[level-membership-for-emergency-medicine-category]Chapter 157
Heavy Metals
Iron
Iron, which is essential to the function of hemoglobin, myoglobin, many cytochromes, and many catalytic enzymes, can be extremely toxic when levels are elevated after an overdose or from accumulation in disease states.1 The acute ingestion of iron is especially hazardous to children. Ingestions of pediatric multivitamin formulations are the most common iron exposures. These occur in children younger than 6 years and are minimally toxic. Life-threatening toxicity is associated with ingestion of potent adult preparations, such as prenatal vitamins. Serious iron ingestions in adults are usually associated with suicide attempts.2
Principles of Disease
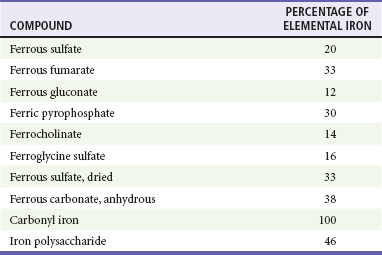
In assessment of the severity of an iron exposure, it is important to refer to the amount of elemental iron ingested because the toxicity of an iron compound depends on the amount of elemental iron it contains (Table 157-1). Different formulations of iron salts contain different percentages of elemental iron. The total amount of elemental iron ingested can be approximated by multiplying the estimated number of tablets by the fraction of elemental iron contained in the tablet. Manufacturers are also required to list the amount of elemental iron per tablet. Ingestions of less than 20 mg/kg of elemental iron usually cause no symptoms. Ingestion of 20 to 60 mg/kg results in mild to moderate symptoms, and ingestion of more than 60 mg/kg may lead to severe morbidity and mortality. Although the dose of elemental iron associated with 50% mortality (LD50) is 200 to 250 mg/kg, doses as small as 130 mg of elemental iron have been lethal in children.3 Newer forms of iron are carbonyl iron and iron polysaccharide; both are non-ionic and associated with lower toxicity. Neither form is directly corrosive, and the conversion to the iron ion, which is responsible for toxicity, is very slow. There are no reported cases of serious toxicity or death from the ingestion of these compounds.4,5
Pathophysiology
Iron has two distinct toxic effects: (1) it causes direct caustic injury to the gastrointestinal mucosa, and (2) it impairs cellular metabolism, primarily of the heart, liver, and central nervous system (CNS). The caustic effects of iron on the gut cause the initial symptoms of vomiting, diarrhea, and abdominal pain. Hemorrhagic necrosis of gastric or intestinal mucosa can lead to bleeding, perforation, and peritonitis.3 Unbound (free) iron moves into cells and localizes near the mitochondrial cristae, resulting in uncoupling of oxidative phosphorylation and impairment of adenosine triphosphate synthesis. Cell membranes are injured by free radical–mediated lipid peroxidation.3,6
Iron increases capillary permeability and both arteriolar dilation and venodilation, resulting in hypotension. Direct myocardial toxicity decreases cardiac output. Hydration of the iron molecule creates an excess of unbuffered protons, worsening metabolic acidosis.3 These effects, combined with severe gastrointestinal fluid losses, can lead to shock, cardiovascular collapse, and death.3
Clinical Features
The clinical effects of acute iron poisoning occur in five stages. Not every patient goes through every phase. Phase I reflects the corrosive effects of iron on the gut. Vomiting occurs within 80 minutes of ingestion in more than 90% of symptomatic cases. Diarrhea, which can be bloody, soon follows. Phase II represents an apparent (but not complete) recovery that lasts less than 24 hours but can extend up to 2 days. Most patients recover after this point. Phase III is characterized by the recurrence of gastrointestinal symptoms, severe lethargy or coma, anion gap metabolic acidosis, leukocytosis, coagulopathy, renal failure, and cardiovascular collapse. Serum iron levels may have fallen to normal during this phase because of distribution of iron into the tissues. Metabolic derangements due to iron poisoning include hypoglycemia, leukocytosis, and severe lactic acidosis from hypoperfusion and interference with cellular respiration. Early coagulation defects are probably related to direct effects of iron on vitamin K–dependent clotting factors.7 Later coagulation defects are due to hepatic failure. Hypoglycemia and elevations of bilirubin and aspartate and alanine transaminases are other markers of hepatotoxicity. Phase IV, characterized by fulminant hepatic failure, occurs 2 to 5 days after ingestion. This is relatively rare, appears to be dose related, and is usually fatal.8 Phase V represents the consequences of healing of the injured gastrointestinal mucosa. It is characterized by pyloric or proximal bowel scarring, which is sometimes associated with obstruction.
Diagnostic Strategies
The presence of gastrointestinal symptoms suggests a potentially serious ingestion, whereas their absence is reassuring.5 A serum iron level measured at its peak, 3 to 5 hours after ingestion, is the most useful laboratory test to evaluate the potential severity of an iron overdose. Sustained-release or enteric-coated preparations may have erratic absorption, so a second level 6 to 8 hours after ingestion should also be checked. Peak serum iron levels below 350 µg/dL are generally associated with minimal toxicity; 350 to 500 µg/dL, with moderate toxicity; and above 500 µg/dL, with potentially severe toxicity (Table 157-2).5 Because iron is rapidly cleared from the serum and deposited in the liver, the level of iron after a substantial ingestion may be deceptively low if it is measured after its peak. TIBC is a crude test and is not useful to gauge the severity of iron poisoning.9
Table 157-2
Toxicity of Iron by Amount Ingested and Peak Serum Levels
| ELEMENTAL IRON (mg/kg) | PEAK SERUM IRON LEVEL (µg/dL) | TOXICITY |
| <20 | 50-150 | None |
| 20-40 | 150-300 | Mild |
| 40-60 | 300-500 | Moderate |
| >60 | >500 | Severe |
When serum iron levels are not immediately available, elevations of the serum glucose level above 150 mg/dL and leukocyte count above 15,000 are 100% specific for prediction of serum iron levels of more than 300 µg/mL. However, a sensitivity of only 50% limits their sole use as indicators of toxicity.10
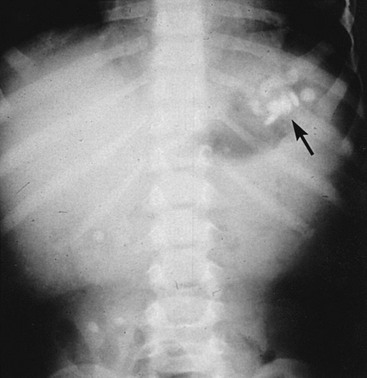
Most tablets that contain a significant amount of elemental iron are radiopaque, although a false-negative radiograph may occur with chewable, liquid, and completely dissolved iron compounds. The presence of tablets on a radiograph correlates with the severity of the ingestion (Fig. 157-1).11,12 Repeated radiographs can also demonstrate the efficacy of gastrointestinal decontamination efforts.
