274 |
The Bradyarrhythmias: Disorders of the Sinoatrial Node |
Electrical activation of the heart normally originates in the sinoatrial (SA) node, the predominant pacemaker. Other subsidiary pacemakers in the atrioventricular (AV) node, specialized conducting system, and muscle may initiate electrical activation if the SA node is dysfunctional or suppressed. Typically, subsidiary pacemakers discharge at a slower rate and, in the absence of an appropriate increase in stroke volume, may result in tissue hypoperfusion.
Spontaneous activation and contraction of the heart are a consequence of the specialized pacemaking tissue in these anatomic locales. As described in Chap. 273e, action potentials in the heart are regionally heterogeneous. The action potentials in cells isolated from nodal tissue are distinct from those recorded from atrial and ventricular myocytes (Fig. 274-1). The complement of ionic currents present in nodal cells results in a less negative resting membrane potential compared with atrial or ventricular myocytes. Electrical diastole in nodal cells is characterized by slow diastolic depolarization (phase 4), which generates an action potential as the membrane voltage reaches threshold. The action potential upstrokes (phase 0) are slow compared with atrial or ventricular myocytes, being mediated by calcium rather than sodium current. Cells with properties of SA and AV nodal tissue are electrically connected to the remainder of the myocardium by cells with an electrophysiologic phenotype between that of nodal cells and that of atrial or ventricular myocytes. Cells in the SA node exhibit the most rapid phase 4 depolarization and thus are the dominant pacemakers in a normal heart.
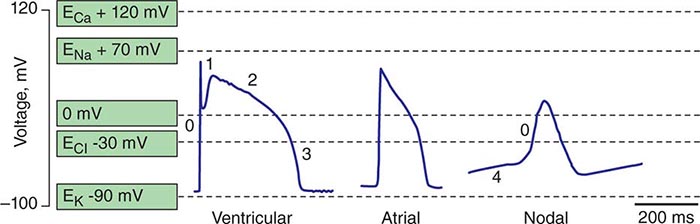
FIGURE 274-1 Action potential profiles recorded in cells isolated from sinoatrial or atrioventricular nodal tissue compared with those of cells from atrial or ventricular myocardium. Nodal cell action potentials exhibit more depolarized resting membrane potentials, slower phase 0 upstrokes, and phase 4 diastolic depolarization.
Bradycardia results from a failure of either impulse initiation or impulse conduction. Failure of impulse initiation may be caused by depressed automaticity resulting from a slowing or failure of phase 4 diastolic depolarization (Fig. 274-2), which may result from disease or exposure to drugs. Prominently, the autonomic nervous system modulates the rate of phase 4 diastolic depolarization and thus the firing rate of both primary (SA node) and subsidiary pacemakers. Failure of conduction of an impulse from nodal tissue to atrial or ventricular myocardium may produce bradycardia as a result of exit block. Conditions that alter the activation and connectivity of cells (e.g., fibrosis) in the heart may result in failure of impulse conduction.
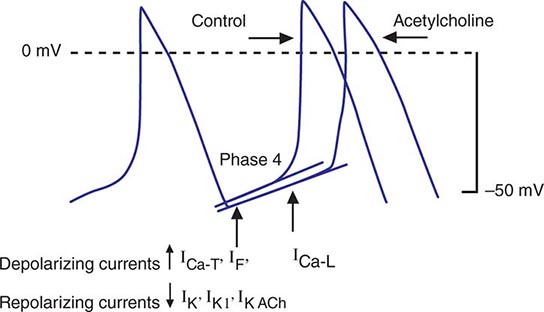
FIGURE 274-2 Schematics of nodal action potentials and the currents that contribute to phase 4 depolarization. Relative increases in depolarizing L- (ICa-L) and T- (ICa-T) type calcium and pacemaker currents (If) along with a reduction in repolarizing inward rectifier (IK1) and delayed rectifier (IK) potassium currents result in depolarization. Activation of ACh-gated (IKACh) potassium current and beta blockade slow the rate of phase 4 and decrease the pacing rate. (Modified from J Jalife et al: Basic Cardiac Electrophysiology for the Clinician, Blackwell Publishing, 1999.)
SA node dysfunction and AV conduction block are the most common causes of pathologic bradycardia. SA node dysfunction may be difficult to distinguish from physiologic sinus bradycardia, particularly in the young. SA node dysfunction increases in frequency between the fifth and sixth decades of life and should be considered in patients with fatigue, exercise intolerance, or syncope and sinus bradycardia.
Permanent pacemaking is the only reliable therapy for symptomatic bradycardia in the absence of extrinsic and reversible etiologies such as increased vagal tone, hypoxia, hypothermia, and drugs (Table 274-1). Approximately 50% of the 150,000 permanent pacemakers implanted in the United States and 20–30% of the 150,000 of those in Europe were implanted for SA node disease.
|
ETIOLOGIES OF SA NODE DYSFUNCTION |

STRUCTURE AND PHYSIOLOGY OF THE SA NODE
The SA node is composed of a cluster of small fusiform cells in the sulcus terminalis on the epicardial surface of the heart at the right atrial–superior vena caval junction, where they envelop the SA nodal artery. The SA node is structurally heterogeneous, but the central prototypic nodal cells have fewer distinct myofibrils than does the surrounding atrial myocardium, no intercalated disks visible on light microscopy, a poorly developed sarcoplasmic reticulum, and no T-tubules. Cells in the peripheral regions of the SA node are transitional in both structure and function. The SA nodal artery arises from the right coronary artery in 55–60% and the left circumflex artery in 40–45% of persons. The SA node is richly innervated by sympathetic and parasympathetic nerves and ganglia.
Irregular and slow propagation of impulses from the SA node can be explained by the electrophysiology of nodal cells and the structure of the SA node itself. The action potentials of SA nodal cells are characterized by a relatively depolarized membrane potential (Fig. 274-1) of –40 to –60 mV, slow phase 0 upstroke, and relatively rapid phase 4 diastolic depolarization compared with the action potentials recorded in cardiac muscle cells. The relative absence of inward rectifier potassium current (IK1) accounts for the depolarized membrane potential; the slow upstroke of phase 0 results from the absence of available fast sodium current (INa) and is mediated by L-type calcium current (ICa-L); and phase 4 depolarization is a result of the aggregate activity of a number of ionic currents. Prominently, both L- and T-type (ICa-T) calcium currents, the pacemaker current (so-called funny current, or If) formed by hyperpolarization-activated cyclic nucleotide-gated channels, and the electrogenic sodium-calcium exchanger provide depolarizing current that is antagonized by delayed rectifier (IKr) and acetylcholine-gated (IKACh) potassium currents. ICa-L, ICa-T, and If are modulated by β-adrenergic stimulation and IKACh by vagal stimulation, explaining the exquisite sensitivity of diastolic depolarization to autonomic nervous system activity. The slow conduction within the SA node is explained by the absence of INa and poor electrical coupling of cells in the node, resulting from sizable amounts of interstitial tissue and a low abundance of gap junctions. The poor coupling allows for graded electrophysiologic properties within the node, with the peripheral transitional cells being silenced by electrotonic coupling to atrial myocardium.
ETIOLOGY OF SA NODAL DISEASE
SA nodal dysfunction has been classified as intrinsic or extrinsic. The distinction is important because extrinsic dysfunction is often reversible and generally should be corrected before pacemaker therapy is considered (Table 274-1). The most common causes of extrinsic SA node dysfunction are drugs and autonomic nervous system influences that suppress automaticity and/or compromise conduction. Other extrinsic causes include hypothyroidism, sleep apnea, and conditions likely to occur in critically ill patients such as hypothermia, hypoxia, increased intracranial pressure (Cushing’s response), and endotracheal suctioning via activation of the vagus nerve.
Intrinsic sinus node dysfunction is degenerative and often is characterized pathologically by fibrous replacement of the SA node or its connections to the atrium. Acute and chronic coronary artery disease (CAD) may be associated with SA node dysfunction, although in the setting of acute myocardial infarction (MI; typically inferior), the abnormalities are transient. Inflammatory processes may alter SA node function, ultimately producing replacement fibrosis. Pericarditis, myocarditis, and rheumatic heart disease have been associated with SA nodal disease with sinus bradycardia, sinus arrest, and exit block. Carditis associated with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and mixed connective tissue disorders (MCTDs) may also affect SA node structure and function. Senile amyloidosis is an infiltrative disorder in patients typically in the ninth decade of life; deposition of amyloid protein in the atrial myocardium can impair SA node function. Some SA node disease is iatrogenic and results from direct injury to the SA node during cardiothoracic surgery.
Rare heritable forms of sinus node disease have been described, and several have been characterized genetically. Autosomal dominant sinus node dysfunction in conjunction with supraventricular tachycardia (i.e., tachycardia-bradycardia variant of sick-sinus syndrome [SSS2]) has been linked to mutations in the pacemaker current (If) subunit gene HCN4 on chromosome 15. An autosomal recessive form of SSS1 with the prominent feature of atrial inexcitability and absence of P waves on the electrocardiogram (ECG) is caused by mutations in the cardiac sodium channel gene, SCN5A, on chromosome 3. Variants in myosin heavy chain 6 (MYH6) increase the susceptibility to SSS (SSS3). SA node dysfunction associated with myopia has been described but not genetically characterized. There are several neuromuscular diseases, including Kearns-Sayre syndrome (ophthalmoplegia, pigmentary degeneration of the retina, and cardiomyopathy) and myotonic dystrophy, that have a predilection for the conducting system and SA node.
SSS in both the young and the elderly is associated with an increase in fibrous tissue in the SA node. The onset of SSS may be hastened by coexisting disease, such as CAD, diabetes mellitus, hypertension, and valvular diseases and cardiomyopathies.
CLINICAL FEATURES OF SA NODE DISEASE
SA node dysfunction may be completely asymptomatic and manifest as an ECG anomaly such as sinus bradycardia; sinus arrest and exit block; or alternating supraventricular tachycardia, usually atrial fibrillation, and bradycardia. Symptoms associated with SA node dysfunction, in particular tachycardia-bradycardia syndrome, may be related to both slow and fast heart rates. For example, tachycardia may be associated with palpitations, angina pectoris, and heart failure, and bradycardia may be associated with hypotension, syncope, presyncope, fatigue, and weakness. In the setting of SSS, overdrive suppression of the SA node may result in prolonged pauses and syncope upon termination of the tachycardia. In many cases, symptoms associated with SA node dysfunction result from concomitant cardiovascular disease. A significant minority of patients with SSS develop signs and symptoms of heart failure that may be related to slow or fast heart rates.
One-third to one-half of patients with SA node dysfunction develop supraventricular tachycardia, usually atrial fibrillation or atrial flutter. The incidence of persistent atrial fibrillation in patients with SA node dysfunction increases with advanced age, hypertension, diabetes mellitus, left ventricular dilation, valvular heart disease, and ventricular pacing. Remarkably, some symptomatic patients may experience an improvement in symptoms with the development of atrial fibrillation, presumably from an increase in their average heart rate. Patients with the tachycardia-bradycardia variant of SSS, similar to patients with atrial fibrillation, are at risk for thromboembolism, and those at greatest risk, including patients ≥65 years and patients with a prior history of stroke, valvular heart disease, left ventricular dysfunction, or atrial enlargement, should be treated with anticoagulants. Up to one-quarter of patients with SA node disease will have concurrent AV conduction disease, although only a minority will require specific therapy for high-grade AV block.
The natural history of SA node dysfunction is one of varying intensity of symptoms even in patients who present with syncope. Symptoms related to SA node dysfunction may be significant, but overall mortality usually is not compromised in the absence of other significant comorbid conditions. These features of the natural history need to be taken into account in considering therapy for these patients.
ELECTROCARDIOGRAPHY OF SA NODE DISEASE
The electrocardiographic manifestations of SA node dysfunction include sinus bradycardia, sinus pauses, sinus arrest, sinus exit block, tachycardia (in SSS), and chronotropic incompetence. It is often difficult to distinguish pathologic from physiologic sinus bradycardia. By definition, sinus bradycardia is a rhythm driven by the SA node with a rate of <60 beats/min; sinus bradycardia is very common and typically benign. Resting heart rates <60 beats/min are very common in young healthy individuals and physically conditioned subjects. A sinus rate of <40 beats/min in the awake state in the absence of physical conditioning generally is considered abnormal. Sinus pauses and sinus arrest result from failure of the SA node to discharge, producing a pause without P waves visible on the ECG (Fig. 274-3). Sinus pauses of up to 3 s are common in awake athletes, and pauses of this duration or longer may be observed in asymptomatic elderly subjects. Intermittent failure of conduction from the SA node produces sinus exit block. The severity of sinus exit block may vary in a manner similar to that of AV block (Chap. 275). Prolongation of conduction from the sinus node will not be apparent on the ECG; second-degree SA block will produce intermittent conduction from the SA node and a regularly irregular atrial rhythm.
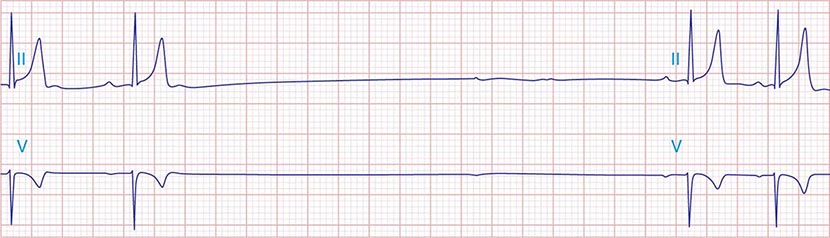
FIGURE 274-3 Sinus slowing and pauses on the electrocardiogram (ECG). The ECG is recorded during sleep in a young patient without heart disease. The heart rate before the pause is slow, and the PR interval is prolonged, consistent with an increase in vagal tone. The P waves have a morphology consistent with sinus rhythm. The recording is from a two-lead telemetry system in which the tracing labeled II mimics frontal lead II and V represents Modified Central Lead 1, which mimics lead V1 of the standard 12-lead ECG.
Type I second-degree SA block results from progressive prolongation of SA node conduction with intermittent failure of the impulses originating in the sinus node to conduct to the surrounding atrial tissue. Second-degree SA block appears on the ECG as an intermittent absence of P waves (Fig. 274-4). In type II second-degree SA block, there is no change in SA node conduction before the pause. Complete or third-degree SA block results in no P waves on the ECG. Tachycardia-bradycardia syndrome is manifest as alternating sinus bradycardia and atrial tachyarrhythmias. Although atrial tachycardia, atrial flutter, and atrial fibrillation may be observed, the latter is the most common tachycardia. Chronotropic incompetence is the inability to increase the heart rate in response to exercise or other stress appropriately and is defined in greater detail below.
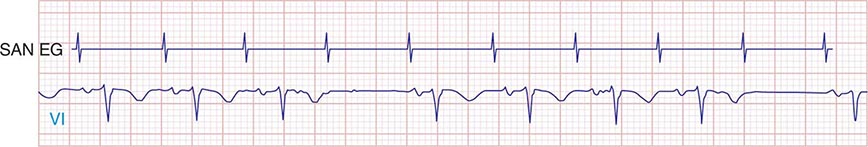
FIGURE 274-4 Mobitz type I SA nodal exit block. A theoretical SA node electrogram (SAN EG) is shown. Note that there is grouped beating producing a regularly irregular heart rhythm. The SA node EG rate is constant with progressive delay in exit from the node and activation of the atria, inscribing the P wave. This produces subtly decreasing P-P intervals before the pause, and the pause is less than twice the cycle length of the last sinus interval.
DIAGNOSTIC TESTING
SA node dysfunction is most commonly a clinical or electrocardiographic diagnosis. Sinus bradycardia or pauses on the resting ECG are rarely sufficient to diagnose SA node disease, and longer-term recording and symptom correlation generally are required. Symptoms in the absence of sinus bradyarrhythmias may be sufficient to exclude a diagnosis of SA node dysfunction.
