tissue eosinophilia188,189 (Fig. 15-49). Phenotypically, there may be an aberrant loss of a pan T cell marker (CD2, CD3, CD5, or CD7), which may be easiest to detect on flow cytometry, where the tumor cells can be studied for three or four markers at the same time. However, immunohistochemistry on serial sections from paraffin blocks can also show this phenotypic alteration.188,189
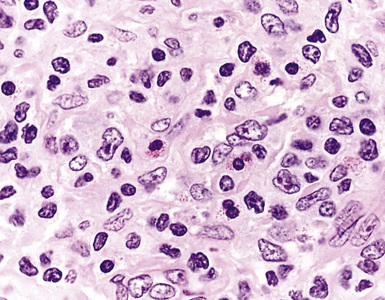
Specific T lineage lymphomas have distinctive clinicopathologic profiles (Box 15-13), some principally marrow-based and others principally nodal and parenchymally based, but primary or predominant lung or mediastinal involvement is uncommon. Of these, T cell lymphoblastic lymphoma and T-ALCL are the only two malignancies encountered in chest or lung biopsy specimens with any frequency.
T Cell Lymphoblastic Lymphoma/T Cell Acute Lymphoblastic Leukemia
T cell LBL often presents in adolescent boys with a mediastinal mass, hepatosplenomegaly, and generalized lymphadenopathy. The liver, spleen, marrow, and blood are often involved.5
In fully involved tissues, the architecture is diffuse and the blasts are intermediate in size (approximately 1.5 times the size of a benign lymph node), although there may be a surprising degree of variation in cell size, with some blasts approximatley twice the size of a benign lymph node. The nuclear contours may be smooth or convoluted, and the blasts may have fine or evenly condensed chromatin, often with a distinct nucleolus (Fig. 15-50). With good fixation and thinly sectioned slides, the neoplastic and blastic nature of the infiltrate is seldom in doubt, although with therapy, necrosis and apoptosis may make the diagnosis more difficult.
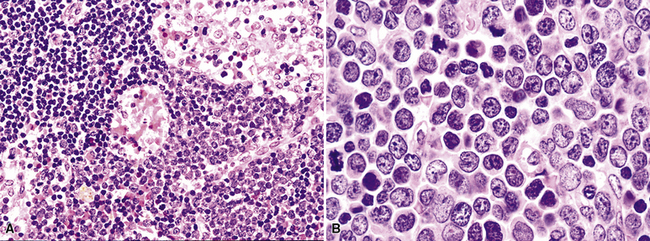
In most cases, the leukemic cells are positive for TdT, CD2, and CD7, with variable expression of CD34, CD5, CD1a, and CD10 (Box 15-14 and Fig. 15-51). Surface expression of CD3 may be slight to absent, so the cells may appear to be negative on flow cytometry, although immunohistochemical stains show cytoplasmic positivity. There are few to no cytokeratin-positive cells commingled with the blastic infiltrate, and no lobulation is seen on routine or special stains.
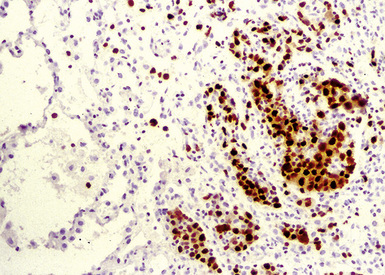
Differential diagnostic considerations for T-LBL in the mediastinum include benign thymus and lymphocyte-rich thymoma, and an adequate sample size is the only way to assess for effacement of the thymic architecture, which would favor T-LBL. Because benign thymic tissues can have an identical phenotype, attempting an initial diagnosis of T-LBL based on the findings of cytology or needle-core biopsy alone can be treacherous. Cytokeratin stains show an intact lobulated array of thymic epithelial elements in benign thymus and lymphocyte-rich thymomas, but these are absent or rare in T-LBL (Fig. 15-52). Neuroendocrine carcinomas, including carcinoids and small cell carcinoma, can mimic T-LBL, although immunophenotypic studies readily show the correct diagnosis. B lineage lymphoblastic lymphoma and acute myeloid leukemia are histologically indistinguishable, but the former are positive for PAX5, CD19, CD79a, or CD22, and the latter are positive for CD13, CD33, and myeloperoxidase.
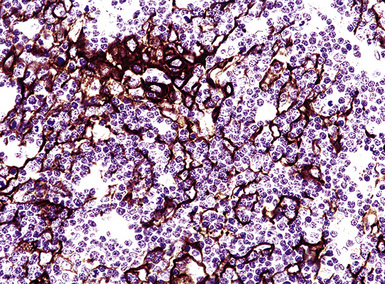
T Cell Anaplastic Large Cell Lymphoma
The lung is a common site of secondary involvement by systemic T cell anaplastic large cell lymphoma (T-ALCL), and several cases of primary pulmonary ALCL of T or null cell type have also been reported.190–195 Presentation is via mass effect, with cough, hemoptysis, or symptoms referable to pleural effusions.195–197
Microscopically, ALCL is formed of diffuse sheets of large and very large cells with pleomorphic nuclei and abundant cytoplasm, and the similarity to carcinoma or melanoma may be striking. Careful attention to the periphery may show a dishesive array of intra-alveolar tumor cells, a helpful cue that the disease may be of hematolymphoid origin (Box 15-15 and Fig. 15-53). There are multiple histopathologic variants or patterns (Box 15-16), and more than one pattern can be seen in a single patient or a single biopsy specimen. The uniting elements are the very large hallmark cells (Fig. 15-54), which are especially numerous in the common or pleomorphic form.197 Hallmark cells have reniform, or U-shaped, nuclei, and abundant amounts of cytoplasm compared with other lymphomas. In some cases, the nuclear indentation is in the plane of view, such that the nucleus appears as an O (donut cells). Nucleoli are distinct but generally are not the size of a small lymphocyte and lack the perinucleolar halo characteristic of RS cells. The remainder of the cellularity includes medium and large cells with round, irregular, lobated, and folded nuclear shapes, dispersed chromatin, distinct nucleoli, and moderate or abundant amounts of glassy eosinophilic cytoplasm193,195,197 (Fig. 15-55).
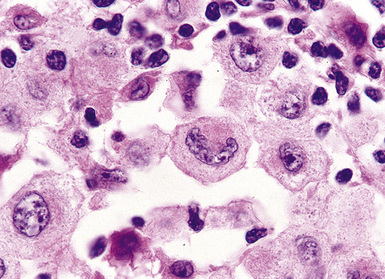
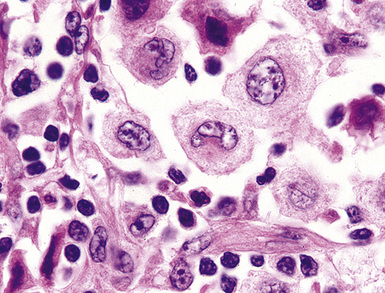
Figure 15-54 The hallmark cells of T cell anaplastic large-cell lymphoma are large and have centrally placed, reniform nuclei.
