[level-membership-for-critical-care-medicine-category]
Objectives

Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at http://evolve.elsevier.com/Urden/priorities/.
Understanding the pathology of a disease, the areas of assessment on which to focus, and the usual medical management allows the critical care nurse to more accurately anticipate and plan nursing interventions. This chapter focuses on hematological and oncological disorders commonly seen in the critical care environment.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a syndrome that arises as a complication of other serious or life-threatening conditions. Although DIC is not seen often, it can seriously hamper diagnostic and treatment efforts for the critically ill patient. An understanding of the etiological and pathophysiological mechanisms of DIC can assist in anticipating the syndrome’s occurrence, recognizing its signs and symptoms, and prompting intervention. Also known as consumptive coagulopathy, DIC is characterized by bleeding and thrombosis, both of which result from depletion of clotting factors, platelets, and red blood cells (RBCs). If not treated quickly, DIC will progress to multiple organ failure and death.1
Etiology
Many clinical events can prompt the development of DIC in the critically ill patient, but the exact underlying trigger may not be identifiable (Box 27-1). There are, however, some commonly known conditions associated with the development of DIC.
The most common precipitating events for DIC are sepsis and trauma.1 In sepsis, endotoxins serve as a trigger for activation of tissue factor and the extrinsic coagulation pathway. Metabolic acidosis and hypoperfusion associated with shock syndromes can result in increased formation of free radicals and damage to tissues. Tissue factor is activated, resulting in DIC.2 Massive trauma or burns are frequently associated with DIC. Direct tissue damage activates the extrinsic coagulation pathway, and damage to endothelial surfaces activates the intrinsic pathway.2 Obstetric emergencies, such as abruptio placentae, retained placenta, or incomplete abortion, are also associated with the development of DIC. Tissue factor is concentrated in the placenta, and damage or disruption of this structure can activate coagulation pathways, resulting in coagulopathy.3
Pathophysiology
Regardless of the cause, the common thread in the development of DIC is damage to the endothelium, which results in activation of the coagulation mechanism (Figure 27-1).1 The extrinsic coagulation pathway plays a major role in the development of DIC. Direct damage to the endothelium results in the release of tissue factor and activation of this pathway. The secondary surge of thrombin formation as a result of activation of the intrinsic coagulation pathway leads to the massive disruption of the delicate balance that is hemostasis. Excessive thrombin formation results in rapid consumption of coagulation factors and depletion of regulatory substances—protein C, protein S, and antithrombin.4 With no checks and balances, thrombi continue to form along damaged epithelial walls, resulting in occlusion of the vessels. As occlusion reaches a critical level, tissue ischemia ensues, leading to further tissue damage and perpetuating the process. Eventually, end-organ function is affected by the ischemia, and failure is evident.1
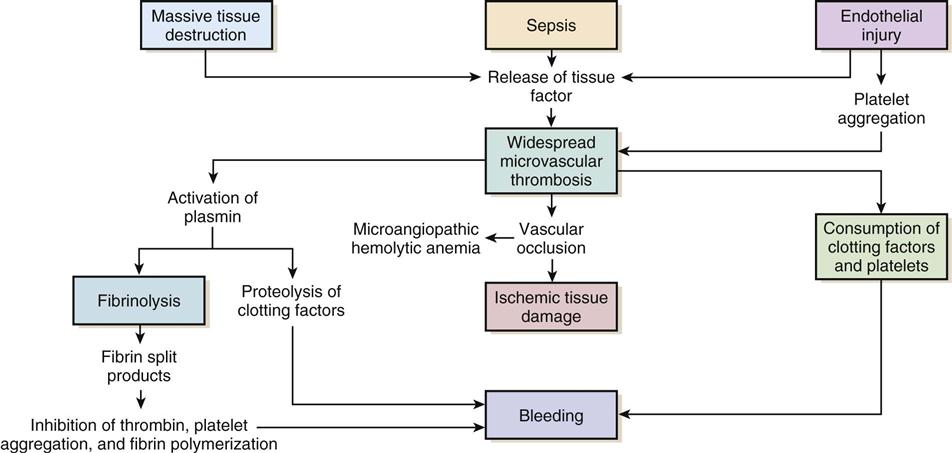
In response to the formation of clots, the fibrinolytic system is activated. As plasmin breaks down the fibrin clots, fibrin split products are released, and they act as anticoagulants.2,3 Coupled with depletion of circulating clotting factors, activation of fibrinolysis results in excessive bleeding. The end result is shock and further tissue ischemia that aggravate end-organ dysfunction and failure. Death is imminent if this destructive cycle is not interrupted.5
Assessment and Diagnosis
Favorable outcomes for patients with DIC depend on accurate and timely diagnosis of the condition. Realization of the role underlying pathology plays, recognition of clinical manifestations, and assessment of appropriate laboratory values are key steps in this process.
Clinical Manifestations
Clinical manifestations are related to the two primary pathophysiological mechanisms of DIC: the formation of thrombi and bleeding. Thrombi in peripheral capillaries can lead to cyanosis, particularly in the fingers, toes, ears, and nose. In severe, untreated cases, this peripheral ischemia may progress to gangrene.2,4–6 As the condition progresses, ischemia worsens, and end organs are affected. The result of this more central ischemia can be respiratory insufficiency and failure, acute tubular necrosis, bowel infarction, and ischemic stroke. The tissue damage that results perpetuates the anomalies of DIC.1
As coagulation factors are depleted, bleeding from intravenous and other puncture sites is observed. Ecchymoses may result from even routine interventions such as the use of a manual blood pressure cuff, bathing, or turning.2 Bloody drainage may also occur from surgical sites, drains, and urinary catheters. With progression of DIC, the patient is at risk for severe gastrointestinal or subarachnoid hemorrhage.2,6 Table 27-1 lists many of the common signs and symptoms of DIC.
TABLE 27-1
COMMON SIGNS AND SYMPTOMS OF DISSEMINATED INTRAVASCULAR COAGULATION
| SYSTEM | SIGNS RELATED TO HEMORRHAGE | SIGNS RELATED TO THROMBI |
| Integumentary | Bleeding from gums, venipunctures, and old surgical sites; epistaxis; ecchymoses | Peripheral cyanosis, gangrene |
| Cardiopulmonary | Hemoptysis | Dysrhythmias, chest pain, acute myocardial infarction, pulmonary embolus, respiratory failure |
| Renal | Hematuria | Oliguria, acute tubular necrosis, renal failure |
| Gastrointestinal | Abdominal distention, hemorrhage | Diarrhea, constipation, bowel infarct |
| Neurological | Subarachnoid hemorrhage | Altered level of consciousness, ischemic stroke |
Laboratory Findings
Laboratory tests used to diagnose DIC essentially assess the four basic characteristics of this syndrome: (1) increased coagulant activity, (2) increased fibrinolytic activity, (3) impaired regulatory function, and (4) end-organ failure.
