CHAPTER 128 Hemangioblastomas
Hemangioblastomas originate nearly exclusively from central nervous system tissues and are rarely found in the peripheral nervous system. They occur sporadically (60% to 75% of cases) or in the context of the neoplasia syndrome, von Hippel-Lindau disease (25% to 40%; [VHL]).1,2 They are World Health Organization (WHO) grade I CNS neoplasms.3 Advances in imaging and development of serial surveillance protocols in VHL patients with CNS hemangioblastomas have given insights into the molecular biology, pathogenesis, natural history, and clinical findings associated with these tumors. These findings have refined indications for treatment and helped define optimal management of hemangioblastomas in sporadic and VHL-related cases. Here, we describe the epidemiology, imaging features, clinical findings, histopathology, pathogenesis, and treatment options of hemangioblastomas.
Epidemiology
Hemangioblastomas are found more frequently (1.5 to 2 times) in males than in females.2–5 Hemangioblastoma-related symptoms typically occur earlier in VHL patients (30 to 40 years) compared with sporadic patients (40 to 50 years).4,6,7 Hemangioblastomas are distributed in a highly conserved region-specific manner within the CNS that includes the brainstem, spinal cord, and cerebellum (about 90% of hemangioblastomas are found in these regions).3,8 Five percent to 10% of hemangioblastomas are found in the supratentorial compartment. When hemangioblastomas are found supratentorially, they most frequently originate in the region of the pituitary stalk and tuber cinereum (30% of supratentorial hemangioblastomas).8 Although hemangioblastomas represent about 2% of all intracranial neoplasms, they account for 7% to 12% of posterior fossa tumors.3,4,9 Two percent to 10% of all primary spinal cord tumors are hemangioblastomas.3,5
Imaging Findings
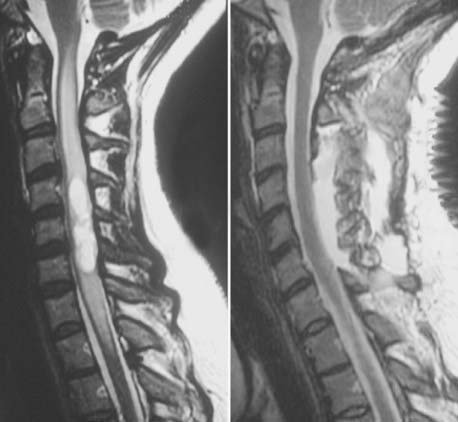
The most sensitive and accurate imaging modality for discovery and tracking changes in CNS hemangioblastomas is contrast-enhanced magnetic resonance imaging (MRI). Because hemangioblastomas are highly vascular tumors, they vividly and discretely enhance on T1-weighted MRI sequences following gadolinium-based contrast infusion (Fig. 128-1). Even small tumors (2 to 3 mm in diameter) can be detected and precisely measured to determine changes in size or other radiographic characteristics over serial studies. Because hemangioblastomas are frequently associated with peritumoral edema and cysts, imaging designed to detect changes in interstitial fluid is often a sensitive approach for tumor detection. Peritumoral edema and cysts are best detected and followed using fluid-attenuated inversion recovery or T2-weighted MRI sequences (Fig. 128-2). Prominent flow voids are often identified with larger hemangioblastomas on T1- and T2-weighted MRI sequences. Infrequently, arteriography is used to supplement MRI findings. Angiography of vessels supplying and draining a hemangioblastoma reveals a highly vascular tumor nodule with prolonged contrast staining that can be associated with an avascular peritumoral cyst.
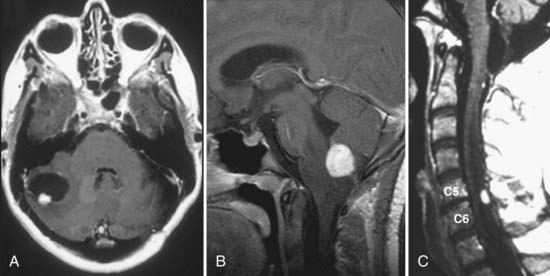
Clinical Findings
Clinical findings associated with CNS hemangioblastomas are related to mass effect (tumor mass or peritumoral edema– or cyst-related mass effect) and their anatomic location. Cerebellar hemangioblastomas frequently occur in the posterior and medial portions (70% to 80% of all cerebellar hemangioblastomas) of the cerebellum.2,8,10 Tumors in this anatomic region most frequently cause headache (70% to 80% of patients), gait ataxia (55% to 65%), dysmetria (30% to 65%), hydrocephalus (20% to 30%), or nausea and vomiting (5% to 30%).6,10,11 Brainstem hemangioblastomas frequently occur in the posterior medulla at the obex.8,12,13 Tumors in this anatomic region most frequently cause hypesthesia (40% to 55%), gait ataxia (20% to 30%), dysphagia (20% to 30%), hyperreflexia (20% to 25%), headache (10% to 20%), or disorders of appetite and feeding (2% to 5%).13,14 Spinal cord hemangioblastomas usually arise posterior to the dentate ligament (96% of all spinal cord hemangioblastomas) at the dorsal root entry zone (66% of all posterior spinal cord hemangioblastomas).8,15 Spinal hemangioblastomas most frequently cause hypesthesia (80% to 90%), weakness (60% to 70%), gait ataxia (50% to 65%), hyperreflexia (40% to 60%), or pain (10% to 30%).