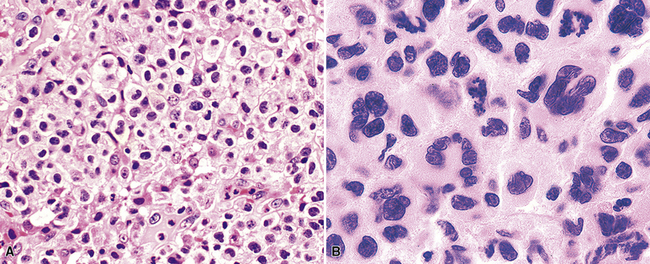
In general, ALCL is reliably CD30+ (Fig. 15-56A), and the majority of these cases are positive for a pan T cell marker, such as CD2, CD3, or betaF1, although some may be negative for all of these antigens (“null cell phenotype”) or negative for leukocyte common antigen. ALK expression can be documented immunohistochemically in many but not all cases of ALCL, and it correlates with the presence of the t(2;5) translocation involving the nucleophosmin and ALK genes.198 Although there are exceptions,199 ALK expression is associated with better overall survival5,198 and should be tested in every lymphoma that shows anaplastic morphologic features. B cell lineage and Hodgkin markers, including PAX5, are negative (see Fig. 15-56B).
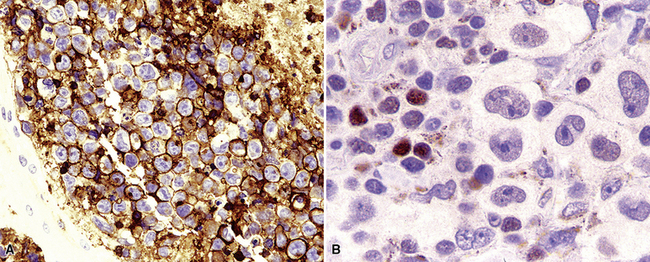
Differential considerations may include metastatic carcinoma, melanoma, and epithelioid sarcoma, and so a panel of stains including pancytokeratin, S-100, MART1, actin, desmin, and vimentin will be helpful. Some cases have a degree of morphologic overlap with classic Hodgkin lymphoma, and PAX5, which has weak to moderate intensity relative to small resting B cells, helps to resolve this gray zone differential. Exceptional cases of T-ALCL are rich in neutrophils, raising a differential with suppurative inflammation; the presence of CD30+ large cells effectively excludes a reactive process. There is also the potential for morphologic overlap with true histiocytic malignancies, which are negative for CD3, CD30, and ALK, and positive for CD68, CD31 (PECAM-1), and CD163. The rare B lineage ALCL, which tends to be negative for CD20, enters into differential consideration, but most cases are negative for CD30, a helpful cue, and are positive for CD138 or MUM1.200 Despite ALK expression, these cases of B lineage ALCL have a more aggressive clinical course than ALK+ T and null cell ALCL, and should therefore be distinguished with the appropriate stains.
Myeloid Proliferations
Extramedullary Myeloid Tumors
Extramedullary myeloid tumors (EMTs; “granulocytic sarcomas”) are masses composed of neoplastic (clonal) precursors of the granulocytic lineaege. Cell types include blasts, promyelocytes, and myelocytes, as well as monocytes and precursor monocytoid forms. Myeloid malignancies, such as idiopathic myelofibrosis (agnogenic myeloid metaplasia), may, instead of producing discrete tumefactive masses, produce a radiologic picture similar to that of interstitial lung disease.201–203 Both the clinical context and the histologic composition are important: In the pediatric setting, most extramedullary accumulations of hematopoiesis are benign and arise because peripheral demand for red cells and neutrophils is in excess of what the marrow space can provide.204 However, in an adult with an established diagnosis of a chronic myeloproliferative disorder, the same histologic features suggest a neoplasm and should raise concern that the disease is progressing to a more aggressive phase.205–207
By definition, EMTs arise outside the bone marrow. Although they usually develop during the course of systemic myeloproliferative disease, either as a complication or as a transformation from the chronic to the acute phase,201,202,208 on occasion, they are a presenting sign or the first sign of relapse.206,207 The underlying condition may be myelodysplasia or acute or chronic leukemia, including hyereosinophilic syndrome/chronic eosinophilic leukemia.208–215 Therefore, examination of the peripheral blood is a necessary part of the workup (Fig. 15-57). When the peripheral blood is free of blasts but the bone marrow meets the criteria for acute leukemia, the process is often referred to as “aleukemic leukemia.”209,210
The demographic features of EMTs match those of myeloid malignancy in general. Most patients with both of those conditions are adults, with a median age of 60 years at presentation, simply because nonlymphocytic leukemia is uncommon in pediatric practice. The overall incidence of EMT is estimated at 0.7 cases per million children in the general population per year and 2 cases per million adults per year.208 Organs that normally harbor hematopoietic cells (bone, spleen, and lymph nodes) are often involved, but a significant proportion of EMTs occur elsewhere—including the lungs.209,211 Associated pulmonary symptoms and signs include pleural effusions, chest pain, and cough. Gross examination of some excised EMTs shows a greenish color that formed the basis for one of the earliest names for such lesions, “chloroma.” The greenish color occurs when these tumors are exposed to room air and is caused by the breakdown of cytoplasmic enzymes, including myeloperoxidase. Otherwise, the gross appearance of EMT is that of a soft tan or off-white lesion, indistinguishable from non-Hodgkin lymphoma.
Histologically, three morphologic patterns of EMT can be recognized: blastic, “intermediate,” and differentiated.208 In the first pattern, sheets of round or polyhedral cells that infiltrate around or sometimes efface the architecture of the native tissue are seen. The neoplastic cells generally have medium or large vesicular nuclei, sometimes with folded nuclear membranes, discernible nucleoli, and amphophilic or basophilic cytoplasm (Fig. 15-58A). In poorly fixed tissue, these subtle features may be concealed by artifact. A few small scattered cells with sparse cytoplasmic granulation are always present. Background inflammatory cells in the blastic form of EMT are banal, usually comprising mature lymphocytes and histiocytes. This “hiatus” between the tumor cells and the reactive elements is a helpful clue to the nonlymphoid nature of the lesion in cases initially believed to be lymphoma. Compared with blastic EMT, the intermediate morphologic features show a balanced mixture of immature (blastic) and maturing cells (promyelocytes, myelocytes, and eosinophilic myelocytes; see Fig. 15-58B). The “differentiated” histologic pattern of EMT may be the easiest to overlook or to interpret as reflecting a non-neoplastic, inflammatory process.
