Chapter 19 Heart Failure
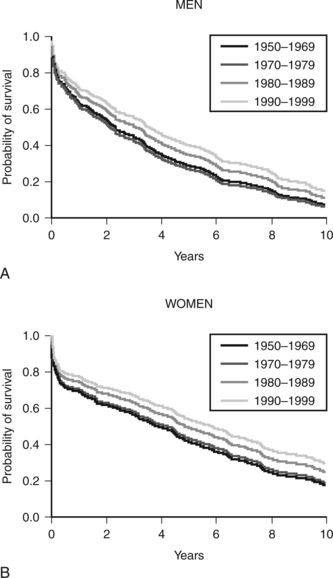
Heart failure is a clinical syndrome that occurs as the end result of any structural or functional cardiac disorder that impairs the ability of the ventricles to fill with or eject blood. In Western countries coronary artery disease and hypertension are its most common causes. In the United States heart failure affects more than 5 million people and results in significant impairment of quality of life, frequent hospitalizations, and poor long-term survival rates (Fig. 19-1).1 The number of deaths from heart failure is increasing despite advances in diagnosis and treatment. This increase is due in part to an aging population and to improved survival after acute myocardial infarction.2
ACUTE HEART FAILURE
Pathophysiology
Impaired left ventricular function leads to increased left ventricular end-diastolic pressure (LVEDP) and reduced stroke volume. Increased LVEDP causes increased pulmonary capillary hydrostatic pressure, which results in the increased filtration of protein-poor fluid into the pulmonary interstitium (Equation 1-12). Moderately increased left atrial pressure (18 to 25 mmHg) results in interstitial edema, which is defined as fluid accumulation in the peribronchovascular spaces; severely increased left atrial pressure (>25 mmHg) results in alveolar edema, which is defined as fluid accumulation in the alveoli.3 Interstitial edema reduces lung compliance and leads to increased work of breathing. Alveolar edema causes increased work of breathing and intrapulmonary shunting, leading to hypoxemia. Acute pulmonary edema can occur in the absence of increased circulating volume or pathologic fluid retention.
Systemic hypoperfusion leads to lactic acidosis and oliguric renal failure. Severe acidemia aggravates myocardial dysfunction, whereas renal dysfunction exacerbates fluid retention. Over a period of days fluid retention leads to elevated systemic venous pressure and dependent edema, even in the absence of right ventricular dysfunction. Catecholamines and other substances that evolve during cardiogenic shock have important effects on intermediary metabolism (see Chapter 2) that result in hyperglycemia, lipolysis, and increased protein catabolism.
Systemic inflammation also occurs with heart failure, cardiogenic shock, and cardiac arrest. Patients with severe heart failure have increased levels of inducible nitric oxide synthetase and various inflammatory cytokines, such as tumor necrosis factor-α and interleukins 1 and 6.4 The inducible form of nitric oxide synthetase is involved in the regulation of vascular tone in various disease states and, along with tumor necrosis factor-α, it mediates myocardial depression. In patients resuscitated from cardiac arrest, systemic inflammation may produce a hemodynamic state that resembles septic shock, with pathologic vasodilation and capillary leak.5 In addition, there is commonly fever, leukocytosis, and elevations in acute-phase reactants.
Based on a simple pump-failure model of cardiogenic shock, patients would be expected to have profoundly depressed ventricular function (i.e., ejection fraction <20%) and elevated systemic vascular resistance. However, many of the patients with cardiogenic shock in the SHOCK trial registry (see later discussion) did not have elevated systemic vascular resistance, and their average ejection fraction was 30%—that is, only moderate to severely depressed.6 These findings may be explained as being a severe systemic inflammatory response to myocardial infarction, shock, and cardiac arrest.
Classification and Clinical Presentation
Acute heart failure may be classified on the basis of the presenting clinical syndrome, the hemodynamic profile, and the severity of symptoms. The European Society of Cardiology system recognizes clinical syndromes7: (1) mild acute decompensated heart failure characterized by fluid overload with or without hypoperfusion; (2) hypertensive acute heart failure in which there is hypertension, preserved left ventricular systolic function, and acute pulmonary edema; (3) acute heart failure with pulmonary edema; (4) cardiogenic shock; (5) high output failure; (6) right heart failure. Acute heart failure may also be classified on the basis of hemodynamic profile. Patients are categorized as “warm” or “cold” on the basis of their systemic perfusion and as “wet” or “dry” on the basis of the presence of congestion (either systemic or pulmonary). Class I is warm and dry, Class II is warm and wet, Class III is cold and dry, and Class IV is cold and wet. After myocardial infarction, the Killip classification is widely used for grading heart failure (Table 19-1).8
Table 19-1 Killip Classification of Heart Failure Following Myocardial Infarction
| Class I | No signs of heart failure |
|---|---|
| Class II | Crackles, S3 gallop, elevated jugular venous pressure |
| Class III | Pulmonary edema |
| Class IV | Cardiogenic shock |
From Killip T 3rd, Kimball JT: Treatment of myocardial infarction in a coronary care unit: a two-year experience with 250 patients. Am J Cardiol 20:457-464, 1967.
Pulmonary Edema
Pulmonary edema may be cardiogenic or noncardiogenic. A pulmonary artery wedge pressure (PAWP) greater than 18 mmHg defines cardiogenic pulmonary edema. Pulmonary edema that occurs following a myocardial infarction is typically due to left ventricular systolic dysfunction. It may or may not be associated with systemic hypoperfusion. In contrast, “flash” pulmonary edema is commonly associated with preserved systolic function,9,10 and the trigger for decompensation is often diastolic dysfunction secondary to a hypertensive crisis.9 Other causes of cardiogenic pulmonary edema include acute aortic and mitral valve disease, arrhythmias, atrial myxoma, and high-output states, such as anemia and thyrotoxicosis.