Peritumoral Cyst Formation
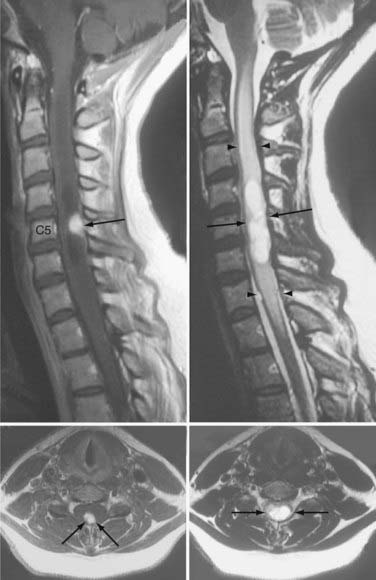
About 80% of CNS hemangioblastomas are associated with a peritumoral cyst (a cyst that forms at the margin of the tumor). Although signs and symptoms can be related specifically to the mass effect of a hemangioblastoma, neurological dysfunction is most frequently caused by the combined mass effect of the tumor and an associated peritumoral cyst. About 70% of symptomatic cerebellar and brainstem hemangioblastomas are associated with a peritumoral cyst, and more than 90% of symptomatic spinal cord hemangioblastomas are associated with a peritumoral cyst (syringomyelia).15,16 When peritumoral cysts occur with hemangioblastomas, the cyst typically accounts for the bulk of the mass burden. Wanebo and colleagues8 compared tumor volume to associated peritumoral cyst volume and found that cerebellar hemangioblastomas had average cyst volume that was 4 times larger than the associated tumor and that was 12 times larger in the brainstem than the associated tumor in symptomatic VHL patients. Alternatively, asymptomatic cerebellar, brainstem, or spinal cord hemangioblastomas are infrequently associated with peritumoral cysts (only 5% to 10% of tumors with cysts).
Because development and growth of peritumoral cyst frequently underlie the formation of signs and symptoms associated with hemangioblastomas, understanding the mechanism underlying peritumoral cyst development and propagation in CNS hemangioblastomas is critical (Fig. 128-3). Recent studies have shown that development of a peritumoral cyst follows a defined sequence initiated by increased hemangioblastoma vascular permeability.17,18 This results in extravasation of a plasma ultrafiltrate into the interstitial spaces of the tumor, and high interstitial tumor pressure drives this fluid into the surrounding CNS parenchyma. This results in edema formation as the resorptive capacity of the peritumoral tissue is exceeded. As the mismatch between fluid production and tissue resorption increases, the surrounding tissues swell owing to solid stresses (stretching) that favor cyst formation. Formation of the cyst alters interstitial flow patterns so that most excess interstitial fluid flows into the peritumoral cyst (path of least resistance), resulting in cyst expansion.
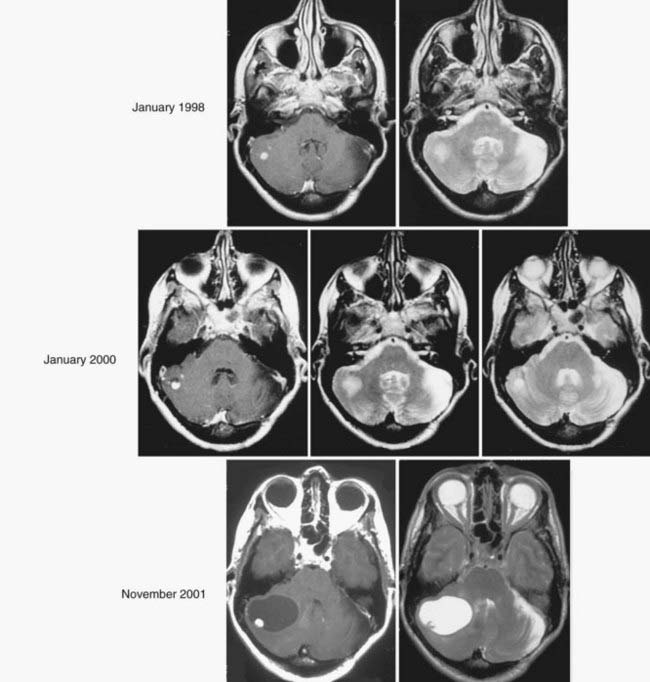
Based on deeper understanding of the mechanism of peritumoral cyst development and propagation, critical clinical insights have been established. Because the hemangioblastoma is the source of extravasated plasma ultrafiltrate, peritumoral edema and cysts resolve after tumor removal, and treatment does not require cyst wall resection or fenestration. Likewise, increases in tumor vascular permeability can be deleterious because they lead to increased plasma leakage into the surrounding CNS parenchyma and favor edema and cyst formation. Studies have demonstrated that radiation can at least transiently induce increases in vascular permeability, leading to peritumoral edema and cyst formation.19–21 Subsequently, judicious use of irradiation of hemangioblastomas associated with peritumoral edema and cysts is warranted.21–23 Alternatively, a reduction in hemangioblastoma vascular permeability may be beneficial. Anti–vascular endothelial growth factor (VEGF) tumor treatments have been associated with edema reduction and symptom improvement, despite having no effect on tumor size.24,25 Finally, peritumoral cysts stop growing once the surface area of the cyst wall is sufficiently large to absorb the excess fluid and become quiescent. Subsequently, imaging evidence of edema or cyst formation in asymptomatic patients is not an absolute indication for surgery.26
Von Hippel-Lindau Disease
VHL is an autosomal dominant neoplasia syndrome that has a prevalence of 1 in 39,000 live births.27 VHL is the result of a germline mutation of the VHL gene.28,29 Although this disorder has variable expression, it has greater than 90% penetrance by 65 years of age.30 Patients with VHL are predisposed to develop specific CNS and visceral lesions (Table 128-1).6,31,32 CNS manifestations include hemangioblastomas (the most common manifestation of VHL) and endolymphatic sac tumors (ELSTs).33,34 Visceral manifestations include renal cysts, renal carcinomas, pancreatic cysts, pancreatic neuroendocrine tumors, pheochromocytomas, and cystadenomas of the adnexal organs (broad ligament in females and epididymis in males). Before the advent of routine surveillance and better defined treatment recommendations for VHL-associated lesions (Table 128-2), the median survival of VHL patients was less than 50 years of age.32,35 Currently, the primary cause of death is related to renal cell carcinomas or CNS hemangioblastomas.32,35