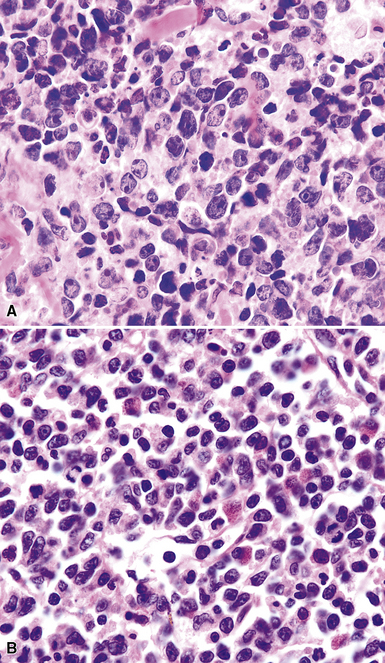
If air-dried touch preparations of fresh tumor have been prepared, histochemical stains for myeloperoxidase, Sudan B black, and alpha naphthyl acetate and other esterases can be performed. The enzymes being assessed are contained in primary myeloid granules; hence, the fewer of those structures that the cells of any given EMT possess, the less likely they are to manifest histochemical positivity. Overall, reactivity with von Leder stain is seen in fewer than 30% of blastic EMTs, 30% to 50% of “intermediate” tumors, and more than 50% of differentiated lesions. On tissue section, the chloroacetate esterase (von Leder) stain can be performed, as well as immunostains for CD45, CD34, CD15, CD68, CD117, CD163, TdT, lysozyme, and myeloperoxidase.216–219
Because of the primitive nature of the cells, the target antigens are usually present at low density, and so, on immunostains, CD45 may be very weak. If the process is monocytic in lineage, CD34 is absent. The main problem in this context is that “myeloid” antibodies (CD15, CD117, CD163, lysozyme, and myeloperoxidase) are rarely included in the first-round panels performed to diagnose and classify presumed cases of lymphoma. Weak or apparently negative CD45 results on immunostains are interpreted as excluding hematologic malignancy. Therefore, as sensitive as some “myeloid” antibodies may be for the recognition of granulocytic precursors, they are of little practical use if not used. A very helpful study by Goldstein and colleagues216 proposed that, with permissive morphologic features, a CD45± CD3–, CD20– profile should address the possibiltiy of myeloid neoplasia, especially if the lesional cells are CD43+.
1 Adam P., Katzenberger T., Seeberger H., et al. A case of diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol. 2003;27:1473-1476.
2 Laurent C., Do C., Gascoyne R.D., et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: a rare clinicopathologic entity with poor prognosis. J Clin Oncol. 2009;27(25):4211-4216. 1
3 Gascoyne R., Lamant L., Martin-Subero J.I., et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of six cases. Blood. 2003;102:2568-2573.
4 Reichard K.K., McKenna R.W., Kroft S.H. ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature. Mod Pathol. 2007;20(3):310-319.
5 Swerdlow S.H., Campo E., Harris N.L., et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. Lyon: IARC, 2008.
6 Dargent J.L., Lespagnard L., Sirtaine N., et al. Plasmablastic microlymphoma occurring in human herpesvirus 8 (HHV-8)-positive multicentric Castleman’s disease and featuring a follicular growth pattern. APMIS. 2007;115(7):869-874.
7 Seliem R.M., Griffith R.C., Harris N.L., et al. HHV-8+, EBV+ multicentric. Am J Surg Pathol. 2007;31(9):1439-1445.
8 Ehinger M., Linderoth J., Christensson B., et al. A subset of CD5- diffuse large B-cell lymphomas expresses nuclear cyclin D1 with aberrations at the CCND1 locus. Am J Clin Pathol. 2008;129(4):630-638.
9 Le Gouill S., Talmant P., Touzeau C., et al. The clinical presentation and prognosis of diffuse large B-cell lymphoma with t(14;18) and 8q24/c-MYC rearrangement. Haematologica. 2007;92(10):1335-1342.
10 Hunt K.E., Reichard K.K. Diffuse large B-cell lymphoma. Arch Pathol Lab Med. 2008;132(1):118-124.
11 Muris J.J., Meijer C.J., Vos W., et al. Immunohistochemical profiling based on bcl-2, CD10 and MUM1 expression improves risk stratification in patients with primary nodal diffuse large B cell lymphoma. J Pathol. 2006;208(5):714-723.
12 Haarer C.F., Roberts R.A., Frutiger Y.M., et al. Immunohistochemical classification of de novo, transformed, and relapsed diffuse large B-cell lymphoma into germinal center B-cell and nongerminal center B-cell subtypes correlates with gene expression profile and patient survival. Arch Pathol Lab Med. 2006;130(12):1819-1824.
13 Lawrie C.H., Soneji S., Marafioti T., et al. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer. 2007;121(5):1156-1161.
14 Zhang W., Li L., Li X., et al. Unravelling the hidden heterogeneities of diffuse large B-cell lymphoma based on coupled two-way clustering. BMC Genomics. 2007;8:332.
15 Sjo L.D., Poulsen C.B., Hansen M., et al. Profiling of diffuse large B-cell lymphoma by immunohistochemistry: identification of prognostic subgroups. Eur J Haematol. 2007;79(6):501-507.
16 Ponzoni M., Arrigoni G., Doglioni C. New transcription factors in diagnostic hematopathology. Adv Anat Pathol. 2007;14:25-35.
17 Ehinger M., Linderoth J., Christensson B., et al. A subset of CD5- diffuse large B-cell lymphomas expresses nuclear cyclin D1 with aberrations at the CCND1 locus. Am J Clin Pathol. 2008;129(4):630-638.
18 Mhawech-Fauceglia P., Saxena R., Zhang S., et al. Pax-5 immunoexpression in various types of benign and malignant tumours: a high-throughput tissue microarray analysis. J Clin Pathol. 2007;60(6):709-714.
19 Feldman A.L., Dogan A. Diagnostic uses of PAX5 in immunohistochemistry. Adv Anat Pathol. 2007;14:323-334.
20 Yaziji H., Barry T. Diagnostic immunohistochemistry: what can go wrong? Adv Anat Pathol. 2006;13(5):238-246.
21 Higgins R.A., Blankenship J.E., Kinney M.C. Application of immunohistochemistry in the diagnosis of non-Hodgkin and Hodgkin lymphoma. Arch Pathol Lab Med. 2008;132:441-461.
22 Garcia C.F., Swerdlow S.H. Best practices in contemporary diagnostic pathology: panel approach to hematolymphoid proliferations. Arch Pathol Lab Med. 2009;133(5):756-765.
23 Chu P.G., Arber D.A., Weiss L.M. Expression of T/NK cell and plasma cell antigens in non-hematopoietic epithelioid neoplasms: an immunohistochemical study of 447 Cases. Am J Clin Pathol. 2003;120:64-70.
24 Olsen R.J., Chun-che C., Herrick J.L., et al. Acute leukemia immunohistochemistry. Arch Pathol Lab Med. 2008;132:462-475.
25 Higgins R.A., Blankenship J.E., Kinney M.C. Application of immunohistochemistry in the diagnosis of non-Hodgkin and Hodgkin lymphoma. Arch Pathol Lab Med. 2008;132(3):441-461.
26 Rao D.S., Said J.W. Small lymphoid proliferations in extranodal locations. Arch Pathol Lab Med. 2007;131(3):383-396.
27 Chuang S.S., Ye H., Du M.Q., et al. Histopathology and immunohistochemistry in distinguishing Burkitt lymphoma from diffuse large B-cell lymphoma with very high proliferation index and with or without a starry-sky pattern: a comparative study with EBER and FISH. Am J Clin Pathol. 2007;128(4):558-564.
28 Yatabe Y., Suzuki R., Tobinai K., et al. Significance of cyclin D1 overexpression for the diagnosis of mantle cell lymphoma: a clinicopathologic comparison of cyclin D1-positive MCL and cyclin D1-negative MCL-like B-cell lymphoma. Blood. 2000;95:2253-2261.
29 Ehinger M., Linderoth J., Christensson B., et al. A subset of CD5- diffuse large B-cell lymphomas expresses nuclear cyclin D1 with aberrations at the CCND1 locus. Am J Clin Pathol. 2008;129(4):630-638.