Cardiogenic Shock
Cardiogenic shock is hypotension (systolic blood pressure below 90 mmHg for more than 30 minutes or systolic blood pressure above 90 mmHg with vasoactive support) in association with tissue hypoperfusion or, if a pulmonary artery catheter (PAC) is in situ, a cardiac output of less than 2.2 l/min/m2.11
The main cause of cardiogenic shock is left ventricular systolic dysfunction secondary to acute STEMI. Cardiogenic shock occurs in about 4% of patients following STEMI.12 Risk factors for cardiogenic shock include extensive infarction, anterior infarction, previous myocardial infarction, low ejection fraction, and multivessel coronary artery disease. Shock is more common in myocardial infarctions that occur in the elderly, in diabetics, and in patients who have histories of cerebral or peripheral vascular disease. Only very rarely does cardiogenic shock develop following non-ST-segment elevation myocardial infarction. Most patients who develop cardiogenic shock do so within 24 hours of the myocardial infarction. In the trial known as should we emergently revascularize Occluded Coronaries in cardiogenic shock (the SHOCK trial; see subsequent discussion), 11% of patients had shock at the time of hospital admission and 75% had developed shock by 24 hours.11
Shock that occurs beyond 24 hours should raise the possibility of a mechanical complication of myocardial infarction, such as papillary muscle rupture, ventricular septal rupture, or ventricular free wall rupture. Another cause of shock following myocardial infarction is right ventricular failure. Approximately 25% of patients with inferior myocardial infarction have right ventricular infarction,13 and a proportion of these patients develop right ventricular failure. Cardiogenic shock can also occur for reasons unrelated to myocardial infarction, such as pericardial tamponade, fulminant myocarditis, or acute valvular dysfunction.
Clinical signs of tissue hypoperfusion include cool clammy skin, peripheral cyanosis, oliguria, tachycardia, confusion, and drowsiness. Patients with right ventricular failure may have distended neck veins with visible pulsations (V waves; see Figure 8-11) that are indicative of tricuspid regurgitation. Biochemical abnormalities include hyperglycemia, lactic acidosis, increased blood urea nitrogen and creatinine, and elevations in hepatic transaminases.
Cardiac arrest, typically ventricular fibrillation, occurs in about 3% of patients with STEMI12 and may be the presenting feature. At least 25% of patients who subsequently develop cardiogenic shock will have had a cardiac arrest, usually at presentation.11 Following cardiac arrest, patients may suffer hypoxic ischemic encephalopathy (HIE; see Chapter 37). Cardiogenic shock is the leading cause of death after acute myocardial infarction and is an important cause of multiple organ dysfunction syndrome (MODS).
Diagnosis
The diagnosis of acute heart failure rests on clinical assessment, electrocardiography, chest radiography, echocardiography, and laboratory tests. Diagnostic assessment should be directed at confirming the diagnosis of heart failure and identifying the cause. The differential diagnoses of pulmonary edema and cardiogenic shock are listed in Table 19-2.
Table 19-2 Differential Diagnosis of Noncardiogenic Pulmonary Edema and Cardiogenic Shock
| Pulmonary Edema Without Shock |
| ARDS |
| Transfusion-related acute lung injury |
| Neurogenic pulmonary edema |
| Cardiogenic Shock |
| Massive pulmonary embolism |
| Occult hypovolemic shock |
| Septic-shock-induced severe myocardial depression |
| Anaphylaxis |
| Aortic dissection with tamponade and/or acute aortic regurgitation |
| Pericardial tamponade (e.g., due to malignant effusion) |
| Mechanical complication of myocardial infarction: |
| Acute mitral regurgitation |
| Ventricular septal rupture |
| Ventricular free wall rupture |
ARDS, acute respiratory distress syndrome.
Clinical Assessment
Symptoms are generally nondiscriminatory. An exception is a history of paroxysmal nocturnal dyspnea, which implies a cardiac cause for shortness of breath. Risk factors for and a previous history of coronary artery disease should be sought. Chest pain may be suggestive of myocardial infarction or point to another diagnosis (see Table 18-4). Systemic conditions that are associated with heart failure, such as collagen vascular diseases, pregnancy, and thyrotoxicosis, may be identified. Features suggestive of acute cardiomyopathy are outlined subsequently.
On physical examination, the finding of an S3 gallop is a specific, but insensitive, sign of heart failure.14 Both S3 gallop and an elevated jugular venous pressure are independently associated with adverse outcome.15 Crackles and wheezes are common but nondiscriminatory. A murmur may indicate valvular dysfunction or a mechanical complication of myocardial infarction.
Electrocardiogram and Chest Radiograph
On the chest radiograph, useful information is obtained from the cardiac silhouette and the pulmonary vasculature. With de novo heart failure, cardiac size is not enlarged. Thus, an enlarged cardiac silhouette indicates a chronic process or a pericardial collection. The shape of the cardiac contour may be useful in differentiating a pericardial fluid collection from left or right ventricular dilatation. Features suggestive of a specific valve abnormality, such as mitral stenosis, may be apparent. The chest radiograph may demonstrate interstitial or alveolar edema (see Chapter 6). Features suggestive of a cardiogenic cause may also be apparent (see Table 27-5). Radiographic signs of pulmonary edema may develop very rapidly but their resolution lags behind clinical recovery, often by several days.
Echocardiography
Echocardiography is the single most useful test in the evaluation of patients with heart failure.2 Two-dimensional and Doppler imaging allows for the evaluation of global and regional left ventricular systolic function, left ventricular diastolic function, right ventricular function, and valvular function. Uncommon conditions such as constrictive pericarditis, pericardial tamponade, congenital heart disease, and restrictive or obstructive cardiomyopathy may be identified. In most circumstances, transthoracic echocardiography (TTE) is adequate, although transesophageal echocardiography may be required in patients who are mechanically ventilated because image quality is often poor with TTE.16
Laboratory Tests
The plasma level of BNP is elevated in patients with raised LVEDP and may be used to help determine the cause of acute dyspnea. Heart failure is “very improbable” when the plasma BNP concentration is below 100 pg/ml and “very probable” when it is above 500 pg/ml; when accompanied by a history of heart failure or strong clinical suspicion, heart failure is “very probable” when the BNP concentration is between 100 and 500 pg/ml.17 However, BNP levels must be interpreted with caution because they also are elevated in a range of other conditions encountered in the ICU, such as aortic stenosis,18 pulmonary embolus,19 cardiac surgery sequelae,20 septic shock,21–23 and acute respiratory distress syndrome.24 BNP levels are commonly elevated above 500 pg/ml in patients who are critically unwell, independent of any cardiac dysfunction.23 BNP levels are also increased in renal failure, and a cutoff value of 200 pg/ml to exclude heart failure is recommended when the glomerular filtration rate is less than 60 ml/min.17 Conversely, BNP levels may be normal at the time of admission in patients with flash pulmonary edema.