Electrocardiographic recording plays a central role in the diagnosis and management of SA node dysfunction. Despite the limitations of the resting ECG, longer-term recording employing Holter or event monitors may permit correlation of symptoms with the cardiac rhythm. Many contemporary event monitors may be automatically triggered to record the ECG when certain programmed heart rate criteria are met. Implantable ECG monitors permit long-term recording (12–18 months) in particularly challenging patients.
Failure to increase the heart rate with exercise is referred to as chronotropic incompetence. This is alternatively defined as failure to reach 85% of predicted maximal heart rate at peak exercise or failure to achieve a heart rate >100 beats/min with exercise or a maximal heart rate with exercise less than two standard deviations below that of an age-matched control population. Exercise testing may be useful in discriminating chronotropic incompetence from resting bradycardia and may aid in the identification of the mechanism of exercise intolerance.
Autonomic nervous system testing is useful in diagnosing carotid sinus hypersensitivity; pauses >3 s are consistent with the diagnosis but may be present in asymptomatic elderly subjects. Determining the intrinsic heart rate (IHR) may distinguish SA node dysfunction from slow heart rates that result from high vagal tone. The normal IHR after administration of 0.2 mg/kg propranolol and 0.04 mg/kg atropine is 117.2 – (0.53 × age) in beats/min; a low IHR is indicative of SA disease.
Electrophysiologic testing may play a role in the assessment of patients with presumed SA node dysfunction and in the evaluation of syncope, particularly in the setting of structural heart disease. In this circumstance, electrophysiologic testing is used to rule out more malignant etiologies of syncope, such as ventricular tachyarrhythmias and AV conduction block. There are several ways to assess SA node function invasively. They include the sinus node recovery time (SNRT), defined as the longest pause after cessation of overdrive pacing of the right atrium near the SA node (normal: <1500 ms or, corrected for sinus cycle length, <550 ms), and the sinoatrial conduction time (SACT), defined as one-half the difference between the intrinsic sinus cycle length and a noncompensatory pause after a premature atrial stimulus (normal <125 ms). The combination of an abnormal SNRT, an abnormal SACT, and a low IHR is a sensitive and specific indicator of intrinsic SA node disease.
275 |
The Bradyarrhythmias: Disorders of the Atrioventricular Node |
Impulses generated in the sinoatrial (SA) node or in ectopic atrial loci are conducted to the ventricles through the electrically and anatomically complex atrioventricular (AV) node. As described in Chap. 274, the electrophysiologic properties of nodal tissue are distinct from atrial and ventricular myocardium. Cells located in the AV node sit at a relatively higher resting membrane potential than surrounding atrial and ventricular myocytes, exhibit spontaneous depolarization during phase 4 of the action potential, and have slower phase 0 depolarization (mediated by calcium influx in nodal tissue) than that seen in ventricular tissue (mediated by sodium influx).
Bradycardia may occur when conduction across the AV node is compromised, resulting in ineffective ventricular rates, with the possibility of attendant symptoms, including fatigue, syncope, and (if subsidiary pacemaker activity is insufficient) even death. It is important to recognize that in the setting of disturbed AV conduction, SA activation and atrial systole may occur at normal or even accelerated rates, while ventricular activation is either slowed or nonexistent. Transient AV conduction block is common in the young and is most likely the result of high vagal tone found in up to 10% of young adults. Acquired and persistent failure of AV conduction is decidedly rare in healthy adult populations, with an estimated incidence of 200 per million population per year. In the setting of myocardial ischemia, aging and fibrosis, or cardiac infiltrative diseases, however, persistent AV block is much more common.
As with symptomatic bradycardia arising from SA node dysfunction, permanent pacing is the only reliable therapy for symptoms arising from AV conduction block. Approximately 50% of the 150,000 permanent pacemakers implanted in the United States and 70–80% of those in Europe are implanted for disorders of AV conduction.
STRUCTURE AND PHYSIOLOGY OF THE AV NODE
The AV conduction axis is structurally complex, involving the atria and ventricles as well as the AV node. Unlike the SA node, the AV node is a subendocardial structure originating in the transitional zone, which is composed of aggregates of cells in the posterior-inferior right atrium. Superior, medial, and posterior transitional atrionodal bundles converge on the compact AV node. The compact AV node (~1 × 3 × 5 mm) is situated at the apex of the triangle of Koch, which is defined by the coronary sinus ostium posteriorly, the septal tricuspid valve annulus anteriorly, and the tendon of Todaro superiorly. The compact AV node continues as the penetrating AV bundle where it immediately traverses the central fibrous body and is in close proximity to the aortic, mitral, and tricuspid valve annuli; thus, it is subject to injury in the setting of valvular heart disease or its surgical treatment. The penetrating AV bundle continues through the annulus fibrosis and emerges along the ventricular septum adjacent to the membranous septum as the bundle of His. The right bundle branch (RBB) emerges from the distal AV bundle in a band that traverses the right ventricle (moderator band). In contrast, the left bundle branch (LBB) is a broad subendocardial sheet of tissue on the septal left ventricle. The Purkinje fiber network emerges from the RBB and LBB and extensively ramifies on the endocardial surfaces of the right and left ventricles, respectively.
The blood supply to the penetrating AV bundle is from the AV nodal artery and first septal perforator of the left anterior descending coronary artery. The bundle branches also have a dual blood supply from the septal perforators of the left anterior descending coronary artery and branches of the posterior descending coronary artery. The AV node is highly innervated with postganglionic sympathetic and parasympathetic nerves. The bundle of His and distal conducting system are minimally influenced by autonomic tone.
The cells that constitute the AV node complex are heterogeneous with a range of action potential profiles. In the transitional zones, the cells have an electrical phenotype between those of atrial myocytes and cells of the compact node (see Fig. 274-1). Atrionodal transitional connections may exhibit decremental conduction, defined as slowing of conduction with increasingly rapid rates of stimulation. Fast and slow AV nodal pathways have been described, but it is controversial whether these two types of pathway are anatomically distinct or represent functional heterogeneities in different regions of the AV nodal complex. Myocytes that constitute the compact node are depolarized (resting membrane potential ~–60 mV) and exhibit action potentials with low amplitudes, slow upstrokes of phase 0 (<10 V/s), and phase 4 diastolic depolarization; high-input resistance; and relative insensitivity to external [K+]. The action potential phenotype is explained by the complement of ionic currents expressed. AV nodal cells lack a robust inward rectifier potassium current (IK1) and fast sodium current (INa); L-type calcium current (ICa-L) is responsible for phase 0; and phase 4 depolarization reflects the composite activity of the depolarizing currents—funny current (If), ICa-L, T-type calcium current (ICa-T), and sodium calcium exchanger current (INCX)—and the repolarizing currents—delayed rectifier (IKr) and acetylcholine-gated (IKACh) potassium currents. Electrical coupling between cells in the AV node is tenuous due to the relatively sparse expression of gap junction channels (predominantly connexin-40) and increased extracellular volume.
The His bundle and the bundle branches are insulated from ventricular myocardium. The most rapid conduction in the heart is observed in these tissues. The action potentials exhibit very rapid upstrokes (phase 0), prolonged plateaus (phase 2), and modest automaticity (phase 4 depolarization). Gap junctions, composed largely of connexin-40, are abundant, but bundles are poorly connected transversely to ventricular myocardium.
ETIOLOGY OF AV CONDUCTION DISEASE
Conduction block from the atrium to the ventricle can occur for a variety of reasons in a number of clinical situations, and AV conduction block may be classified in a number of ways. The etiologies may be functional or structural, in part analogous to extrinsic and intrinsic causes of SA nodal dysfunction. The block may be classified by its severity from first to third degree or complete AV block or by the location of block within the AV conduction system. Table 275-1 summarizes the etiologies of AV conduction block. Those that are functional (autonomic, metabolic/endocrine, and drug-related) tend to be reversible. Most other etiologies produce structural changes, typically fibrosis, in segments of the AV conduction axis that are generally permanent. Heightened vagal tone during sleep or in well-conditioned individuals can be associated with all grades of AV block. Carotid sinus hypersensitivity, vasovagal syncope, and cough and micturition syncope may be associated with SA node slowing and AV conduction block. Transient metabolic and endocrinologic disturbances as well as a number of pharmacologic agents also may produce reversible AV conduction block.
|
ETIOLOGIES OF ATRIOVENTRICULAR BLOCK |
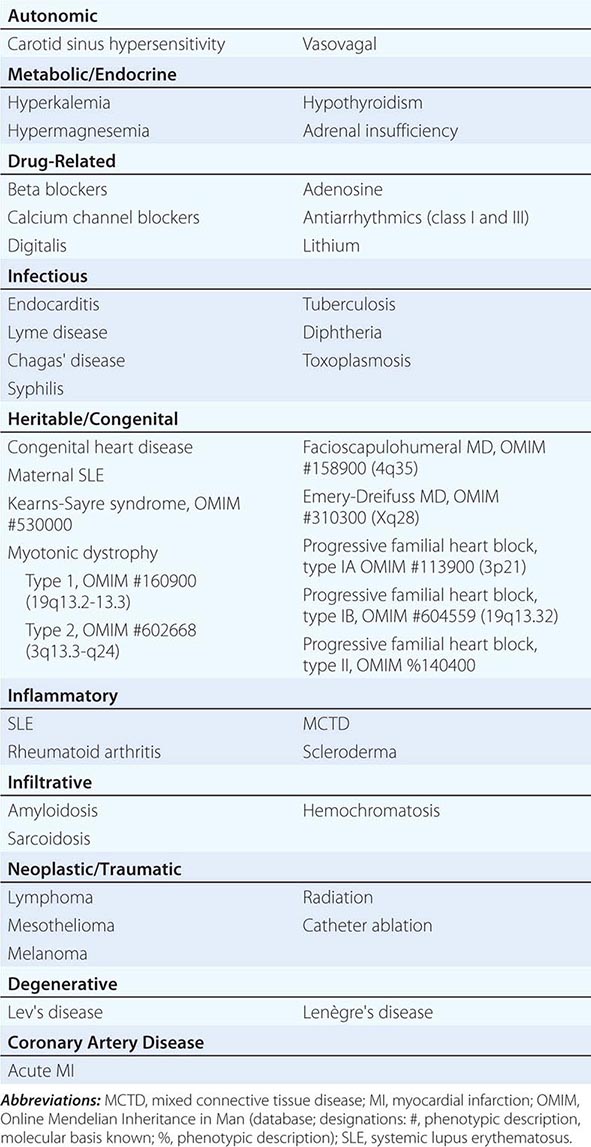
Several infectious diseases have a predilection for the conducting system. Lyme disease may involve the heart in up to 50% of cases; 10% of patients with Lyme carditis develop AV conduction block, which is generally reversible but may require temporary pacing support. Chagas’ disease, which is common in Latin America, and syphilis may produce more persistent AV conduction disturbances. Some autoimmune and infiltrative diseases may produce AV conduction block, including systemic lupus erythematosus (SLE), rheumatoid arthritis, mixed connective tissue disease, scleroderma, amyloidosis (primary and secondary), sarcoidosis, and hemochromatosis; rare malignancies also may impair AV conduction.
Idiopathic progressive fibrosis of the conduction system is one of the more common and degenerative causes of AV conduction block. Aging is associated with degenerative changes in the summit of the ventricular septum, central fibrous body, and aortic and mitral annuli and has been described as “sclerosis of the left cardiac skeleton.” The process typically begins in the fourth decade of life and may be accelerated by atherosclerosis, hypertension, and diabetes mellitus. Accelerated forms of progressive familial heart block have been identified in families with mutations in the cardiac sodium channel gene (SCN5A) and other loci that have been mapped to chromosomes 1 and 19.
AV conduction block has been associated with heritable neuromuscular diseases, including the nucleotide repeat disease myotonic dystrophy, the mitochondrial myopathy Kearns-Sayre syndrome (Chap. 462e), and several of the monogenic muscular dystrophies. Congenital AV block may be observed in complex congenital cardiac anomalies (Chap. 282), such as transposition of the great arteries, ostium primum atrial septal defects (ASDs), ventricular septal defects (VSDs), endocardial cushion defects, and some single-ventricle defects. Congenital AV block in the setting of a structurally normal heart has been seen in children born to mothers with SLE. Iatrogenic AV block may occur during mitral or aortic valve surgery, rarely in the setting of thoracic radiation, and as a consequence of catheter ablation. AV block is a decidedly rare complication of the surgical repair of VSDs or ASDs but may complicate repairs of transposition of the great arteries.
Coronary artery disease may produce transient or persistent AV block. In the setting of coronary spasm, ischemia, particularly in the right coronary artery distribution, may produce transient AV block. In acute myocardial infarction (MI), AV block transiently develops in 10–25% of patients; most commonly, this is first-or second-degree AV block, but complete heart block (CHB) may also occur. Second-degree and higher-grade AV block tends to occur more often in inferior than in anterior acute MI; however, the level of block in inferior MI tends to be in the AV node with more stable, narrow escape rhythms. In contrast, acute anterior MI is associated with block in the distal AV nodal complex, His bundle, or bundle branches and results in wide complex, unstable escape rhythms and a worse prognosis with high mortality rates.
ELECTROCARDIOGRAPHY AND ELECTROPHYSIOLOGY OF AV CONDUCTION BLOCK
AV conduction block typically is diagnosed electrocardiographically, which characterizes the severity of the conduction disturbance and allows one to draw inferences about the location of the block. AV conduction block manifests as slow conduction in its mildest forms and failure to conduct, either intermittent or persistently, in more severe varieties. First-degree AV block (PR interval >200 ms) is a slowing of conduction through the AV junction (Fig. 275-1). The site of delay is typically in the AV node but may be in the atria, bundle of His, or His-Purkinje system. A wide QRS is suggestive of delay in the distal conduction system, whereas a narrow QRS suggests delay in the AV node proper or, less commonly, in the bundle of His. In second-degree AV block there is an intermittent failure of electrical impulse conduction from atrium to ventricle. Second-degree AV block is subclassified as Mobitz type I (Wenckebach) or Mobitz type II. The periodic failure of conduction in Mobitz type I block is characterized by a progressively lengthening PR interval, shortening of the RR interval, and a pause that is less than two times the immediately preceding RR interval on the electrocardiogram (ECG). The ECG complex after the pause exhibits a shorter PR interval than that immediately preceding the pause (Fig. 275-2). This ECG pattern most often arises because of decremental conduction of electrical impulses in the AV node.
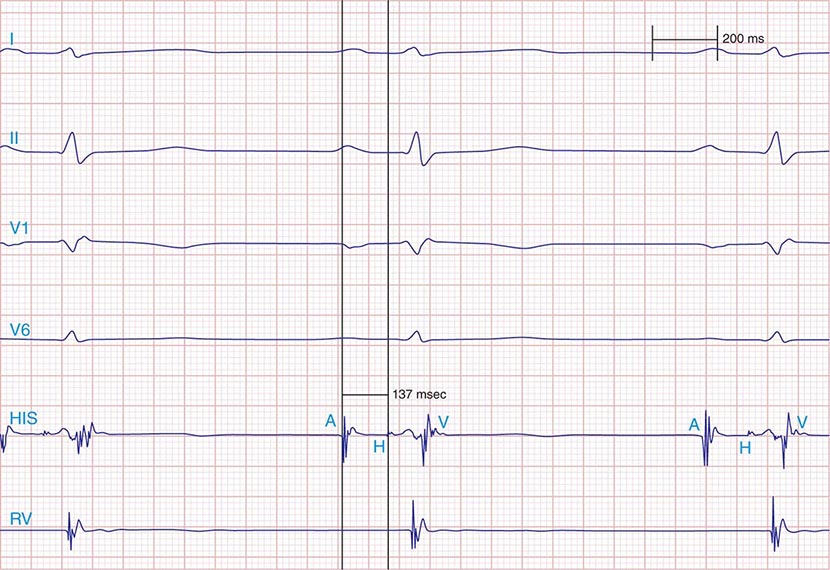
FIGURE 275-1 First-degree AV block with slowing of conduction in the AV node as indicated by the prolonged atrial-to-His bundle electrogram (AH) interval, in this case 157 ms. The His bundle-to-earliest ventricular activation on the surface ECG (HV) interval is normal. The normal HV interval suggests normal conduction below the AV node to the ventricle. I and V1 are surface ECG leads, and HIS is the recording of the endocavitary electrogram at the His bundle position. A, H, and V are labels for the atrial, His bundle, and right ventricular electrograms, respectively.