Continuous activation of the coagulation pathways results in consumption of coagulation factors. Because of this, the prothrombin time (PT), the activated partial thromboplastin time (aPTT), and the international normalized ratio (INR) values are elevated. Although the platelet count may fall within normal ranges, serial examination reveals a declining trend in values. An unexpected drop of at least 50% in the platelet count, particularly in the presence of known contributing factors and associated signs and symptoms, strongly indicates DIC.7 Fibrinogen levels drop as more and more clots are formed. Thrombus formation in small vessels narrows the vessel lumen, forcing RBCs to squeeze through. The resulting damage and fragmentation of these cells can be seen on microscopic examination of blood samples. Damaged, fragmented RBCs are called schistocytes.2,6,8
In response to the excess clotting activity, the fibrinolytic process accelerates, and levels of by-products increase. This is reflected in markedly elevated levels of fibrin degradation products. Another key laboratory test used to evaluate the degree of clot dissolution—and therefore the severity of the coagulopathy—is the D-dimer level.7 D-dimers exclusively indicate clot degradation because, unlike fibrin degradation products, which also result from the breakdown of free circulating fibrin, D-dimers result only from dissolution of clots.2 With progression of the coagulopathy, normal regulatory mechanisms are disrupted, as reflected in decreasing levels of inhibitory factors such as protein C, factor V, and antithrombin III.2,6
Unchecked DIC resulting in occlusion of vessels and tissue ischemia leads to end-organ dysfunction.1 Respiratory failure, indicated by abnormal arterial blood gas (ABG) levels; liver failure, indicated by increasing liver enzymes; and renal impairment, indicated by rising blood urea nitrogen (BUN) and creatinine levels, are common findings in advanced DIC.5
No single laboratory study can confirm the diagnosis of DIC, but several key results are strong indicators of the condition (Table 27-2). The International Society of Thrombosis and Hemostasis emphasizes early detection of DIC through observation of abnormal trends in laboratory values.4
TABLE 27-2
KEY LABORATORY STUDIES IN DISSEMINATED INTRAVASCULAR COAGULATION
| TEST | VALUE |
| Prothrombin time (PT) | >12.5 sec |
| Platelets | <50,000/mm3, or at least 50% drop from baseline |
| Activated partial thromboplastin time (aPTT) | >40 sec |
| D-dimer | >250 ng/mL |
| Fibrin degradation products (FDP) | >40 mcg/mL |
| Fibrinogen | <100 mg/dL |
Medical Management
Without question, the primary intervention in DIC is prevention. Being aware of the conditions that commonly contribute to the development of DIC and treating them vigorously and without delay provide the best defense against this devastating condition.2,4,6,8 After DIC is identified, maintaining organ perfusion and slowing consumption of coagulation factors are paramount to achieving a favorable outcome.1,2
Multiple organ dysfunction syndrome (MODS) frequently results from DIC and exacerbates the underlying pathology. It is essential to prevent end-organ ischemia and damage by supporting blood pressure and circulating volume. Administration of intravenous fluids and inotropic agents, and, if overt hemorrhaging is evident, infusion of packed RBCs are appropriate interventions to replace blood volume and essential, oxygen-carrying RBCs.
In the presence of severe platelet depletion (<50,000/ mm3) and severe hemorrhage, platelet transfusions are often indicated.4,6 However, caution must be used when administering platelets because antiplatelet antibodies may be formed. These antibodies may become activated during future platelet transfusions and elicit DIC.2
Replacement of clotting factors in the patient with DIC is thought by some authorities to perpetuate the coagulopathy; however, there is little scientific evidence to support this theory.1 Fibrinogen levels less than 100 mg/dL indicate the appropriateness of administering cryoprecipitate. A prolonged PT indicates the need for fresh-frozen plasma.2,4,6
Slowing consumption of coagulation factors by inhibiting the processes involved in clot formation is another strategy used in treating DIC. The use of heparin, particularly low-molecular-weight heparin, to prevent formation of future clots is controversial.5 It is contraindicated in patients with DIC associated with recent surgery or with gastrointestinal or central nervous system (CNS) bleeding. However, heparin has been beneficial in obstetric emergencies such as retained placenta or incomplete abortion, severe arterial occlusions, or MODS caused by microemboli.2,6
The use of recombinant activated protein C is gaining popularity in treating DIC, especially in the setting of severe sepsis. Activated protein C acts as an anticoagulant and works to restore normal inhibition of coagulation pathways. However, it has been associated with an increased incidence of intracerebral bleeding and must be used with caution in patients with severely decreased platelets.2,4
Thrombin production in DIC surpasses that of antithrombins and other regulatory factors that would normally be present to inactivate thrombin and its subsequent actions. The use of antithrombin III has recently been approved in the United States. Ongoing research is yielding mixed results in the treatment of DIC.4 One interesting area of research is the use of protease inhibitors. Protease molecules normally inhibit the conversion of fibrinogen to fibrin in the coagulation mechanism, but in DIC, this inhibitory mechanism is impaired. The introduction of protease inhibitors by intravenous infusion may be advantageous in arresting DIC.2
Nursing Management
Nursing management of the patient with DIC incorporates a variety of nursing diagnoses (see the Nursing Diagnosis Priorities Box on Disseminated Intravascular Coagulation). Assessment and monitoring are the primary weapons in the critical care nurse’s arsenal against DIC. Knowing the diseases and conditions that are most often associated with DIC and understanding the pathophysiological mechanisms involved enables the critical care nurse to anticipate its development and intervene quickly. Nursing priorities are directed toward (1) supporting the patient’s vital functions, (2) initiating bleeding precautions, (3) providing comfort and emotional support, and (4) maintaining surveillance for complications.
Supporting Patient’s Vital Functions
Frequent assessments should include parameters for neurological status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.6
The critical care nurse must recognize and support the patient’s vital physiological functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.
Initiating Bleeding Precautions
Awareness of the patient’s bleeding potential necessitates adjustments to normal nursing interventions. The nurse avoids unnecessary venipunctures that may result in bleeding, bruising, or hematomas by drawing blood from and administering medications through existing arterial or venous lines. The use of manual or automatic blood pressure cuffs is avoided whenever possible. If tracheal or oral suctioning is necessary, the use of low-level suction is recommended.6 Meticulous skin care is advised, keeping the skin moist and using specialty mattresses and beds as appropriate to prevent breakdown. Gentle care should be used when bathing or turning the patient to prevent bruising or hematoma formation.