As an alternative to BNP, the plasma concentration of N-BNP, the N-terminus of pro-BNP, may be used, in which case the recommended lower cutoff value to exclude heart failure is 125 pg/ml in patients younger than 75 years of age and 450 pg/ml in patients older than 75 years.17 An elevated troponin level supports a diagnosis of myocardial infarction but, as with BNP, elevated troponin levels can occur in a range of conditions (see Table 18-7) other than acute coronary syndromes.
Features Suggestive of Acute Cardiomyopathy
Severe acute heart failure is occasionally caused by cardiomyopathy. The diagnosis is suggested by the abrupt onset of severe biventricular failure in a young patient in association with ventricular arrhythmias and widespread repolarization (ST segment) abnormalities on the ECG. A history of recent viral illness or drug ingestion (e.g., alcohol, cocaine, antiretroviral drugs, doxorubicin) suggests the diagnosis. The main differential diagnosis is myocardial ischemia and infarction. Biventricular involvement and a normal coronary angiogram are strongly suggestive of acute cardiomyopathy. The diagnosis may be confirmed by right ventricular endomyocardial biopsy.
Management of Acute Heart Failure
Treatment of Acute Pulmonary Edema
Pharmacotherapy
Pharmacotherapy25 for acute pulmonary edema consists of diuretics, morphine, and vasodilators. Intravenous furosemide is the most commonly used initial therapy for acute heart failure,26 and it provides relief from pulmonary congestion. Furosemide must be used with caution because it can cause hypokalemia, hypomagnesemia, hypovolemia, and renal dysfunction. Morphine reduces the sensation of dyspnea and acts as a weak vasodilator, thereby reducing preload and afterload. Morphine must be used cautiously in patients with low cardiac output or reduced level of consciousness. Small intravenous boluses (1 to 3 mg) should be titrated to effect.
Venodilatation decreases systemic venous return and therefore reduces left ventricular preload, whereas arteriolar dilatation reduces left ventricular afterload. Both effects reduce left atrial pressure. Intravenous nitroglycerin dilates both veins and arterioles and is useful for treating cardiogenic pulmonary edema. Higher doses (>1 μg/kg/min) than are required to treat angina may be needed. Nitroprusside, a potent arteriolar dilator, is also effective in reducing left atrial pressure but can cause marked hypotension, even at low doses. It is particularly useful in patients with acute pulmonary edema secondary to hypertensive crisis. It should be administered only to patients with invasive blood pressure monitoring. Nesiritide, an analogue of BNP, is a mixed vein and arteriole vasodilator that has a mild diuretic effect.27 In patients with acute heart failure, nesiritide is more effective than nitroglycerin in improving symptoms and reducing left atrial pressure.28,29 Vasodilators should be avoided in patients with cardiogenic shock.
Ventilatory Support
Patients with persistent hypoxemia (SaO2 <90%) or marked respiratory distress despite high-flow face-mask oxygen should be given continuous positive airways pressure (CPAP), which is administered by using a tight-fitting face mask. CPAP increases functional residual capacity, reduces work of breathing, improves oxygenation, and reduces left ventricular afterload. Initial CPAP settings of 10 cm H2O with 100% oxygen are appropriate. The inspired oxygen concentration should be titrated to achieve an SaO2 above 90% with a stable arterial carbon dioxide tension and pH. Patients should be closely monitored for deteriorating hemodynamics, worsening gas exchange, and exhaustion. Criteria for endotracheal intubation are outlined in Table 27-3.
If endotracheal intubation is required, it may be fraught with problems for two reasons. First, copious quantities of frothy edema fluid may fill the pharynx, making laryngoscopy extremely difficult. If this occurs, an assistant can place the tip of a suction catheter directly into the laryngeal inlet during laryngoscopy to allow visualization of the vocal cords. Second, the loss of sympathetic tone associated with general anesthesia may precipitate severe hypotension. Vasopressors should be drawn up in preparation for this state. Invasive ventilation for patients with acute pulmonary edema is described in Chapter 29.
Treatment of Hypoperfusion and Shock
Monitoring and Investigations
In addition to the monitors listed earlier, a central venous catheter should be placed in patients with cardiogenic shock to measure central venous pressure and superior vena caval oxygen saturation (SSVCO2; see Chapter 20) and to administer vasoactive drugs. Note, however, that central venous pressure may be a very misleading measure of preload in patients with acute heart failure (see Chapter 8). A PAC is not indicated routinely but should be considered in patients who do not respond in predictable ways to standard treatment.7 In the absence of a central venous catheter, inodilator therapy (e.g., dobutamine, milrinone) may be administered via a peripheral intravenous cannula.
Hemodynamic Support and Fluids
Inotropic drugs improve cardiac output and reduce pulmonary congestion in patients with left ventricular systolic dysfunction, but they must be used with caution because they increase myocardial oxygen consumption and add to myocyte calcium loading, both of which exacerbate myocardial ischemia. There is no role for inotropic drugs in patients with acute heart failure who are not hypotensive or who have preserved left ventricular systolic function; available evidence indicates either no benefit or increased mortality rates when this strategy is used.30 Such patients should be treated with vasodilators and diuretics as described earlier. A possible exception to this is the inodilator levosimendan, which in randomized trials has been associated with improved survival after acute heart failure, including that due to acute myocardial ischemia.31
Modest pulmonary hypertension (a mean pulmonary artery pressure (mPAP) of 25 to 35 mmHg) is common with cardiogenic shock because of the combined effects of increased left atrial pressure and elevated pulmonary vascular resistance (due to pulmonary edema and systemic inflammation). Severe pulmonary hypertension (mPAP >35 mmHg) is indicative of chronically elevated pulmonary vascular resistance. The treatment of pulmonary hypertension and right ventricular dysfunction are discussed in Chapters 24 and Chapter 20 respectively.