TABLE 128-1 Approximate Distribution, Mean Age of Onset, Range of Onset Age, and Frequency of Lesions Associated with von Hippel-Lindau Disease
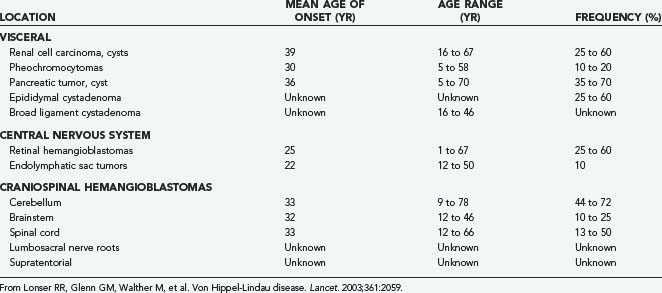
TABLE 128-2 Time Periods Often Recommended for Various Screening Tests in At-Risk Individuals with von Hippel-Lindau Disease
| TEST | START AGE (FREQUENCY) |
|---|---|
| Ophthalmoscopy | Infancy (yearly) |
| Plasma or 24-hr urinary catecholamines and metanephrines | 2 years of age (yearly and when blood pressure is elevated) |
| Magnetic resonance imaging of craniospinal axis* | 11 years of age (yearly) |
| Computed tomography and magnetic resonance imaging of internal auditory canals* | Onset of symptoms (hearing loss, tinnitus, vertigo, or unexplained balance difficulties) |
| Ultrasound of abdomen | 8 years of age (yearly; magnetic resonance imaging as clinically indicated) |
| Computed tomography of abdomen* | 18 years of age or earlier if clinically indicated (yearly) |
| Audiologic function tests | When clinically indicated |
* Precontrast and postcontrast studies are often recommended.
Adapted from Choyke PL, Glenn GM, Walther MM, et al. Von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology. 1995;194:629.
Clinical criteria or genetic testing can be used to establish the diagnosis of VHL. Patients with a family history of VHL and CNS hemangioblastoma (including retinal hemangioblastomas), clear cell renal carcinoma, pheochromocytoma, or ELST are diagnosed with the disease. Twenty percent of VHL patients do not have a family history but fulfill the clinical diagnostic criteria for VHL by having two or more CNS hemangioblastomas or one CNS hemangioblastoma and a VHL-associated visceral tumor.35,36 At-risk patients, including patients with isolated CNS hemangioblastoma37 and undiagnosed family members of VHL patients, now frequently undergo testing for a germline VHL mutation. Blood samples can tested for the VHL mutation at a number reference laboratories internationally. The detection rate of VHL mutations in patients with a family history of VHL is nearly 100%.38 However, mutations in de novo patients without a family history may result in a disease mosaicism, whereby some tissues carry the mutation.39 These patients may test negative if their peripheral blood leukocytes do not carry the VHL gene mutation.
Natural History
Understanding the natural history of CNS hemangioblastomas is critical for optimizing treatment and for determining the efficacy of various therapies. It is especially important for VHL patients with CNS hemangioblastomas because they may have multiple hemangioblastomas along the neuraxis at any point in time and may develop new tumors over their lifetime. Wanebo and colleagues8 examined the natural history of 665 CNS hemangioblastomas in 160 consecutive VHL patients. Symptoms in patients with CNS hemangioblastomas were related to tumor size and cyst-associated mass effect. As a result, symptom formation in patients with CNS hemangioblastomas was associated with lesions with peritumoral cysts and with more rapid rates of hemangioblastoma growth. However, the pattern of growth of CNS hemangioblastomas is variable and is often marked by prolonged (years) periods of quiescence.8 Ammerman and colleagues,16 by examining patients with VHL and hemangioblastomas who had been followed with serial MRI for at least 10 years, identified certain thresholds of tumor size and threshold rates of growth that were associated with development of symptoms, but because of the relatively small number of patients, no recommendations resulted for management of individual lesions. Thus, neither tumor size nor rate of growth currently permits an argument for early intervention, a circumstance in which a tumor may be smaller and potentially more easily amenable to surgery or medical therapy. Thus, in the setting of VHL, it is probably best to reserve treatment of hemangioblastomas until they produce symptoms.