Providing Emotional Support
The development of DIC in the already critically ill patient can be stressful for the patient and his or her significant others. It is imperative to provide psychosocial support throughout this crisis. Calm reassurance and uncomplicated explanations of the care the patient is receiving can help to allay much of the anxiety experienced. The critical care nurse must answer all questions and provide information in terms best understood by all parties. The use of an interpreter when English is not the primary language can enhance understanding and help avoid misconceptions. Providing spiritual support as requested may also be of assistance.
Please see the Collaborative Management Box on Disseminated Intravascular Coagulation for more information.
Heparin-Induced Thrombocytopenia
One form of thrombocytopenia seen in critical care patients is heparin-induced thrombocytopenia (HIT). There are two distinct types of HIT. The most common form is non-immune-mediated HIT, formally known as type 1 HIT.9 Seen in up to 30% of patients receiving heparin therapy, this nonautoimmune condition manifests within a few days of initiation of therapy. Platelet depletion is moderate, counts are usually less than 100,000/mm3, and the condition is transient, often resolving spontaneously. Discontinuation of heparin is not required. The second form is immune-mediated HIT,9 formally known as type 2 HIT, which is less commonly encountered but has more severe consequences.10–12 This discussion is limited to immune-mediated HIT.
Etiology
Immune-mediated HIT is a response to the administration of heparin therapy. It has been observed in 1% to 3% of patients treated with unfractionated heparin and has occurred after exposure to low-molecular-weight heparin (LMWH), although to a lesser degree.9 The disorder is characterized by severe thrombocytopenia during heparin therapy. Diagnostically it is identified by a platelet count less than 50,000/mm3 or at least a 50% decrease from the baseline platelet count from the initiation of therapy. Onset usually occurs 5 to 14 days from the first exposure to heparin, but the onset can occur within hours of a reexposure to heparin.11–13 Depending on the source of the disorder, reported mortality rates are as high as 30%.9
Pathophysiology
The thrombocytopenia that occurs with immune-mediated HIT is related to the formation of heparin-antibody complexes. These complexes release a substance known as platelet factor 4 (PF4). PF4 attracts heparin molecules, forming immunogenic complexes that adhere to platelet and endothelial surfaces (Figure 27-2). Activation of platelets stimulates the release of thrombin and the subsequent formation of platelet clumps.12
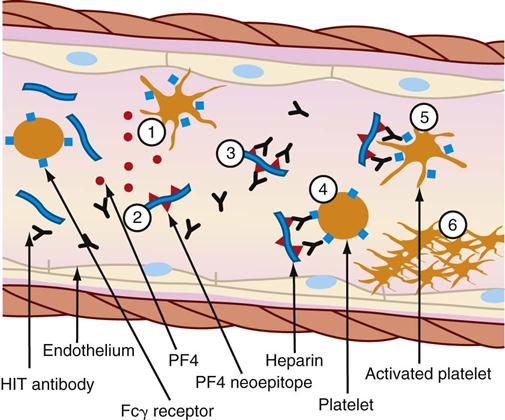
(1) Activated platelets release procoagulant proteins from α-granules, including platelet factor 4 (PF4). Administered heparin binds PF4 (2), which undergoes a conformation change and expresses a new antigen (neoepitope). Individuals with HIT produce an immunoglobulin G (IgG) antibody that specifically reacts (3) with multiple identical neoepitopes on the heparin-PF4 complex. The reaction forms heparin-PF4-IgG immune complexes. Platelets express FcγRIIa receptors (Fcγ receptor) that react (4) with the Fc portion of IgG in immune complexes. Cross-linking of Fc receptors (5) results in FcγRlIa-dependent platelet activation. The activated platelets mediate a series of events that lead to further activation of the coagulation cascade, resulting in thrombin generation. Further release of PF4 from newly activated platelets leads to a cycle of continuing platelet activation and (6) formation of a primary clot. The reaction can be enhanced by the release of platelet-derived microparticles that are rich in surface phosphatidylserine and increase activation of coagulation and by the binding of heparin-PF4 complexes and HIT-IgG to the vascular endothelium (not shown.) (From McCance KL, Huether SE, Brashers VL, Rote NS: Pathophysiology: The biologic basis for disease in adults and children, ed 6, St Louis, 2010, Mosby.)
Patients with immune-mediated HIT are at greater risk for thrombosis than bleeding. Vessel occlusion can result in the need for limb amputation, stroke, acute myocardial infarction, and even death.10–12 The resultant formation of fibrin-platelet-rich thrombi is the primary characteristic of HIT that distinguishes it from other forms of thrombocytopenia and gives rise to its more descriptive name: white clot syndrome.10
Assessment and Diagnosis
HIT can be associated with severe consequences. Rapid recognition of risk factors and subsequent development of signs and symptoms is essential in treating this condition.