Digoxin has inotropic and antiarrhythmic properties, but in patients with myocardial infarction, it has been associated with an increased incidence of life-threatening ventricular arrhythmias.32 It is not recommended for treating acute heart failure.7
Metabolic Disturbance
Lactic acidosis is a marker of tissue hypoperfusion. It resolves if cardiac function recovers. In general, no specific treatment is indicated. However, profound acidemia (pH ≤7.0) exacerbates ventricular dysfunction and inhibits the actions of inotropic drugs33,34; continuous hemofiltration may be considered in this circumstance.
Mechanical Cardiac Support
For patients with progressive cardiovascular decline and evolving MODS despite high dose combination inotropic therapy and an IABP, support with a VAD or extracorporeal membrane oxygenation (ECMO) may be considered on a case-by-case basis. Usually, support with a left VAD is appropriate, but careful evaluation of the right ventricle and pulmonary function is required, because occasionally a biventricular assist device or ECMO is required (see Chapter 22).
Support of Other Organ Systems
Brain
Patients who suffer an out-of-hospital cardiac arrest should undergo therapeutic cooling to 32° to 34°C for 12 to 24 hours followed by passive rewarming to 36°C to protect against HIE.35 There may also be a role for therapeutic hypothermia in patients who suffer a prolonged in-hospital cardiac arrest. This is discussed in greater detail in Chapter 37.
Kidneys
Acute renal failure is to be expected with cardiogenic shock, and it can exacerbate pulmonary and systemic congestion, metabolic acidemia, and hyperkalemia. Oliguria and azotemia are normal responses to hypoperfusion and shock (see Chapter 1) and are not in themselves an indication for diuretic or renal replacement therapy. Diuretics may be used initially in patients with pulmonary congestion but if they are ineffective, early institution of renal replacement therapy, usually with continuous hemofiltration, is appropriate.
Adjuvant Treatments
Glucose Control
As described earlier, hyperglycemia is common in patients with acute heart failure. In medical patients who require prolonged ICU stay, there is some evidence of improved outcome—but some increase in morbidity—with tight glucose control.36,37 As a minimum, hyperglycemia (glucose <180 mg/dl or 10 mmol/l) should be avoided. This may require high doses of insulin because shock is associated with marked insulin resistance.
Corticosteroids
Patients resuscitated from cardiac arrest have an inadequate stress release of cortisol.38 Furthermore, a subgroup of patients with shock due to sepsis benefit from corticosteroid administration (see Chapter 36).39 Although there is no direct evidence of improved outcome from cardiogenic shock, in patients who are critically unwell and are receiving high-dose inotropic support, the administration of corticosteroids in a replacement dose (e.g., hydrocortisone 50 mg 6 hourly) may be beneficial.
Statins
In addition to their role in the treatment of dyslipidemia, statins have antiinflammatory effects, and their use has been linked to improved outcome after acute myocardial infarction40 and septic shock.41 In the absence of hepatic ischemia, it is reasonable to administer a statin to all patients with postmyocardial infarction cardiogenic shock.
Treating the Causes
Myocardial Infarction
Patients with cardiogenic shock secondary to STEMI should receive urgent catheter-based or surgical revascularization.7,42 This recommendation is based primarily on the results of the SHOCK trial11 and subsequent follow-up studies.43 In the SHOCK trial, patients with cardiogenic shock were randomized to medical treatment, including thrombolysis and IABP, or coronary revascularization, either by percutaneous coronary intervention (PCI) or coronary artery bypass graft surgery (CABG). The 6-month11 and 1-year43 survival rates were better in patients treated with PCI or CABG. The benefits of revascularization extended to 12 to 18 hours after the onset of shock, even if shock presented up to 36 hours following myocardial infarction. Although no survival benefit was observed in patients over 75 years of age, that finding may have been the result of a higher baseline risk and a small sample size in this group. Thus, urgent PCI or CABG may still be indicated in selected patients over 75.44 The choice between PCI and CABG depends on the findings at angiography (see Table 9-1). Patients with respiratory distress or hemodynamic instability should be intubated and ventilated prior to angiography. If not already in situ, a central venous catheter and IABP may be inserted in the angiography suite at the completion of the procedure.
Patients who present within 12 hours of a myocardial infarction to a hospital without facilities for urgent coronary angiography should undergo thrombolysis (see Chapter 18). Despite this recommendation, there is scant evidence that thrombolysis improves outcomes after cardiogenic shock. For patients who suffer a mechanical complication of myocardial infarction, urgent surgical repair may be lifesaving. However, even with appropriate treatment, mortality rates are very high (see Chapter 9).
Acute Myocarditis
Patients with myocarditis have been treated with a range of immunosuppressant regimens. Therapies that have been used include high-dose methylprednisolone, azathioprine, cyclosporine, intravenous immunoglobulin, and plasmapheresis. To date, none of these therapies has been shown to improve survival rates.45,46
Weaning Acute Support
Myocardial stunning and systemic inflammation usually have resolved after a week. If at this time echocardiography demonstrates severely impaired ventricular function and the patient’s clinical condition remains very poor—that is, the patient is inotrope- and ventilator-dependent, is intolerant of weaning, and has MODS—it may be helpful to perform a limited bedside dobutamine stress echocardiogram to assess myocardial viability. The absence of viability suggests that further improvement in cardiac function will be limited or nonexistent. This information can then be used to guide further management: either referral for transplantation or a VAD or compassionate end-of-life care.