30 Amin H.M., Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood.. 2007;110(7):2259-2267.
31 Sica G., Vazquez M.F., Altorki N., et al. PAX-5 expression in pulmonary neuroendocrine neoplasms: its usefulness in surgical and fine-needle aspiration biopsy specimens. Am J Clin Pathol. 2008;129(4):556-562.
32 Torlakovic E., Slipicevic A., Robinson C., et al. Pax-5 expression in nonhematopoietic tissues. Am J Clin Pathol. 2006;126(5):798-804.
33 Chu P.G., Loera S., Huang Q., Weiss L.M. Lineage determination of CD20- B-cell neoplasms: an immunohistochemical study. Am J Clin Pathol. 2006;126(4):534-544.
34 Feldman A.L., Dogan A. Diagnostic uses of Pax5 immunohistochemistry. Adv Anat Pathol. 2007;14(5):323-334.
35 Kaleem Z. Flow cytometric analysis of lymphomas: current status and usefulness. Arch Pathol Lab Med. 2006;130(12):1850-1858.
36 Kurtin P.J. How do you distinguish benign from malignant extranodal small B cell proliferations? Am J Clin Pathol. 1999;111(suppl 1):S119-S126.
37 Jennings C.D., Foon K.A. Recent advances in flow cytometry: application to the diagnosis of hematologic malignancy. Blood. 1997;90:2863-2892.
38 Beck R.C., Stahl S., O’Keefe C.L., et al. Detection of mature T-cell leukemias by flow cytometry antibodies using anti–T-cell receptor V-beta. Am J Clin Pathol. 2003;120:785-794.
39 Delgado J., Matutes E., Morilla A.M., et al. Diagnostic significance of CD20 and FMC7 expression in B cell disorders. Am J Clin Pathol. 2003;120:754-759.
40 Zaer F.S., Brayland R.C., Aanxer D.S., et al. Multi-parametric flow cytometry in the diagnosis and characterization of low grade pulmonary mucosa-associated lymphoid tissue lymphomas. Mod Pathol. 1998;11:525-532.
41 Onciu M., Berrak S.G., Medeiros L.J., et al. Discrepancies in the immunophenotype of lymphoma cells in samples obtained simultaneously from different anatomic sites. Am J Clin Pathol. 2002;117:644-650.
42 Ho A.K., Hill S., Preobrazhensky S.N., et al. Small B cell neoplasms with typical mantle cell lymphoma phenotype, often include chronic lymphocytic leukemias. Am J Clin Pathol. 2009;131:27-32.
43 Joao C., Farinha P., da Silva M.G., et al. Cytogenetic abnormalities in MALT lymphomas and their precursor lesions from different organs. A fluorescence in situ hybridization (FISH) study. Histopathology. 2007;50(2):217-224.
44 Remstein E.D., Dogan A., Einerson R.R., et al. The incidence and anatomic site specificity of chromosomal translocations in primary extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) in North America. Am J Surg Pathol. 2006;30(12):1546-1553.
45 Remstein E.D., Kurtin P.J., Einerson R.R., et al. Primary pulmonary MALT lymphomas show frequent and heterogeneous cytogenetic abnormalities, including aneuploidy and translocations involving API2 and MALT1 and IGH and MALT1. Leukemia. 2004;18(1):156-160.
46 Einerson R.R., Kurtin P.J., Dayharsh G.A., et al. FISH is superior to PCR in detecting t(14;18)(q32;q21)-IgH/bcl-2 in follicular lymphoma using paraffin-embedded tissue samples. Am J Clin Pathol. 2005;124(3):421-429.
47 Ye H., Remstein E.D., Bacon C.M., et al. Chromosomal translocations involving BCL6 in MALT lymphoma. Haematologica. 2008;93(1):145-146.
48 Lin P., Medeiros L.J. High-grade B-cell lymphoma/leukemia associated with t(14;18) and 8q24/MYC rearrangement: a neoplasm of germinal center immunophenotype with poor prognosis. Haematologica. 2007;92(10):1297-1301.
49 Nomura K., Yoshino T., Nakamura S., et al. Detection of t(11;18)(q21;q21) in marginal zone lymphoma of mucosa-associated lymphocytic tissue type on paraffin-embedded tissue sections by using fluorescence in situ hybridization. Cancer Genet Cytogenet. 2003;140(1):49-54.
50 Stachurski D., Miron P.M., Al-Homsi S., et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma with a complex karyotype and cryptic 3′ ALK gene insertion to chromosome 4 q22–24. Hum Pathol. 2007;38(6):940-945.
51 Morris S.W., Xue L., Ma Z., Kinney M.C. Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of non-Hodgkin’s lymphoma. Br J Haematol. 2001;113(2):275-295.
52 Muller-Hermelink H.K. Genetic and molecular genetic studies in the diagnosis of B cell lymphomas: marginal zone lymphomas. Hum Pathol. 2003;34(4):336-340.
53 Reddy A., Lyall E.G., Crawford D.H. Epstein-Barr virus and lymphoid interstitial pneumonitis: an association revisited. Pediatr Infect Dis J. 1998;17:82-83. 1998
54 Hunt J.L. Molecular pathology in anatomic pathology practice: a review of basic principles. Arch Pathol Lab Med. 2008;132:248-260.
55 Nicholson A.G., Wotherspoon A.C., Diss C., et al. Pulmonary B cell non-Hodgkin’s lymphomas. The value of immunohistochemistry and gene analysis in diagnosis. Histopathology. 1995;26:395-403.
56 Allen T.C., Cagle P.T., Popper H.H. Basic concepts of molecular pathology. Arch Pathol Lab Med. 2008;132:1551-1556.
57 Tashiro K., Ohshima K., Suzumiya J., et al. Clonality of primary pulmonary lymphoproliferative disorders; using in situ hybridization and polymerase chain reaction for immunoglobulin. Leuk Lymphoma. 1999;36(1–2):157-167.
58 Hewitt S.M., Lewis F.A., Cao Y., et al. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2008;132:1929-1935.
59 Kurosu K., Yumoto N., Mikata A., et al. Monoclonality of B-cell lineage in primary pulmonary lymphoma demonstrated by immunoglobulin heavy chain gene sequence analysis of histologically non-definitive transbronchial biopsy specimens. J Pathol. 1996;178(3):316-322.
60 Swerdlow S.H. Genetic and molecular genetic studies in the diagnosis of atypical lymphoid hyperplasias versus lymphoma. Hum Pathol. 2003;34(4):346-351.
61 Zhang S., Abreo F., Lowery-Nordberg M., et al. The role of fluorescence in situ hybridization and polymerase chain reaction in the diagnosis and classification of lymphoproliferative disorders on fine-needle aspiration. Cancer Cytopathol. 2010;118:105-112.
62 Muller-Hermelink H.K. Genetic and molecular genetic studies in the diagnosis of B cell lymphomas: marginal zone lymphomas. Hum Pathol. 2003;34(4):336-340.
63 Nathwani B.N., Sasu S.J., Ahsanuddin A.N., et al. The critical role of histology in an era of genomics and proteomics: a commentary and reflection. Adv Anat Pathol. 2007;14(6):375-400.
64 Colby T.V., Leslie K.O., Yousem S.A. Lungs. In: Mills S.E., editor. Histology for Pathologists. Philadelphia: Lippincott Williams and Wilkins; 2007:51-61.
65 Harris N.L. Extranodal lymphoid infiltrates and mucosa-associated lymphoid tissue: a unifying concept. Am J Surg Pathol. 1991;15:879-884.