Clinical Manifestations
Common signs and symptoms are listed in Table 27-3. The clinical manifestations of HIT are related to the formation of thrombi and subsequent vessel occlusion.9 Most thrombotic events are venous, although venous and arterial thrombosis can occur. Thrombotic events typically include deep vein thrombosis, pulmonary embolism, limb ischemia thrombosis, thrombotic stroke, and myocardial infarction.9 The presence of blanching and the loss of peripheral pulses, sensation, or motor function in a limb indicate peripheral vascular thrombi. Neurological signs and symptoms such as confusion, headache, and impaired speech can signal the onset of cerebral artery occlusion and stroke. Acute myocardial infarction may be heralded by dyspnea, chest pain, pallor, and alterations in blood pressure. Thrombi in the pulmonary vasculature may be evidenced by pleuritic pain, rales, and dyspnea.10,13
TABLE 27-3
HEPARIN-INDUCED THROMBOCYTOPENIA: SIGNS, SYMPTOMS, AND LABORATORY DATA
| SYSTEM OR STUDY | SIGNS AND SYMPTOMS |
| Cardiac | Chest pain, diaphoresis, pallor, alterations in blood pressure, dysrhythmias |
| Vascular | Arterial: pain, pallor, pulselessness, paresthesia, paralysis |
| Venous: pain, tenderness, unilateral leg swelling, warmth, erythema, a palpable cord, pain on passive dorsiflexion of the foot, and spontaneous maintenance of the relaxed foot in abnormal plantar flexion (Homans’ sign) | |
| Pulmonary | Dyspnea, pleuritic pain, rales, chest pain, chest wall tenderness, back pain, shoulder pain, upper abdominal pain, syncope, hemoptysis, shortness of breath, wheezing |
| Renal | Thirst, decreased urine output, dizziness, orthostatic hypotension |
| Gastrointestinal | Abdominal pain, vomiting, bloody diarrhea, abnormal bowel sounds |
| Neurological | Confusion, headache, impaired speech patterns, hemiparesis or hemiplegia, vision disturbances, dysarthria, aphasia, ataxia, vertigo, nystagmus, sudden decrease in consciousness |
| Laboratory | Platelets <50,000/mm3 or sudden drop of 30% to 50% from baseline; positive results for HIPA, SRA, ELISA |
Laboratory Findings
The key indicator for identifying HIT is the platelet count. General consensus in the literature considers a platelet count of less than 100,000/mm3 or a sudden drop of 50% from the patient’s baseline after initiation of heparin therapy to strongly indicate HIT.10–13
Two types of assays have become available to assist in confirming the diagnosis of HIT: activation assays, based on platelet aggregation or the release of granular contents such as serotonin, and assays that identify the HIT antigen. Activation assays are highly sensitive in detecting the presence of HIT. The most common assay used is heparin-induced platelet aggregation (HIPA). Serotonin release assay (SRA) is used by a few institutions. The enzyme-linked immunosorbent assay (ELISA) identifies the presence of the HIT antigen.11,12
Medical Management
Evidenced-based guidelines for the prevention and management of the patient with HIT are listed in the Evidence-Based Collaborative Practice Box on Treatment and Prevention of Heparin-Induced Thrombocytopenia.14 When a decrease in the platelet count is detected, heparin therapy should be discontinued immediately, and the patient should be tested for the presence of heparin antibodies.11–14 If the original indication for heparin still exists or new thromboses occur, an alternative form of anticoagulation is usually necessary.14
Direct thrombin inhibitors (DTIs) are being used with increasing frequency to treat HIT. DTIs bind directly to the thrombin molecule, thereby inhibiting its action.14 Two such drugs are lepirudin and argatroban.9 Warfarin, although commonly used to treat deep vein thrombosis, is not indicated as a sole agent in treating HIT because of its prolonged onset of action. Studies have shown that the use of warfarin without concomitant use of DTIs can significantly increase the incidence of thrombosis in patients with HIT.9 Comparative information on these medications is provided in Table 27-4.9,14
TABLE 27-4
PHARMACOLOGICAL MANAGEMENT: HEPARIN-INDUCED THROMBOCYTOPENIA
| DRUG | DOSAGE | ACTIONS | SPECIAL CONSIDERATIONS |
| Lepirudin (Refludan) | Loading dose: 0.4 mg/kg IV bolus IV infusion: 0.15 mg/kg/hr |
Used to inhibit free and clot-bound thrombin; a recombinant form of leech-derived hirudin | Monitor aPTT; maintain INR 1.5-2.5 times normal. |
| Reduce dosage in patients with known or suspected renal insufficiency. | |||
| Side effects include bleeding. | |||
| Can develop antilepirudin antibodies that enhance anticoagulant effect. | |||
| Argatroban | Loading dose: None IV infusion: 2 mcg/kg/min not to exceed 10 mcg/kg/min |
Used to inhibit thrombin | Obtain baseline aPTT 2 hr after therapy started. |
| Monitor aPTT; maintain INR 1.5-3.0 times initial baseline. | |||
| Reduce dosage in patients with known or suspected hepatic impairment. | |||
| Bivalirudin | Loading dose: 0.75 mg/kg IV bolus IV infusion: 1.75 mg/kg/hr |
Used to inhibit thrombin | Monitor aPTT; maintain INR 1.5-2.5 times initial baseline. |
| Adjust dose in presence of renal failure. |

aPTT, activated partial thromboplastin time; INR, international normalized ratio; IV, intravenous.
Nursing Management
Nursing management of the patient with HIT incorporates a variety of nursing diagnoses (see the Nursing Diagnosis Priorities Box on Heparin-Induced Thrombocytopenia). Nursing priorities are directed toward (1) decreasing the incidence of heparin exposure, (2) maintaining surveillance for complications, (3) providing comfort and emotional support, and (4) initiating patient education. The critical care nurse plays a pivotal role in prevention and detection of heparin-induced thrombocytopenia. Initial assessment is crucial to identifying those patients at risk for HIT. Ascertaining a medical history that includes previous heparin therapy, deep vein thrombosis, or cardiovascular surgery that included the use of cardiopulmonary bypass can alert the nurse to potential problems.
Decreasing the Incidence of Heparin Exposure
Ensuring that all heparin has been removed from the patient’s hemodynamic pressure monitoring system, avoiding the use of heparin-coated catheters, and discontinuing heparin flushes to maintain the patency of other intravenous lines are essential elements of nursing management.
Maintaining Surveillance for Complications
Patients with HIT remain at high risk for thrombotic complications for several days or weeks after cessation of heparin. Vigilant monitoring, early recognition of signs and symptoms, deep vein thrombosis prevention strategies, and prompt notification of the physician are key roles of the critical care nurse.
Initiating Patient Education
Prevention of subsequent episodes in patients sensitized to heparin includes education of the patient and family (see the Patient Education Box on Heparin-Induced Thrombocytopenia). Education should include measures to avoid future exposure to heparin. The use of medical alert bracelets and listing heparin allergies in the medical record are necessary to avoid this serious complication in the future.
Please see the Collaborative Management Box on Heparin-Induced Thrombocytopenia for more information.