Management
Decontamination may prevent absorption of additional iron from the gastrointestinal tract and can be performed concomitantly with interventions that address drug that has already been absorbed (see later section on deferoxamine). Activated charcoal does not bind iron, and neither gastric lavage nor ipecac effectively removes large numbers of pills. Iron tablets clump together as their outer coatings dissolve, often forming large pharmacobezoars. Whole-bowel irrigation is generally the preferred method of decontamination for significant iron ingestions.13 This recommendation is supported by the position statement of the American Academy of Clinical Toxicology and the European Association of Poison Centres and Clinical Toxicologists; it is based on case reports and the lack of effective alternative decontamination techniques for this potentially deadly toxin.14
For significant ingestions (>20 mg/kg of elemental iron), especially when tablets are identified on the abdominal radiograph, whole-bowel irrigation with a polyethylene glycol–electrolyte lavage solution (PEG-ELS; CoLyte, NuLytely, or GoLytely) should be initiated.14 The solution is either taken orally or, more often, administered through a small nasogastric tube.15 The usual rate of administration of PEG-ELS is 20 to 40 mL/kg/hr in young children and 1.5 to 2 L/hr in teenagers or adults, continued until the rectal effluent is clear and there is no radiographic evidence of pill fragments. This technique has been used in children, adolescents, and pregnant women without serious complications or electrolyte disturbances.13,16,17 Common side effects include nausea, vomiting, abdominal cramping, and bloating. Whole-bowel irrigation is contraindicated in the presence of bowel obstruction, perforation, or ileus.14
Hemodialysis and hemoperfusion are not effective in the removal of iron because of its large volume of distribution. Early exchange transfusions have been recommended for severely symptomatic patients with serum iron levels exceeding 1000 µg/dL.18,19
Deferoxamine
Deferoxamine chelates iron to form the water-soluble compound ferrioxamine, which is renally excreted and can be dialyzed; 100 mg will chelate 9.35 mg of elemental iron.3 Deferoxamine may also limit the entrance of iron into the cell and chelate intracellular iron. Because of its short half-life, it is administered as a continuous infusion at 15 mg/kg/hr for up to 24 hours.20 The maximum rate of administration is 35 mg/kg/hr. Rapid administration of deferoxamine can lead to hypotension, which resolves by reducing the initial rate of the infusion and slowly increasing it to the desired rate. Pregnancy is not a contraindication to deferoxamine.4,21,22 However, the pre-pregnancy weight should be used to calculate the dose. Deferoxamine can falsely lower serum iron levels, so measurements should be taken before its administration.23
The presence of ferrioxamine turns the urine a “vin rosé” color, which reflects the excretion of chelated iron. The deferoxamine challenge test, which relied on detection of this color change, has been used to detect the presence of free iron in the serum. However, the color change is difficult to detect, especially when the urine is dilute, with false-negative results even in cases of serious poisoning.24
Deferoxamine has been associated with acute respiratory distress syndrome and also with Yersinia sepsis. The pulmonary complications are usually related to high-dose deferoxamine for durations longer than 24 hours.20 The presence of fever and sepsis in a patient who has recently been treated for iron poisoning should prompt suspicion for Yersinia sepsis.25,26
Disposition
The asymptomatic patient who is reliably known to have ingested less than 20 mg/kg of elemental iron and has a normal abdominal radiograph can be observed without further therapy. The patient can be discharged if he or she remains asymptomatic after 6 hours of observation.5
The patient who has ingested more than 20 mg/kg of elemental iron or has pills visible on an abdominal radiograph should receive whole-bowel irrigation.23 Follow-up abdominal radiographs can verify adequate gastrointestinal decontamination. The serum iron concentration should be checked 3 to 5 hours after ingestion. A second iron level 6 to 8 hours after ingestion should be decreasing. If peak levels are less than 300 µg/dL and are not rising and the patient is asymptomatic during 6 hours of observation, the patient can be discharged home. Moderate gastrointestinal toxicity can be expected with peak levels of 300 to 500 µg/dL. Deferoxamine should be used if these patients have symptoms of systemic toxicity evidenced by metabolic acidosis, repetitive vomiting, toxic appearance, lethargy, hypotension, or signs of shock. Patients with a serum iron level above 500 µg/dL require chelation with deferoxamine regardless of symptoms.4 If serum iron levels are not available, a normal serum glucose level and leukocyte count suggest a low iron level; if there are no signs or symptoms of toxicity during the 6-hour postingestion period and the abdominal radiographs do not show pills in the gastrointestinal tract, the patient can be sent home.
Lead
Lead poisoning is a disease of industrialization and has become the most common environmental toxicologic problem in the United States.27 Most exposures are from inhalation of lead dust or fumes or ingestion of contaminated substances, such as paint chips. Less often, it results from direct skin contact with organic lead compounds or from retained bullets in or near joints.28 Approximately 3 to 4 million children (1 in 20) in the United States have toxic blood lead levels (BLLs), defined currently as levels exceeding 10 µg/dL.29 Although the addition of lead to household paint and gasoline was banned in the United States in the 1970s, residual lead-based paint is still found in 30 million homes.29 Other sources of toxic lead ingestions include curtain weights, buckshot, fishing weights, lead-contaminated soil or water, bootleg whiskey (“moonshine”), food or beverages stored or prepared in lead-soldered cans, lead-glazed pottery, and lead crystal decanters.30 Herbal and folk remedies, toys, and numerous products imported from Asia and Mexico can also contain dangerous amounts of lead.31,32 This has resulted in multiple drug and toy recalls in the United States. Lead found in buckshot can cause toxic effects in those who eat wild game.33
Principles of Disease
Pathophysiology
There is no known physiologic need for lead. Lead binds to sulfhydryl groups and other ligands and interferes with critical enzymatic reactions.12 Its toxic effects are most prominent in the hematopoietic, neurologic, and renal systems.34
Anemia, the classic manifestation of hematopoietic toxicity, may be either normochromic or hypochromic. The severity of the anemia correlates directly with the BLL. Inhibition of heme biosynthesis results in the accumulation of heme precursors, such as D-aminolevulinic acid and protoporphyrin. In the peripheral nervous system, segmental demyelination and degeneration of motor axons result in peripheral neuropathies. Wristdrop and footdrop are characteristic of adult lead poisoning. Lead toxicity also causes a host of neuropsychiatric disorders. In children, elevated BLL is associated with decreased intelligence (IQ) scores, hyperactivity, decreased attention span, overaggressive behavior, learning disabilities, and criminal behavior.35–38 It has also been associated with subclinical sensorineural hearing loss.39 Lead nephropathy is characterized by fibrosis in the proximal tubules, with relative sparing of the glomeruli. Hyperuricemic gout (“saturnine gout”) can result from increased reuptake of uric acid by the tubular cells.40 Lead poisoning also correlates with hypertension.41 Adults and children with severe acute toxicity may present with lead encephalopathy associated with increased capillary permeability and cerebral edema.42,43
Clinical Features
Acute exposure to lead can result in symptomatic poisoning. “Lead colic” is characterized by cramping abdominal pain with nausea, vomiting, constipation, and, occasionally, diarrhea.43 Other characteristic symptoms and signs of acute toxicity include fatigue, anemia, peripheral neuropathy, renal impairment, and hepatic and CNS dysfunction. The CNS toxicity may be manifested as mild headache or personality changes to full-blown encephalopathy with coma, convulsions, and papilledema. Permanent neurologic and behavioral sequelae may occur.
Diagnostic Strategies
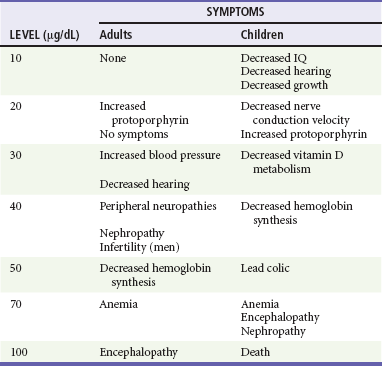
Although capillary lead levels correlate well with BLLs, the most informative biomarker is a BLL.44–46 The Centers for Disease Control and Prevention has defined a chronic BLL of more than 10 µg/dL as toxic for a child. Acute exposure can result in levels above 100 µg/dL (Table 157-3). Other ancillary data include findings on complete blood cell count, serum glucose level, blood urea nitrogen and creatinine concentrations, electrolyte levels, and urinalysis. A peripheral smear may show basophilic stippling. Markers of hepatic injury may be elevated after acute exposure. Because lead-containing paints and objects are radiopaque when lead is present in sufficient quantities, radiographs can confirm acute ingestion and also monitor the effectiveness of whole-bowel irrigation. In cases of altered mental status, seizures, or coma, a computed tomography scan of the head will show cerebral edema associated with acute lead encephalopathy and rule out other causes of these symptoms. In children, plain radiographs of the wrist and knees may show increased metaphyseal activity manifested as “lead bands” or “lead lines” that are characteristic of chronic exposures.
Management
Acute lead encephalopathy can be rapidly fatal. The initial goals in management are identification and treatment of all life-threatening conditions, followed by prevention of further exposure to lead, minimization of ingested lead absorption, enhancement of its elimination, and prevention or reversal of pathologic cell changes. Standard measures to control cerebral edema, including intubation and neurosurgical consultation for invasive monitoring of intracranial pressure, are indicated. When a severe poisoning is associated with recent ingestion or if radiopacities are seen on the radiograph, decontamination with whole-bowel irrigation should be considered.47,48 This intervention has not been prospectively studied but has been successful in case reports.14 Activated charcoal does not adsorb lead and should not be used.