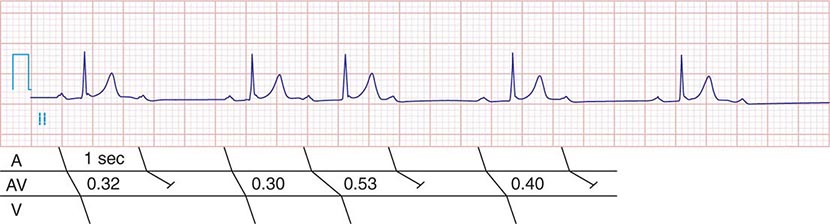
FIGURE 275-2 Mobitz type I second-degree AV block. The PR interval prolongs before the pause, as shown in the ladder diagram. The ECG pattern results from slowing of conduction in the AV node.
It is important to distinguish type I from type II second-degree AV nodal block because the latter has more serious prognostic implications. Type II second-degree AV block is characterized by intermittent failure of conduction of the P wave without changes in the preceding PR or RR intervals. When AV block is 2:1, it may be difficult to distinguish type I from type II block. Type II second-degree AV block typically occurs in the distal or infra-His conduction system, is often associated with intraventricular conduction delays (e.g., bundle branch block), and is more likely to proceed to higher grades of AV block than is type I second-degree AV block. Second-degree AV block (particularly type II) may be associated with a series of nonconducted P waves, referred to as paroxysmal AV block (Fig. 275-3), and implies significant conduction system disease and is an indication for permanent pacing. Complete failure of conduction from atrium to ventricle is referred to as complete or third-degree AV block. AV block that is intermediate between second degree and third degree is referred to as high-grade AV block and, as with CHB, implies advanced AV conduction system disease. In both cases, the block is most often distal to the AV node, and the duration of the QRS complex can be helpful in determining the level of the block. In the absence of a preexisting bundle branch block, a wide QRS escape rhythm (Fig. 275-4B) implies a block in the distal His or bundle branches; in contrast, a narrow QRS rhythm implies a block in the AV node or proximal His and an escape rhythm originating in the AV junction (Fig. 275-4A). Narrow QRS escape rhythms are typically faster and more stable than wide QRS escape rhythms and originate more proximally in the AV conduction system.
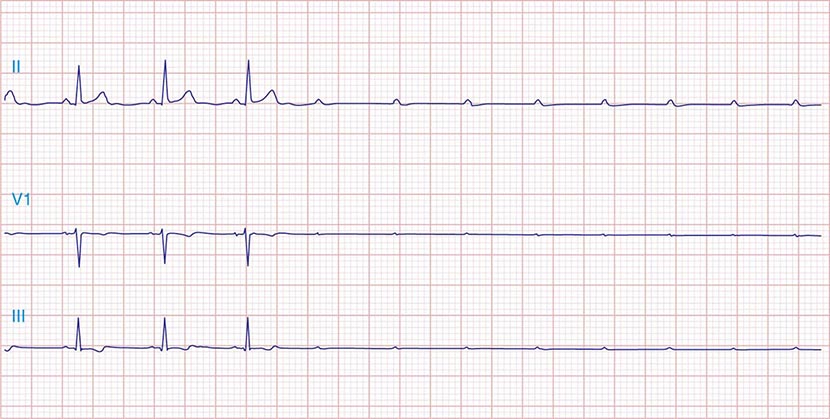
FIGURE 275-3 Paroxysmal AV block. Multiple nonconducted P waves after a period of sinus bradycardia with a normal PR interval. This implies significant conduction system disease, requiring permanent pacemaker implantation.
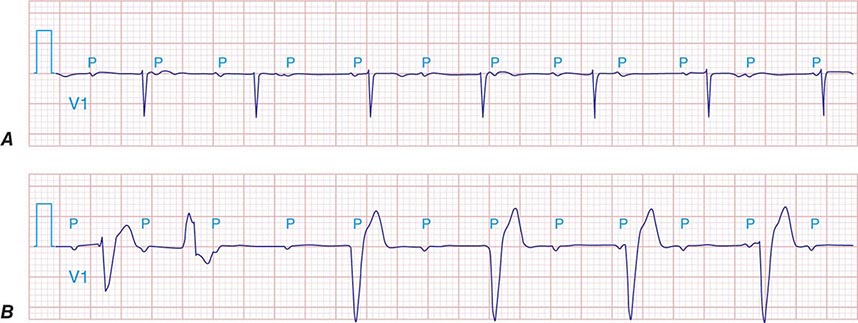
FIGURE 275-4 High-grade AV block. A. Multiple nonconducted P waves with a regular narrow complex QRS escape rhythm probably emanating from the AV junction. B. A wide complex QRS escape and a single premature ventricular contraction. In both cases, there is no consistent temporal relationship between the P waves and QRS complexes.
DIAGNOSTIC TESTING
Diagnostic testing in the evaluation of AV block is aimed at determining the level of conduction block, particularly in asymptomatic patients, since the prognosis and therapy depend on whether the block is in or below the AV node. Vagal maneuvers, carotid sinus massage, exercise, and administration of drugs such as atropine and isoproterenol may be diagnostically informative. Owing to the differences in the innervation of the AV node and infranodal conduction system, vagal stimulation and carotid sinus massage slow conduction in the AV node but have less of an effect on infranodal tissue and may even improve conduction due to a reduced rate of activation of distal tissues. Conversely, atropine, isoproterenol, and exercise improve conduction through the AV node and impair infranodal conduction. In patients with congenital CHB and a narrow QRS complex, exercise typically increases heart rate; by contrast, those with acquired CHB, particularly with wide QRS, do not respond to exercise with an increase in heart rate.
Additional diagnostic evaluation, including electrophysiologic testing, may be indicated in patients with syncope and suspected high-grade AV block. This is particularly relevant if noninvasive testing does not reveal the cause of syncope or if the patient has structural heart disease with ventricular tachyarrhythmias as a cause of symptoms. Electrophysiologic testing provides more precise information regarding the location of AV conduction block and permits studies of AV conduction under conditions of pharmacologic stress and exercise. Recording of the His bundle electrogram by a catheter positioned at the superior margin of the tricuspid valve annulus provides information about conduction at all levels of the AV conduction axis. A properly recorded His bundle electrogram reveals local atrial activity, the His electrogram, and local ventricular activation; when it is monitored simultaneously with recorded body surface electrocardiographic traces, intraatrial, AV nodal, and infranodal conduction times can be assessed (Fig. 275-1). The time from the most rapid deflection of the atrial electrogram in the His bundle recording to the His electrogram (AH interval) represents conduction through the AV node and is normally <130 ms. The time from the His electrogram to the earliest onset of the QRS on the surface ECG (HV interval) represents the conduction time through the His-Purkinje system and is normally ≤55 ms.
Rate stress produced by pacing can unveil abnormal AV conduction. Mobitz I second-degree AV block at short atrial paced cycle lengths is a normal response. However, when it occurs at atrial cycle lengths >500 ms (<120 beats/min) in the absence of high vagal tone, it is abnormal. Typically, type I second-degree AV block is associated with prolongation of the AH interval, representing conduction slowing and block in the AV node. AH prolongation occasionally is due to the effect of drugs (beta blockers, calcium channel blockers, digitalis) or increased vagal tone. Atropine can be used to reverse high vagal tone; however, if AH prolongation and AV block at long pacing cycle lengths persists, intrinsic AV node disease is likely. Type II second-degree block is typically infranodal, often in the His-Purkinje system. Block below the node with prolongation of the HV interval or a His bundle electrogram with no ventricular activation (Fig. 275-5) is abnormal unless it is elicited at fast pacing rates or short coupling intervals with extra stimulation. It is often difficult to determine the type of second-degree AV block when 2:1 conduction is present; however, the finding of a His bundle electrogram after every atrial electrogram indicates that block is occurring in the distal conduction system.
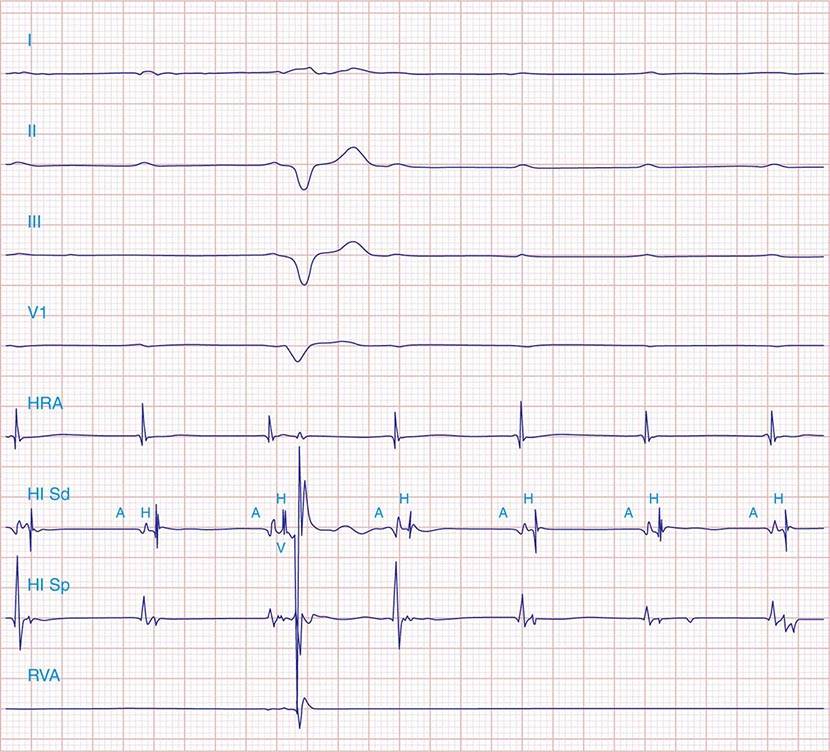
FIGURE 275-5 High-grade AV block below the His. The AH interval is normal and is not changing before the block. Atrial and His bundle electrograms are recorded consistent with block below the distal AV junction. I, II, III, and V1 are surface ECG leads. HISp, HISd, and RVA are the proximal HIS, distal HIS, and right ventricular apical electrical recordings, respectively. A, H, and V represent the atrial, His, and ventricular electrograms on the His bundle recording, respectively. (Tracing courtesy of Dr. Joseph Marine; with permission.)
Intracardiac recording at electrophysiologic study that reveals prolongation of conduction through the His-Purkinje system (i.e., long HV interval) is associated with an increased risk of progression to higher grades of block and is generally an indication for pacing. In the setting of bundle branch block, the HV interval may reveal the condition of the unblocked bundle and the prognosis for developing more advanced AV conduction block. Prolongation of the HV interval in patients with asymptomatic bundle branch block is associated with an increased risk of developing higher-grade AV block. The risk increases with greater prolongation of the HV interval such that in patients with an HV interval >100 ms, the annual incidence of complete AV block approaches 10%, indicating a need for pacing. In patients with acquired CHB, even if intermittent, there is little role for electrophysiologic testing, and pacemaker implantation is almost always indicated.
276 |
Supraventricular Tachyarrhythmias |
Supraventricular tachyarrhythmias originate from or are dependent on conduction through the atrium or atrioventricular (AV) node to the ventricles. Most produce narrow QRS-complex tachycardia (QRS duration <120 ms) characteristic of ventricular activation over the Purkinje system. Conduction block in the left or right bundle branch or activation of the ventricles from an accessory pathway produces a wide QRS complex during supraventricular tachycardia that must be distinguished from ventricular tachycardia (Chap. 277). Supraventricular tachyarrhythmia may be divided into physiologic sinus tachycardia and pathologic tachycardia (Table 276-1). The prognosis and treatment vary considerably depending on the mechanism and underlying heart disease.
|
SUPRAVENTRICULAR TACHYCARDIA |
Abbreviations: AV, atrioventricular; VT, ventricular tachycardia.
Supraventricular tachycardia can be of brief duration, termed nonsustained, or can be sustained such that an intervention, such as cardioversion or drug administration, is required for termination. Episodes that occur with sudden onset and termination are referred to as paroxysmal. Paroxysmal supraventricular tachycardia (PSVT) refers to a family of tachycardias including AV node reentry, AV reentry using an accessory pathway, and atrial tachycardia.
CLINICAL PRESENTATION
Symptoms of supraventricular arrhythmia vary depending on the rate, duration, associated heart disease, and comorbidities and include palpitations, chest pain, dyspnea, diminished exertional capacity, and occasionally syncope. Rarely, a supraventricular arrhythmia precipitates cardiac arrest in patients with the Wolff-Parkinson-White syndrome or severe heart disease, such as hypertrophic cardiomyopathy.
Diagnosis requires obtaining an electrocardiogram (ECG) at the time of symptoms. For transient arrhythmia, ambulatory ECG recording is warranted (see Table 277-1). Exercise testing is useful for assessing exercise-related symptoms. Occasionally an invasive electrophysiology study is warranted to provoke the arrhythmia with pacing, confirm the mechanism, and often, perform catheter ablation.
Physiologic Sinus Tachycardia The sinus node is comprised of a group of cells dispersed within the superior aspect of the thick ridge of muscle known as the crista terminalis where the posterior smooth atrial wall derived from the sinus venosus meets the trabeculated anterior portion of the right atrium (Fig. 276-1). Sinus p waves are characterized by a frontal plane axis directed inferiorly and leftward, with positive p waves in leads II, III, and aVF; a negative p wave in aVR; and an initially positive biphasic p wave in V1. Normal sinus rhythm has a rate of 60–100 beats/min. Sinus tachycardia (>100 beats/min) typically occurs in response to sympathetic stimulation and vagal withdrawal, whereby the rate of spontaneous depolarization of the sinus node increases and the focus of earliest activation within the node typically shifts more leftward and closer to the superior septal aspect of the crista terminalis, thus producing taller p waves in the inferior limb leads when compared to normal sinus rhythm.
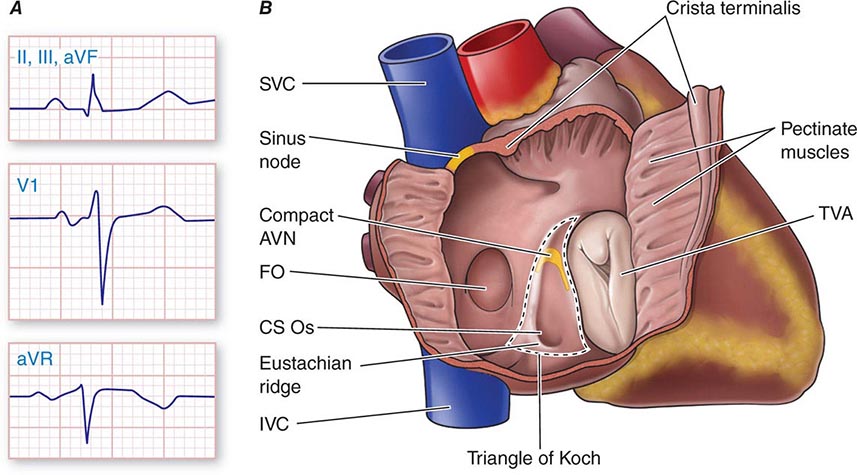
FIGURE 276-1 Right atrial anatomy pertinent to normal sinus rhythm and supraventricular tachycardia. A. Typical P-wave morphology during normal sinus rhythm based on standard 12-lead electrocardiogram. There is a positive P wave in leads II, III, and aVF; biphasic, initially positive P wave in V1; and negative P wave in aVR. B. Right atrial anatomy seen from a right lateral perspective with the lateral wall opened to view the septum. AVN, atrioventricular node; CS Os, coronary sinus ostium; FO, fossa ovalis; IVC, inferior vena cava; SVC, superior vena cava; TVA, tricuspid valve annulus.