Tumor Lysis Syndrome
Tumor lysis syndrome (TLS) refers to a variety of metabolic disturbances that may be seen with the treatment of cancer. A potentially lethal complication of various forms of cancer treatment, TLS occurs when large numbers of neoplastic cells are rapidly killed, resulting in the release of large amounts of potassium, phosphate, and uric acid into the systemic circulation. It occurs in 5% to 20%15 of cancer patients and is most commonly seen in patients with lymphoma, leukemia, or multiple metastatic conditions.15–17
Etiology
Although most often associated with the use of chemotherapeutic drugs, biological agents, and irradiation used in the treatment of malignant disorders, TLS can in rare instances occur spontaneously. The development of TLS has been linked to other pathophysiological conditions such as elevated WBC counts, large tumors, multiple organ involvement by malignancy, and renal insufficiency.18
Pathophysiology
The primary mechanism involved in the development of TLS is the destruction of massive numbers of malignant cells by chemotherapy or radiation therapy (Figure 27-3). Massive destruction of cells releases large amounts of potassium, phosphorus, and nucleic acids, leading to severe metabolic disturbances, such as hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia (Table 27-5). Vomiting, diarrhea, and other insensible fluid losses from fever or tachypnea also contribute to these electrolyte disturbances.17 Death of patients with TLS is most often caused by complications of renal failure or cardiac arrest.18
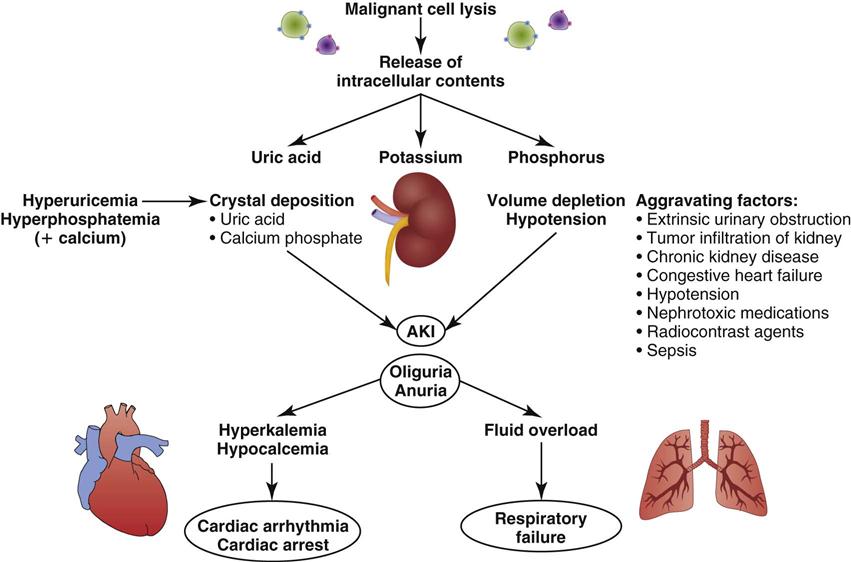
AKI, acute kidney injury. (From Abu-Alfa AK, Younes A: Tumor lysis syndrome and acute kidney injury: evaluation, prevention and management, Am J Kidney Dis 55[5 Suppl 3]:S1, 2010.)
TABLE 27-5
ELECTROLYTE ABNORMALITIES ENCOUNTERED IN TUMOR LYSIS SYNDROME AND THEIR CLINICAL CONSEQUENCES
| ELECTROLYTE | PATHOPHYSIOLOGY | CLINICAL CONSEQUENCE | TREATMENT OPTIONS |
| Potassium | Rapid expulsion of intracellular K+ into the circulation due to cell lysis | Adverse skeletal and cardiac manifestations (e.g., ventricular dysrhythmias, weakness, paresthesias) | Insulin/glucose, sodium bicarbonate, inhaled beta-agonist, K+-binding resins, dialysis, calcium gluconate |
| Phosphate | Release of intracellular PO4− due to cell lysis | Muscle cramps, tetany, dysrhythmias, seizures | Dialysis, phosphate binders |
| May be compounded by renal dysfunction | |||
| Calcium | Precipitation of the calcium phosphate complex because of the rapid increase in the phosphorous concentration | Muscle cramps, tetany, dysrhythmias, seizures, renal failure (acute nephrocalcinosis) | Calcium gluconate (treatment should be reserved for those with neuromuscular irritability) |
| Uric acid | Cell lysis leads to increased levels of purine nucleic acids into the circulation that are metabolized to uric acid | Renal failure (uric acid nephropathy) | Hydration, dialysis, xanthine oxidase inhibitors, alkalization of urine, urate oxidase |
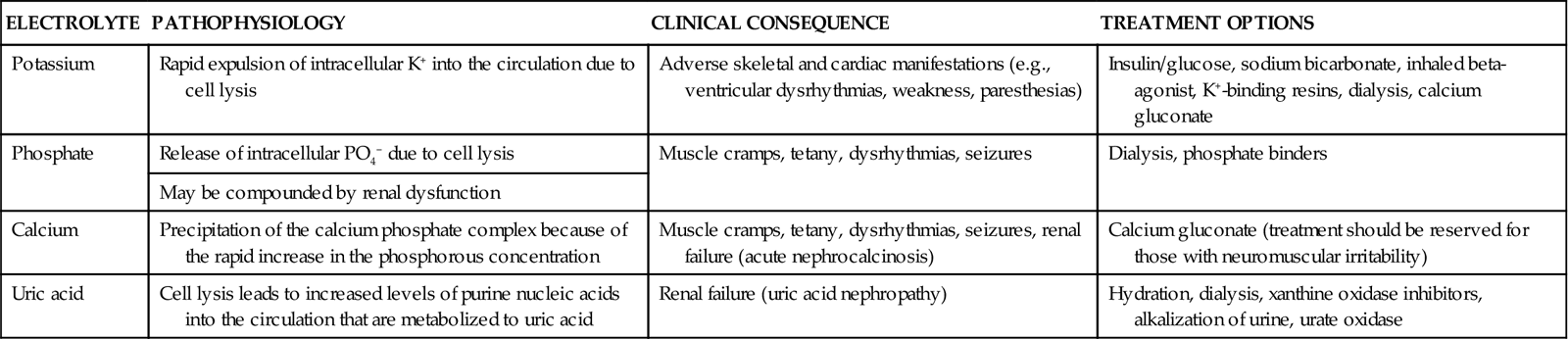
From Davidson MB, et al: Pathophysiology, clinical consequences, and treatment of tumor lysis syndrome, Am J Med 116(8):546, 2004.
Hyperuricemia
Hyperuricemia occurs 48 to 72 hours after the initiation of anticancer therapy.17 Tumor cells undergo rapid growth and development, and large amounts of nucleic acids are present within them. When therapy is initiated, tumor cell destruction releases nucleic acids, which are metabolized into uric acid. Metabolic acidosis ensues, resulting in crystallization of the uric acid in the distal tubules of the kidney and leading to obstruction of urine flow. Glomerular filtration rates drop as the kidneys are unable to clear the increasing amounts of uric acid. Consequently, renal insufficiency and acute renal failure eventually occur.19 Acute renal failure is discussed further in Chapter 20.