Chelation Therapy
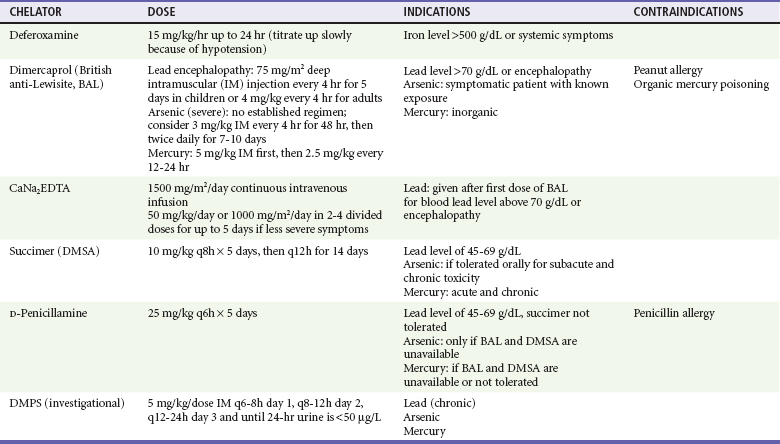
The use of chelation in cases of acute lead poisoning is guided by the patient’s clinical status and the BLL. Any patient with a BLL above 70 µg/dL or with signs suggestive of encephalopathy will require admission for parenteral chelation therapy. For these seriously poisoned patients, dimercaprol (or British anti-Lewisite [BAL]) should be the first chelator given. The dosage for treatment of lead encephalopathy is 75 mg/m2 deep intramuscular injection every 4 hours for 5 days for children or 4 mg/kg every 4 hours for adults. Dimercaprol forms complexes that undergo both renal and biliary excretion. Adverse reactions to dimercaprol include nausea, vomiting, urticaria, pyrexia, hypertension, and hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency.49 Because dimercaprol is diluted in peanut oil, it is contraindicated in patients allergic to peanuts.49 Dimercaprol is followed by calcium disodium ethylenediaminetetraacetic acid (CaNa2EDTA), a highly effective lead chelator. Because of concerns that the chelated lead can cross the blood-brain barrier and worsen encephalopathy, the first dose of CaNa2EDTA is administered with the second dose of dimercaprol. The dosage of CaNa2EDTA for patients with acute lead encephalopathy is 1500 mg/m2/day (approximately 50-75 mg/kg/day) given by continuous intravenous infusion. Adverse reactions include renal tubular injury and chelation of other metals, especially iron and zinc. CaNa2EDTA should be given only with adequate urine flow or with hemodialysis in the patient with renal failure.50 It is important that CaNa2EDTA not be confused with sodium EDTA. The administration of sodium EDTA has been associated with hypocalcemia and death from arrhythmias.51
Serum lead levels of 45 to 69 µg/dL in patients without vomiting or CNS symptoms can be managed in the outpatient setting with oral succimer (2,3-dimercaptosuccinic acid [DMSA]; Chemet). The initial dose of DMSA is 10 mg/kg every 8 hours for 5 days, then 10 mg/kg every 12 hours for 14 days. The most common adverse reactions include nausea, vomiting, diarrhea, and transient elevations in liver transaminase levels. Although DMSA has been approved only for children, it is effective and also used for adults.44,52,53 Oral D-penicillamine is less efficacious than succimer and has more adverse reactions; it should be used only in patients who do not tolerate succimer. The usual oral dose of D-penicillamine is 25 mg/kg every 6 hours for 5 days. Penicillin allergy is a contraindication to the use of D-penicillamine. Another oral chelator is 2,3-dimercapto-1-propanesulfonic acid (DMPS), which, like succimer, is similar to BAL. At this time, it is experimental, although it can be considered in chronic lead poisoning.54,55 Table 157-4 summarizes available chelating agents with indications and doses.
Table 157-4
Evidence indicates no need for chelation for children with a BLL lower than 45 µg/dL.43 A BLL between 20 and 44 µg/dL in a patient who is asymptomatic or minimally symptomatic requires a medical and environmental evaluation. Children with lead levels of 10 to 19 µg/dL require family counseling about the symptoms and sources of lead exposure and careful follow-up, with frequent screening of BLLs. The key to management of chronic lead toxicity is the identification and reduction of sources of primary exposure, which can often be arranged with the local public health department. Any patient treated on an outpatient basis should be discharged to a lead-free environment.
Disposition
Patients who have ingested a single lead foreign body (e.g., fishing sinker) will usually pass it harmlessly.56 If the foreign body remains in the gastrointestinal tract after 2 weeks, removal should be considered to prevent systemic lead toxicity. Gunshot victims with too many lead buckshot pellets to remove may be at risk for elevated BLL and require follow-up lead levels. Asymptomatic children and adults should be counseled on how to avoid further exposure and have a repeated BLL checked in 2 weeks to ensure decreasing levels.
Patients who are significantly symptomatic with CNS symptoms after an acute lead exposure and children with a BLL of 69 µg/dL or higher require hospitalization and chelation therapy. Patients discharged home with oral chelation therapy should not return to a contaminated environment. The health department should conduct an environmental assessment so that the primary source of lead exposure can be identified and further exposure prevented. Follow-up should be arranged with an experienced pediatrician, toxicologist, or occupational medicine physician.57
Arsenic
Arsenic, a tasteless, odorless substance that looks like sugar, has an infamous history as an agent of homicide. Arsenic has also been implicated in many epidemic poisonings. Currently, arsenic exposure is primarily environmental and occupational. It is found in smelters and electric power plants that burn arsenic-rich coal. It is used in industry as a wood preservative and in the production of glass and microcircuits. Inorganic arsenicals are also used in rodenticides, fungicides, insecticides, paint, and tanning agents and as defoliants in the cotton industry. Arsenic is still used for medicinal purposes in the treatment of trypanosomiasis, amebiasis, and leukemia. It has also been found as a contaminant in herbal remedies and drugs such as opium.58 There are widespread reports of chronic poisoning associated with arsenic-contaminated drinking water in underdeveloped countries.59 Arsenic poisoning should be suspected if compatible symptoms occur with the use of these products or these possible exposures.
Principles of Disease
Arsenic has no metabolic or biologic function. The elemental metal (As) is poorly water soluble and is considered nontoxic. Of the two inorganic forms, trivalent arsenite (As3+) is highly lipid soluble and is 5 to 10 times more toxic than the pentavalent arsenate (As5+) form. The more toxic trivalent arsenite form has a lower gastrointestinal absorption but is well absorbed by the skin. The pentavalent arsenate form, although less toxic, is water soluble and readily absorbed from the gastrointestinal tract. Absorbed arsenic is bound by hemoglobin, leukocytes, and plasma proteins. It is cleared from the intravascular compartment within 24 hours and concentrates in the liver, kidneys, spleen, lungs, and gastrointestinal tract. Arsenic crosses the placenta and can also accumulate in the fetus. Its affinity for sulfhydryl groups in keratin makes arsenic detectable in the hair, skin, and nails.60 Arsine (AsH3), a colorless and almost odorless gas, is extremely toxic.61 It is immediately lethal at 250 ppm.
The excretion of arsenic and its metabolites occurs mainly through the kidneys.
Pathophysiology
Arsenic binds avidly to sulfhydryl groups, inhibiting critical enzymes such as lactate dehydrogenase and glyceraldehyde-3-phosphate dehydrogenase, a critical step in glycolysis. It also disrupts oxidative phosphorylation by replacing phosphorus in the formation of high-energy phosphate bonds (arsenolysis).59 Arsine causes massive hemolysis, but the exact mechanism for this is poorly understood.62,63
Clinical Features
Acute exposure to arsine gas is characterized by severe hemolysis that is also associated with renal tubular injury. Gastrointestinal symptoms are common, and CNS and liver dysfunction can occur; the mortality rate is 25 to 30%.64 Exchange transfusions and plasma exchange have been used to remove arsine, which is tightly bound to the erythrocytes.64 Urinary alkalinization can be used to decrease renal deposition of hemoglobin and subsequent renal failure.