Sinus tachycardia is considered physiologic when it is an appropriate response to exercise, stress, or illness. Sinus tachycardia can be difficult to distinguish from focal atrial tachycardia (see below) that originates from a focus near the sinus node. A causative factor (such as exertion) and a gradual increase and decrease in rate favors sinus tachycardia, whereas an abrupt onset and offset favor atrial tachycardia. The distinction can be difficult and occasionally requires extended ECG monitoring or even invasive electrophysiology study. Treatment for physiologic sinus tachycardia is aimed at the underlying condition (Table 276-2).
|
COMMON CAUSES OF PHYSIOLOGIC SINUS TACHYCARDIA |
Nonphysiologic Sinus Tachycardia Inappropriate sinus tachycardia is an uncommon condition in which the sinus rate increases spontaneously at rest or out of proportion to physiologic stress or exertion. Affected individuals are often women in the third or fourth decade of life. Fatigue, dizziness, and even syncope may accompany palpitations, which can be disabling. Additional symptoms of chest pain, headaches, and gastrointestinal upset are common. It must be distinguished from appropriate sinus tachycardia and from focal atrial tachycardia, as discussed above. Misdiagnosis of physiologic sinus tachycardia with an anxiety disorder is common. Therapy is often ineffective or poorly tolerated. Careful titration of beta blockers and/or calcium channel blockers may reduce symptoms. Clonidine and serotonin reuptake inhibitors have also been used. Ivabradine, a drug that blocks the If current causing sinus node depolarization, is promising but is not approved for use in the United States. Catheter ablation of the sinus node has been used, but long-term control of symptoms is usually poor, and it often leaves young individuals with a permanent pacemaker.
When symptomatic sinus tachycardia occurs with postural hypotension, the syndrome is called postural orthostatic tachycardia syndrome (POTS). Symptoms are often similar to those in patients with inappropriate sinus tachycardia. POTS is sometimes due to autonomic dysfunction following a viral illness and may resolve spontaneously over 3–12 months. Volume expansion with salt supplementation, oral fludrocortisone, compression stockings, and the α-agonist midodrine, often in combination, can be helpful. Exercise training has also been purported to improve symptoms.
Focal Atrial Tachycardia Focal atrial tachycardia (AT) can be due to abnormal automaticity, triggered automaticity, or a small reentry circuit confined to the atrium or atrial tissue extending into a pulmonary vein, the coronary sinus, or vena cava. It can be sustained, nonsustained, paroxysmal, or incessant. Focal AT accounts for approximately 10% of PSVT referred for catheter ablation. Nonsustained AT is commonly observed on 24-h ambulatory ECG recordings, and the prevalence increases with age. Tachycardia can occur in the absence of structural heart disease or can be associated with any form of heart disease that affects the atrium. Sympathetic stimulation is a promoting factor such that AT can be a sign of underlying illness. AT with AV block can occur in digitalis toxicity. Symptoms are similar to other supraventricular tachycardias (SVTs). Incessant AT can cause tachycardia-induced cardiomyopathy.
AT typically presents as an SVT either with 1:1 AV conduction or with AV block that can be Wenckebach type conduction or fixed (e.g., 2:1 or 3:1) block. Because it is not dependent on AV nodal conduction, AT will not terminate with AV block, and the atrial rate will not be affected, which distinguishes AT from most AV nodal–dependent SVTs, such as AV nodal reentry and AV reentry using an accessory pathway (see below). An accelerated warm-up phase after initiation or cool-down phase prior to termination also favors AT rather than AV nodal–dependent SVT. P waves are often discrete, with an intervening isoelectric segment, in contrast to atrial flutter and macroreentrant AT (see below). When 1:1 conduction to the ventricles is present, the arrhythmia can resemble sinus tachycardia typically with a P-R interval shorter than the R-P interval (Fig. 276-2). It can be distinguished from sinus tachycardia by the p-wave morphology, which usually differs from sinus p waves depending on the location of the focus. Focal AT tends to originate in areas of complex atrial anatomy, such as the crista terminalis, valve annuli, atrial septum, and atrial muscle extending along cardiac thoracic veins (superior vena cava, coronary sinus, and pulmonary veins) (Fig. 276-3), and the location can often be estimated by the P-wave morphology. AT from the right atrium has a positive P-wave morphology in lead I and biphasic P-wave morphology in lead V1. AT from the atrial septum will frequently have a narrower P-wave duration than sinus rhythm. AT from the left atrium will usually have a monophasic, positive P wave in lead V1. AT that originates from superior atrial locations, such as the superior vena cava or superior pulmonary veins, will be positive in the inferior limb leads II, III, and aVF, whereas AT from a more inferior location, such as the ostium of the coronary sinus, will inscribe negative P waves in these same leads. When the focus is in the superior aspect of the crista terminalis, close to the sinus node, however, the p wave will resemble that of sinus tachycardia. Abrupt onset and offset then favor AT rather than sinus tachycardia. Depending on the atrial rate, the P wave may fall on top of the t wave or, during 2:1 conduction, may fall coincident with the QRS. Maneuvers that increase AV block, such as carotid sinus massage, Valsalva maneuver, or administration of AV nodal–blocking agents, such as adenosine, are useful to create AV block that will expose the p wave (Fig. 276-4).
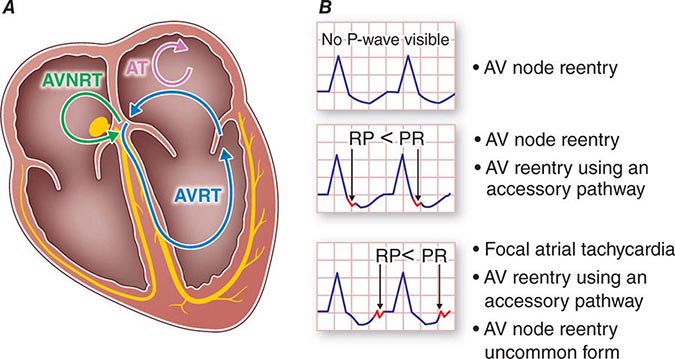
FIGURE 276-2 Common mechanisms underlying paroxysmal supraventricular tachycardia along with typical R-P relationships. A. Schematic showing a four-chamber view of the heart with atrioventricular node in green and an accessory pathway between the left atrium and left ventricle in yellow. Atrial tachycardia (AT; red circuit) is confined completely to atrial tissue. Atrioventricular nodal reentry tachycardia (AVNRT; blue circuit) uses atrioventricular (AV) nodal and perinodal atrial tissue. Atrioventricular reentry tachycardia (AVRT; black circuit) uses atrial and ventricular tissue, accessory pathway, AV node, and specialized conduction fibers (His-Purkinje) as part of the reentry circuit. B. Typical relation of the p wave to QRS, commonly described as the R-P to P-R relationships for the different tachycardia mechanisms.
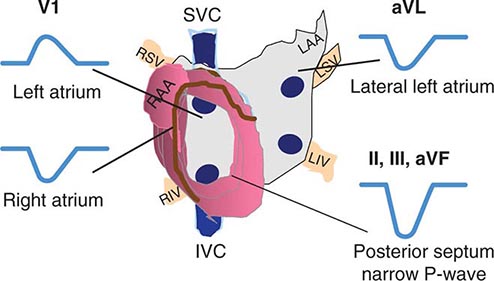
FIGURE 276-3 Location of focal atrial tachycardia focus estimated by P-wave morphology. LAA, left atrial appendage; LIV, left inferior pulmonary vein; LSV, left superior pulmonary vein; RAA, right atrial appendage; RIV, right inferior pulmonary vein; RSV, right superior pulmonary vein; SVC, superior vena cava.
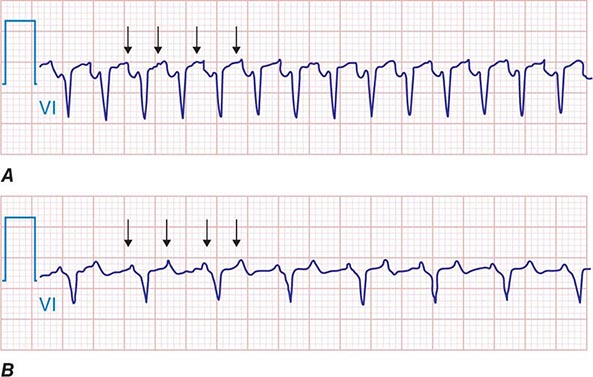
FIGURE 276-4 Atrial tachycardia (AT) with 1:1 and 2:1 atrioventricular (AV) conduction. Arrows indicate p waves. A. AT with 1:1 AV relationship and R-P > P-R. B. Same AT with 2:1 AV relationship after AV nodal–blocking agent administered. (Adapted from F Marchlinski: The tachyarrhythmias. In Longo DL et al [eds]: Harrison’s Principles of Internal Medicine, 18th ed. New York, McGraw-Hill, 2012, pp 1878–1900.)
Acute management of sudden-onset, sustained AT is the same as for PSVT (see below), but the response to pharmacologic therapy is variable, likely depending on the mechanism. For AT due to reentry, administration of adenosine or vagal maneuvers may transiently increase AV block without terminating tachycardia. Some ATs terminate with a sufficient dose of adenosine, consistent with triggered activity as the mechanism. Cardioversion can be effective in some, but fails in others, suggesting automaticity as the mechanism. Beta blockers and calcium channel blockers may slow the ventricular rate by increasing AV block, which can improve tolerance of the arrhythmias. Potential precipitating factors and intercurrent illness should be sought and corrected. Underlying heart disease should be considered and excluded.
For patients with recurrent episodes, beta blockers, the calcium channel blockers diltiazem or verapamil, and the antiarrhythmic drugs flecainide, propafenone, disopyramide, sotalol, and amiodarone can be effective, but potential toxicities and adverse effects often warrant avoiding these agents (Tables 276-3, 276-4, and 276-5). Catheter ablation targeting the AT focus is effective in more than 80% of patients and is recommended for recurrent symptomatic AT when drugs fail or are not desired or for incessant AT causing tachycardia-induced cardiomyopathy.
|
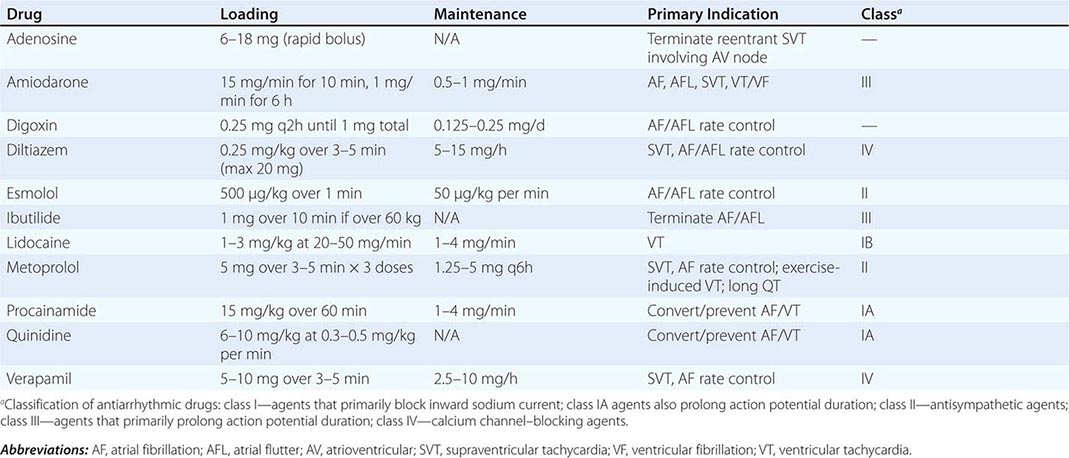
COMMONLY USED ANTIARRHYTHMIC AGENTS—INTRAVENOUS DOSE RANGE/PRIMARY INDICATION |
|
COMMONLY USED ANTIARRHYTHMIC AGENTS: CHRONIC ORAL DOSING/PRIMARY INDICATIONS |
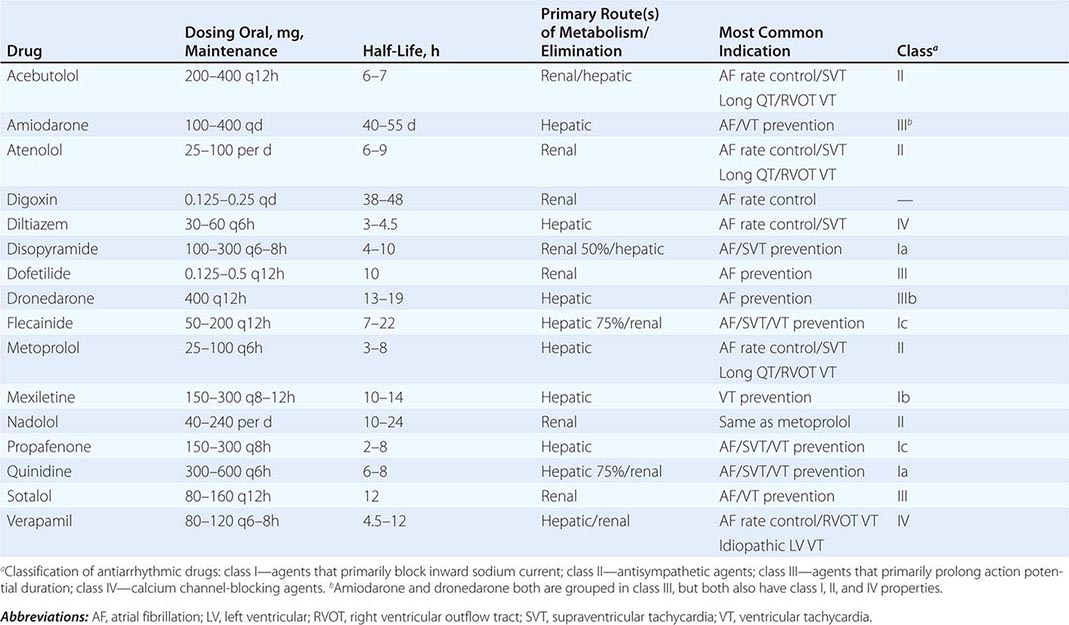
|
COMMON AND PROARRHYTHMIC TOXICITIES OF ANTIARRHYTHMIC AGENTS |
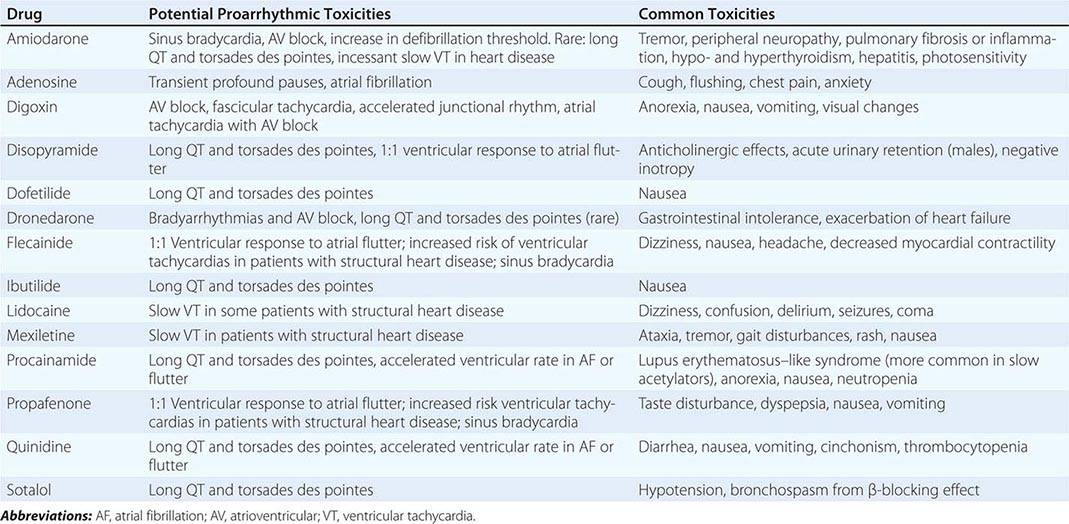
Atrioventricular Nodal Reentry Tachycardia AV nodal reentry tachycardia (AVNRT) is the most common form of PSVT, representing approximately 60% of cases referred for catheter ablation. It most commonly manifests in the second to fourth decades of life, often in women. It is often well tolerated, but rapid tachycardia, particularly in the elderly, may cause angina, pulmonary edema, hypotension, or syncope. It is not usually associated with structural heart disease.