Pathologic Findings
Hemangioblastomas are bright red or red-orange and are invariably associated with intense vascularity (Fig. 128-4). Typically, large abnormal veins are found draining these tumors during resection. They are well-circumscribed, encapsulated tumors that occasionally have intratumoral cysts but more frequently are associated with peritumoral cysts whose walls are composed of compressed brain tissue and reactive gliosis. Histologically, the tumors are characterized by proliferation of stromal cells and endothelial cells. The endothelial cells surrounded by pericytes form extensive vascular channels that surround the bland, polygonal, vacuolated stromal cells (see Fig. 128-4).40
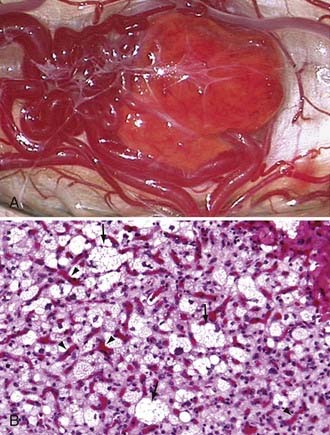
Because the stromal cells have numerous lipid-containing vacuoles that have a clear cell morphology similar to renal cell carcinoma, these tumors often appear histologically similar to renal cell carcinoma metastases. Further, because VHL patients often have contemporaneous renal cell carcinomas that can metastasize to the CNS or metastasize to hemangioblastomas,3,41 immunohistochemical differentiation between renal cell carcinoma and hemangioblastoma may be necessary. Jarrell and colleagues41 reported that 8% of a series of CNS hemangioblastomas removed from consecutive VHL patients harbored either a renal cell carcinoma or pancreatic neuroendocrine tumor that had metastasized to a coexisting hemangioblastoma.
Pathogenesis
Based on embryologic studies, as well as morphologic and molecular analyses of CNS hemangioblastomas, the origin of these tumors has recently been established. From her detailed analysis of chick embryo development, Sabin hypothesized, in 1917, that a single multipotent embryologic precursor cell was responsible for forming both blood and vessels.42 After comprehensive histomorphologic studies in VHL patients, Lindau suggested, in 1931, that hemangioblastomas arise from a congenital analage and have an embryologic cell origin.43 In 1960, Stein and colleagues44 described islands of blood formation and vessel formation in CNS hemangioblastomas and, based on Sabin’s embryologic studies, speculated that these tumors may be the result of arrested defective embryonic development. In 2003, Vortmeyer and colleagues45 discovered that hemangioblastomas contain regions of extramedullary hematopoiesis and that the hematopoietic cells in these regions of the tumor are VHL gene deficient. The same group also detected numerous microscopic nests of hemangioblastomas (tumorlets) in the same unique CNS distribution that characterizes hemangioblastomas in VHL patients at autopsy.46
Developmental data, combined with evidence of intratumoral hematopoiesis and vessel formation, as well as microscopic primordial hemangioblastomas in patients with VHL, suggested that hemangioblastomas may derive from an embryologic cell capable of blood and vessel formation. Until Choi and colleagues,47 in 1998, discovered a common embryologic precursor cell capable of blood and endothelial cell formation, this hypothesis was not testable. Using embryonic stem cell lineage studies, Choi and colleagues47 characterized a multipotent embryonic precursor for both hematopoietic and endothelial cells that is transiently present during mesodermal development. They defined this multipotent embryologic cell as a hemangioblast. Subsequent investigations uniquely characterized the embryonic hemangioblast as a cell that coexpresses a unique set of molecules, including brachyury, Flk-1 (VEGF receptor-2) and Scl (stem cell leukemia).48
To establish whether the neoplastic cell of origin in hemangioblastomas is the embryologically arrested hemangioblast, the expression of brachyury, Flk-1, and Scl was recently characterized from the neoplastic stromal cells of CNS hemangioblastomas from VHL patients.49 Consistent with the embryologic hemangioblast and a cell of mesodermal origin, the neoplastic stromal cells in hemangioblastomas were found to coexpress brachyury (which is normally expressed only during mesoderm development), Flk-1, and Scl. Analogous to embryologic hemangioblasts, the hemangioblastoma-derived hemangioblasts demonstrated self-renewal and could be differentiated into separate hematopoietic and endothelial lineages using distinctly controlled microenvironments. The hematopoietic and endothelial progeny were confirmed to derive from tumor hemangioblasts by demonstration of loss of the region of chromosome 3 containing the VHL allele in the differentiated endothelial and blood components.
These findings have several important implications for understanding hemangioblastoma development and treatment. First, they imply that loss of the wild-type allele, which is necessary for hemangioblastoma formation in VHL,50 occurs during embryogenesis because the hemangioblast normally exists only during development of the embryo. Second, they suggest that the developmentally arrested hemangioblast may be reactivated to proliferate under suitable conditions. Third, the highly conserved anatomic distribution of hemangioblastomas precisely recapitulates the topographic distribution that would be expected if the embryologic hemangioblast were indeed the cell of origin for these tumors.49,51 Finally, the identification of the hemangioblast as the neoplastic cell of origin may permit development of new approaches for treatment targeting unique developmental proteins and pathways or via differentiation strategies.
Management
Von Hippel-Lindau versus Sporadic Disease
The management strategy for hemangioblastomas in patients with sporadic tumors differs from the strategy in VHL patients. Because most VHL patients have multiple CNS hemangioblastomas and because the growth rate of these tumors is unpredictable, treatment of individual tumors is generally postponed until the tumor becomes symptomatic. Using this strategy, most VHL patients maintain excellent neurological function over long-term evaluation, and unnecessary surgical resection can be avoided.16 Alternatively, sporadic patients with hemangioblastomas are investigated because they have symptoms, and the diagnosis of hemangioblastoma cannot be confirmed until resection. Thus, to relieve symptoms and to provide a diagnosis in sporadic patients, surgery is required.