Acute gastrointestinal effects (nausea, vomiting, abdominal pain, and diarrhea) predominate as the initial manifestations of acute exposure to arsenic salts.65,66 These symptoms can be so severe as to be manifested with hematemesis and hematochezia. Within 30 to 60 minutes of exposure, patients complain of a metallic or garlicky taste. The patient can also have encephalopathy with seizures and coma, respiratory failure associated with acute respiratory distress syndrome, and dysrhythmias associated with cardiac conduction disturbances.67–69 In cases of severe poisoning, cardiovascular collapse and death ensue.70 Less common complications include hepatitis, rhabdomyolysis, hemolytic anemia, renal failure, unilateral facial nerve palsy, pancreatitis, pericarditis, pleuritis, and fetal demise (Box 157-1).66 The early arsenic poisoning syndrome may be misdiagnosed as gastroenteritis or sepsis.
Diagnostic Strategies
Normal arsenic levels are 5 µg/L or less in blood or less than 50 µg/day in a 24-hour urine collection, which is the best way to diagnose the poisoning. Any urine level above 100 µg/day or 50 µg/L necessitates treatment. A spot urine sample may be falsely low because urinary excretion of arsenic is intermittent. Seafood contains arsenobetaine, which can increase urinary arsenic excretion to as high as 1700 µg/L.71 Arsenobetaine, however, does not result in arsenic toxicity. For this reason, patients should refrain from eating seafood, specifically shellfish, before testing, or the laboratory should be asked to speciate the type of arsenic measured.72
Management
The initial management should address life-threatening conditions with supportive management of shock, dysrhythmias, and seizures. Activated charcoal does not adsorb arsenic and is of no value. Although there is no evidence for improved outcomes, orogastric lavage or whole-bowel irrigation should be considered for recent (<1 hour) ingestions or if radiopaque material is visualized on an abdominal radiograph. This recommendation is based on two case reports demonstrating clearance of radiopaque material on the abdominal radiographs of patients who had ingested arsenic. There are no studies demonstrating clinical efficacy of this intervention. Hemodialysis removes arsenic in the setting of acute renal failure.69 Exchange transfusions or plasma exchange can be considered early after an arsine exposure.64
With a known history of exposure in a symptomatic patient, chelation should start as early as possible without waiting for laboratory confirmation of the arsenic levels, which may take some time. Intramuscular dimercaprol is the preferred chelator in patients who are critically ill as described for lead poisoning. DMSA is a water-soluble analogue of dimercaprol that can be given orally.72,73 The use of DMSA is often limited by the violent gastroenteritis resulting from arsenic poisoning. D-Penicillamine has a high side effect profile, and its ability to chelate arsenic is inferior to that of dimercaprol and DMSA. Therefore, it should be used only when both dimercaprol and DMSA are unavailable. All patients receiving chelation for acute arsenic toxicity should be admitted. Chelation is not useful for arsine exposures. Workers should modify their habits to avoid further absorption, and repeated monthly 24-hour urine collections can follow arsenic excretion.
Table 157-4 summarizes available chelating agents with indications and doses.
Mercury
Mercury is a silver-white metal, familiar to most as the only metal that is liquid at room temperature. It has a long history of medicinal uses as an antiparasitic, a diuretic, a cathartic, an antiseptic, and a preservative in many vaccines. Significant poisoning in the home has occurred when relatively small amounts of spilled mercury, such as that contained in a sphygmomanometer, were aerosolized by vacuuming (which should be avoided) or when mercury was heated on the kitchen stove to extract gold from ore.74,75 Various other sources of mercury have also been implicated in intoxication. Because of many industrial uses that include the manufacture of fluorescent lights, batteries, polyvinyl chloride, and latex paint, mercury is a common pollutant of air and water. This has led to restrictions in the consumption of fish caught in many local waters, especially by pregnant women and children.76,77
Principles of Disease
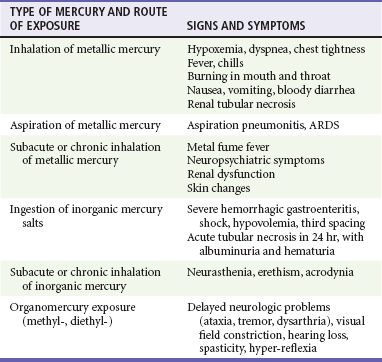
The most familiar form of mercury is elemental or metallic mercury, also known as quicksilver. A common route of exposure to elemental mercury is the inhalation of volatilized vapor.78 Aspiration of elemental mercury and intentional subcutaneous and intravenous injections also cause poisoning.79 After inhalation, 74% of the metallic mercury is retained in the lungs. This can result in severe pneumonitis and acute respiratory distress syndrome.80 Aspiration of elemental mercury results in primary pulmonary toxicity in addition to CNS and renal toxicities.81 Elemental mercury is not absorbed by the gastrointestinal tract, so ingestion does not normally lead to systemic toxicity unless it becomes trapped in diverticula. Mercury is absorbed through the skin at 1% of the rate of inhaled mercury and is not a concern.
The organic mercury compounds are categorized as either short chain (alkyl) or long chain (aryl). The major route of exposure to this type of mercury is through ingestion, but these compounds are also readily absorbed through the skin. These organic forms classically result in delayed neurotoxicity with prominent ataxia, tremor, dysarthria, and tunnel vision.76,82
Pathophysiology
Mercury has no known physiologic role. Mercury binds covalently to sulfhydryl groups, disturbing multiple cellular enzyme functions. Nephrotoxicity results from both direct damage and an immune reaction in the kidney.74 Inorganic salts have a direct corrosive effect on the gastrointestinal tract with third spacing and hemorrhage. Skin changes associated with mercury poisoning are also caused by an immune reaction. Mercury increases catecholamine levels by inhibition of catechol-O-methyltransferase, resulting in hypertension and tachycardia.83,84 Atrophy of the cerebellum, postcentral gyri, and calcarine areas of the brain correlates with the symptoms of ataxia and sensory and visual field disturbances.85
Clinical Features
The clinical manifestations of mercury poisoning depend on the acuity of the exposure, the route of exposure, and the chemical form of mercury. Inhalation of elemental mercury vapor results in the rapid onset of shortness of breath, fever, and chills that progress to pneumonitis and respiratory distress.75,80,86 Aspiration of liquid metallic mercury during medical procedures results in the rapid onset of tracheobronchial hemorrhage.
Acute ingestion of inorganic salts typically causes a corrosive gastroenteritis with third spacing and hemorrhage. Patients may have a metallic taste in the mouth and a grayish discoloration of the mucous membranes. Massive fluid loss results in shock and acute tubular necrosis. The manifestations of subacute or chronic inorganic mercury poisoning are neurologic (e.g., neurasthenia and erethism), renal (ranging from proteinuria to the nephrotic syndrome), and gastrointestinal (e.g., metallic taste, gingivostomatitis, loose teeth sensation, burning sensation in mouth, hypersalivation, and nausea).74,87
Exposure to organic mercury compounds is not associated with acute toxicity. Neurologic symptoms develop during weeks to months. The neurologic and teratogenic effects of chronic exposure to methylmercury were well illustrated by the tragic poisoning of the population of Minamata, Japan, by eating of fish caught in mercury-polluted water. Slowly progressive fatal CNS injury was described after a minor topical exposure to dimethylmercury in a laboratory researcher (Table 157-5).76,82,88
Diagnostic Strategies
Measurement of 24-hour urine mercury levels is the most helpful test in confirming exposure and monitoring the effectiveness of chelation. For organic mercury compounds, which undergo little urinary excretion, serum levels should be used to confirm the diagnosis. “Normal” mercury levels are considered to be less than 10 µg/L in the blood or less than 20 µg/L in the urine. Blood levels above 35 µg/L and urine levels above 150 µg/L require intervention. No information is available on chelation at levels between 20 and 150 µg/L. Fish can be contaminated with mercury, especially larger fish and those from certain bodies of water known by state health departments to be most polluted. Individuals eating these locally caught fish will have elevated mercury levels. Metallic mercury is radiopaque on plain radiographs, which can be ordered in cases of injection or ingestion of metallic mercury.79
Management
Initial management in the acutely poisoned patient is supportive. Gastric lavage with protein-containing solutions (e.g., milk and egg whites) has been suggested; however, no data exist to support this practice, and it is not recommended. Similarly, whole-bowel irrigation or any other gastric decontamination is without evidence of benefit. Charcoal adsorbs very little mercury and is not recommended unless another serious coingestant is suspected. Ingested metallic mercury is generally harmless unless its passage is impaired by entrapment in a diverticulum or the appendix.89
For acute inhalational exposures, the patient should be removed from the source and supportive management provided. There is no role for prophylactic antibiotics or steroids. Suction and postural drainage are indicated in cases of acute aspiration of metallic mercury. Self-injection of metallic mercury often requires surgical débridement of infiltrated tissue.79
Help in the management of mercury spills can be obtained from the local hazardous materials team and the local health department.78 Information on handling of mercury spills or compact fluorescent light bulb breakage can also be found at the Environmental Protection Agency websites at www.epa.gov/hg/spills/index.htm and www.epa.gov/cfl/cflcleanup.html, respectively. Sand or mercury decontamination kits that contain calcium polysulfide, which converts mercury to mercuric sulfide, should be used for elemental mercury spills. Absorbable surfaces such as carpets should be removed. Attempts to remove mercury by vacuuming should be avoided as this can volatilize the mercury and precipitate acute inhalational toxicity. Small spills, such as the contents of a home thermometer or a fluorescent bulb (amount = 30 mL or two tablespoons), can be scooped up with a stiff card or aspirated into a dropper placed onto a damp paper towel, sealed in a plastic bag, and disposed of as hazardous waste. Mercury placed in household trash is incinerated, is spread as an airborne contaminant, contributes to soil and water pollution, and ultimately accumulates in the food chain.