Outcome
Acute heart failure carries a poor prognosis. In one large series, 13.5% of patients hospitalized with acute heart failure died between admission and 12-week follow-up.47 The presence of hypotension (systolic blood pressure <115 mmHg) and elevated blood urea nitrogen (>43 mg/dl or 15.3 mmol/l) and creatinine (>2.75 mg/dl or 243 μmol/l) are predictors of death.48 Patients with all three risk factors on admission have an in-hospital mortality rate of about 20%.48 The mortality rate after severe cardiogenic shock associated with myocardial infarction is about 50%.11 The mode of death is unsupportable cardiac failure, MODS, or HIE. However, of survivors, many make a good recovery and are in New York Heart Association (NYHA) functional class I or II.6
CHRONIC HEART FAILURE
Patients with chronic heart failure are admitted to the ICU for the management of acute decompensation (the causes are listed in Table 19-3) and for various cardiac operations, such as CABG surgery, ventricular remodeling surgery, mitral valve surgery, insertion of a mechanical device, and heart transplantation. Heart failure is a complicating factor in about 30% of general ICU admissions.49
Table 19-3 Causes of Acute Exacerbations of Chronic Heart Failure
| Progression of chronic heart failure |
| Acute coronary syndrome |
| Accelerated hypertension or poorly controlled hypertension |
| Acute valve dysfunction or chronic progression of existing severe valve lesion |
| Arrhythmia |
| Infections, particularly pneumonia or septicemia |
| Major surgery |
| Renal dysfunction |
| Severe brain injury |
| Exacerbation of COPD |
| Aortic dissection |
| Drugs (e.g., nonsteroidal antiinflammatory drugs, alcohol) |
| Poor compliance with medical therapy |
| Excess fluid or salt intake |
| High output syndromes (e.g., anemia, thyrotoxicosis, shunts) |
COPD, chronic obstructive pulmonary disease.
Modified from Nieminen MS, Bohm M, Cowie MR, et al: Executive summary of the guidelines on the diagnosis and treatment of acute heart failure: the Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J 26:384-416, 2005.
Classification and Clinical Presentation
Chronic heart failure may be classified on the basis of cause, structural abnormality, clinical syndrome, symptom severity, and disease progression. With respect to cause, the majority of heart failure is due to coronary artery disease, hypertension, valvular dysfunction, and cardiomyopathy. Structurally, heart failure may be classified as left ventricular, right ventricular, or biventricular. Left ventricular dysfunction may be systolic or diastolic. Clinically, patients with heart failure present with: (1) a syndrome of reduced exercise tolerance; (2) a syndrome of fluid retention; (3) no symptoms or symptoms relating to another condition (e.g., myocardial infarction or stroke).2 Systemic congestion can be difficult to diagnose clinically in patients with chronic heart failure: in one study, physical findings of congestion were detected in only half the patients found to be hypervolemic by using iodine 131-tagged albumin.50 Anemia is a useful marker of chronic volume overload.
The severity of symptoms may be categorized according to the NYHA functional classification (Table 19-4). This classification is widely used and is helpful prognostically, but it has a number of limitations. First, the severity of symptoms does not always correlate with the severity of the underlying cardiac dysfunction. Second, even though chronic heart failure is a progressive condition, symptoms do not usually follow an inexorable pattern of decline but fluctuate, even in the absence of obvious precipitating factors or changes in medication. Thus, it is common for patients to be assigned to more than one NYHA class or to be reassigned from one class to another. NYHA functional class does not determine an evidenced-based approach to therapy.
Table 19-4 New York Heart Association Functional Classification
| Class I | Symptoms on levels of exertion that limit normal individuals |
| Class II | Symptoms on ordinary exertion |
| Class III | Symptoms at less than ordinary exertion |
| Class IV | Symptoms at rest |
The American College of Cardiology and American Heart Association have classified chronic heart failure as a progressive disorder based on the underlying cardiac disease (see Table 19-5).2 This classification system takes into account risk factors for heart failure and the presence of structural heart disease and provides a rational basis for medical intervention.
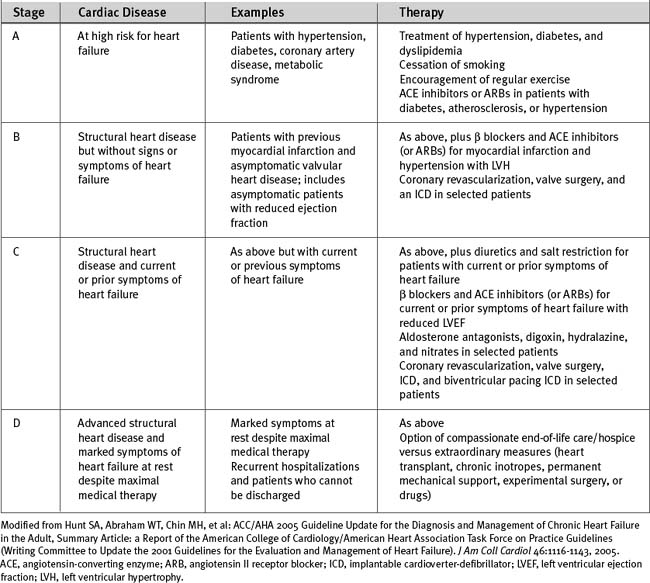
Diastolic Heart Failure
Heart failure is often thought of as being synonymous with impaired left ventricular systolic function and is graded by the severity of the reduction in ejection fraction. However, between 20% and 60% of patients with signs and symptoms of heart failure have normal or near-normal ejection fraction.2 The primary abnormality in many of these patients is thought to be reduced ventricular compliance (i.e., diastolic dysfunction) resulting in increased diastolic ventricular pressure. In many cases there is pathologic fluid retention. This syndrome occurs more commonly in the elderly, in women, and in patients with hypertension.51–53 Patients typically present with exercise-related dyspnea and may experience episodes of acute decompensation that manifests as flash pulmonary edema. Diagnosis is based on the finding of normal or near-normal ejection fraction and the absence of valvular pathology in a patient with symptoms and signs of pulmonary congestion. There may be echocardiographic evidence of raised left atrial pressure and abnormal diastolic function, either prolonged relaxation or reduced left ventricular compliance (see Chapter 7). Underlying coronary artery disease is very common,10 and patients may subsequently develop impaired systolic function.