Hyperuricemia associated with TLS can be potentiated by several other factors, including elevated uric acid levels before the initiation of therapy. Other causes of increased uric acid production are elevated WBC counts, destruction of WBCs, and enlargement of the lymph nodes, spleen, or liver.17
Hyperkalemia
Hyperkalemia occurs within 6 to 72 hours after the initiation of chemotherapy. This is the most deleterious of all the manifestations of TLS.17 In addition to the release of nucleic acids, tumor cell destruction also results in the release of potassium. Renal insufficiency related to hyperuricemia prevents adequate excretion of potassium, and levels rise. The resultant hyperkalemia may have a profound effect on intracellular and extracellular fluid levels.18 Left untreated, hyperkalemia can have devastating consequences, including cardiac arrest and death.17
Hyperphosphatemia and Hypocalcemia
Hyperphosphatemia and hypocalcemia occur 24 to 48 hours after the initiation of therapy.17 Phosphorus levels also rise as a consequence of tumor cell destruction. Calcium ions then bind with the excess phosphorus, creating calcium phosphate salts and bringing about hypocalcemia. These salts precipitate in the kidney tubules, worsening renal insufficiency. Hypocalcemia causes tetany and cardiac dysrhythmias, which can result in cardiac arrest and death.17,18
Assessment and Diagnosis
Detection and recognition of TLS is accomplished through assessment of clinical manifestations, evaluation of laboratory findings, and other diagnostic tests. Table 27-6 summarizes common findings in TLS.17–19
TABLE 27-6
COMMON FINDINGS IN TUMOR LYSIS SYNDROME
| DIAGNOSTIC PARAMETER | FINDINGS |
| Clinical | Weight gain, edema, diarrhea, lethargy, muscle cramps, nausea and vomiting, paresthesia, weakness, oliguria, uremia, seizures |
| Laboratory | ↑Potassium, phosphorus, uric acid, BUN, Cr |
| ↓Calcium, creatinine clearance, pH, bicarbonate, PaCO2 | |
| Diagnostic | Positive Chvostek’s and Trousseau’s signs, hyperactive deep tendon reflexes, dysrhythmias, ECG changes |

Clinical Manifestations
Clinical manifestations are related to the metabolic disturbances associated with TLS.16 The patient’s history reveals an unexplained weight gain after initiation of chemotherapy or radiation therapy. The weight gain is associated with fluid retention due to electrolyte disturbances. Other early signs heralding the onset of TLS include diarrhea, lethargy, muscle cramps, nausea, vomiting, paresthesia, and weakness.20
Laboratory Findings
Laboratory findings demonstrate electrolyte disturbances such as elevated potassium and phosphorus levels and a decreased calcium level. Uric acid levels are increased. Elevated levels of BUN and creatinine and a decreased creatinine clearance also indicate TLS. Metabolic acidosis is confirmed by the presence of decreased pH, bicarbonate levels, and partial pressure of carbon dioxide (PaCO2) on arterial blood gas measurements.20
Other Diagnostic Tests
Physical examination reveals positive Chvostek’s and Trousseau’s signs related to hypocalcemia. Hyperactive deep tendon reflexes indicate hyperkalemia and hypocalcemia. Potassium and calcium disturbances result in changes that can be seen on the electrocardiogram (ECG), such as peaked or inverted T waves, altered QT intervals, widened QRS complexes, and dysrhythmias.17
Medical Management
Medical interventions are aimed at maintaining adequate hydration, treating metabolic imbalances, and preventing life-threatening complications.15–17 Administration of intravenous fluids may be necessary early in the course of treatment if inadequate hydration exists. The administration of isotonic saline (0.9% normal saline) reduces serum concentrations of uric acid, phosphate, and potassium.16,17 The use of nonthiazide diuretics to maintain adequate urine output may be required. If renal failure occurs, hemodialysis should be considered.16–18
Electrolytes and arterial blood gases are closely monitored. Dietary restrictions of potassium and phosphorus may be necessary. The goals in treating hyperuricemia are to inhibit uric acid formation and to increase renal clearance.17 This can be accomplished through the administration of sodium bicarbonate to increase the pH of the urine to above 7.0, which increases the solubility of uric acid, preventing subsequent crystallization. Allopurinol administration can also inhibit uric acid formation (see the Priority Medications Box on Allopurinol).16
 Priority Medications
Priority Medications
Allopurinol
Drug Class: Antiinflammatory agent
Drug Action: Allopurinol decreases the formation of uric acid by competitively inhibiting xanthine oxidase and thereby blocking the metabolism of hypoxanthine and xanthine to uric acid.
Drug Delivery and Drug Dosage: For the treatment of tumor lysis syndrome, allopurinol may be given orally or intravenously, though the intravenous route is generally reserved for those patient who cannot tolerate oral administration. The oral dose is 600-800 mg/day in 2-3 divided doses. The intravenous dose is 200-400 mg/m2/day as a single dose or in 2-4 equally divided doses. The maximum dose is 600 mg/day.
Priority Nursing Considerations: Patients receiving allopurinol should have their serum uric acid concentrations closely monitored. When given orally, allopurinol should be administered after meals and with plenty of fluids (unless contraindicated). To prevent kidney stones, the patient’s urine output should be maintained at least at 2 L a day.
Clinical Examples: Allopurinol is used for the prevention of acute hyperuricemia during the treatment of leukemia, lymphoma, or solid tumors that may cause tumor lysis syndrome. Allopurinol is also used for the treatment of gout.
If potassium levels rise dangerously, Kayexalate (sodium polystyrene sulfonate) may be given orally, or if the patient is unable to tolerate oral medications due to nausea and vomiting, rectal instillation may be used. If the patient is oliguric, glucose and insulin infusions may be given to facilitate lowering the potassium levels. A 10% solution of calcium gluconate may be administered to stabilize cardiac tissue membranes to prevent life-threatening dysrhythmias.21 Phosphorus-binding antacids can be used for treating hyperphosphatemia. Stool softeners may be necessary to treat the constipation often associated with the administration of these antacids. Calcium gluconate may be required to replace calcium, but it should be used judiciously.17
Nursing Management
Nursing management of the patient with TLS incorporates a variety of nursing diagnoses (see the Nursing Diagnosis Priorities Box on Tumor Lysis Syndrome). Nursing priorities are directed toward (1) monitoring fluid and electrolytes, (2) providing comfort and emotional support, (3) maintaining surveillance for complications, and (4) initiating patient education.
Monitoring Fluid and Electrolytes
Assessment and continued monitoring of the patient is an important role of the critical care nurse when caring for the patient with TLS. Recognizing critical laboratory changes or development of symptoms and notifying the physician in a timely manner are essential.21 Insertion of a urinary catheter and maintenance of the intravenous line site are necessary to ensure adequate intake and output. Vital signs should be monitored frequently, and weight should be monitored daily.20
Maintaining Surveillance for Complications
Nursing interventions are aimed at preventing complications. Seizure precautions should be instituted, especially if calcium levels are disrupted. Insertion of a nasogastric tube is appropriate if nausea or vomiting occurs. Dietary adjustments are necessary, such as potassium and phosphorus restrictions in the presence of elevated serum levels and providing additional fiber to combat the constipation associated with the administration of antacids.