66 Douglas K.M., Raza K., Stevens R., et al. Bronchial MALT lymphoma in longstanding rheumatoid arthritis. Rheumatology. 2005;44(5):687-689.
67 Koss M.N. Malignant and benign lymphoid lesions in the lung. Ann Diagn Pathol. 2004;8:167-187.
68 Begueret H., Vergier B., Parrens M., et al. Primary lung small B-cell lymphoma versus lymphoid hyperplasia: evaluation of diagnostic criteria in 26 Cases. Am J Surg Pathol. 2002;26(1):76-81.
69 Nicholson A.G., Wotherspoon A.C., Diss T.C., et al. Reactive pulmonary lymphoid disorders. Histopathology. 1995;26:405-412.
70 Yousem S.A., Colby T.V., Carrington C.B. Follicular bronchitis—bronchiolitis. Hum Pathol. 1985;16:700-706.
71 Romero S., Barroso E., Gil J., et al. Follicular bronchiolitis: clinical and pathologic findings in six patients. Lung. 2003;181(6):309-319.
72 Kinnane B.T., Mansell A.L., Zwerdling R.G., et al. Follicular bronchitis in the pediatric population. Chest. 1993;104:1183-1186.
73 Exley C.M., Suvarna S.K., Matthews S. Follicular bronchiolitis as a presentation of HIV. Clin Radiol. 2006;61(8):710-713.
74 Lee H.K., Kim D.S., Yoo B., et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest. 2005;127(6):2019-2027.
75 Couture C., Colby T.V. Histopathology of bronchiolar disorders. Semin Respir Crit Care Med. 2003;24(5):489-498.
76 Ryu J.H. Classification and approach to bronchiolar diseases. Curr Opin Pulm Med. 2006;12(2):145-151.
77 Visscher D.W., Myers J.L. Bronchiolitis: the pathologist’s perspective. Proc Am Thorac Soc. 2006;3(1):41-47.
78 Collard H.R., Cool C.D., Leslie K.O., et al. Organizing pneumonia and lymphoplasmacytic inflammation predict treatment response in idiopathic pulmonary fibrosis. Histopathology. 2007;50(2):258-265.
79 Tansey D., Wells A.U., Colby T.V., et al. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology. 2004;44(6):585-596.
80 Abbondanzo S.L., Rush W., Bijwaard K.E., Koss M.N. Nodular lymphoid hyperplasia of the lung: clinicopathologic study of 14 cases. Am J Surg Pathol. 2000;24:587-597.
81 Sakurai H., Hada M., Oyama T. Nodular lymphoid hyperplasia of the lung: a very rare disease entity. Ann Thorac Surg. 2007;83(6):2197-2199.
82 Begueret H., Vergier B., Parrens M., et al. Primary lung small cell lymphoma versus lymphoid hyperplasia: evaluation of diagnostic criteria in 26 cases. Am J Surg Pathol. 2002;26:76-81.
83 Kobzik L. Benign pulmonary lesions that may be misdiagnosed as malignant. Semin Diagn Pathol. 1990;7:129-138.
84 Isaacson P., Wotherspoon A., Pan L. Follicular colonization in B-cell lymphoma of mucosa-associated lymphoid tissue. Am J Surg Pathol. 1991;15:819-828.
85 Ito I., Nagai S., Kitaichi M., et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiological and pathologic study. Am J Respir Crit Care Med. 2005;171:632-638.
86 Joshi V.V., Oleske J.M., Minnefor A.B., et al. Pathologic pulmonary findings in children with the acquired immunodeficiency syndrome: a study of ten cases. Hum Pathol. 1985;16:241-246.
87 Joshi V.V., Gagnon G.A., Chadwick E.G., et al. The spectrum of mucosa associated lymphoid tissue lesions in pediatric patients infected with HIV: A clinicopathologic study of six cases. Am J Clin Pathol. 1997;107:592-600.
88 Sacco O., Fregonese B., Picco P., et al. Common variable immunodeficiency presenting in a girl as lung infiltrates and mediastinal adenopathies leading to severe “superior vena cava” syndrome. Eur Respir J. 1996;9(9):1958-1961.
89 Swigris J.J., Berry G.J., Raffin T.A., et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122:2150-2164.
90 Fishback N., Koss M. Update on lymphoid interstitial pneumonitis. Curr Opin Pulm Med. 1996;2:429-433.
91 Cha S.I., Fessler M.B., Cool C.D., et al. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J. 2006;28(2):364-369.
92 Silva C.I., Flint J.D., Levy R.D., Muller N.L. Diffuse lung cysts in lymphoid interstitial pneumonia: high-resolution CT and pathologic findings. J Thorac Imaging. 2006;21(3):241-244.
93 Meyers J.L., Peiper S.C., Katzenstein A.-L. Pulmonary involvement in infectious mononucleosis: histopathologic features and detection of Epstein-Barr virus related DNA sequences. Mod Pathol. 1989;2:444-448.
94 Marzouk K., Corate L., Saleh S., Sharma O.P. Epstein-Barr-virus-induced interstitial lung disease. Curr Opin Pulm Med. 2005;11(5):456-460.
95 Castleman B., Iverson L., Menendez P. Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer. 1956;9:822-930.
96 Gupta N.K., Torigian D.A., Gefter W.B., et al. Mediastinal Castleman disease mimicking mediastinal pulmonary sequestration. J Thorac Imaging. 2005;20(3):229-232. Symposium: Multislice CT Part I
97 Pham T.T., Harrell J.H., Herndier B., Yi E.S. Endotracheal hyaline vascular Castleman disease: a case report. Chest. 2007;131(2):590-592.
98 Ng S.H., Ko S.F., Lin J.W., et al. Paracardiac pleural Castleman disease: radiographic and MR findings. Br J Radiol. 2004;77(917):433-435.
99 Cronin D.M.P., Warnke R.A. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16:236-246.
100 Weiss L.M. Castleman Disease. In: Lymph Nodes. New York: Cambridge University Press; 2008:25-32.
101 Harris N.L., Bhan A.K. “Plasmacytoid T cells’ in Castleman’s disease. Immunohistologic phenotype. Am J Surg Pathol. 1987;11:109-113.
102 Menke D.M., Tiemann M., Camoriano J.K., et al. Diagnosis of Castleman’s disease by identification of an immunophenotypically aberrant population of mantle zone B lymphocytes in paraffin embedded lymph node biopsies. Am J Clin Pathol. 1996;105(3):268-276.
103 Zarate-Osorno A., Medeiros L.J., Danon A.D., Neiman R.S. Hodgkin’s disease with co-existent Castleman-like histologic features: a report of 3 cases. Arch Pathol Lab Med. 1994;118(3):270-274.
104 Nonaka D., Rodriguez J., Rollo J.L., Rosai J. Undifferentiated large cell carcinoma of the thymus associated with Castleman disease-like reaction a distinctive type of thymic neoplasm characterized by an indolent behavior. Am J Surg Pathol. 2005;29:490-495.
105 Casper C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol. 2005;129(1):3-17.
106 Keller A.R., Hochholzer L., Castleman B. Hyaline vascular and plasma cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670-683.
107 Mandler R.N., Kerrigan D.P., Smart J., et al. Castleman’s disease in POEMS syndrome with elevated interleukin-6. Cancer. 1992;69:2697-2703.