Chelation Therapy
Chelating agents have thiol groups that compete with the enzyme sulfhydryl groups that bind mercury. BAL is used for clinically significant acute inorganic mercury intoxication. Because it increases brain mercury levels in patients with methylmercury poisoning, BAL is contraindicated for patients poisoned with organic mercury compounds.90 Although DMSA is not currently approved by the U.S. Food and Drug Administration for this indication, it is used for both acute and chronic mercury poisoning and may be the best chelator for methylmercury. D-Penicillamine is also used but should be administered only after thorough gastrointestinal decontamination because mercury absorption from the intestinal lumen is enhanced by the penicillamines. DMPS may also be considered if other options are unavailable or contraindicated. Table 157-4 summarizes available chelating agents with indications and doses.







References
1. van Eijk, HG, de Jong, G. The physiology of iron, transferrin, and ferritin. Biol Trace Elem Res. 1992;35:13–24.
2. Fine, JS. Iron poisoning. Curr Probl Pediatr. 2000;30:71–90.
3. Robotham, JL, Lietman, PS. Acute iron poisoning. A review. Am J Dis Child. 1980;134:875–879.
4. Madiwale, T, Liebelt, E. Iron: Not a benign therapeutic drug. Current Opin Pediatr. 2006;18:174–179.
5. Manoguerra, AS, et al. Iron ingestion: An evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2005;43:553–570.
6. Aisen, P, Cohen, G, Kang, JO. Iron toxicosis. Int Rev Exp Pathol. 1990;31:1–46.
7. Evensen, SA, Forde, R, Opedal, I, Stormorken, H. Acute iron intoxication with abruptly reduced levels of vitamin K–dependent coagulation factors. Scand J Haematol. 1982;29:25–30.
8. Robertson, A, Tenenbein, M. Hepatotoxicity in acute iron poisoning. Hum Exp Toxicol. 2005;24:559–562.
9. Siff, JE, Meldon, SW, Tomassoni, AJ. Usefulness of the total iron binding capacity in the evaluation and treatment of acute iron overdose. Ann Emerg Med. 1999;33:73–76.
10. Lacouture, PG, Wason, S, Temple, AR, Wallace, DK, Lovejoy, FH, Jr. Emergency assessment of severity in iron overdose by clinical and laboratory methods. J Pediatr. 1981;99:89–91.
11. Chyka, PA, Butler, AY. Assessment of acute iron poisoning by laboratory and clinical observations. American J Emerg Med. 1993;11:99–103.
12. Ng, RC, Perry, K, Martin, DJ. Iron poisoning: Assessment of radiography in diagnosis and management. Clin Pediatr. 1979;18:614–616.
13. Tennebein, M, Wiseman, N, Yatscoff, RW. Gastrotomy and whole bowel irrigation in iron poisoning. Pediatr Emerg Care. 1991;7:286–288.
14. Position paper: Whole bowel irrigation. J Toxicol Clin Toxicol. 2004;42:843–854.
15. Tenenbein, M. Whole bowel irrigation in iron poisoning. J Pediatr. 1987;111:142–145.
16. Velez, LI, Gracia, R, Mills, LD, Shepherd, G, Feng, SY. Iron bezoar retained in colon despite 3 days of whole bowel irrigation. J Toxicol Clin Toxicol. 2004;42:653–656.
17. Kaczorowski, JM, Wax, PM. Five days of whole-bowel irrigation in a case of pediatric iron ingestion. Ann Emerg Med. 1996;27:258–263.
18. De Palma, G, et al. Toxicokinetics and toxicodynamics of elemental mercury following self-administration. Clin Toxicol (Phila). 2008;46:869–876.
19. Movassaghi, N, Purugganan, GG, Leikin, S. Comparison of exchange transfusion and deferoxamine in the treatment of acute iron poisoning. J Pediatr. 1969;75:604–608.
20. Tenenbein, M, Kowalski, S, Sienko, A, Bowden, DH, Adamson, IY. Pulmonary toxic effects of continuous desferrioxamine administration in acute iron poisoning. Lancet. 1992;339:699–701.
21. Khoury, S, Odeh, M, Oettinger, M. Deferoxamine treatment for acute iron intoxication in pregnancy. Acta Obstet Gynecol Scand. 1995;74:756–757.
22. Tran, T, Wax, JR, Philput, C, Steinfeld, JD, Ingardia, CJ. Intentional iron overdose in pregnancy—management and outcome. J Emerg Med. 2000;18:225–228.
23. Wythe, E, Osterloh, J, Becker, C. The reliability of serum iron levels after deferoxamine treatment: An in vitro study. Vet Hum Toxicol. 1986;28:478.
24. Proudfoot, AT. Antidotes: Benefits and risks. Toxicol Lett. 1995;82-83:779–783.
25. Gallant, T, Freedman, MH, Vellend, H, Francombe, WH. Yersinia sepsis in patients with iron overload treated with deferoxamine. N Engl J Med. 1986;314:1643.
26. Mazzoleni, G, deSa, D, Gately, J, Riddell, RH. Yersinia enterocolitica infection with ileal perforation associated with iron overload and deferoxamine therapy. Dig Dis Sci. 1991;36:1154–1160.
27. Warniment, C, Tsang, K, Galazka, SS. Lead poisoning in children. American Fam Physician. 2010;81:751–757.
28. Akhtar, AJ, Funnyé, AS, Akanno, J. Gunshot-induced plumbism in an adult male. J Natl Med Assoc. 2003;95:986–990.
29. Markowitz, G, Rosner, D. “Cater to the children”: The role of the lead industry in a public health tragedy, 1900-1955. Am J Public Health. 2000;90:36–46.
30. Morgan, BW, Barnes, L, Parramore, CS, Kaufmann, RB. Elevated blood lead levels associated with the consumption of moonshine among emergency department patients in Atlanta, Georgia. Ann Emerg Med. 2003;42:351–358.
31. Tait, PA, Vora, A, James, S, Fitzgerald, DJ, Pester, BA. Severe congenital lead poisoning in a preterm infant due to a herbal remedy. Med J Aust. 2002;177:193–195.
32. Lin, CG, Schaider, LA, Brabander, DJ, Woolf, AD. Pediatric lead exposure from imported Indian spices and cultural powders. Pediatrics. 2010;125:e828–e835.
33. Renner, R. Sick picnic—evaluating the legacy of lead ammunitions. Environ Sci Technol. 2010;44:853–854.
34. Garza, A, Vega, R, Soto, E. Cellular mechanisms of lead neurotoxicity. Med Sci Monit. 2006;12:RA57–RA65.
35. Lidsky, TI, Schneider, JS. Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain. 2003;126(Pt 1):5–19.
36. Canfield, RL, et al. Intellectual impairment in children with blood lead concentrations below 10 µg per deciliter. N Engl J Med. 2003;348:1517–1526.