The mechanism is reentry involving the AV node and likely the perinodal atrium, made possible by the existence of multiple pathways for conduction from the atrium into the AV node (Fig. 276-5). In the most common form, a slowly conducting AV nodal pathway extends from the compact AV node near the bundle of His, inferiorly along the tricuspid annulus, adjacent to the coronary sinus os. The reentry wavefront propagates up this slow pathway to the compact AV node and then exits from the fast pathway at the top of the AV node. The path back to the slow pathway to complete the circuit is not defined. The conduction time from the compact AV node region to the atrium is similar to that from the compact node to the His bundle and ventricles, such that atrial activation occurs at about the same time as ventricular activation. The p wave is therefore inscribed during, slightly before, or slightly after the QRS and can be difficult to discern. Often the P wave is seen at the end of the QRS complex as a pseudo-r′ in lead V1 and pseudo-S waves in leads II, III, and aVF (Fig. 276-5A). The rate can vary with sympathetic tone. Simultaneous atrial and ventricular contraction results in atrial contraction against a closed tricuspid valve that produces cannon a waves visible in the jugular venous pulse and that the patient often perceives as a fluttering sensation in the neck. Elevated venous pressures may also lead to release of natriuretic peptides that cause posttachycardia diuresis. Less frequently, the AV nodal reentry circuit revolves in the opposite direction and gives rise to a tachycardia with an R-P interval longer than the P-R interval, similar to AT. The p wave will have the morphology noted above, and in contrast to ATs, maneuvers or medications that produce AV block terminate the arrhythmia.
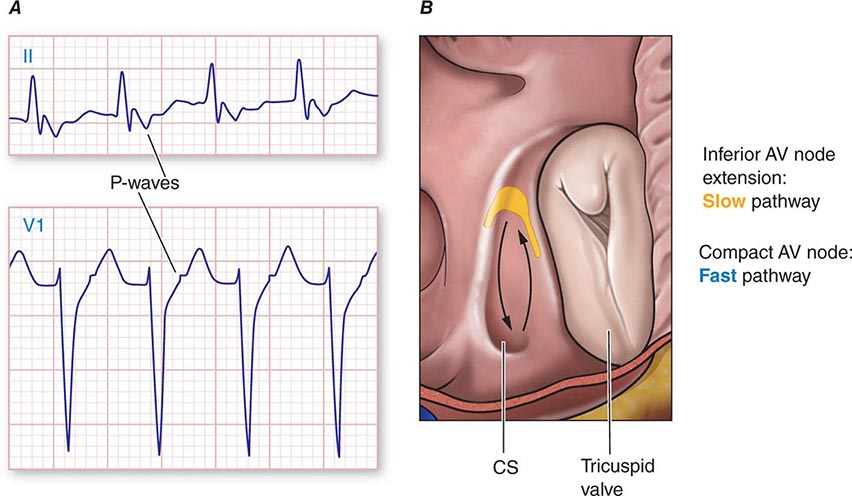
FIGURE 276-5 Atrioventricular (AV) node reentry. A. Leads II and V1 are shown. P waves are visible at the end of the QRS complex and are negative in lead II, and may give the impression of S waves in the inferior limb leads II, III, and aVF and an R’ in lead V1. B. Stylized version of the AV nodal reentry circuit within the triangle of Koch (Fig. 276-1) that involves AV node and its extensions along with perinodal atrial tissue.
Acute treatment is the same as for PSVT (discussed below). Whether ongoing therapy is warranted depends on the severity of symptoms and frequency of episodes. Reassurance and instruction as to performance of the Valsalva maneuver to terminate episodes are sufficient for many patients. Administration of an oral beta blocker, verapamil, or diltiazem at the onset of an episode has been used to facilitate termination. Chronic therapy with these medications or flecainide is an option if prophylactic therapy is needed. Catheter ablation of the slow AV nodal pathway is recommended for patients with recurrent or severe episodes or when drug therapy is ineffective, not tolerated, or not desired by the patient. Catheter ablation is curative in over 95% of patients. The major risk is heart block requiring permanent pacemaker implantation, which occurs in less than 1% of patients.
Junctional Tachycardia Junctional ectopic tachycardia (JET) is due to automaticity within the AV node. It is rare in adults and more frequently encountered as an incessant tachycardia in children, often in the perioperative period of surgery for congenital heart disease. It presents as a narrow QRS tachycardia, often with ventriculoatrial (VA) block, such that AV dissociation is present. JET can occur as a manifestation of increased adrenergic tone and may be seen after administration of isoproterenol. It may also occur for a short period of time after ablation for AVNRT.
Accelerated junctional rhythm is a junctional automatic rhythm between 50 and 100 beats/min. Initiation may occur with gradual acceleration in rate, suggesting an automatic focus, or after a premature ventricular contraction, suggesting a focus of triggered automaticity. VA conduction is usually present, with p-wave morphology and timing such that it resembles slow AVNRT.
ACCESSORY PATHWAYS AND THE WOLFF-PARKINSON-WHITE SYNDROME
Accessory pathways (APs) occur in 1 in 1500–2000 people and are associated with a variety of arrhythmias including narrow-complex PSVT, wide-complex tachycardias, and, rarely, sudden death. Most patients have structurally normal hearts, but APs are associated with Ebstein’s anomaly of the tricuspid valve and forms of hypertrophic cardiomyopathy including PRKAG2 mutations, Danon’s disease, and Fabry’s disease.
APs are abnormal connections that allow conduction between the atrium and ventricles across the AV ring (Fig. 276-6). They are present from birth and are due to failure of complete partitioning of atrium and ventricle by the fibrous AV rings. They occur across either an AV valve annulus or the septum, most frequently between the left atrium and free wall of the left ventricle, followed by posteroseptal, right free wall, and anteroseptal locations. If the AP conducts from atrium to ventricle (antegrade) with a shorter conduction time than the AV node and His bundle, then the ventricles are preexcited during sinus rhythm, and the ECG shows a short P-R interval (<0.12 s), slurred initial portion of the QRS (delta wave), and prolonged QRS duration produced by slow conduction through direct activation of ventricular myocardium over the AP (Fig. 276-6A). The morphology of the QRS and delta wave is determined by the AP location (Fig. 276-7) and the degree of fusion between the excitation wavefronts from conduction over the AV node and conduction over the AP. Right-sided pathways preexcite the right ventricle, producing a left bundle branch block–like configuration in lead V1, and often show marked preexcitation because of relatively close proximity of the AP to the sinus node (Fig. 276-7). Left-sided pathways preexcite the left ventricle and may produce a right bundle branch–like configuration in lead V1 and a negative delta wave in aVL, indicating initial depolarization of the lateral portion of the left ventricle that can mimic q waves of lateral wall infarction (Fig. 276-7). Preexcitation due to an AP at the diaphragmatic surface of the heart, typically in the paraseptal region, produces delta waves that are negative in leads III and aVF, mimicking the q waves of inferior wall infarction (Fig. 276-7). Preexcitation can be intermittent and disappear during exercise as conduction over the AV node accelerates and takes over ventricular activation completely.
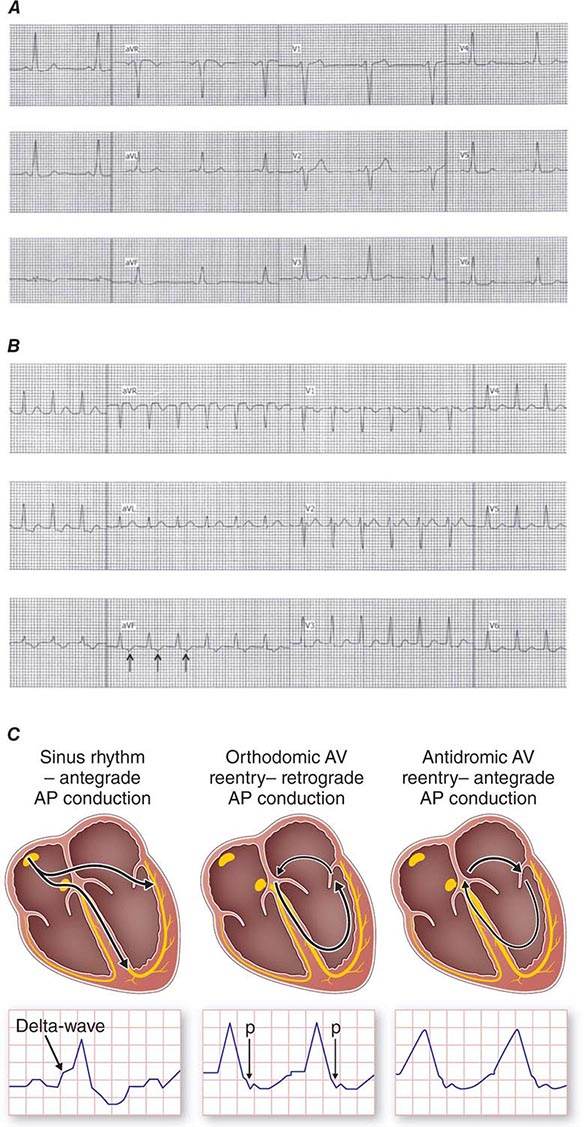
FIGURE 276-6 Wolff-Parkinson-White (WPW) syndrome. A. A 12-lead electrocardiogram in sinus rhythm (SR) of a patient with WPW demonstrating short P-R interval, delta waves, and widened QRS complex. This patient had an anteroseptal location of the AP. B. Orthodromic AV reentry in a patient with WPW syndrome using a posteroseptal AP. Note the P waves in the ST segment (arrows) seen in lead III and normal appearance of QRS complex. C. Three most common rhythms associated with WPW syndrome: sinus rhythm demonstrating antegrade conduction over the AP and AV node; orthodromic AVRT using retrograde conduction over the AP and antegrade conduction over the AV node; and antidromic AVRT using retrograde conduction over the AV node and antegrade conduction over the AP. AP, accessory pathway; AV, atrioventricular; AVRT, atrioventricular reentry tachycardia; WPW, Wolff-Parkinson-White.
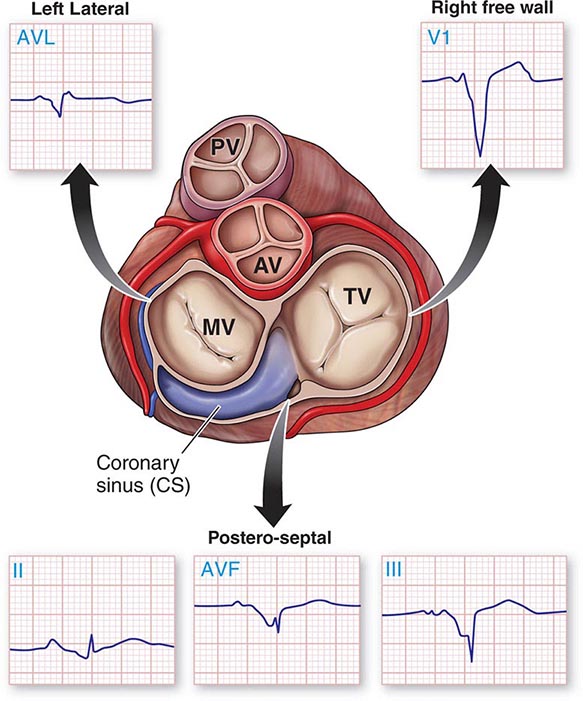
FIGURE 276-7 Potential locations for accessory pathways in patients with Wolff-Parkinson-White Syndrome and typical QRS appearance of delta waves that can mimic underlying structural heart disease such as myocardial infraction of bundle branch block. AV, aortic valve; MV, mitral valve; PV, pulmonary valve; TV, tricuspid valve.
Wolff-Parkinson-White (WPW) syndrome is defined as a preexcited QRS during sinus rhythm and episodes of PSVT. There are a number of variations of APs, which may not cause preexcitation and/or arrhythmias. Concealed APs allow only retrograde conduction, from ventricle to atrium, so no preexcitation is present during sinus rhythm, but SVT can occur. Fasciculoventricular connections between the His bundle and ventricular septum produce preexcitation but do not cause arrhythmia, nor do fibers such as atrio-Hisian connections, probably because the circuit is too short to promote reentry. Atriofascicular pathways, also known as Mahaim fibers, probably represent a duplicate AV node and His-Purkinje system that connect the right atrium to fascicles of the right bundle branch and conduct slowly only in the anterograde direction.
AV Reentry Tachycardia The most common tachycardia caused by an AP is the PSVT designated orthodromic AV reentry. The circulating reentry wavefront propagates from the atrium anterogradely over the AV node and His-Purkinje system to the ventricles and then reenters the atria via retrograde conduction over the AP (Fig. 276-6B). The QRS is narrow or may have typical right or left bundle branch block, but without preexcitation during tachycardia. Because excitation through the normal AV conduction system and AP are necessary, AV or VA block results in tachycardia termination. During sinus rhythm, preexcitation is seen if the pathway also allows anterograde conduction (Fig. 276-6A). Most commonly, during tachycardia the R-P interval is shorter than the P-R interval and can resemble AVNRT (Fig. 276-1). Unlike typical AVNRT, P-wave timing is never simultaneous with a narrow QRS complex because the ventricles must be activated before the reentry wavefront reaches the AP and conducts back to the atrium. The morphology of the P wave is determined by the pathway location, but can be difficult to assess because it is usually inscribed during the ST segment. The p wave in posteroseptal APs is negative in leads II, III, and aVF, similar to that of AV nodal reentry, but P-wave morphology differs from AV nodal reentry for pathways in other locations (Fig. 276-7).
Occasionally, an AP conducts extremely slowly in the retrograde direction, which results in tachycardia with a long R-P interval, similar to most ATs. These pathways are usually located in the septal region and have negative p waves in leads II, III, and aVF. Slow conduction facilitates reentry, often leading to nearly incessant tachycardia, known as paroxysmal junctional reciprocating tachycardia (PJRT). Tachycardia-induced cardiomyopathy can occur. Without an invasive electrophysiology study, it may be difficult to distinguish this form of orthodromic AV reentry from atypical AV nodal reentry or AT.