Screening for Von Hippel-Lindau Disease
Because of the frequent association of hemangioblastomas with VHL, screening for VHL in apparently sporadic patients (those without a family history of VHL and imaging evidence of a single CNS hemangioblastoma) is important. VHL mutation analysis of peripheral blood should be performed in all patients with isolated CNS hemangioblastomas (patients with a single hemangioblastoma, negative retinal examination, and negative abdominal imaging).37 Because of the possibility of false-negative mutation analysis (i.e., mosaicism), baseline-screening studies for CNS and visceral manifestations of VHL should be performed. About 5% of patients with apparently isolated CNS hemangioblastomas and no detectable VHL mutation on genetic testing develop tumors within the VHL spectrum.37 Thus, younger patients with negative mutation analysis and normal baseline screening need long-term surveillance and screening of at-risk relatives. Older patients (>50 years old) with negative mutation analysis and normal results of baseline screening are at low risk for VHL.
Surgical Resection
The treatment of choice for CNS hemangioblastomas is complete resection (see “Surgical Technique” later). Microsurgical resection is curative, and most craniospinal hemangioblastomas can be completely and safely resected. When the tumor is associated with a cyst (regardless of anatomic location), the gliotic cyst walls are left undisturbed because they are not neoplastic, and simply removing the source of the cyst (the tumor) inactivates the cyst and the cyst will collapse. This is the case, regardless of the location of the tumor along the craniospinal axis. When complete resection of the tumor is accomplished, recurrence is rare, and stabilization or improvement of symptoms occurs in more than 90% of patients.10,13,15,52,53
Preoperative Embolization
To minimize blood loss and facilitate resection, preoperative embolization of hemangioblastomas has been investigated. Although some centers find that preoperative embolization of hemangioblastomas enhances the ease of resection by reduction of tumor vascularity, other centers have found preoperative embolization or even arteriography unnecessary for safe and effective resection. Moreover, hemorrhage after embolization of CNS hemangioblastomas that results in morbidity or mortality occurs in some patients.54 Although preoperative arteriography can be useful to delineate the feeding vessels of large complex tumors, the additional risk of embolization is usually unnecessary for the safe removal of CNS hemangioblastomas if the basic tenets of microsurgical resection are followed.
Radiation Therapy
Because of the potential morbidity that can be associated with resection of large craniospinal hemangioblastomas and their well-defined boundaries on MRI, stereotactic radiosurgery and conventional conformal external-beam irradiation therapy have been investigated as potential therapeutic options. Small tumors that are not associated with peritumoral cysts are likely to respond best to radiosurgery. Reported local control rates for stereotactic radiosurgery range from 85% to 95% at 2 years and 65% to 86% at 5 years.23,55–57 Far less data on the outcome of the use of fractionated external-beam irradiation therapy are available. Recently, Koh and colleagues58 reported a disease-free survival rate of 57% at 5 years and 30% at 10 years in 18 sporadic or VHL patients with CNS hemangioblastomas treated with external-beam irradiation. However, it must be emphasized that the definition of the successful use of radiosurgery or external-beam irradiation has been based in large part on stability of hemangioblastoma size on serial imaging. However, serial MRI observation of untreated hemangioblastomas associated with VHL indicates that many of these benign tumors remain stable in size for extended intervals (years).8,16 Thus, caution is necessary in interpreting the results of radiotherapy because many of the treated tumors have been in a quiescent phase, and the absence of enlargement on serial MRI may not have been the result of treatment. Indeed, some studies have shown delayed progression of hemangioblastomas several years after radiotherapy treatment.10,16 Studies with longer term assessment and more patients are necessary to determine accurately the effectiveness of radiation in the treatment of CNS hemangioblastomas.
Surgical Technique
Cerebellar Hemangioblastomas
Cerebellar hemangioblastomas are frequently (74% of cerebellar hemangioblastomas) located in the posterior and medial portions of the cerebellum. Subsequently, a midline suboccipital approach can most often be used to resect these tumors, as described previously.10 Cerebellar hemangioblastomas located in the lateral, anterior, or anteromedial regions of the cerebellum can be resected by a variety of approaches.59–63 Optimal visualization, direct access to the tumor, and surgeon preference determine the selection of surgical approach to hemangioblastomas in these regions. Regardless of the approach used, the biologic features of hemangioblastoma dictate the basic tenets of removal from the surrounding cerebellar tissues, and the technique described next is applicable to tumors in all regions of the cerebellum.
Two maneuvers can be used to reduce the tumor mass, minimize cerebellar manipulation and retraction, and enhance the ease of removal for large tumors. First, the central portion of the tumor can be coagulated using bipolar forceps with broad tips and removed in a piecemeal manner with microscissors or ultrasonic aspiration to permit manipulation of the tumor margin and to gain access to the ventral edge of the tumor. Typically, this maneuver is used in later stages of the dissection when the blood supply has been reduced to minimize bleeding. Second, the tumor can be shrunk using bipolar coagulation of the exposed capsule with broad-tipped bipolar forceps.53 Coagulation of the hemangioblastoma surface turns the capsule tan-white. Subsequently, cauterization of the tumor surface at the point of deepest dissection at the tumor-cerebellum interface is usually avoided so that the bright red color of the tumor remains visually distinct from the immediately surrounding cerebellar tissue as dissection is carried deeper.