Medical Treatment of Chronic Heart Failure
The evidenced-based treatment of chronic heart failure is summarized in Tables 19-5 and 19-6.2
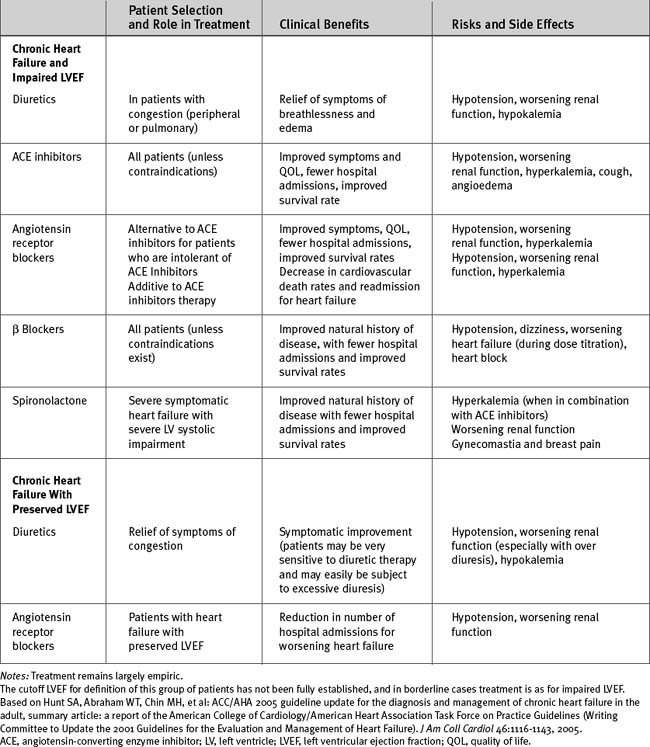
Diuretics
Diuretics remain an important component of therapy for chronic heart failure. However, diuretic use has been linked with an increased incidence of fatal arrhythmias in patients with reduced ejection fraction54 and has been associated with increased mortality rates in patients with acute heart failure.55 Thus, diuretics should not be used as monotherapy, and the dosage should be titrated to the minimum level required to control the symptoms of congestion.
Inhibition of the Renin-Angiotensin-Aldosterone System
ACE Inhibitors and Angiotensin Receptor Blockers.
In patients with chronic heart failure, treatment with ACE inhibitors leads to improvement in symptoms, increased exercise capacity, inhibition of ventricular remodeling, and decreased mortality rates.56,57 ACE inhibitors are indicated for all stages of heart failure, from patients with asymptomatic left ventricular dysfunction to those with severe NYHA class IV symptoms. The effects of ACE inhibitors are a class effect, and no specific agent is recommended. Generally, higher dosages should be aimed for (see Table 3-4). The combination of an ACE inhibitor and an angiotensin-receptor blocker (ARB) results in a lower incidence of death and hospital admission but in a higher risk for developing hypotension and renal impairment than is found when patients are treated with an ACE inhibitor alone.58 ARBs may also be used as an alternative to ACE inhibitors in patients who are intolerant of ACE inhibitors59 and for treating heart failure with preserved left ventricular systolic function (see subsequent material).
Aldosterone Antagonists: Spironolactone and Eplerenone.
Aldosterone levels remain elevated in patients with heart failure despite ACE inhibitor therapy, and those levels contribute to the progression of the disease process. Treatment with spironolactone improves survival rates in patients with chronic heart failure,60 whereas treatment with eplerenone improves survival rates in patients with left ventricular dysfunction following myocardial infarction.61
The aim when using aldosterone antagonists is to alter the natural history of the heart failure rather than to provide short-term benefit in terms of diuresis. Low-dose spironolactone (e.g., 25 mg/day) is indicated in addition to ACE inhibitor and loop diuretic therapy in patients with severe heart failure secondary to left ventricular systolic dysfunction. Contraindications and precautions are listed in Chapter 3.
Inhibition of the Sympathetic Nervous System
Traditionally, β blockers were contraindicated in patients with heart failure, and this remains true in acute heart failure, when activation of the sympathetic nervous system is needed to maintain blood flow to essential organs. However, in chronic heart failure, activation of the sympathetic nervous system continues despite ACE inhibitor and diuretic therapy and contributes to the disease’s progression. Antagonism of the sympathetic nervous system with β blockers leads to a reduction in hospitalizations and improved survival rates in patients with chronic heart failure.62–64 However, β blockers do not improve—and may worsen—symptoms in the short term.
Titration of Heart Failure Treatment
Titration of pharmacotherapy for chronic heart failure is based primarily on the clinical assessment of signs and symptoms and on functional capacity. Recently, BNP levels have been proposed as a method of guiding therapy, and in one small study this approach resulted in a reduction in adverse cardiovascular events.65 However, targeting a specific BNP level in an individual is problematic because some patients maintain high BNP levels despite optimal therapy and minimal symptoms, whereas others have BNP levels within the normal range despite advanced heart failure.2,17 Measurement of ejection fraction is useful following a major change in a patient’s clinical status, but it is not indicated as routine.2
Treatment of Diastolic Heart Failure
The evidence-based recommendations for the treatment of chronic heart failure pertain largely to patients with impaired left ventricular systolic function. Therapy for heart failure with preserved ejection fraction is largely empiric and involves treatment of hypertension, rate control of atrial fibrillation, diuretics for congestion, and revascularization for coronary artery disease. Recently, treatment with the ARB candesartan has been shown to reduce hospital admissions for worsening heart failure in patients with preserved left ventricular ejection fraction.66
Surgical and Device Therapy for Chronic Heart Failure
Despite appropriate pharmacotherapy, the prognosis for patients with heart failure remains poor (see Figure 19-1). For certain patients, surgical or device-based therapy is an alternative.