Preexcited Tachycardias Preexcitated tachycardia occurs when the ventricles are activated by antegrade conduction over the AP (Fig. 276-6C). The most common is antidromic AV reentry in which activation propagates from atrium to ventricle via the AP and then conducts retrogradely to the atria via the His-Purkinje system and the AV node (or rarely a second AP). The wide QRS complex is produced entirely via ventricular excitation over the AP because there is no contribution of ventricular activation over more rapidly conducting specialized His-Purkinje fibers. This tachycardia is often indistinguishable from monomorphic ventricular tachycardia. The presence of preexcitation in sinus rhythm suggests the diagnosis.
Preexcitated tachycardia also occurs if an AP allows antegrade conduction to the ventricles during AT, atrial flutter, atrial fibrillation (Fig. 276-8), or AV nodal reentry. Atrial fibrillation and atrial flutter are potentially life threatening if the AP allows very rapid repetitive conduction. Approximately 25% of APs causing preexcitation allow minimum R-to-R intervals of less than 250 ms during atrial fibrillation are therefore associated with a risk of inducing ventricular fibrillation and sudden death. Preexcited atrial fibrillation presents as a wide-complex, very irregular rhythm. During atrial fibrillation, the ventricular rate is determined by the conduction properties of the AP and AV node. The QRS complex can appear quite bizarre and change on a beat-to-beat basis due to the variability in the degree of fusion from activation over the AV node and AP, or all beats may be due to conduction over the AP (Fig. 276-8). Ventricular activation from the Purkinje system may depolarize the ventricular end of the AP and prevent 1:1 atrial wavefront conduction over the AP. Slowing AV nodal conduction can thereby facilitate AP conduction and dangerously accelerate the ventricular rate. Administration of AV nodal–blocking agents including oral or intravenous verapamil, diltiazem, beta blockers, intravenous adenosine, and intravenous amiodarone are contraindicated. Preexcited tachycardias should be treated with electrical cardioversion or intravenous procainamide or ibutilide, which may terminate or slow the ventricular rate.
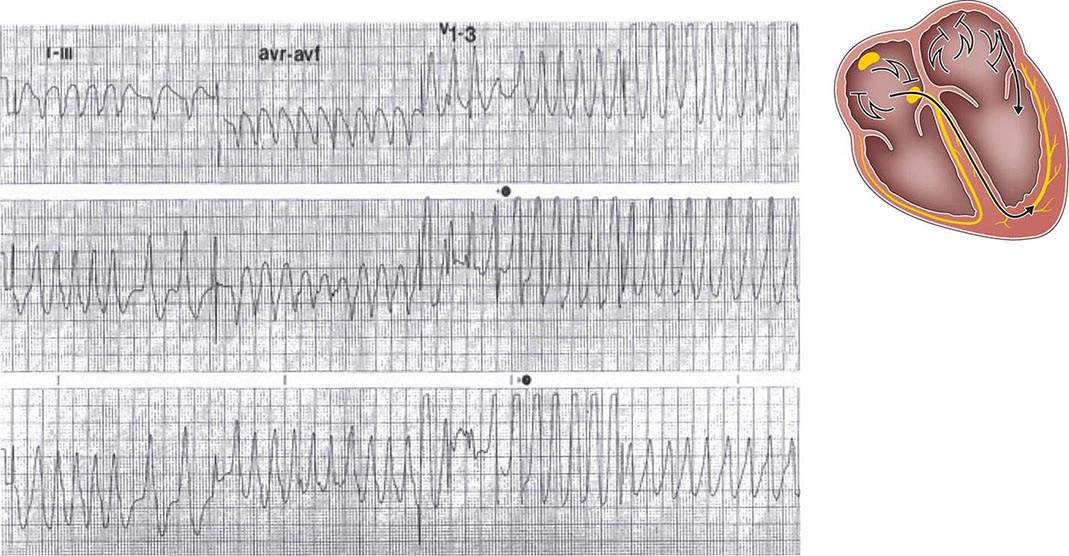
FIGURE 276-8 Preexcited atrial fibrillation (AF) due to conduction over a left free wall accessory pathway (AP). The electrocardiogram shows rapid irregular QRS complexes that represent fusion between conduction over the atrioventricular node and left free wall AP. Shortest R-R intervals between preexcited QRS complexes of less than 250 ms, as in this case, indicate a risk of sudden death with this arrhythmia.
Management of Patients with Accessory Pathways Acute management of orthodromic AV reentry is discussed below for PSVT. Patients with WPW syndrome may have wide-complex tachycardia due to antidromic AV reentry, orthodromic AV with bundle branch block, or a preexcited tachycardia, and treatment depends on the underlying rhythm.
Initial patient evaluation should include assessment for aggravating factors, including intercurrent illness and factors that increase sympathetic tone. Examination should focus on excluding underlying heart disease. An echocardiogram is reasonable to exclude Ebstein’s anomaly and hypertrophic cardiomyopathy.
Patients with preexcitation who have symptoms of arrhythmia are at risk for developing atrial fibrillation and sudden death if they have an AP with high-risk properties. The risk of cardiac arrest is in the range of 2 per 1000 patients in adults but is likely greater in children. An invasive electrophysiology study is warranted to determine if the AP is high enough risk to warrant potentially curative catheter ablation. For patients with concealed APs or known low-risk APs causing orthodromic AV reentry, chronic therapy is guided by symptoms and frequency of events. Vagal maneuvers may terminate episodes, as may a dose of beta blocker, verapamil, or diltiazem taken at the onset of an episode. Chronic therapy with these agents or flecainide can reduce the frequency of episodes in some patients. Catheter ablation is warranted for recurrent arrhythmias when drugs are ineffective, not tolerated, or not desired by the patient or if the AP is considered high risk (Fig. 276-8). Efficacy is in the range of 95% depending on the location of the AP. Serious complications occur in fewer than 3% of patients, but can include AV block, cardiac tamponade, thromboemboli, coronary artery injury, and vascular access complications. Mortality occurs in less than 1 in 1000 patients.
Adults who have preexcitation but no arrhythmia symptoms have a risk of sudden death estimated to be 1 per 1000 patient-years. Electrophysiology study is usually advised for people in occupations for which an arrhythmia occurrence would place them or others at risk, such as police, military, and pilots, or for individuals who desire evaluation for risk. Routine follow-up without therapy is reasonable in others. Children are at greater risk of sudden death, approximately 2 per 1000 patient-years.
|
TREATMENT |
PAROXYSMAL SUPRAVENTRICULAR TACHYCARDIA |
Acute management of narrow QRS PSVT is guided by the clinical presentation. Continuous ECG monitoring should be implemented and a 12-lead ECG should always be obtained when possible. In the presence of hypotension with unconsciousness or respiratory distress, QRS-synchronous direct current cardioversion is warranted, but this is rarely needed, because intravenous adenosine works promptly in most situations (see below). For stable individuals, initial therapy takes advantage of the fact that most PSVTs are dependent on AV nodal conduction (AV nodal reentry or orthodromic AV reentry) and therefore likely to respond to sympatholytic and vagotonic maneuvers and drugs (Fig. 276-9). As these are administered, the ECG should be continuously recorded, because the response can establish the diagnosis. AV block with only transient slowing of tachycardia may expose ongoing p waves, indicating AT or atrial flutter as the mechanism.
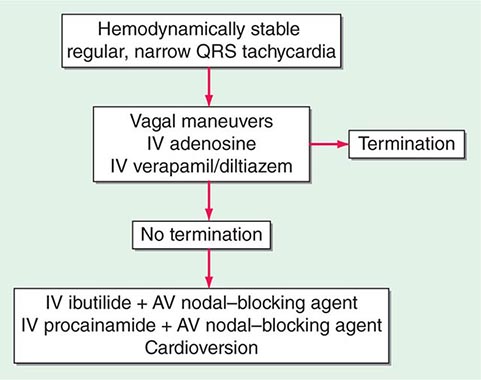
FIGURE 276-9 Treatment algorithm for patients presenting with hemodynamically stable paroxysmal supraventricular tachycardia. AV, atrioventricular.
Carotid sinus massage is reasonable provided the risk of carotid vascular disease is low, as indicated by absence of carotid bruits and no prior history of stroke. A Valsalva maneuver should be attempted in cooperative individuals, and if effective, the patient can be taught to perform this maneuver as needed. If vagal maneuvers fail or cannot be performed, intravenous adenosine will terminate the vast majority of PSVT by transiently blocking conduction in the AV node. Adenosine may produce transient chest pain, dyspnea, and anxiety. It is contraindicated in patients with prior cardiac transplantation due to potential hypersensitivity. It can theoretically aggravate bronchospasm. Adenosine precipitates atrial fibrillation, which is usually brief, in up to 15% of patients, so it should be used cautiously in patients with WPW syndrome in whom AF may produce hemodynamic instability. Intravenous beta blockers and calcium channel blockers (verapamil or diltiazem) are also effective but may cause hypotension before and after arrhythmia termination and have a longer duration of action. These agents can also be given orally and can be taken by the patient on an as-needed basis to slow ventricular rate and facilitate termination by Valsalva maneuver.
The differential diagnosis of wide-complex tachycardia includes ventricular tachycardia (Chap. 277), PSVT with bundle branch block aberrancy, and preexcited tachycardia (see above). In general, these should be managed as ventricular tachycardia until proven otherwise. If the tachycardia is regular and the patient is stable, a trial of intravenous adenosine is reasonable. Very irregular wide-complex tachycardia should be managed with cardioversion, intravenous procainamide, or ibutilide, which presumes preexcited atrial fibrillation or flutter (see above). If the diagnosis of PSVT with aberrancy is unequivocal, as may be the case in patients with prior episodes, treatment for PSVT is reasonable. In all cases, continuous ECG monitoring should be implemented, and emergency cardioversion and defibrillation should be available.
COMMON ATRIAL FLUTTER AND MACROREENTRANT ATRIAL TACHYCARDIAS
Macrorrentrant atrial tachycardia is due to a large reentry circuit, often associated with areas of scar in the atria. Common or typical right atrial flutter is due to a circuit that revolves around the tricuspid valve annulus, bounded anteriorly by the annulus and posteriorly by functional conduction block in the crista terminalis. The wavefront passes through an isthmus between the inferior vena cava and the tricuspid valve annulus, known as the sub-Eustachian or cavotricuspid isthmus, where it is susceptible to interruption by catheter ablation. Thus, common atrial flutter is cavotricuspid isthmus-dependent atrial flutter. This circuit most commonly revolves in a counterclockwise direction (as viewed looking toward the tricuspid annulus from the ventricular aspect), which produces the characteristic negative sawtooth flutter waves in leads II, III, and aVF and positive P waves in lead V1 (Fig. 276-10). When the direction is reversed, clockwise rotation produces the opposite P-wave vector in those leads. The atrial rate is typically 240–300 beats/min but may be slower in the presence of atrial disease or antiarrhythmic drugs. It often conducts to the ventricles with 2:1 AV block, creating a regular tachycardia at 150 beats/min, with p waves that may be difficult to discern. Maneuvers that increase AV nodal block will typically expose flutter waves, allowing diagnosis.
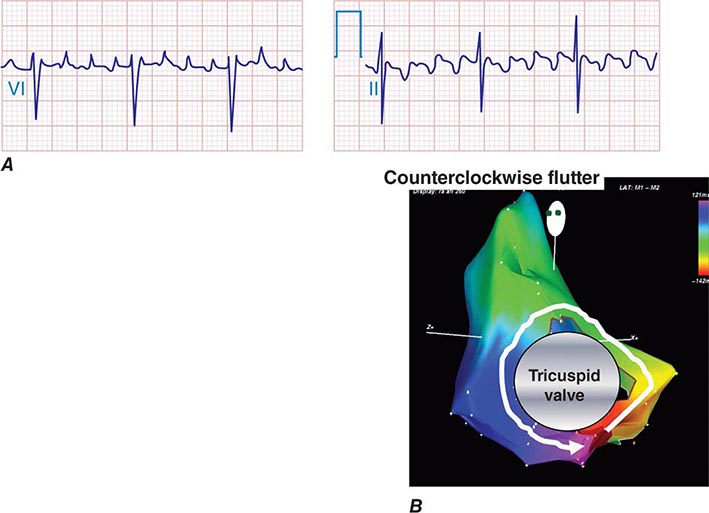
FIGURE 276-10 A. Common right atrial flutter, also known as cavotricuspid isthmus flutter, showing positive P waves in lead V1 and negative “sawtooth” pattern in lead II typical of counterclockwise rotation relative to the tricuspid valve annulus. (Adapted from F Marchlinski: The tachyarrhythmias. In Longo DL et al [eds]: Harrison’s Principles of Internal Medicine, 18th ed. New York, McGraw-Hill, 2012, pp 1878–1900.)B. A right atrial map of common counterclockwise flutter is shown. Colors indicate activation time, progressing from red to yellow to green, blue, and purple. The reentry path parallels the tricuspid annulus.
Common right atrial flutter often occurs in association with atrial fibrillation and with atrial scar from senescence or prior cardiac surgery. Some patients with atrial fibrillation that is treated with an antiarrhythmic drug, particularly flecainide, propafenone, or amiodarone, will present with atrial flutter rather than fibrillation.
Macroreentrant ATs that are not dependent on conduction through the cavotricuspid isthmus are referred to as atypical atrial flutters. They can occur in either atrium and are usually associated with areas of scar. Left atrial flutter and perimitral left atrial flutter are commonly seen after extensive left atrial ablation for atrial fibrillation or atrial surgery. The clinical presentation is similar to common atrial flutter, but with different P-wave morphologies. They can be difficult to distinguish from focal AT, and in most cases, the mechanism can only be confirmed by an electrophysiology study.
|
ATRIAL FLUTTER AND MACROREENTRANT ATRIAL TACHYCARDIAS |
Initial management of atrial flutter is similar to that for atrial fibrillation, discussed in more detail below. Electrical cardioversion is warranted for hemodynamic instability or severe symptoms. Otherwise, rate control can be achieved with administration of AV nodal–blocking agents, but this is often more difficult than for atrial fibrillation. The risk of thromboembolic events is felt to be similar to that associated with atrial fibrillation. Anticoagulation is warranted prior to conversion for episodes more than 48 h in duration and chronically for patients at increased risk of thromboembolic stroke based on the CHA2DS2-VASc scoring system (Table 276-6).
|
CHA2DS2-VASc RISK ASSESSMENT AND ORAL ANTICOAGULANTS |
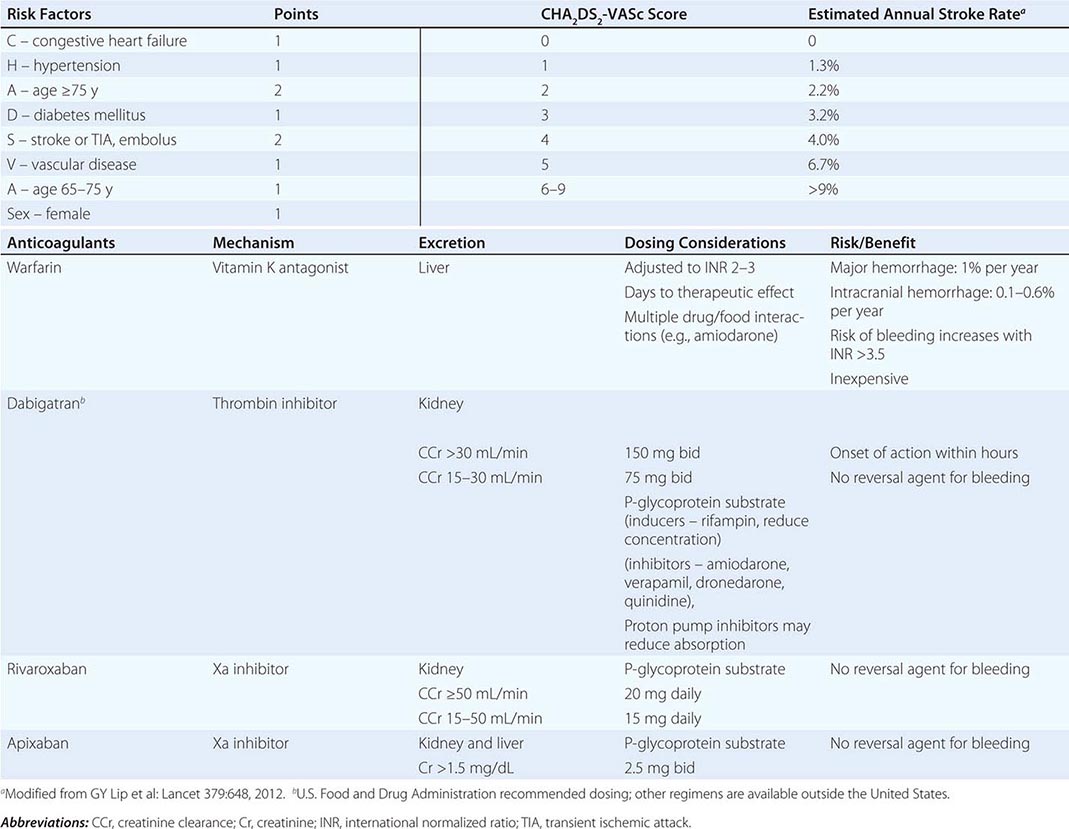
For a first episode of atrial flutter, conversion to sinus rhythm with no antiarrhythmic drug therapy is reasonable. For recurrent episodes, antiarrhythmic drug therapy with sotalol, dofetilide, disopyramide, and amiodarone may be considered, but more than 70% of patients experience recurrences. For recurrent episodes of common atrial flutter, catheter ablation of the cavotricuspid isthmus abolishes the arrhythmia in over 90% of patients with a low risk of complications that are largely related to vascular access and infrequent heart block. Approximately 50% of patients presenting with atrial flutter develop atrial fibrillation within the next 5 years.