108 Guihot A., Couderc L.J., Agbalika F., et al. Pulmonary manifestations of multicentric Castleman’s disease in HIV infection: a clinical, biological and radiological study. Eur Respir J. 2005;26(1):118-125.
109 Dispenzieri A., Kyle R.A., Lacy M.Q., et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101:2496-2506.
110 Seliem R.M., Griffith R.C., Harris N.L., et al. HHV-8+, EBV+ multicentric plasmablastic microlymphoma in an HIV+ man: the spectrum of HHV-8+ lymphoproliferative disorders expands. Am J Surg Pathol. 2007;31(9):1439-1445.
111 Amin H.M., Medeiros L.J., Manning J.T., et al. Dissolution of the lymphoid follicle is a feature of the HHV8+ variant of plasma cell Castleman’s disease. Am J Surg Pathol. 2003;27:91-100.
112 Dham A., Peterson B.A. Castleman disease. Curr Opin Hematol. 2007;14(4):354-359.
113 Li C.-F., Ye H., Liu H., et al. Fatal HHV-8-associated hemophagocytic syndrome in an HIV-negative immunocompetent patient with plasmablastic variant of multicentric Castleman disease (plasmablastic microlymphoma). Am J Surg Pathol. 2006;30(1):123-127.
114 Herrada J., Cabanillas F., Rice L., et al. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128:657-662.
115 Graham B.B., Mathisen D.J., Mark E.J., Takvorian R.W. Primary pulmonary lymphoma. Ann Thorac Surg. 2005;80(4):1248-1253.
116 Habermann T.M., Ryu J.H., Inwards D.J., Kurtin P.J. Primary pulmonary lymphoma. Semin Oncol. 1999;26:307-315.
117 L’Hoste R.J., Fillipa D.A., Leiberman P.H., Bretsky S. Primary pulmonary lymphomas. Cancer. 1984;54:1397-1406.
118 Rush W.L., Andriko J.A., Taubenberger J.K., et al. Primary anaplastic large cell lymphoma of the lung: clinicopathologic study of five patients. Mod Pathol. 2000;13:1285-1292.
119 Koss M.N. Malignant and benign lymphoid lesions of the lung. Ann Diagn Pathol. 2004;8(3):167-187.
120 Zinzani P.L., Magagnoli M., Ascani S., et al. Nongastrointestinal mucosa-associated lymphoid tissue (MALT) lymphomas: clinical and therapeutic features of 24 localized patients. Ann Oncol. 1997;8:883-886.
121 Kurtin P.J., Myers J.L., Adlakha H., et al. Pathologic and clinical features of primary pulmonary extranodal marginal zone B-cell lymphoma of MALT type. Am J Surg Pathol. 2001;25(8):997-1008.
122 Ferreri A.J., Zucca E. Marginal-zone lymphoma. Crit Rev Oncol Hematol. 2007;63(3):245-256.
123 Arkenau H.T., Gordon C., Cunningham D., et al. Mucosa associated lymphoid tissue lymphoma of the lung: the Royal Marsden Hospital experience. Leuk Lymphoma. 2007;48(3):547-550.
124 Remstein E.D., Dogan A., Einerson R.R., et al. The incidence and anatomic site specificity of chromosomal translocations in primary extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) in North America. Am J Surg Pathol. 2006;30(12):1546-1553.
125 O’Donnell P.G., Jackson S.A., Tung K.T., et al. Radiological appearances of lymphomas arising from mucosa-associated lymphoid tissue (MALT) in the lung. Clin Radiol. 1998;53:258-263.
126 Mitchell A., Meunier C., Ouellette D., Colby T. Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue with initial presentation in the pleura. Chest. 2006;129:791-794.
127 McCluggage W.G., McManus K., Qureshi R., et al. Low-grade B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) of thymus. Hum Pathol. 2000;31(2):255-259.
128 Burke J.S. Are there site-specific differences among the MALT lymphomas—morphologic, clinical? Am J Clin Pathol. 1999;111(1 suppl 1):S133-S143.
129 Ferry J.A. Extranodal lymphomas. Arch Pathol Lab Med. 2008;132:565-578.
130 Bacon C.M., Du M.Q., Dogan A. Mucosa-associated lymphoid tissue (MALT) lymphoma: a practical guide for pathologists. J Clin Pathol. 2007;60(4):361-372.
131 Woehrer S., Streubel B., Chott A., et al. Transformation of MALT lymphoma to pure plasma cell histology following treatment with the anti-CD20 antibody rituximab. Leuk Lymphoma. 2005;46(11):1645-1649.
132 Fairweather P.M., Williamson R., Tsikleas G. Pulmonary extranodal marginal zone lymphoma with massive crystal storing histiocytosis. Am J Surg Pathol. 2006;30(2):262-267.
133 Ionescu D.N., Pierson D.M., Qing G., et al. Pulmonary crystal-storing histiocytoma. Arch Pathol Lab Med. 2005;129(9):1159-1163.
134 Prasad M.L., Charney D.A., Sarlin J., et al. Pulmonary immunocytoma with massive crystal storing histiocytosis: a case report with review of the literature. Am J Surg Pathol. 1998;22:1148-1153.
135 Jones D., Renshaw A.A. Recurrent crystal storing histiocytosis of the lung in a patient without a clonal lymphoproliferative disorder. Arch Pathol Lab Med. 1996;120:978-980.
136 Lim J.K., Lacy M.Q., Kurtin P.J., et al. Pulmonary marginal zone lymphoma of MALT type as a cause of localised pulmonary amyloidosis. J Clin Pathol. 2001;54(8):642-646.
137 Khoor A., Myers J.L., Tazelaar H.D., Kurtin P.J. Amyloid-like pulmonary nodules, including localized light-chain deposition: clinicopathologic analysis of three cases. Am J Clin Pathol. 2004;121:200-204.
138 Satani T., Yokose T., Kaburagi T., et al. Amyloid deposition in primary pulmonary marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue. Pathol Int. 2007;57(11):746-750.
139 Dacic S., Colby T.V., Yousem S.A. Nodular amyloidoma and primary pulmonary lymphoma with amyloid production: a differential diagnostic problem. Mod Pathol. 2000;13(9):934-940.
140 Bhargava P., Rushin J.M., Rusnock E.J., et al. Pulmonary light chain deposition disease: report of five cases and review of the literature. Am J Surg Pathol. 2007;31:267-276.
141 Jourdan F., Molina T.J., Le Tourneau A., et al. Florid marginal zone differentiation in follicular lymphoma mimicking marginal zone lymphoma of MALT type in the lung. Histopathology. 2006;49:426-445.
142 Zettl A., Rudiger T., Marx A., et al. Composite marginal zone B-cell lymphoma and classical Hodgkin’s lymphoma: a clinicopathological study of 12 cases. Histopathology. 2005;46(2):217-228.
143 Fiche M., Capron F., Berger F., et al. Primary pulmonary non-Hodgkin’s lymphomas. Histopathology. 1995;26:529-537.
144 Habermann T.M., Ryu J.H., Inwards D.J., Kurtin P.J. Primary pulmonary lymphoma. Semin Oncol. 1999;26:307-315.
145 Lim M.S., Beaty M., Sorbara L., et al. T-cell/histiocyte-rich large B-cell lymphoma: a heterogeneous entity with derivation from germinal center B cells. Am J Surg Pathol. 2002;26(11):1458-1466.