Heart Failure Surgery
As outlined in Chapter 9, patients with depressed left ventricular function secondary to hibernating myocardium—as identified by a viability study—benefit from myocardial revascularization. Many such patients have extensive three-vessel coronary artery disease, and CABG surgery, rather than PCI, is the preferred method of revascularization. For patients with severe ventricular dilatation (end-diastolic volume index >60 ml/m2) and an anteroseptal scar, surgery to remodel the left ventricle should be considered at the time of CABG surgery (see Chapter 9). If appropriate, mitral valve surgery for mitral regurgitation or resection of endocardium and cryotherapy for arrhythmias may also be performed at that time.
Cardiac Resynchronization
Approximately one third of patients with cardiomyopathy have intraventricular conduction delay, such as left or right bundle branch block.67 Abnormal intraventricular conduction with or without delayed atrioventricular conduction is associated with a lack of functional synergy between the left and right ventricles. Echocardiographic findings include paradoxic ventricular septal motion, presystolic mitral regurgitation, and shortened diastolic filling time.67 Biventricular pacing improves ventricular function in patients with advanced heart failure and intraventricular conduction delay. Pacing wires are inserted into the right atrium and right ventricle and—via the coronary sinus and cardiac vein—the lateral left ventricular wall. The pacemaker is programmed to provide the appropriate atrioventricular delay and coordinated contraction between the right and left ventricles; hence the term cardiac resynchronization. In appropriate patients, biventricular pacing reverses ventricular remodeling, improves symptoms, and enhances survival rates.68,69 Patients should be considered for biventricular pacing if they fulfill the following criteria: (1) left ventricular ejection fraction less than or equal to 35%; (2) NYHA class III or IV symptoms despite optimal medical therapy; (3) sinus rhythm; (4) QRS duration of greater than 0.12 sec.2
Implantable Cardioverter-Defibrillators
Implantable cardioverter-defibrillators are used for patients at high risk for lethal ventricular arrhythmias such as those who have survived ventricular tachycardia or ventricular fibrillation. However, they also improve survival rates in patients with prior myocardial infarction and reduced ejection fraction who have not had documented arrhythmias or arrhythmias induced during electrophysiologic studies.68,70
1 Levy D, Kenchaiah S, Larson MG, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397-1402.
2 Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult, summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol. 2005;46:1116-1143.
3 Staub NC. Pulmonary edema. Physiol Rev. 1974;54:678-811.
4 Baig MK, Mahon N, McKenna WJ, et al. The pathophysiology of advanced heart failure. Am Heart J. 1998;135:S216-S230.
5 Adrie C, Laurent I, Monchi M, et al. Postresuscitation disease after cardiac arrest: a sepsis-like syndrome ? Curr Opin Crit Care. 2004;10:208-212.
6 Hochman JS. Cardiogenic shock complicating acute myocardial infarction: expanding the paradigm. Circulation. 2003;107:2998-3002.
7 Nieminen MS, Bohm M, Cowie MR, et al. Executive summary of the guidelines on the diagnosis and treatment of acute heart failure: the Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J. 2005;26:384-416.
8 Killip T3rd, Kimball JT. Treatment of myocardial infarction in a coronary care unit: a two-year experience with 250 patients. Am J Cardiol. 1967;20:457-464.
9 Gandhi SK, Powers JC, Nomeir AM, et al. The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med. 2001;344:17-22.
10 Kramer K, Kirkman P, Kitzman D, et al. Flash pulmonary edema: association with hypertension and reoccurrence despite coronary revascularization. Am Heart J. 2000;140:451-455.
11 Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. N Engl J Med. 1999;341:625-634.
12 Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622-1632.
13 Isner JM, Roberts WC. Right ventricular infarction complicating left ventricular infarction secondary to coronary heart disease: frequency, location, associated findings and significance from analysis of 236 necropsy patients with acute or healed myocardial infarction. Am J Cardiol. 1978;42:885-894.
14 Patel R, Bushnell DL, Sobotka PA. Implications of an audible third heart sound in evaluating cardiac function. West J Med. 1993;158:606-609.
15 Drazner MH, Rame JE, Stevenson LW, et al. Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure. N Engl J Med. 2001;345:574-581.
16 Heidenreich PA, Stainback RF, Redberg RF, et al. Transesophageal echocardiography predicts mortality in critically ill patients with unexplained hypotension. J Am Coll Cardiol. 1995;26:152-158.
17 Silver MA, Maisel A, Yancy CW, et al. BNP Consensus Panel 2004: a clinical approach for the diagnostic, prognostic, screening, treatment monitoring, and therapeutic roles of natriuretic peptides in cardiovascular diseases. Congest Heart Fail. 2004;10:1-30.
18 Gerber IL, Stewart RA, Legget ME, et al. Increased plasma natriuretic peptide levels reflect symptom onset in aortic stenosis. Circulation. 2003;107:1690-1884.
19 Kucher N, Printzen G, Goldhaber SZ. Prognostic role of brain natriuretic peptide in acute pulmonary embolism. Circulation. 2003;107:2545-2547.
20 Sinha AM, Breithardt OA, Schmid M, et al. Brain natriuretic peptide release in cardiac surgery patients. Thorac Cardiovasc Surg. 2005;53:138-143.
21 Bhalla V, Bhalla MA, Maisel AS. Evolution of B-type natriuretic peptide in evaluation of intensive care unit shock. Crit Care Med. 2004;32:1787-1789.
22 Tung RH, Garcia C, Morss AM, et al. Utility of B-type natriuretic peptide for the evaluation of intensive care unit shock. Crit Care Med. 2004;32:1643-1647.
23 Maeder M, Ammann P, Kiowski W, et al. B-type natriuretic peptide in patients with sepsis and preserved left ventricular ejection fraction. Eur J Heart Fail. 2005;7:1164-1167.
24 Maeder M, Ammann P, Rickli H, et al. Elevation of B-type natriuretic peptide levels in acute respiratory distress syndrome. Swiss Med Week. 2003;133:515-518.
25 Stough WG, O’Connor CM, Gheorghiade M. Overview of current noninodilator therapies for acute heart failure syndromes. Am J Cardiol. 2005;96:41G-46G.