37. Nevin, R. Understanding international crime trends: The legacy of preschool lead exposure. Environ Res. 2007;104:315–336.
38. Chiodo, LM, et al. Blood lead levels and specific attention effects in young children. Neurotoxicol Teratol. 2007;29:538–546.
39. Park, SK, et al. Cumulative lead exposure and age-related hearing loss: The VA Normative Aging Study. Hear Res. 2010;269:48–55.
40. Ekong, EB, Jaar, BG, Weaver, VM. Lead-related nephrotoxicity: A review of the epidemiologic evidence. Kidney Int. 2006;70:2074–2084.
41. Cheng, Y, Schwartz, J, Sparrow, D, et al. Bone lead and blood lead levels in relation to baseline blood pressure and the prospective development of hypertension: The Normative Aging Study. Am J Epidemiol. 2001;153:164–171.
42. Nevin, R. How lead exposure relates to temporal changes in IQ, violent crime, and unwed pregnancy. Environ Res. 2000;83:1–22.
43. al Khayat, A, Menon, NS, Alidina, MR. Acute lead encephalopathy in early infancy—clinical presentation and outcome. Ann Trop Paediatr. 1997;17:39–44.
44. Graziano, JH. Validity of lead exposure markers in diagnosis and surveillance. Clin Chem. 1994;40(Pt 2):1387–1390.
45. Gordon, JN, Taylor, A, Bennett, PN. Lead poisoning: Case studies. Br J Clin Pharmacol. 2002;53:451–458.
46. Centers for Disease Control and Prevention. Interpreting and managing blood lead levels <10 µg/dL in children and reducing childhood exposures to lead. Recommendations of CDC’s Advisory Committee on Childhood Lead Poisoning Prevention. MMWR Recomm Rep. 2007;56:1–14.
47. McNutt, TK, Chambers-Emerson, J, Dethlefsen, M, Shah, R. Bite the bullet: Lead poisoning after ingestion of 206 lead bullets. Vet Hum Toxicol. 2001;43:288–289.
48. Roberge, RJ, Martin, TG. Whole bowel irrigation in an acute oral lead intoxication. Am J Emerg Med. 1992;10:577–583.
49. Vilensky, JA, Redman, K. British anti-Lewisite (dimercaprol): An amazing history. Ann Emerg Med. 2003;41:378–383.
50. Chisolm, JJ, Jr. Safety and efficacy of meso-2,3-dimercaptosuccinic acid (DMSA) in children with elevated blood lead concentrations. J Toxicol Clin Toxicol. 2000;38:365–375.
51. Centers for Disease Control and Prevention. Deaths associated with hypocalcemia from chelation therapy—Texas, Pennsylvania, and Oregon, 2003-2005. MMWR Morb Mortal Wkly Rep. 2006;55:204–207.
52. Torres-Alanis, O, Garza-Ocanas, L, Pineyro-Lopez, A. Effect of meso-2,3-dimercaptosuccinic acid on urinary lead excretion in exposed men. Hum Exp Toxicol. 2002;21:573–577.
53. Safety and efficacy of succimer in toddlers with blood lead levels of 20-44 µg/dL. Treatment of Lead-Exposed Children (TLC) Trial Group. Pediatr Res. 2000;48:593–599.
54. Chisolm, JJ, Jr. BAL, EDTA, DMSA and DMPS in the treatment of lead poisoning in children. J Toxicol Clin Toxicol. 1992;30:493–504.
55. Aposhian, HV, et al. Human studies with the chelating agents, DMPS and DMSA. J Toxicol Clin Toxicol. 1992;30:505–528.
56. Durback, LF, Wedin, GP, Seidler, DE. Management of lead foreign body ingestion. J Toxicol Clin Toxicol. 1989;27:173–182.
57. American Academy of Pediatrics Committee on Environmental Health. Lead poisoning: From screening to primary prevention. Pediatrics. 1993;92:176–183.
58. Saper, RB, et al. Heavy metal content of ayurvedic herbal medicine products. JAMA. 2004;292:2868–2873.
59. Lubin, JH, Beane Freeman, LE, Cantor, KP. Inorganic arsenic in drinking water: An evolving public health concern. J Natl Cancer Inst. 2007;99:906–907.
60. Daniel, CR, 3rd., Piraccini, BM, Tosti, A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258–261.
61. Song, Y, et al. Severe acute arsine poisoning treated by plasma exchange. Clin Toxicol (Phila). 2007;45:721–727.
62. Pakulska, D, Czerczak, S. Hazardous effects of arsine: A short review. Int J Occup Med Environ Health. 2006;19:36–44.
63. Rael, LT, Ayala-Fierro, F, Bar-Or, R, Carter, DE, Barber, DS. Interaction of arsine with hemoglobin in arsine-induced hemolysis. Toxicol Sci. 2006;90:142–148.
64. Bonastre Blanco, E, Domingo Garau, A, Cols Roig, M, Panzino Occhiuzzo, F, Vilar Escrigas, P. Accidental iron poisoning [in Spanish]. An Pediatr (Barc). 2010;73:373–375.
65. Cullen, NM, Wolf, LR, St Clair, D. Pediatric arsenic ingestion. Am J Emerg Med. 1995;13:432–435.
66. Roses, OE, et al. Mass poisoning by sodium arsenite. J Toxicol Clin Toxicol. 1991;29:209–213.
67. Beckman, KJ, Bauman, JL, Pimental, PA, Garrard, C, Hariman, RJ. Arsenic-induced torsade de pointes. Crit Care Med. 1991;19:290–292.
68. Bolliger, CT, van Zijl, P, Louw, JA. Multiple organ failure with the adult respiratory distress syndrome in homicidal arsenic poisoning. Respiration. 1992;59:57–61.
69. Mathieu, D, et al. Massive arsenic poisoning—effect of hemodialysis and dimercaprol on arsenic kinetics. Intensive Care Med. 1992;18:47–50.
70. Hantson, P, Haufroid, V, Buchet, JP, Mahieu, P. Acute arsenic poisoning treated by intravenous dimercaptosuccinic acid (DMSA) and combined extrarenal epuration techniques. J Toxicol Clin Toxicol. 2003;41:1–6.
71. Arbouine, MW, Wilson, HK. The effect of seafood consumption on the assessment of occupational exposure to arsenic by urinary arsenic speciation measurements. J Trace Elem Electrolytes Health Dis. 1992;6:153–160.
72. Kales, SN, Huyck, KL, Goldman, RH. Elevated urine arsenic: Un-speciated results lead to unnecessary concern and further evaluations. J Anal Toxicol. 2006;30:80–85.
73. Shum, S, Whitehead, J, Vaughn, L, Shum, S, Hale, T. Chelation of organoarsenate with dimercaptosuccinic acid. Vet Hum Toxicol. 1995;37:239–242.
74. Kazantzis, G. Mercury exposure and early effects: An overview. Med Lav. 2002;93:139–147.
75. Rennie, AC, McGregor-Schuerman, M, Dale, IM, Robinson, C, McWilliam, R. Mercury poisoning after spillage at home from a sphygmomanometer on loan from hospital. BMJ. 1999;319:366–367.
76. Kulig, K. A tragic reminder about organic mercury. N Engl J Med. 1998;338:1692–1694.
77. Kondro, W. Mercury disposal sole health concern with fluorescent lights. CMAJ. 2007;177:136–137.
78. Cherry, D, Lowry, L, Velez, L, Cotrell, C, Keyes, DC. Elemental mercury poisoning in a family of seven. Fam Community Health. 2002;24:1–8.
79. Soo, YO, Wong, CH, Griffith, JF, Chan, TY. Subcutaneous injection of metallic mercury. Hum Exp Toxicol. 2003;22:345–348.
80. Solis, MT, Yuen, E, Cortez, PS, Goebel, PJ. Family poisoned by mercury vapor inhalation. Am J Emerg Med. 2000;18:599–602.
81. Lech, T, Goszcz, H. Poisoning from aspiration of elemental mercury. Clin Toxicol (Phila). 2006;44:333–336.
82. Nierenberg, DW, et al. Delayed cerebellar disease and death after accidental exposure to dimethylmercury. N Engl J Med. 1998;338:1672–1676.