146 Colomo L., Loong F., Rives S., et al. Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am J Surg Pathol. 2004;28(6):736-747.
147 Yamaguchi M., Seto M., Okamoto M., et al. De novo CD5+ diffuse large B cell lymphoma: a clinicopathological study of 109 patients. Blood. 2002;99:815-821.
148 Oyama T., Ichimura K., Suzuki R., et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathological study of 22 patients. Am J Surg Pathol. 2003;27(1):16-26.
149 Nakamura S., Jaffe E.S., Swerdlow S.H. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow S.H., Campo E., Harris N.L., et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC Press; 2008:243-244.
150 Calaminici M., Piper K., Lee A.M., Norton A.J. CD23 expression in mediastinal large B-cell lymphomas. Histopathology. 2004;45:619-624.
151 Pileri S.A., Gaidano G., Zintzani P.L., et al. Primary mediastinal B-cell lymphoma: high frequency of BCL-6 mutations and consistent expression of the transcription factors OCT-2, BOB.1, and PU.1 in the absence of immunoglobulins. Am J Pathol. 2003;162(1):243-253.
152 Nicholson A.G., Wotherspoon A.C., Diss T.C., et al. Lymphomatoid granulomatosis: evidence that some cases represent Epstein–Barr virus-associated B-cell lymphoma. Histopathology. 1996;29:317-324.
153 Meyers J.L., Kurtin P.J., Katzenstein A.LA., et al. Lymphomatoid granulomatosis: evidence of immunophenotypic diversity and relationship to Epstein–Barr virus infection. Am J Surg Pathol. 1995;19:1300-1312.
154 Guinee D.G., Perkins S.L., Travis W.D., et al. Proliferation and cellular phenotype in lymphomatoid granulomatosis: implications of a higher proliferation index in B cells. Am J Surg Pathol. 1998;22:1093-1100.
155 Haque A.K., Myers J.L., Hudnall S.D., et al. Pulmonary lymphomatoid granulomatosis in acquired immunodeficiency syndrome: lesions with Epstein-Barr virus infection. Mod Pathol. 1998;11(4):347-356.
156 Koss M.N., Hochholzer L., Langloss J.M., et al. Lymphomatoid granulomatosis: a clinicopathologic and immunopathologic study of 42 patients. Pathology. 1986;18:283-288.
157 Hochberg E.P., Gilman M.D., Hasserjian R.P. Case 17–2006: A 34-year-old man with cavitary lung lesions. N Engl J Med. 2006;354:2485-2493.
158 Morice W.G., Kurtin P.J., Myers J.L. Expression of cytolytic lymphocyte-associated antigens in pulmonary lymphomatoid granulomatosis. Am J Clin Pathol. 2002;118(3):391-398.
159 Ng S-B, Khoury J.D. Epstein-Barr virus in lymphoproliferative processes: an update for the diagnostic pathologist. Adv Anat Pathol. 2009;16:40-55.
160 Yousem S.A., Weis L.M., Colby T.V. Primary pulmonary Hodgkin’s disease: a clinicopathologic study of 15 cases. Cancer. 1986;57:1217-1224.
161 Radin A.I. Primary pulmonary Hodgkin’s disease. Cancer. 1990;65:550-563.
162 Harris N.L. Hodgkin’s lymphomas: classification, diagnosis, and grading. Semin Hematol. 1999;36:220-232.
163 Pileri S., Ascani S., Leoncini L., et al. Hodgkin’s lymphoma: the pathologist’s viewpoint. J Clin Pathol. 2002;55:162-176.
164 Browne P., Petrosyan K., Hernandez A., Chan J.A. The B-cell transcription factors BSAP, Oct-2, and BOB.1 and the pan-B-cell markers CD20, CD22, and CD79a are useful in the differential diagnosis of classic Hodgkin lymphoma. Am J Clin Pathol. 2003;120:767-777.
165 Calvo KR, Traverse-Glehen A, Pittaluga S, Jaffe ES. Molecular profiling provides evidence of primary mediastinal large B cell lymphoma as a distinct entity related to classic Hodgkin lymphoma: implications for mediastinal gray zone lymphomas as an intermediate form of B-cell Lymphoma. Adv Anat Pathol. 11(5):227–238.
166 Uherova P., Valdez R., Ross C.W., et al. Nodular lymphocyte predominant Hodgkin lymphoma. An immunophenotypic reappraisal based on a single-institution experience. Am J Clin Pathol. 2003;119(2):192-198.
167 Brousset P., Chittal S.M., Schlafer D., Delsol G. T cell rich B cell large cell lymphoma in the lung. Histopathology. 1995;26:371-373.
168 Chilosi M., Zinzani P.L., Poletti V. Lymphoproliferative lung disorders. Semin Respir Crit Care Med. 2005;26(5):490-501.
169 Ho A.K., Hill S., Preobrazhensk S.N., et al. Small B-cell neoplasms with typical mantle cell lymphoma immunophenotypes often include chronic lymphocytic leukemias. Am J Clin Pathol. 2009;131:27-32.
170 Swerdlow S.H., Williams M.E. From centrocytic to mantle cell lymphoma: a clinicopathologic and molecular review of 3 decades. Hum Pathol. 2002;33:7-20.
171 Gandhi M.K., Marcus R.E. Follicular lymphoma: time for a re-think? Blood Rev. 2005;19:165-178.
172 Hummel M., Bentink S., Berger H., et al. for the Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche Krebshilfe. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419-2430.
173 Leoncini L., Delsol G., Gascoyne R.D., et al. Aggressive B-cell lymphomas: a review based on the workshop of the XI Meeting of the European Association for Haematopathology. Histopathology. 2005;46:241-255.
174 McClure R.F., Remstein E.D., Macon W.R., et al. Adult B-cell lymphomas with Burkitt-like morphology are phenotypically and genotypically heterogeneous with aggressive clinical behavior. Am J Surg Pathol. 2005;29(12):1652-1660.
175 Keller C.E., Nandula S., Fisher J., et al. The spectrum of B-cell non-Hodgkin lymphomas with dual IgH–BCL2 and BCL6 translocations. Am J Clin Pathol. 2008;130:193-201.
176 Hussong J.W., Perkins S.L., Schnitzer B., et al. Extramedullary plasmacytoma. A form of marginal zone cell lymphoma? Am J Clin Pathol. 1999;111:111-116.
177 Koss M.N., Hochholzer L., Moran C.A., Frizzera G. Pulmonary plasmacytomas: a clinicopathologic and immunohistochemical study of five cases. Ann Diagn Pathol. 1998;2(1):1-11.
178 Wise J.N., Schaefer R.F., Read R.C. Primary pulmonary plasmacytoma: a case report. Chest. 2001;120:1405-1407.
179 Banerjee S.S., Verma S., Shanks J.H. Morphological variants of plasma cell tumours. Histopathology. 2004;44:2-8.
180 Niitsu N., Kohri M., Hayama M., et al. Primary pulmonary plasmacytoma involving bilateral lungs and marked hypergammaglobulinemia: differentiation from extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue. Leuk Res. 2005;29(11):1361-1364.
181 Chetty R., Close P.M., Timme A.H., et al. Primary biphasic lymphoplasmacytic lymphoma of the lung. A mucosa associated lymphoid tissue lymphoma with compartmentalization of plasma cells in the lung and lymph node. Cancer. 1992;69:1124-1129.