26 Komajda M, Follath F, Swedberg K, et al. The EuroHeart Failure Survey programme —a survey on the quality of care among patients with heart failure in Europe. Part 2: treatment. Eur Heart J. 2003;24:464-474.
27 Keating GM, Goa KL. Nesiritide: a review of its use in acute decompensated heart failure. Drugs. 2003;63:47-70.
28 Elkayam U, Akhter MW, Singh H, et al. Comparison of effects on left ventricular filling pressure of intravenous nesiritide and high-dose nitroglycerin in patients with decompensated heart failure. Am J Cardiol. 2004;93:237-240.
29 Publication Committee for the VMAC Investigators. Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531-1540.
30 Bayram M, de Luca L, Massie MB, et al. Reassessment of dobutamine, dopamine, and milrinone in the management of acute heart failure syndromes. Am J Cardiol. 2005;96:47G-58G.
31 Mebazaa A, Barraud D, Welschbillig S. Randomized clinical trials with levosimendan. Am J Cardiol. 2005;96:74G-79G.
32 McClements BM, Adgey AA. Value of signal-averaged electrocardiography, radionuclide ventriculography, Holter monitoring and clinical variables for prediction of arrhythmic events in survivors of acute myocardial infarction in the thrombolytic era. J Am Coll Cardiol. 1993;21:1419-1427.
33 Huang YG, Wong KC, Yip WH, et al. Cardiovascular responses to graded doses of three catecholamines during lactic and hydrochloric acidosis in dogs. Br J Anaesth. 1995;74:583-590.
34 Toller W, Wolkart G, Stranz C, et al. Contractile action of levosimendan and epinephrine during acidosis. Eur J Pharmacol. 2005;507:199-209.
35 Nolan JP, Morley PT, Vanden Hoek TL, et al. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118-121.
36 van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449-461.
37 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
38 Schultz CH, Rivers EP, Feldkamp CS, et al. A characterization of hypothalamic-pituitary-adrenal axis function during and after human cardiac arrest. Crit Care Med. 1993;21:1339-1347.
39 Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862-871.
40 Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol. 2005;96:611-616.
41 Almog Y, Shefer A, Novack V, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110:880-885.
42 Eagle KA, Guyton RA, Davidoff R, et al. ACC/AHA 2004 guideline update for coronary artery bypass graft surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1999 Guidelines for Coronary Artery Bypass Graft Surgery). Circulation. 2004;110:e340-e437.
43 Hochman JS, Sleeper LA, White HD, et al. One-year survival following early revascularization for cardiogenic shock. JAMA. 2001;285:190-192.
44 Dzavik V, Sleeper LA, Picard MH, et al. Outcome of patients aged 75 years in the SHould we emergently revascularize Occluded Coronaries in cardiogenic shocK (SHOCK) trial: do elderly patients with acute myocardial infarction complicated by cardiogenic shock respond differently to emergent revascularization ? Am Heart J. 2005;149:1128-1134.
45 Robinson J, Hartling L, Vandermeer B, et al. Intravenous immunoglobulin for presumed viral myocarditis in children and adults. Cochrane Database of Systematic Reviews. 2005:CD 004370.
46 Vallejo J, Mann DL. Antiinflammatory therapy in myocarditis. Curr Opin Cardiol. 2003;18:189-193.
47 Cleland JG, Swedberg K, Follath F, et al. The EuroHeart Failure survey programme —a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis. Eur Heart J. 2003;24:442-463.
48 Fonarow GC, Adams KFJr, Abraham WT, et al. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA. 2005;293:572-580.
49 Bossone E, DiGiovine B, Watts S, et al. Range and prevalence of cardiac abnormalities in patients hospitalized in a medical ICU. Chest. 2002;122:1370-1376.
50 Androne AS, Katz SD, Lund L, et al. Hemodilution is common in patients with advanced heart failure. Circulation. 2003;107:226-229.
51 Dougherty AH, Naccarelli GV, Gray EL, et al. Congestive heart failure with normal systolic function. Am J Cardiol. 1984;54:778-782.
52 Ramachandran SV, Benjamin EJ, Levy D. Prevalence, clinical features and prognosis of diastolic heart failure: an epidemiologic perspective. J Am Coll Cardiol. 1995;26:1565-1574.
53 Tresch DD, McGough MF. Heart failure with normal systolic function: a common disorder in older people. J Am Geriatr Soc. 1995;43:1035-1042.
54 Cooper HA, Dries DL, Davis CE, et al. Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation. 1999;100:1311-1315.
55 Philbin EF, Cotto M, Rocco TAJr, et al. Association between diuretic use, clinical response, and death in acute heart failure. Am J Cardiol. 1997;80:519-522.
56 CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429-1435.
57 SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685-691.
58 McMurray JJ, Ostergren J, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767-771.
59 Granger CB, McMurray JJ, Yusuf S, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772-776.
60 RALES Investigators. Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol. 1996;78:902-907.
61 Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309-1321.
62 CIBIS II Investigators. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9-13.
63 MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in congestive heart failure. Lancet. 1999;353:2001-2007.
64 Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651-1658.
65 Troughton RW, Frampton CM, Yandle TG, et al. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet. 2000;355:1126-1130.
66 Yusuf S, Pfeffer MA, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362:777-781.
67 Wilensky RL, Yudelman P, Cohen AI, et al. Serial electrocardiographic changes in idiopathic dilated cardiomyopathy confirmed at necropsy. Am J Cardiol. 1988;62:276-283.
68 Bristow MR, Saxon LA, Boehmer J. Comparison of medical therapy, pacing and defibrillation in heart failure (COMPANION) Investigators. Cardiac resynchronization therapy with or without implantable defibrillator in advanced heart failure. N Engl J Med. 2004;350:2140-2150.
69 Sutton MG, Plappert T, Hilpisch KE, et al. Sustained reverse left ventricular structural remodeling with cardiac resynchronization at one year is a function of etiology: quantitative Doppler echocardiographic evidence from the Multicenter InSync Randomized Clinical Evaluation (MIRACLE). Circulation. 2006;113:266-272.
70 Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.