Spinal Cord Hemangioblastomas
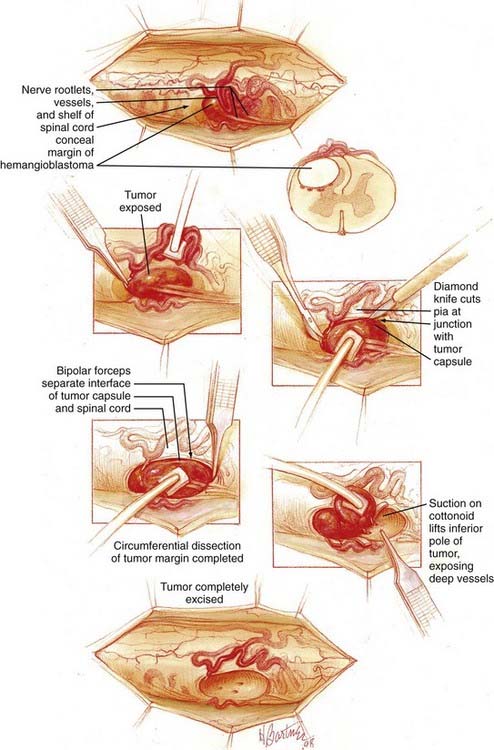
 Because most (96% of spinal cord hemangioblastomas) spinal cord hemangioblastomas are located posterior to the dentate ligament,64 a direct posterior approach is most often used to remove these tumors, as described previously (Fig. 128-5 and Video 128-1).15,65 Hemangioblastomas located in the anterior (anterior to the dentate ligament) or anterolateral region of the spinal cord may be accessed by a direct anterior or anterolateral approach.66 These direct approaches to anteriorly located tumors allow for minimal rotation and manipulation of the spinal cord during resection and have been shown to improve postoperative outcome of hemangioblastomas located anterior to the dentate ligament.66 Occasionally, anteriorly located tumors associated with sufficient preexisting rotation of the spinal cord can be best accessed from a posterior or posterolateral approach.
Because most (96% of spinal cord hemangioblastomas) spinal cord hemangioblastomas are located posterior to the dentate ligament,64 a direct posterior approach is most often used to remove these tumors, as described previously (Fig. 128-5 and Video 128-1).15,65 Hemangioblastomas located in the anterior (anterior to the dentate ligament) or anterolateral region of the spinal cord may be accessed by a direct anterior or anterolateral approach.66 These direct approaches to anteriorly located tumors allow for minimal rotation and manipulation of the spinal cord during resection and have been shown to improve postoperative outcome of hemangioblastomas located anterior to the dentate ligament.66 Occasionally, anteriorly located tumors associated with sufficient preexisting rotation of the spinal cord can be best accessed from a posterior or posterolateral approach.
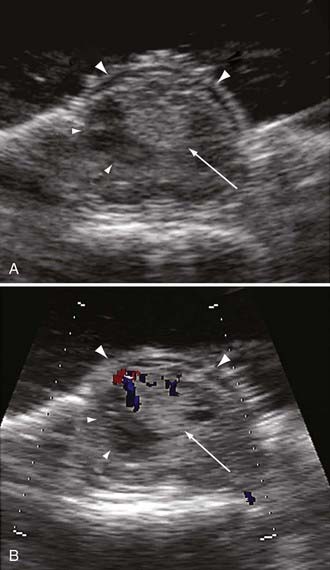
After bony removal is complete, intraoperative ultrasound is used to confirm the adequacy of exposure of the hemangioblastoma within the bony opening. Hemangioblastomas are highly echogenic lesions with high velocity blood flow that can be identified by Doppler analysis (Fig. 128-6). Using loupe magnification, the dura is sharply incised in the midline while preserving the underlying arachnoid. The dura is reflected and tacked laterally using 4-0 silk sutures to the paraspinous musculature. Tacking these sutures as low as possible to the paraspinous muscles keeps them out of the surgical field during the intradural portion of the procedure. Using the operative microscope, the arachnoid is opened with microforceps and microscissors. The opened arachnoid is retracted and tacked to the dural margins with titanium vascular clips.
During the dissection of hemangioblastomas with associated syringomyelia, it is not necessary to enter the syrinx cavity. We often avoid opening into or draining associated hemangioblastoma-associated syrinxes because this can magnify physiologic pulsations of the spinal cord, making resection more difficult. Because the syrinx is caused by the tumor, complete tumor removal will eliminate the syrinx regardless of whether the syrinx is drained (Fig. 128-7).64 Thus, the focus of the surgery is limited to removal of the tumor. After circumferential dissection is carried as deep as direct visualization of the tumor capsule permits, the dissection is continued at the rostral and caudal poles of the tumor. Gentle retraction and elevation of the poles of the hemangioblastoma permit the deep margin of the tumor to be separated from the spinal cord.
Brainstem Hemangioblastomas
Because most brainstem hemangioblastomas (60%) are located in the region of the medullary obex, a midline suboccipital-cervical approach is used to gain access to the tumor.13 Patients are induced and placed under general anesthesia. They are secured in three-point skull fixation and placed prone on gel rolls extending from the shoulders to the anterior iliac crests. The head and neck are flexed, and the midline incision site extending from the inion to the level of the spinous process of the fourth cervical vertebral body is prepared and draped. The dermis of the incision site is infiltrated with 0.25% bupivacaine with epinepherine (1 : 200,000). The skin is incised, and the nuchal musculature is opened in the midline and stripped laterally in the subperiosteal plane over the suboccipital region as well as the first and second cervical laminae. Using a high-speed drill and ronguers, a suboccipital craniectomy is created, and a laminectomy of the ring of the first cervical vertebra is used to expose the lower portion of the cerebellum or cervicomedullary junction, or both.