182 Lin P., Medeiros L.J. Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia: an evolving concept. Adv Anat Pathol. 2005;12:246-255.
183 Kazzaz B., Dewar A., Corrin B. An unusual pulmonary plasmacytoma. Histopathology. 1992;21:285-287.
184 Randhawa P.S., Yousem S.A., Paradis I.L., et al. The clinical spectrum, pathology and clonal analysis of Epstein–Barr virus-associated lymphoproliferative disorders in heart–lung transplant recipients. Am J Clin Pathol. 1989;92:177-185.
185 Harris N.L., Ferry J.A., Swerdlow S.H. Post-transplant lymphoproliferative disorders: summary of Society for Hematopathology workshop. Semin Diagn Pathol. 1997;14:8-14.
186 Swerdlow S.H. Post-transplant lymphoproliferative disorders: a working classification. Curr Diagn Pathol. 1997;4:28-36.
187 Rizvi M.A., Evens A.M., Tallman M.S., et al. T cell non-Hodgkin lymphoma: a review. Blood. 2006;107:1255-1264.
188 Harris N.L., Jaffe E.S., Stein H., et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1999;84:1361-1392.
189 Jaffe E.S., Krenacs L., Kumar S., et al. Extranodal peripheral T-cell and NK-cell neoplasms. Am J Clin Pathol. 1999;111(suppl 1):S46-S55.
190 Amin H.M., Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110(7):2259-2267.
191 Sevilla D.W., Choi J.K., Gong J.Z. Mediastinal adenopathy, lung infiltrates, and hemophagocytosis: unusual manifestation of pediatric anaplastic large cell lymphoma: report of two cases. Am J Clin Pathol. 2007;127(3):458-464.
192 Rush W.L., Andriko J.A., Taubenberger J.K., et al. Primary anaplastic large cell lymphoma of the lung: a clinicopathologic study of five patients. Mod Pathol. 2000;13:1285-1292.
193 Medeiros L.J., Elenitoba-Johnson K.S. Anaplastic large cell lymphoma. Am J Clin Pathol. 2007;127(5):707-722.
194 Chott A., Kaserer K., Augustin I., et al. Ki-1-positive large cell lymphoma. A clinicopathologic study of 41 cases. Am J Surg Pathol. 1998;14:439-448.
195 d’Amore E.S., Menin A., Bonoldi E., et al. Anaplastic large cell lymphomas: a study of 75 pediatric patients. Pediatr Dev Pathol. 2007;10(3):181-191.
196 Campo E., Chott A., Kinney M.C., et al. Update on extranodal lymphomas. Conclusions of the Workshop held by the EAHP and the SH in Thessaloniki, Greece. Histopathology. 2006;48:481-504.
197 Stein H., Foss H.D., Drkop H., et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96(12):3681-3695.
198 Kesler M.V., Paranjape G.S., Asplund S.L., et al. Anaplastic large cell lymphoma: a flow cytometric analysis of 29 cases. Am J Clin Pathol. 2007;128(2):314-322.
199 Grewal J.S., Smith L.B., Winegarden J.D.3rd, et al. Highly aggressive ALK-positive anaplastic large cell lymphoma with a leukemic phase and multi-organ involvement: a report of three cases and a review of the literature. Ann Hematol. 2007;86(7):499-508.
200 Stachurski D., Miron P.M., Al-Homsi S., et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma with a complex karyotype and cryptic 3′ ALK gene insertion to chromosome 4 q22-24. Hum Pathol. 2007;38(6):940-945.
201 Coyne J.D., Burton I.E. Interstitial pneumonitis due to extramedullary haematopoiesis (EMH) in agnogenic myeloid metaplasia. Histopathology. 1999;34(3):275-276.
202 Koh T.T., Colby T.V., Muller N.L. Myeloid leukemias and lung involvement. Semin Respir Crit Care Med. 2005;26(5):514-519.
203 Moran C.A., Suster S. Unusual non-neoplastic lesions of the lung. Semin Diagn Pathol. 2007;24(3):199-208.
204 O’Malley D.P. Benign extramedullary myeloid tumors. Mod Pathol. 2007;20:405-415.
205 Koch C.A., Li C.Y., Mesa R.A., et al. Nonhepatosplenic extramedullary hematopoiesis: associated diseases, pathology, clinical course, and treatment. Mayo Clin Proc. 2003;78:1223-1233.
206 Ginsberg J.P., Orudjev E., Bunin N., et al. Isolated extramedullary relapse in acute myeloid leukemia: a retrospective analysis. Med Pediatr Oncol. 2002;38:387-390.
207 Campidelli C., Agostinelli C., Stitson R., Pileri S.A. Myeloid sarcoma: extramedullary manifestation of myeloid disorders. Am J Clin Pathol. 2009;132:426-437.
208 Nappi O., Boscaino A., Wick M.R. Extramedullary hematopoietic proliferations, extraosseous plasmacytomas, and ectopic splenic implants (splenosis). Semin Diagn Pathol. 2003;20:338-356.
209 Takasugi J.E., Godwin J.D., Marglin S.I., Petersdorf S.H. Intrathoracic granulocytic sarcomas. J Thorac Imaging. 1996;11:223-230.
210 Nieman R.S., Barcos M., Berard C. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:426-437.
211 Wong K.F., Chan J.K.C., Chan J.C., Lam S.Y. Acute myeloid leukemia presenting as granulocytic sarcoma of the lung. Am J Hematol. 1993;43:77-78.
212 Byrd J.C., Edenfield W.J., Dow N.S., et al. Extramedullary myeloid cell tumors in myelodysplastic syndromes: not a true indication of impending acute myeloid leukemia. Leuk Lymphoma. 1996;21:153-159.
213 Hicsonmez G., Cetin M., Yenicesu J., et al. Evaluation of children with myelodysplastic syndromes: importance of extramedullary disease as a presenting symptom. Leuk Lymphoma. 2000;42:665-674.
214 Sisack M.J., Dunsmore K., Sidhu-Malik N. Granulocytic sarcoma in the absence of myeloid leukemia. J Am Acad Dermatol. 1997;37:308-311.
215 Meis J.M., Butler J.J., Osborne B.M. Granulocytic sarcoma in non-leukemic patients. Cancer. 1986;58:2697-2709.
216 Goldstein N.S., Ritter J.H., Argenyi Z.B., et al. Granulocytic sarcoma: potential diagnostic clues from immunostaining patterns seen with anti-lymphoid antibodies. Int J Surg Pathol. 1995;2:199-206.
217 Quintanilla-Martinez L., Zukerberg L.R., Ferry J.A., et al. Extramedullary tumors of lymphoid or myeloid blasts. The role of immunohistology in diagnosis and classification. Am J Clin Pathol. 1995;104:431-443.
218 Roth M.J., Medeiros L.J., Elenitoba-Johnson K., et al. Extramedullary myeloid cell tumors. An immunohistochemical study of 29 cases using routinely fixed and processed paraffin-embedded tissue sections. Arch Pathol Lab Med. 1995;119:790-798.
219 Chen J., Yanuck R.R., Abbondanzo S.L., et al. c-Kit (CD117) reactivity in extramedullary myeloid tumor/granulocytic sarcoma. Arch Pathol Lab Med. 2001;125:1448-1452.