MULTIFOCAL ATRIAL TACHYCARDIA
Multifocal AT (MAT) is characterized by at least three distinct P-wave morphologies with rates typically between 100 and 150 beats/min. Unlike atrial fibrillation, there are clear isoelectric intervals between P waves (Fig. 276-11). The mechanism is likely triggered automaticity from multiple atrial foci. It is usually encountered in patients with chronic pulmonary disease and acute illness.
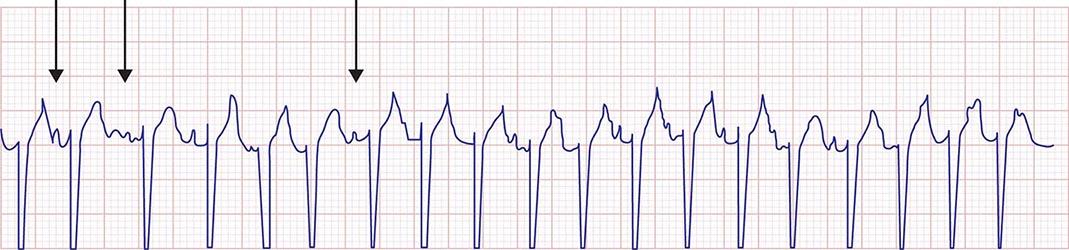
FIGURE 276-11 Multifocal atrial tachycardia. Rhythm strip obtained from a patient with severe pulmonary disease during an acute illness. Arrows note three distinct P-wave morphologies.
ATRIAL FIBRILLATION
Atrial fibrillation (AF) is characterized by disorganized, rapid, and irregular atrial activation with loss of atrial contraction and with an irregular ventricular rate that is determined by AV nodal conduction (Fig. 276-12). In an untreated patient, the ventricular rate also tends to be rapid and variable, between 120 and 160 beats/min, but in some patients, it may exceed 200 beats/min. Patients with high vagal tone or AV nodal conduction disease may have slow rates.
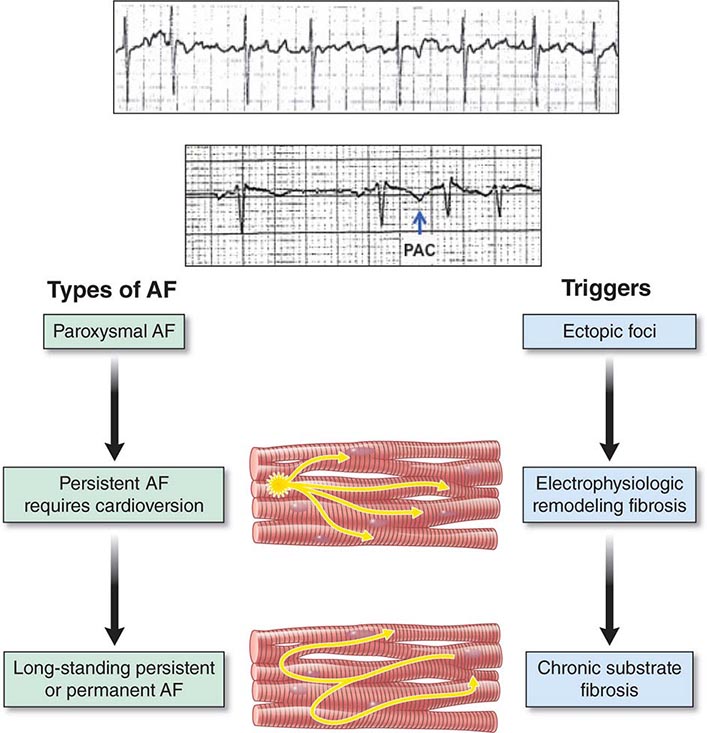
FIGURE 276-12 A rhythm strip of atrial fibrillation (AF) showing no distinct P-wave morphology and irregular ventricular response. Diagram depicts atrial fibrillation types. Paroxysmal AF is initiated by premature beats, as shown in the rhythm strip (arrow) after two sinus beats. Triggering foci are often an important cause of this arrhythmia. Persistent AF is associated with atrial structural and electrophysiologic remodeling, as well as with triggering foci in many patients. Long-standing persistent AF is associated with greater structural remodeling with atrial fibrosis and electrophysiologic remodeling.
AF is the most common sustained arrhythmia and is a major public health problem. Prevalence increases with age, and more than 95% of AF patients are older than 60 years of age. The prevalence by age 80 is approximately 10%. The lifetime risk of developing AF for individuals 40 years old is approximately 25%. AF is slightly more common in men than women and more common in whites than blacks. Risk factors for developing AF in addition to age include hypertension, diabetes mellitus, cardiac disease, and sleep apnea. AF is a marker for heart disease, the severity of heart disease, and age, and it is therefore difficult to determine the extent to which AF itself contributes to associated increased mortality and morbidity. AF is associated with increased risk of developing heart failure. AF increases the risk of stroke by fivefold and is estimated to be the cause of 25% of strokes. It also increases the risk of dementia.
AF is occasionally associated with an acute precipitating factor such as hyperthyroidism, acute alcohol intoxication, or an acute illness including myocardial infarction or pulmonary embolism. AF occurs in up to 30% of patients recovering from cardiac surgery, associated with inflammatory pericarditis.
The clinical type of AF suggests the underlying pathophysiology (Fig. 276-12). Paroxysmal AF is defined as episodes that start and stop spontaneously. It is often initiated by small reentrant or rapidly firing foci in sleeves of atrial muscle along the pulmonary veins. Catheter ablation that isolates these foci usually abolishes the AF. Persistent AF has a longer duration, exceeding 7 days, and, in many cases, will continue unless cardioversion is performed. Cardioversion can be followed by prolonged periods of sinus rhythm. Episodes may be initiated by rapidly firing foci, but persistence of the arrhythmia is likely due to single or multiple areas of reentry facilitated by structural and electrophysiologic atrial abnormalities. In patients with long-standing persistent AF (>1 year), significant structural changes are present in the atrium that support reentry and automaticity, making it difficult to restore and maintain sinus rhythm. Some patients progress over years from paroxysmal to persistent AF. Fibrosis that develops with aging and atrial hypertrophy in response to hypertension and other cardiac disease may be an important promoting factor, although electrophysiologic changes to conduction and refractoriness occur as well in response to chronic tachycardia in the atrium.
Clinical consequences are related to rapid ventricular rates, loss of atrial contribution to ventricular filling, and predisposition to thrombus formation in the left atrial appendage with potential embolization. Presentations vary with the ventricular rate and underlying heart disease and comorbidities. Many patients are asymptomatic. Rapid rates may cause hemodynamic collapse or heart failure exacerbations particularly in patients with impaired cardiac function, hypertrophic cardiomyopathy, and heart failure with preserved systolic function. Exercise intolerance and easy fatigability are common. Occasionally, dizziness or syncope occurs due to pauses when AF terminates to sinus rhythm (Fig. 276-13).
FIGURE 276-13 A continuous rhythm strip is shown. Atrial fibrillation is present at the top and abruptly terminates in the second tracing, with atrial and ventricular standstill for 7.2 s until resumption of sinus rhythm. The patient experienced syncope.
ACKNOWLEDGMENT
Portions of this chapter were retained from the work of the previous author, Francis Marchlinski.
277 |
Ventricular Arrhythmias |
Arrhythmias that originate in the ventricular myocardium or His-Purkinje system include premature ventricular beats, ventricular tachycardias that can be sustained or nonsustained, and ventricular fibrillation. Arrhythmia may emerge from a focus of myocardial or Purkinje cells capable of automaticity, or triggered automaticity, or from reentry through areas of scar or a diseased Purkinje system. Ventricular arrhythmias are often associated with structural heart disease and are an important cause of sudden death (Chap. 327). They also occur in some structurally normal hearts, in which case they are usually benign. Evaluation and management are guided by the risk of arrhythmic death, which is assessed based on symptoms, type of arrhythmia, and associated underlying heart disease.
DEFINITIONS
Ventricular arrhythmias are characterized by their electrocardiographic appearance and duration. Conduction away from the ventricular focus through the ventricular myocardium is slower than activation of the ventricles over the Purkinje system. Hence, the QRS complex during ventricular arrhythmias will be wide, typically >0.12 s.
Premature ventricular beats (also referred to as premature ventricular contractions [PVCs]) are single ventricular beats that fall earlier than the next anticipated supraventricular beat (Fig. 277-1). PVCs that originate from the same focus will have the same QRS morphology and are referred to as unifocal (Fig. 277-1A). PVCs that originate from different ventricular sites have different QRS morphologies and are referred to as multifocal (Fig. 277-1B). Two consecutive ventricular beats are ventricular couplets.
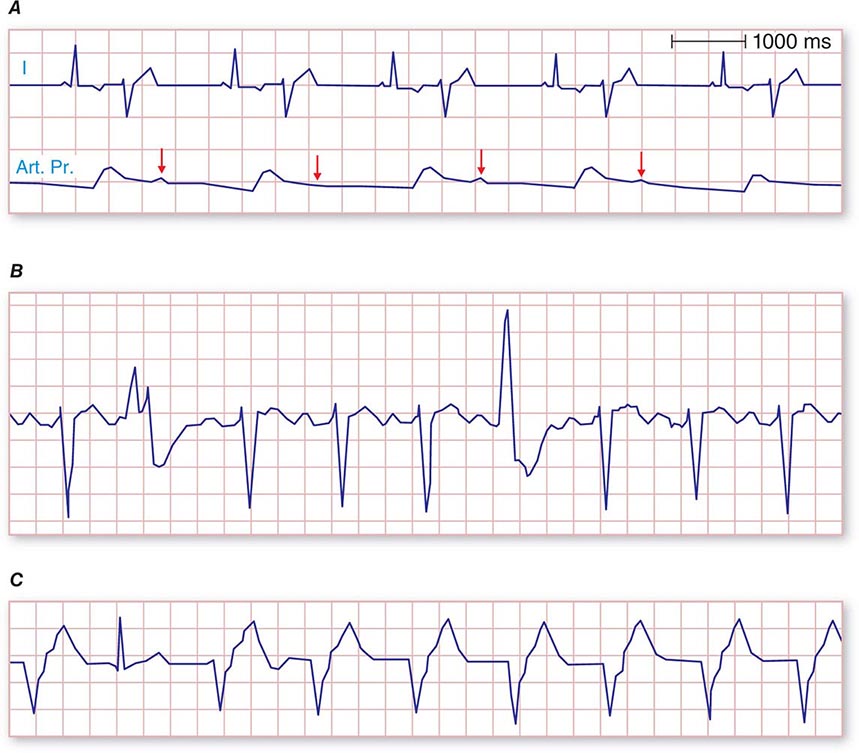
FIGURE 277-1 Examples of types of premature ventricular contractions (PVCs). A. Unifocal PVCs follow every sinus beat in a bigeminal frequency. Trace shows electrocardiogram lead 1 and arterial pressure (Art. Pr.). Sinus rhythm beats are followed by normal arterial waveform. The arterial pressure following premature beats is attenuated (arrows) and imperceptible to palpation. The pulse in this patient is registered at half the heart rate. B. Multifocal PVCs. The two PVCs shown have different morphologies. C. Example of accelerated idioventricular rhythm. The second QRS is a normally conducted beat. All other QRS complexes on this rhythm strip are ventricular due to accelerated idioventricular rhythm.
Ventricular tachycardia (VT) is three or more consecutive beats at a rate faster than 100 beats/min. Three or more consecutive beats at slower rates are designated an idioventricular rhythm (Fig. 277-1C). VT that terminates spontaneously within 30 s is designated nonsustained (Fig. 277-2), whereas sustained VT persists longer than 30 s or is terminated by an active intervention, such as administration of an intravenous medication, external cardioversion, or pacing or a shock from an implanted cardioverter-defibrillator.
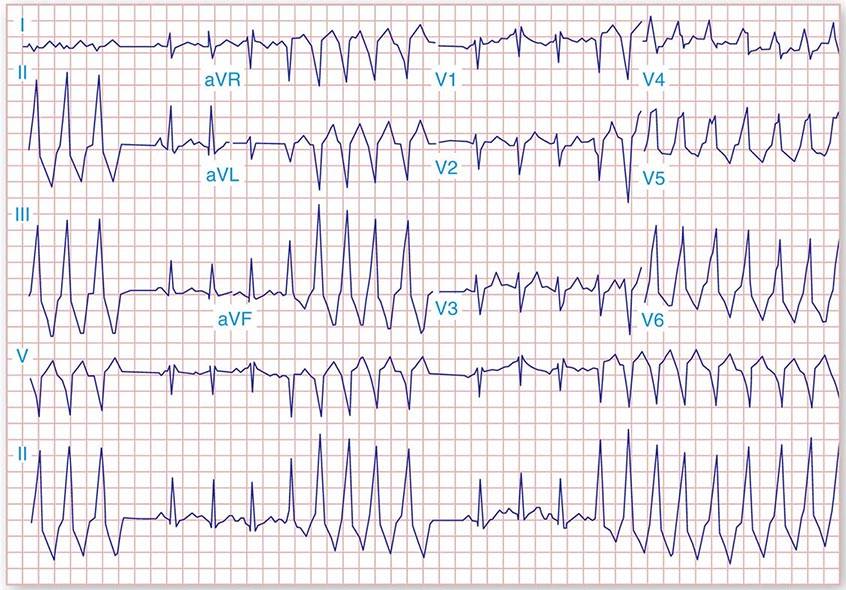
FIGURE 277-2 Repetitive monomorphic nonsustained ventricular tachycardia (VT) of right ventricular outflow tract origin. The VT has a left bundle branch block pattern with inferior axis with tall QRS complexes in the inferior leads.