Ammerman JM, Lonser RR, Dambrosia J, et al. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006;105:248.
Butman JA, Linehan WM, Lonser RR. Neurologic manifestations of von Hippel-Lindau disease. JAMA. 2008;300:1334.
Conway JE, Chou D, Clatterbuck RE, et al. Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery. 2001;48:55.
Cornelius JF, Saint-Maurice JP, Bresson D, et al. Hemorrhage after particle embolization of hemangioblastomas: comparison of outcomes in spinal and cerebellar lesions. J Neurosurg. 2007;106:994.
Iliopoulos O, Kibel A, Gray S, et al. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822.
Jagannathan J, Lonser RR, Smith R, et al. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008;108:210.
Koh ES, Nichol A, Millar BA, et al. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int J Radiat Oncol Biol Phys. 2007;69:1521.
Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68:1.
Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317.
Lonser RR, Butman JA, Oldfield EH. Pathogenesis of tumor-associated syringomyelia demonstrated by peritumoral contrast material leakage. Case illustration. J Neurosurg Spine. 2006;4:426.
Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059.
Lonser RR, Vortmeyer AO, Butman JA, et al. Edema is a precursor to central nervous system peritumoral cyst formation. Ann Neurol. 2005;58:392.
Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106.
Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443.
Malis LI. Atraumatic bloodless removal of intramedullary hemangioblastomas of the spinal cord. J Neurosurg. 2002;97:1.
Melmon KL, Rosen SW. Lindau’s disease. Am J Med. 1964;36:595.
Park DM, Zhuang Z, Chen L, et al. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4:e60.
Patrice SJ, Sneed PK, Flickinger JC, et al. Radiosurgery for hemangioblastoma: results of a multiinstitutional experience. Int J Radiat Oncol Biol Phys. 1996;35:493.
Roonprapunt C, Silvera VM, Setton A, et al. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery. 2001;49:321.
Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417.
Wanebo JE, Lonser RR, Glenn GM, et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:82.
Woodward ER, Wall K, Forsyth J, et al. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain. 2007;130:836.
1 Browne TR, Adams RD, Roberson GH. Hemangioblastoma of the spinal cord: review and report of five cases. Arch Neurol. 1976;33:435.
2 Conway JE, Chou D, Clatterbuck RE, et al. Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery. 2001;48:55.
3 Aldape KD, Plate KH, Vortmeyer AO, et al. Haemangioblastoma. In: Louis DN, Ohgaki H, Wiestler OD, et al, editors. WHO Classification of Tumours of the Central Nervous System. ed 4. Lyon, France: International Agency for Research on Cancer (IARC); 2007:184.
4 Neumann HP, Eggert HR, Weigel K, et al. Hemangioblastomas of the central nervous system. A 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989;70:24.
5 Roonprapunt C, Silvera VM, Setton A, et al. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery. 2001;49:321.
6 Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059.
7 Maher ER, Yates JR, Ferguson-Smith MA. Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J Med Genet. 1990;27:311.
8 Wanebo JE, Lonser RR, Glenn GM, et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:82.
9 Constans JP, Meder F, Maiuri F, et al. Posterior fossa hemangioblastomas. Surg Neurol. 1986;25:269.
10 Jagannathan J, Lonser RR, Smith R, et al. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008;108:210.
11 Richard S, David P, Marsot-Dupuch K, et al. Central nervous system hemangioblastomas, endolymphatic sac tumors, and von Hippel-Lindau disease. Neurosurg Rev. 2000;23:1.
12 Baumgartner JE, Wilson CB. Removal of posterior fossa and spinal hemangioblastomas. In: Wilson CB, editor. Neurosurgical Procedures: Personal Approaches to Classic Operations. Baltimore: Williams and Wilkins; 1992:188.
13 Weil RJ, Lonser RR, DeVroom HL, et al. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:95.
14 Song DK, Lonser RR. Pathological satiety caused by brainstem hemangioblastoma. J Neurosurg Pediatrics. 2008;2:397.
15 Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106.
16 Ammerman JM, Lonser RR, Dambrosia J, et al. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006;105:248.
17 Lohle PN, van Mameren H, Zwinderman KH, et al. On the pathogenesis of brain tumour cysts: a volumetric study of tumour, oedema and cyst. Neuroradiology. 2000;42:639.
18 Lonser RR, Butman JA, Oldfield EH. Pathogenesis of tumor-associated syringomyelia demonstrated by peritumoral contrast material leakage. Case illustration. J Neurosurg Spine. 2006;4:426.
19 Cao Y, Tsien CI, Shen Z, et al. Use of magnetic resonance imaging to assess blood-brain/blood-glioma barrier opening during conformal radiotherapy. J Clin Oncol. 2005;23:4127.
20 Qin D, Ou G, Mo H, et al. Improved efficacy of chemotherapy for glioblastoma by radiation-induced opening of blood-brain barrier: clinical results. Int J Radiat Oncol Biol Phys. 2001;51:959.
21 Shuto T, Inomori S, Fujino H, et al. Cyst formation following gamma knife surgery for intracranial meningioma. J Neurosurg. 2005;102(Suppl):134.
22 Niemela M, Lim YJ, Soderman M, et al. Gamma knife radiosurgery in 11 hemangioblastomas. J Neurosurg. 1996;85:591.
23 Tago M, Terahara A, Shin M, et al. Gamma knife surgery for hemangioblastomas. J Neurosurg. 2005;102(Suppl):171.
24 Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109:1745.