Initiating Patient Education
Education of the patient and family is a primary role of the critical care nurse. All treatments and interventions should be explained before carrying them out, and questions should be answered at a level understandable to the patient and family. Before discharge, potential risk factors and identification of early signs and symptoms should be reviewed.20
See the Collaborative Management Box on Tumor Lysis Syndrome for more information.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
Objectives
Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at http://evolve.elsevier.com/Urden/priorities/.
Understanding the pathology of a disease, the areas of assessment on which to focus, and the usual medical management allows the critical care nurse to more accurately anticipate and plan nursing interventions. This chapter focuses on hematological and oncological disorders commonly seen in the critical care environment.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a syndrome that arises as a complication of other serious or life-threatening conditions. Although DIC is not seen often, it can seriously hamper diagnostic and treatment efforts for the critically ill patient. An understanding of the etiological and pathophysiological mechanisms of DIC can assist in anticipating the syndrome’s occurrence, recognizing its signs and symptoms, and prompting intervention. Also known as consumptive coagulopathy, DIC is characterized by bleeding and thrombosis, both of which result from depletion of clotting factors, platelets, and red blood cells (RBCs). If not treated quickly, DIC will progress to multiple organ failure and death.1
Etiology
Many clinical events can prompt the development of DIC in the critically ill patient, but the exact underlying trigger may not be identifiable (Box 27-1). There are, however, some commonly known conditions associated with the development of DIC.
The most common precipitating events for DIC are sepsis and trauma.1 In sepsis, endotoxins serve as a trigger for activation of tissue factor and the extrinsic coagulation pathway. Metabolic acidosis and hypoperfusion associated with shock syndromes can result in increased formation of free radicals and damage to tissues. Tissue factor is activated, resulting in DIC.2 Massive trauma or burns are frequently associated with DIC. Direct tissue damage activates the extrinsic coagulation pathway, and damage to endothelial surfaces activates the intrinsic pathway.2 Obstetric emergencies, such as abruptio placentae, retained placenta, or incomplete abortion, are also associated with the development of DIC. Tissue factor is concentrated in the placenta, and damage or disruption of this structure can activate coagulation pathways, resulting in coagulopathy.3
Pathophysiology
Regardless of the cause, the common thread in the development of DIC is damage to the endothelium, which results in activation of the coagulation mechanism (Figure 27-1).1 The extrinsic coagulation pathway plays a major role in the development of DIC. Direct damage to the endothelium results in the release of tissue factor and activation of this pathway. The secondary surge of thrombin formation as a result of activation of the intrinsic coagulation pathway leads to the massive disruption of the delicate balance that is hemostasis. Excessive thrombin formation results in rapid consumption of coagulation factors and depletion of regulatory substances—protein C, protein S, and antithrombin.4 With no checks and balances, thrombi continue to form along damaged epithelial walls, resulting in occlusion of the vessels. As occlusion reaches a critical level, tissue ischemia ensues, leading to further tissue damage and perpetuating the process. Eventually, end-organ function is affected by the ischemia, and failure is evident.1
In response to the formation of clots, the fibrinolytic system is activated. As plasmin breaks down the fibrin clots, fibrin split products are released, and they act as anticoagulants.2,3 Coupled with depletion of circulating clotting factors, activation of fibrinolysis results in excessive bleeding. The end result is shock and further tissue ischemia that aggravate end-organ dysfunction and failure. Death is imminent if this destructive cycle is not interrupted.5
Assessment and Diagnosis
Favorable outcomes for patients with DIC depend on accurate and timely diagnosis of the condition. Realization of the role underlying pathology plays, recognition of clinical manifestations, and assessment of appropriate laboratory values are key steps in this process.
Clinical Manifestations
Clinical manifestations are related to the two primary pathophysiological mechanisms of DIC: the formation of thrombi and bleeding. Thrombi in peripheral capillaries can lead to cyanosis, particularly in the fingers, toes, ears, and nose. In severe, untreated cases, this peripheral ischemia may progress to gangrene.2,4–6 As the condition progresses, ischemia worsens, and end organs are affected. The result of this more central ischemia can be respiratory insufficiency and failure, acute tubular necrosis, bowel infarction, and ischemic stroke. The tissue damage that results perpetuates the anomalies of DIC.1
As coagulation factors are depleted, bleeding from intravenous and other puncture sites is observed. Ecchymoses may result from even routine interventions such as the use of a manual blood pressure cuff, bathing, or turning.2 Bloody drainage may also occur from surgical sites, drains, and urinary catheters. With progression of DIC, the patient is at risk for severe gastrointestinal or subarachnoid hemorrhage.2,6 Table 27-1 lists many of the common signs and symptoms of DIC.
TABLE 27-1
COMMON SIGNS AND SYMPTOMS OF DISSEMINATED INTRAVASCULAR COAGULATION
| SYSTEM | SIGNS RELATED TO HEMORRHAGE | SIGNS RELATED TO THROMBI |
| Integumentary | Bleeding from gums, venipunctures, and old surgical sites; epistaxis; ecchymoses | Peripheral cyanosis, gangrene |
| Cardiopulmonary | Hemoptysis | Dysrhythmias, chest pain, acute myocardial infarction, pulmonary embolus, respiratory failure |
| Renal | Hematuria | Oliguria, acute tubular necrosis, renal failure |
| Gastrointestinal | Abdominal distention, hemorrhage | Diarrhea, constipation, bowel infarct |
| Neurological | Subarachnoid hemorrhage | Altered level of consciousness, ischemic stroke |
Laboratory Findings
Laboratory tests used to diagnose DIC essentially assess the four basic characteristics of this syndrome: (1) increased coagulant activity, (2) increased fibrinolytic activity, (3) impaired regulatory function, and (4) end-organ failure.