83. Wossmann, W, Kohl, M, Grüning, G, Bucsky, P. Mercury intoxication presenting with hypertension and tachycardia. Arch Dis Child. 1999;80:556–557.
84. Torres, AD, Rai, AN, Hardiek, ML. Mercury intoxication and arterial hypertension: Report of two patients and review of the literature. Pediatrics. 2000;105:E34.
85. Siegler, RW, Nierenberg, DW, Hickey, WF. Fatal poisoning from liquid dimethylmercury: A neuropathologic study. Human Pathol. 1999;30:720–723.
86. Taueg, C, Sanfilippo, DJ, Rowens, B, Szejda, J, Hesse, JL. Acute and chronic poisoning from residential exposures to elemental mercury—Michigan, 1989-1990. J Toxicol Clin Toxicol. 1992;30:63–67.
87. Rosenman, KD, Valciukas, JA, Glickman, L, Meyers, BR, Cinotti, A. Sensitive indicators of inorganic mercury toxicity. Arch Environ Health. 1986;41:208–215.
88. Clarkson, TW, Magos, L, Myers, GJ. The toxicology of mercury—current exposures and clinical manifestations. N Engl J Med. 2003;349:1731–1737.
89. McKinney, PE. Elemental mercury in the appendix: An unusual complication of a Mexican-American folk remedy. J Toxicol. 1999;37:103–107.
90. Baum, CR. Treatment of mercury intoxication. Curr Opin Pediatr. 1999;11:265–268.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 157
Heavy Metals
Iron
Iron, which is essential to the function of hemoglobin, myoglobin, many cytochromes, and many catalytic enzymes, can be extremely toxic when levels are elevated after an overdose or from accumulation in disease states.1 The acute ingestion of iron is especially hazardous to children. Ingestions of pediatric multivitamin formulations are the most common iron exposures. These occur in children younger than 6 years and are minimally toxic. Life-threatening toxicity is associated with ingestion of potent adult preparations, such as prenatal vitamins. Serious iron ingestions in adults are usually associated with suicide attempts.2
Principles of Disease
In assessment of the severity of an iron exposure, it is important to refer to the amount of elemental iron ingested because the toxicity of an iron compound depends on the amount of elemental iron it contains (Table 157-1). Different formulations of iron salts contain different percentages of elemental iron. The total amount of elemental iron ingested can be approximated by multiplying the estimated number of tablets by the fraction of elemental iron contained in the tablet. Manufacturers are also required to list the amount of elemental iron per tablet. Ingestions of less than 20 mg/kg of elemental iron usually cause no symptoms. Ingestion of 20 to 60 mg/kg results in mild to moderate symptoms, and ingestion of more than 60 mg/kg may lead to severe morbidity and mortality. Although the dose of elemental iron associated with 50% mortality (LD50) is 200 to 250 mg/kg, doses as small as 130 mg of elemental iron have been lethal in children.3 Newer forms of iron are carbonyl iron and iron polysaccharide; both are non-ionic and associated with lower toxicity. Neither form is directly corrosive, and the conversion to the iron ion, which is responsible for toxicity, is very slow. There are no reported cases of serious toxicity or death from the ingestion of these compounds.4,5
Pathophysiology
Iron has two distinct toxic effects: (1) it causes direct caustic injury to the gastrointestinal mucosa, and (2) it impairs cellular metabolism, primarily of the heart, liver, and central nervous system (CNS). The caustic effects of iron on the gut cause the initial symptoms of vomiting, diarrhea, and abdominal pain. Hemorrhagic necrosis of gastric or intestinal mucosa can lead to bleeding, perforation, and peritonitis.3 Unbound (free) iron moves into cells and localizes near the mitochondrial cristae, resulting in uncoupling of oxidative phosphorylation and impairment of adenosine triphosphate synthesis. Cell membranes are injured by free radical–mediated lipid peroxidation.3,6
Iron increases capillary permeability and both arteriolar dilation and venodilation, resulting in hypotension. Direct myocardial toxicity decreases cardiac output. Hydration of the iron molecule creates an excess of unbuffered protons, worsening metabolic acidosis.3 These effects, combined with severe gastrointestinal fluid losses, can lead to shock, cardiovascular collapse, and death.3
Clinical Features
The clinical effects of acute iron poisoning occur in five stages. Not every patient goes through every phase. Phase I reflects the corrosive effects of iron on the gut. Vomiting occurs within 80 minutes of ingestion in more than 90% of symptomatic cases. Diarrhea, which can be bloody, soon follows. Phase II represents an apparent (but not complete) recovery that lasts less than 24 hours but can extend up to 2 days. Most patients recover after this point. Phase III is characterized by the recurrence of gastrointestinal symptoms, severe lethargy or coma, anion gap metabolic acidosis, leukocytosis, coagulopathy, renal failure, and cardiovascular collapse. Serum iron levels may have fallen to normal during this phase because of distribution of iron into the tissues. Metabolic derangements due to iron poisoning include hypoglycemia, leukocytosis, and severe lactic acidosis from hypoperfusion and interference with cellular respiration. Early coagulation defects are probably related to direct effects of iron on vitamin K–dependent clotting factors.7 Later coagulation defects are due to hepatic failure. Hypoglycemia and elevations of bilirubin and aspartate and alanine transaminases are other markers of hepatotoxicity. Phase IV, characterized by fulminant hepatic failure, occurs 2 to 5 days after ingestion. This is relatively rare, appears to be dose related, and is usually fatal.8 Phase V represents the consequences of healing of the injured gastrointestinal mucosa. It is characterized by pyloric or proximal bowel scarring, which is sometimes associated with obstruction.
Diagnostic Strategies
The presence of gastrointestinal symptoms suggests a potentially serious ingestion, whereas their absence is reassuring.5 A serum iron level measured at its peak, 3 to 5 hours after ingestion, is the most useful laboratory test to evaluate the potential severity of an iron overdose. Sustained-release or enteric-coated preparations may have erratic absorption, so a second level 6 to 8 hours after ingestion should also be checked. Peak serum iron levels below 350 µg/dL are generally associated with minimal toxicity; 350 to 500 µg/dL, with moderate toxicity; and above 500 µg/dL, with potentially severe toxicity (Table 157-2).5 Because iron is rapidly cleared from the serum and deposited in the liver, the level of iron after a substantial ingestion may be deceptively low if it is measured after its peak. TIBC is a crude test and is not useful to gauge the severity of iron poisoning.9
Table 157-2
Toxicity of Iron by Amount Ingested and Peak Serum Levels
| ELEMENTAL IRON (mg/kg) | PEAK SERUM IRON LEVEL (µg/dL) | TOXICITY |
| <20 | 50-150 | None |
| 20-40 | 150-300 | Mild |
| 40-60 | 300-500 | Moderate |
| >60 | >500 | Severe |
When serum iron levels are not immediately available, elevations of the serum glucose level above 150 mg/dL and leukocyte count above 15,000 are 100% specific for prediction of serum iron levels of more than 300 µg/mL. However, a sensitivity of only 50% limits their sole use as indicators of toxicity.10
Most tablets that contain a significant amount of elemental iron are radiopaque, although a false-negative radiograph may occur with chewable, liquid, and completely dissolved iron compounds. The presence of tablets on a radiograph correlates with the severity of the ingestion (Fig. 157-1).11,12 Repeated radiographs can also demonstrate the efficacy of gastrointestinal decontamination efforts.