25 Girmens JF, Erginay A, Massin P, et al. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136:194.
26 Lonser RR, Vortmeyer AO, Butman JA, et al. Edema is a precursor to central nervous system peritumoral cyst formation. Ann Neurol. 2005;58:392.
27 Neumann HP, Wiestler OD. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet. 1991;337:1052.
28 Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317.
29 Wait SD, Vortmeyer AO, Lonser RR, et al. Somatic mutations in VHL germline deletion kindred correlate with mild phenotype. Ann Neurol. 2004;55:236.
30 Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443.
31 Butman JA, Linehan WM, Lonser RR. Neurologic manifestations of von Hippel-Lindau disease. JAMA. 2008;300:1334.
32 Richard S, Campello C, Taillandier L, et al. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. French VHL Study Group. J Intern Med. 1998;243:547.
33 Butman JA, Kim HJ, Baggenstos M, et al. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA. 2007;298:41.
34 Lonser RR, Kim HJ, Butman JA, et al. Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med. 2004;350:2481.
35 Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68:1.
36 Melmon KL, Rosen SW. Lindau’s disease. Am J Med. 1964;36:595.
37 Woodward ER, Wall K, Forsyth J, et al. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain. 2007;130:836.
38 Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417.
39 Sgambati MT, Stolle C, Choyke PL, et al. Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet. 2000;66:84.
40 Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28:540.
41 Jarrell ST, Vortmeyer AO, Linehan WM, et al. Metastases to hemangioblastomas in von Hippel-Lindau disease. J Neurosurg. 2006;105:256.
42 Sabin FR. Preliminary note on the differentiation of angioblasts and the method by which they produce blood—vessels, blood-plasma, and red blood—cells as seen in the living chick. Anat Rec. 1917;13:119.
43 Lindau A. Discussion on vascular tumors of the brain and spinal cord. Proc R Soc Med. 1931;24:363.
44 Stein AA, Schilp AO, Whitfield RD. The histogenesis of hemangioblastoma of the brain. A review of twenty-one cases. J Neurosurg. 1960;17:751.
45 Vortmeyer AO, Frank S, Jeong SY, et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel-Lindau disease. Cancer Res. 2003;63:7051.
46 Vortmeyer AO, Yuan Q, Lee YS, et al. Developmental effects of von Hippel-Lindau gene deficiency. Ann Neurol. 2004;55:721.
47 Choi K, Kennedy M, Kazarov A, et al. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725.
48 Huber TL, Kouskoff V, Fehling HJ, et al. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature. 2004;432:625.
49 Park DM, Zhuang Z, Chen L, et al. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4:e60.
50 Iliopoulos O, Kibel A, Gray S, et al. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822.
51 Glasker S, Li J, Xia JB, et al. Hemangioblastomas share protein expression with embryonal hemangioblast progenitor cell. Cancer Res. 2006;66:4167.
52 Lonser RR, Wait SD, Butman JA, et al. Surgical management of lumbosacral nerve root hemangioblastomas in von Hippel-Lindau syndrome. J Neurosurg. 2003;99:64.
53 Malis LI. Atraumatic bloodless removal of intramedullary hemangioblastomas of the spinal cord. J Neurosurg. 2002;97:1.
54 Cornelius JF, Saint-Maurice JP, Bresson D, et al. Hemorrhage after particle embolization of hemangioblastomas: comparison of outcomes in spinal and cerebellar lesions. J Neurosurg. 2007;106:994.
55 Chang SD, Meisel JA, Hancock SL, et al. Treatment of hemangioblastomas in von Hippel-Lindau disease with linear accelerator-based radiosurgery. Neurosurgery. 1998;43:28.
56 Patrice SJ, Sneed PK, Flickinger JC, et al. Radiosurgery for hemangioblastoma: results of a multiinstitutional experience. Int J Radiat Oncol Biol Phys. 1996;35:493.
57 Wang EM, Pan L, Wang BJ, et al. The long-term results of gamma knife radiosurgery for hemangioblastomas of the brain. J Neurosurg. 2005;102(Suppl):225.
58 Koh ES, Nichol A, Millar BA, et al. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int J Radiat Oncol Biol Phys. 2007;69:1521.
59 Ammerman JM, Lonser RR, Oldfield EH. Posterior subtemporal transtentorial approach to intraparenchymal lesions of the anteromedial region of the superior cerebellum. J Neurosurg. 2005;103:783.
60 Baldwin HZ, Spetzler RF, Wascher TM, et al. The far lateral-combined supra- and infratentorial approach: clinical experience. Acta Neurochir (Wien). 1995;134:155.
61 Heros RC. Brain resection for exposure of deep extracerebral and paraventricular lesions. Surg Neurol. 1990;34:188.
62 Kurokawa Y, Uede T, Hashi K. Operative approach to mediosuperior cerebellar tumors: occipital interhemispheric transtentorial approach. Surg Neurol. 1999;51:421.
63 Matsushima T, Rhoton ALJr, de Oliveira E, et al. Microsurgical anatomy of the veins of the posterior fossa. J Neurosurg. 1983;59:63.
64 Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106.
65 Lonser RR, Oldfield EH. Microsurgical resection of spinal cord hemangioblastomas. Neurosurgery. 2005;57:372.
66 Pluta RM, Iuliano B, DeVroom HL, et al. Anterior versus posterior surgical approach for ventral spinal hemangioblastomas in von Hippel-Lindau disease. J Neurosurg. 2003;98:117.