Continuous activation of the coagulation pathways results in consumption of coagulation factors. Because of this, the prothrombin time (PT), the activated partial thromboplastin time (aPTT), and the international normalized ratio (INR) values are elevated. Although the platelet count may fall within normal ranges, serial examination reveals a declining trend in values. An unexpected drop of at least 50% in the platelet count, particularly in the presence of known contributing factors and associated signs and symptoms, strongly indicates DIC.7 Fibrinogen levels drop as more and more clots are formed. Thrombus formation in small vessels narrows the vessel lumen, forcing RBCs to squeeze through. The resulting damage and fragmentation of these cells can be seen on microscopic examination of blood samples. Damaged, fragmented RBCs are called schistocytes.2,6,8
In response to the excess clotting activity, the fibrinolytic process accelerates, and levels of by-products increase. This is reflected in markedly elevated levels of fibrin degradation products. Another key laboratory test used to evaluate the degree of clot dissolution—and therefore the severity of the coagulopathy—is the D-dimer level.7 D-dimers exclusively indicate clot degradation because, unlike fibrin degradation products, which also result from the breakdown of free circulating fibrin, D-dimers result only from dissolution of clots.2 With progression of the coagulopathy, normal regulatory mechanisms are disrupted, as reflected in decreasing levels of inhibitory factors such as protein C, factor V, and antithrombin III.2,6
Unchecked DIC resulting in occlusion of vessels and tissue ischemia leads to end-organ dysfunction.1 Respiratory failure, indicated by abnormal arterial blood gas (ABG) levels; liver failure, indicated by increasing liver enzymes; and renal impairment, indicated by rising blood urea nitrogen (BUN) and creatinine levels, are common findings in advanced DIC.5
No single laboratory study can confirm the diagnosis of DIC, but several key results are strong indicators of the condition (Table 27-2). The International Society of Thrombosis and Hemostasis emphasizes early detection of DIC through observation of abnormal trends in laboratory values.4
TABLE 27-2
KEY LABORATORY STUDIES IN DISSEMINATED INTRAVASCULAR COAGULATION
| TEST | VALUE |
| Prothrombin time (PT) | >12.5 sec |
| Platelets | <50,000/mm3, or at least 50% drop from baseline |
| Activated partial thromboplastin time (aPTT) | >40 sec |
| D-dimer | >250 ng/mL |
| Fibrin degradation products (FDP) | >40 mcg/mL |
| Fibrinogen | <100 mg/dL |
Medical Management
Without question, the primary intervention in DIC is prevention. Being aware of the conditions that commonly contribute to the development of DIC and treating them vigorously and without delay provide the best defense against this devastating condition.2,4,6,8 After DIC is identified, maintaining organ perfusion and slowing consumption of coagulation factors are paramount to achieving a favorable outcome.1,2
Multiple organ dysfunction syndrome (MODS) frequently results from DIC and exacerbates the underlying pathology. It is essential to prevent end-organ ischemia and damage by supporting blood pressure and circulating volume. Administration of intravenous fluids and inotropic agents, and, if overt hemorrhaging is evident, infusion of packed RBCs are appropriate interventions to replace blood volume and essential, oxygen-carrying RBCs.
In the presence of severe platelet depletion (<50,000/ mm3) and severe hemorrhage, platelet transfusions are often indicated.4,6 However, caution must be used when administering platelets because antiplatelet antibodies may be formed. These antibodies may become activated during future platelet transfusions and elicit DIC.2
Replacement of clotting factors in the patient with DIC is thought by some authorities to perpetuate the coagulopathy; however, there is little scientific evidence to support this theory.1 Fibrinogen levels less than 100 mg/dL indicate the appropriateness of administering cryoprecipitate. A prolonged PT indicates the need for fresh-frozen plasma.2,4,6
Slowing consumption of coagulation factors by inhibiting the processes involved in clot formation is another strategy used in treating DIC. The use of heparin, particularly low-molecular-weight heparin, to prevent formation of future clots is controversial.5 It is contraindicated in patients with DIC associated with recent surgery or with gastrointestinal or central nervous system (CNS) bleeding. However, heparin has been beneficial in obstetric emergencies such as retained placenta or incomplete abortion, severe arterial occlusions, or MODS caused by microemboli.2,6
The use of recombinant activated protein C is gaining popularity in treating DIC, especially in the setting of severe sepsis. Activated protein C acts as an anticoagulant and works to restore normal inhibition of coagulation pathways. However, it has been associated with an increased incidence of intracerebral bleeding and must be used with caution in patients with severely decreased platelets.2,4
Thrombin production in DIC surpasses that of antithrombins and other regulatory factors that would normally be present to inactivate thrombin and its subsequent actions. The use of antithrombin III has recently been approved in the United States. Ongoing research is yielding mixed results in the treatment of DIC.4 One interesting area of research is the use of protease inhibitors. Protease molecules normally inhibit the conversion of fibrinogen to fibrin in the coagulation mechanism, but in DIC, this inhibitory mechanism is impaired. The introduction of protease inhibitors by intravenous infusion may be advantageous in arresting DIC.2
Nursing Management
Nursing management of the patient with DIC incorporates a variety of nursing diagnoses (see the Nursing Diagnosis Priorities Box on Disseminated Intravascular Coagulation). Assessment and monitoring are the primary weapons in the critical care nurse’s arsenal against DIC. Knowing the diseases and conditions that are most often associated with DIC and understanding the pathophysiological mechanisms involved enables the critical care nurse to anticipate its development and intervene quickly. Nursing priorities are directed toward (1) supporting the patient’s vital functions, (2) initiating bleeding precautions, (3) providing comfort and emotional support, and (4) maintaining surveillance for complications.
Supporting Patient’s Vital Functions
Frequent assessments should include parameters for neurological status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.6
The critical care nurse must recognize and support the patient’s vital physiological functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.
Initiating Bleeding Precautions
Awareness of the patient’s bleeding potential necessitates adjustments to normal nursing interventions. The nurse avoids unnecessary venipunctures that may result in bleeding, bruising, or hematomas by drawing blood from and administering medications through existing arterial or venous lines. The use of manual or automatic blood pressure cuffs is avoided whenever possible. If tracheal or oral suctioning is necessary, the use of low-level suction is recommended.6 Meticulous skin care is advised, keeping the skin moist and using specialty mattresses and beds as appropriate to prevent breakdown. Gentle care should be used when bathing or turning the patient to prevent bruising or hematoma formation.
Providing Emotional Support
The development of DIC in the already critically ill patient can be stressful for the patient and his or her significant others. It is imperative to provide psychosocial support throughout this crisis. Calm reassurance and uncomplicated explanations of the care the patient is receiving can help to allay much of the anxiety experienced. The critical care nurse must answer all questions and provide information in terms best understood by all parties. The use of an interpreter when English is not the primary language can enhance understanding and help avoid misconceptions. Providing spiritual support as requested may also be of assistance.
Please see the Collaborative Management Box on Disseminated Intravascular Coagulation for more information.