Management
Decontamination may prevent absorption of additional iron from the gastrointestinal tract and can be performed concomitantly with interventions that address drug that has already been absorbed (see later section on deferoxamine). Activated charcoal does not bind iron, and neither gastric lavage nor ipecac effectively removes large numbers of pills. Iron tablets clump together as their outer coatings dissolve, often forming large pharmacobezoars. Whole-bowel irrigation is generally the preferred method of decontamination for significant iron ingestions.13 This recommendation is supported by the position statement of the American Academy of Clinical Toxicology and the European Association of Poison Centres and Clinical Toxicologists; it is based on case reports and the lack of effective alternative decontamination techniques for this potentially deadly toxin.14
For significant ingestions (>20 mg/kg of elemental iron), especially when tablets are identified on the abdominal radiograph, whole-bowel irrigation with a polyethylene glycol–electrolyte lavage solution (PEG-ELS; CoLyte, NuLytely, or GoLytely) should be initiated.14 The solution is either taken orally or, more often, administered through a small nasogastric tube.15 The usual rate of administration of PEG-ELS is 20 to 40 mL/kg/hr in young children and 1.5 to 2 L/hr in teenagers or adults, continued until the rectal effluent is clear and there is no radiographic evidence of pill fragments. This technique has been used in children, adolescents, and pregnant women without serious complications or electrolyte disturbances.13,16,17 Common side effects include nausea, vomiting, abdominal cramping, and bloating. Whole-bowel irrigation is contraindicated in the presence of bowel obstruction, perforation, or ileus.14
Hemodialysis and hemoperfusion are not effective in the removal of iron because of its large volume of distribution. Early exchange transfusions have been recommended for severely symptomatic patients with serum iron levels exceeding 1000 µg/dL.18,19
Deferoxamine
Deferoxamine chelates iron to form the water-soluble compound ferrioxamine, which is renally excreted and can be dialyzed; 100 mg will chelate 9.35 mg of elemental iron.3 Deferoxamine may also limit the entrance of iron into the cell and chelate intracellular iron. Because of its short half-life, it is administered as a continuous infusion at 15 mg/kg/hr for up to 24 hours.20 The maximum rate of administration is 35 mg/kg/hr. Rapid administration of deferoxamine can lead to hypotension, which resolves by reducing the initial rate of the infusion and slowly increasing it to the desired rate. Pregnancy is not a contraindication to deferoxamine.4,21,22 However, the pre-pregnancy weight should be used to calculate the dose. Deferoxamine can falsely lower serum iron levels, so measurements should be taken before its administration.23
The presence of ferrioxamine turns the urine a “vin rosé” color, which reflects the excretion of chelated iron. The deferoxamine challenge test, which relied on detection of this color change, has been used to detect the presence of free iron in the serum. However, the color change is difficult to detect, especially when the urine is dilute, with false-negative results even in cases of serious poisoning.24
Deferoxamine has been associated with acute respiratory distress syndrome and also with Yersinia sepsis. The pulmonary complications are usually related to high-dose deferoxamine for durations longer than 24 hours.20 The presence of fever and sepsis in a patient who has recently been treated for iron poisoning should prompt suspicion for Yersinia sepsis.25,26
Disposition
The asymptomatic patient who is reliably known to have ingested less than 20 mg/kg of elemental iron and has a normal abdominal radiograph can be observed without further therapy. The patient can be discharged if he or she remains asymptomatic after 6 hours of observation.5
The patient who has ingested more than 20 mg/kg of elemental iron or has pills visible on an abdominal radiograph should receive whole-bowel irrigation.23 Follow-up abdominal radiographs can verify adequate gastrointestinal decontamination. The serum iron concentration should be checked 3 to 5 hours after ingestion. A second iron level 6 to 8 hours after ingestion should be decreasing. If peak levels are less than 300 µg/dL and are not rising and the patient is asymptomatic during 6 hours of observation, the patient can be discharged home. Moderate gastrointestinal toxicity can be expected with peak levels of 300 to 500 µg/dL. Deferoxamine should be used if these patients have symptoms of systemic toxicity evidenced by metabolic acidosis, repetitive vomiting, toxic appearance, lethargy, hypotension, or signs of shock. Patients with a serum iron level above 500 µg/dL require chelation with deferoxamine regardless of symptoms.4 If serum iron levels are not available, a normal serum glucose level and leukocyte count suggest a low iron level; if there are no signs or symptoms of toxicity during the 6-hour postingestion period and the abdominal radiographs do not show pills in the gastrointestinal tract, the patient can be sent home.
Lead
Lead poisoning is a disease of industrialization and has become the most common environmental toxicologic problem in the United States.27 Most exposures are from inhalation of lead dust or fumes or ingestion of contaminated substances, such as paint chips. Less often, it results from direct skin contact with organic lead compounds or from retained bullets in or near joints.28 Approximately 3 to 4 million children (1 in 20) in the United States have toxic blood lead levels (BLLs), defined currently as levels exceeding 10 µg/dL.29 Although the addition of lead to household paint and gasoline was banned in the United States in the 1970s, residual lead-based paint is still found in 30 million homes.29 Other sources of toxic lead ingestions include curtain weights, buckshot, fishing weights, lead-contaminated soil or water, bootleg whiskey (“moonshine”), food or beverages stored or prepared in lead-soldered cans, lead-glazed pottery, and lead crystal decanters.30 Herbal and folk remedies, toys, and numerous products imported from Asia and Mexico can also contain dangerous amounts of lead.31,32 This has resulted in multiple drug and toy recalls in the United States. Lead found in buckshot can cause toxic effects in those who eat wild game.33
Principles of Disease
Pathophysiology
There is no known physiologic need for lead. Lead binds to sulfhydryl groups and other ligands and interferes with critical enzymatic reactions.12 Its toxic effects are most prominent in the hematopoietic, neurologic, and renal systems.34
Anemia, the classic manifestation of hematopoietic toxicity, may be either normochromic or hypochromic. The severity of the anemia correlates directly with the BLL. Inhibition of heme biosynthesis results in the accumulation of heme precursors, such as D-aminolevulinic acid and protoporphyrin. In the peripheral nervous system, segmental demyelination and degeneration of motor axons result in peripheral neuropathies. Wristdrop and footdrop are characteristic of adult lead poisoning. Lead toxicity also causes a host of neuropsychiatric disorders. In children, elevated BLL is associated with decreased intelligence (IQ) scores, hyperactivity, decreased attention span, overaggressive behavior, learning disabilities, and criminal behavior.35–38 It has also been associated with subclinical sensorineural hearing loss.39 Lead nephropathy is characterized by fibrosis in the proximal tubules, with relative sparing of the glomeruli. Hyperuricemic gout (“saturnine gout”) can result from increased reuptake of uric acid by the tubular cells.40 Lead poisoning also correlates with hypertension.41 Adults and children with severe acute toxicity may present with lead encephalopathy associated with increased capillary permeability and cerebral edema.42,43
Clinical Features
Acute exposure to lead can result in symptomatic poisoning. “Lead colic” is characterized by cramping abdominal pain with nausea, vomiting, constipation, and, occasionally, diarrhea.43 Other characteristic symptoms and signs of acute toxicity include fatigue, anemia, peripheral neuropathy, renal impairment, and hepatic and CNS dysfunction. The CNS toxicity may be manifested as mild headache or personality changes to full-blown encephalopathy with coma, convulsions, and papilledema. Permanent neurologic and behavioral sequelae may occur.
Diagnostic Strategies
Although capillary lead levels correlate well with BLLs, the most informative biomarker is a BLL.44–46 The Centers for Disease Control and Prevention has defined a chronic BLL of more than 10 µg/dL as toxic for a child. Acute exposure can result in levels above 100 µg/dL (Table 157-3). Other ancillary data include findings on complete blood cell count, serum glucose level, blood urea nitrogen and creatinine concentrations, electrolyte levels, and urinalysis. A peripheral smear may show basophilic stippling. Markers of hepatic injury may be elevated after acute exposure. Because lead-containing paints and objects are radiopaque when lead is present in sufficient quantities, radiographs can confirm acute ingestion and also monitor the effectiveness of whole-bowel irrigation. In cases of altered mental status, seizures, or coma, a computed tomography scan of the head will show cerebral edema associated with acute lead encephalopathy and rule out other causes of these symptoms. In children, plain radiographs of the wrist and knees may show increased metaphyseal activity manifested as “lead bands” or “lead lines” that are characteristic of chronic exposures.
Management
Acute lead encephalopathy can be rapidly fatal. The initial goals in management are identification and treatment of all life-threatening conditions, followed by prevention of further exposure to lead, minimization of ingested lead absorption, enhancement of its elimination, and prevention or reversal of pathologic cell changes. Standard measures to control cerebral edema, including intubation and neurosurgical consultation for invasive monitoring of intracranial pressure, are indicated. When a severe poisoning is associated with recent ingestion or if radiopacities are seen on the radiograph, decontamination with whole-bowel irrigation should be considered.47,48
[/not-level-membership-for-emergency-medicine-category]