Monomorphic VT has the same QRS complex from beat to beat, indicating that the activation sequence is the same from beat to beat and that each beat likely originates from the same source (Fig. 277-3A). The initial site of ventricular activation largely determines the sequence of ventricular activation. Therefore, the QRS morphology of PVCs and monomorphic VT provides an indication of the site of origin within the ventricles (Fig. 277-4). The likely origin often suggests whether an arrhythmia is idiopathic or associated with structural disease. Arrhythmias that originate from the right ventricle or septum result in late activation of much of the left ventricle, thereby producing a prominent S wave in V1 referred to as a left bundle branch block–like configuration. Arrhythmias that originate from the free wall of the left ventricle have a prominent positive deflection in V1, thereby producing a right bundle branch block–like morphology in V1. The frontal plane axis of the QRS is also useful. An axis that is directed inferiorly, as indicated by dominant R waves in leads II, III, and AVF, suggests initial activation of the cranial portion of the ventricle, whereas a frontal plane axis that is directed superiorly (dominant S waves in II, III, and AVF) suggests initial activation at the inferior wall.
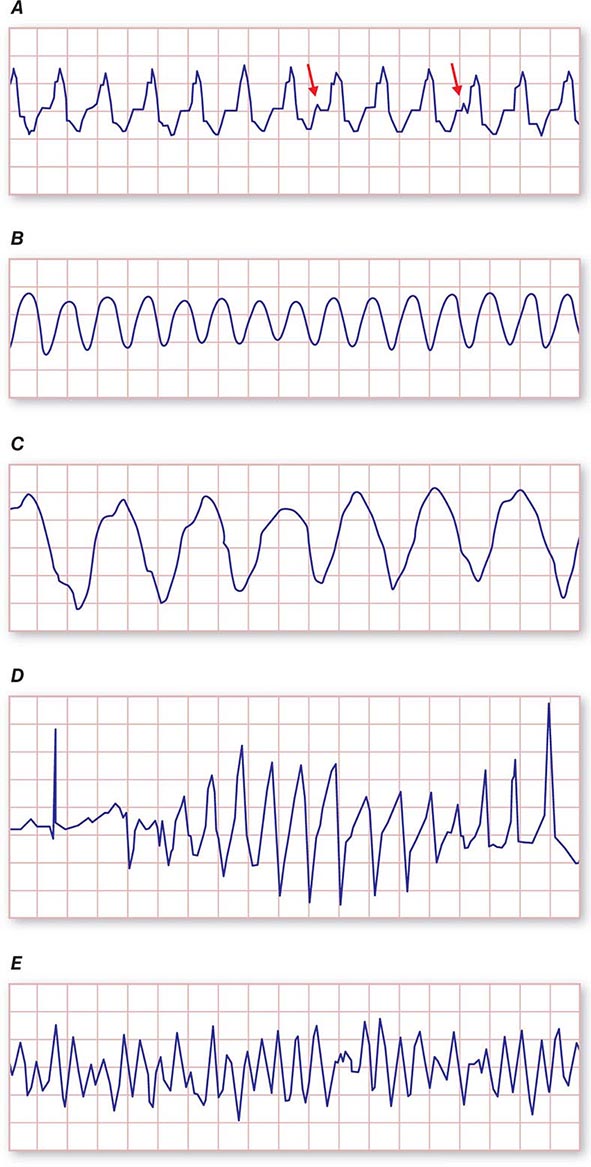
FIGURE 277-3 Examples of types of ventricular tachycardia (VT). A. Monomorphic VT with dissociated P waves (short arrows). B. Ventricular flutter. C. Sinusoidal VT due to electrolyte disturbance or drug effects. D. Polymorphic VT resulting from prolongation of QT interval (torsade de pointes VT). E. Ventricular fibrillation.
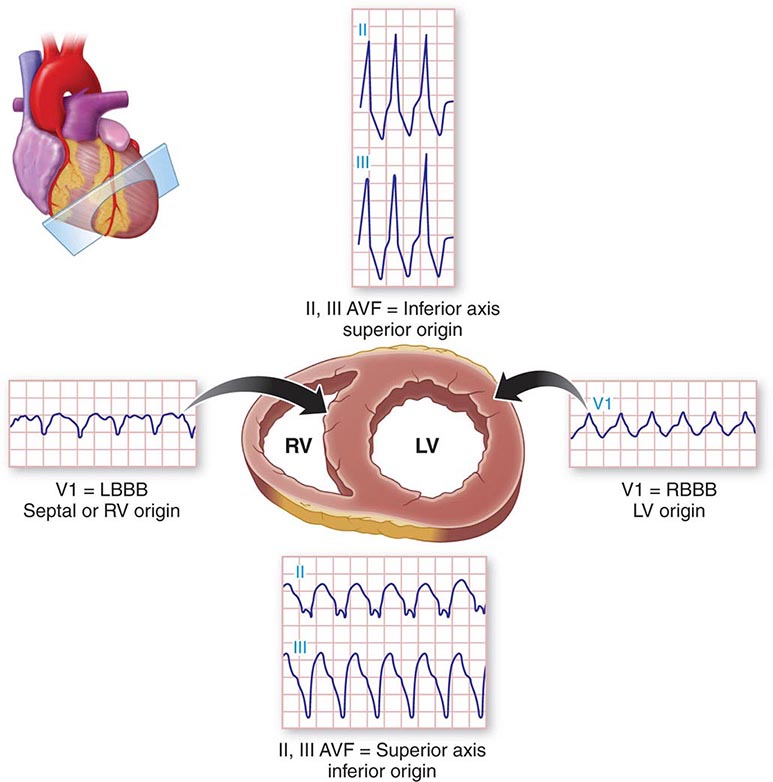
FIGURE 277-4 Site of VT origin based on QRS morphology. LBBB, left bundle branch block; LV, left ventricle; RBBB, right bundle branch block; RV, right ventricle.
Very rapid monomorphic VT has a sinusoidal appearance, also called ventricular flutter, because it is not possible to distinguish the QRS complex from the T wave (Fig. 277-3B). Relatively slow sinusoidal VTs have a wide QRS indicative of slowed ventricular conduction (Fig. 277-3C). Hyperkalemia, toxicity from excessive effects of drugs that blocks sodium channels (e.g., flecainide, propafenone, or tricyclic antidepressants), and severe global myocardial ischemia are causes.
Polymorphic VT has a continually changing QRS morphology indicating a changing ventricular activation sequence. Polymorphic VT that occurs in the context of congenital or acquired prolongation of the QT interval often has a waxing and waning QRS amplitude creating a “twisting about the points” appearance referred to as Torsade de Pointes (Fig. 277-3D).
Ventricular fibrillation (VF) has continuous irregular activation with no discrete QRS complexes (Fig. 277-3E). Monomorphic or polymorphic VT may transition to VF in susceptible patients.
CLINICAL MANIFESTATIONS AND DIAGNOSIS
Common symptoms of ventricular arrhythmias include palpitations, dizziness, exercise intolerance, episodes of lightheadedness, syncope, or sudden death. These arrhythmias can be asymptomatic and encountered unexpectedly as an irregular pulse or heart sounds on examination, or seen on a routine electrocardiogram (ECG), exercise test, or cardiac ECG monitoring.
Syncope is a concerning symptom that can be due to an episode of VT with hypotension. Syncope due to a ventricular arrhythmia often indicates that there is a significant risk for subsequent cardiac arrest and sudden death with arrhythmia recurrence. Although benign causes of syncope, such as reflex-mediated neurocardiogenic (vasovagal) syncope and orthostatic hypotension, are generally more common, it is important to consider the possibility of heart disease or a genetic syndrome causing VT. When these are suspected, hospitalization for further evaluation and monitoring is often appropriate.
Sustained VT may present with cardiac arrest, often with degeneration of the VT to VF. Occasionally a sustained VT will be hemodynamically tolerated and present with diminished exercise capacity or exacerbation of heart failure. Many patients who are at risk for VT have known heart disease and may have an implantable cardioverter-defibrillator (ICD). In patients with an ICD, spontaneous episodes of VT may elicit an episode of transient lightheadedness, palpitations, or syncope that may be followed by a shock from the ICD (see below).
The diagnosis of ventricular arrhythmias is established by recording of the arrhythmia on an ECG or, in some cases, initiation of the arrhythmia during an electrophysiologic study (Table 277-1). A 12-lead ECG of the arrhythmia should be obtained when possible and often provides clues to the potential site of origin and possible presence of underlying heart disease (see above). When the arrhythmia is intermittent with days to weeks between symptoms, prolonged ambulatory monitoring to capture the ECG at the time of symptoms is required to make the diagnosis. Continuous ambulatory monitoring or looping event recording monitors are options. Exercise testing should be considered in patients with exercise-induced symptoms.
|
DIAGNOSTIC TESTS FOR VENTRICULAR ARRHYTHMIAS |
Abbreviation: RV, right ventricle. See text for other abbreviations.
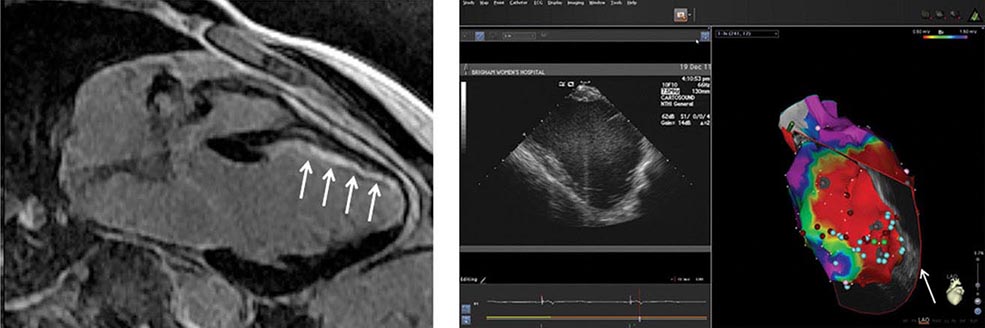
FIGURE 277-5 Imaging studies of the left ventricle (LV) used to assist ablation for ventricle tachycardia (VT). Left panel is a magnetic resonance image of a longitudinal section demonstrating thinning of the anterior wall and late gadolinium enhancement in a subendocardial scar (white arrows). The middle panel shows a two-dimensional image of the LV in long axis corresponding to the sector through the mid LV (arrow, right panel) obtained by an intracardiac echo probe positioned in the right ventricle. An electroanatomic three-dimensional map of the LV in the left anterior oblique projection is displayed in the right panel. The purple color depicts areas of normal voltage (>1.5 mV). Blue, green, and yellow represent progressively lower voltages with the red areas indicating scar (<0.5 mV). Channels of viable myocardium with slow conduction within the scar are identified with the light blue dots. Areas of ablation delivered to regions involved in reentrant VT are indicated by maroon dots.
SPECIFIC ARRHYTHMIAS
PVCs and Nonsustained VT Ventricular extrasystoles (Fig. 277-1A) can be due to automaticity or reentry (Chap. 278e). PVCs can be a sign of increased sympathetic tone; myocardial ischemia; hypoxia; electrolyte abnormalities, particularly hypokalemia; or underlying heart disease. During myocardial ischemia or in association with other heart disease, PVCs can be a harbinger of sustained VT or VF. In patients with heart disease, a higher frequency of ectopy and complexity (couplets and nonsustained VT) are associated with more severe disease and, in those with heart failure, with increased mortality. However, suppression of these arrhythmias with antiarrhythmic drugs does not improve survival. In the absence of cardiac disease, PVCs and nonsustained VT generally have a benign prognosis. PVCs that occur at a bigeminal frequency may not generate sufficient cardiac output for a radial pulse and hence may register at rates half that of the heart rate (Fig. 277-1A). Very frequent PVCs can depress ventricular function (see below).
EVALUATION AND MANAGEMENT When encountered during acute illness or as a new finding, evaluation should focus on detection and correction of potential aggravating factors and causes, specifically myocardial ischemia, ventricular dysfunction, and electrolyte abnormalities, most commonly hypokalemia. Underlying heart disease should be defined.
The ECG characteristics of the arrhythmia are often suggestive of whether structural heart disease is present. PVCs with smooth uninterrupted contours and sharp QRS deflections suggest an ectopic focus in relatively normal myocardium, whereas broad notching and slurred QRS deflections suggest a diseased myocardial substrate. The most frequent site of origin for idiopathic ventricular arrhythmias is the right ventricular outflow tract, giving rise to PVCs or VT that have a left bundle branch block configuration, with an inferiorly directed frontal plane axis as discussed below (Fig. 277-2). However, QRS morphology alone is not reliable as an indicator of disease or subsequent risk. Nonsustained VT is usually monomorphic with rates less than 200 beats/min and typically lasts less than 8 beats (Fig. 277-2). Nonsustained VT that is very rapid, polymorphic, or with a first beat that occurs prior to the peak of the T wave (“short-coupled”) is uncommon and should prompt careful evaluation for underlying disease or genetic syndromes associated with sudden death.
A family history of sudden death should prompt evaluation for genetic syndromes associated with sudden death, including cardiomyopathy, long QT syndrome, and arrhythmogenic right ventricular cardiomyopathy (see below). Any abnormality on the 12-lead ECG warrants further evaluation (Fig. 277-6). Repolarization abnormalities are seen in a number of genetically determined syndromes associated with sudden death, including the long QT syndrome, Brugada syndrome, arrhythmogenic right ventricular cardiomyopathy (ARVC), and hypertrophic cardiomyopathy. An echocardiogram is often necessary to assess ventricular function, wall motion abnormalities, and valvular heart disease. Cardiac magnetic resonance (CMR) imaging is also useful for this purpose and for the detection of ventricular scarring that is the substrate for sustained VT (Fig. 277-5). Exercise stress testing should be performed in patients with effort-related symptoms and in those at risk for coronary artery disease.
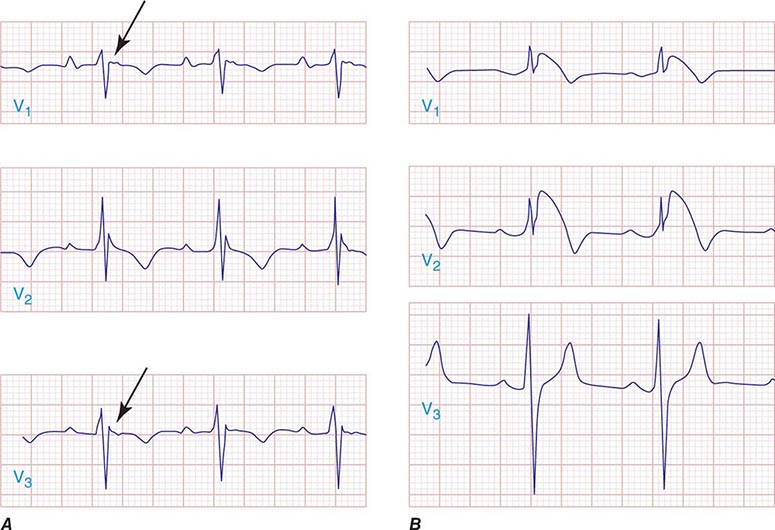
FIGURE 277-6 Precordial chest leads V1–V3 showing typical abnormalities of arrhythmogenic right ventricular cardiomyopathy (ARVC) (A) and Brugada syndrome (B). In ARVC, there is T inversion and delayed ventricular activation manifest as epsilon waves (arrows). Panel B shows ST elevation in V1 and V2 typical of the Brugada syndrome. (Figures reproduced from F Marchlinski: The tachyarrhythmias. In Longo DL et al [eds]: Harrison’s Principles of Internal Medicine, 18th edition. New York, McGraw-Hill, 2012, pp 1878–1900.)
IDIOPATHIC PVCS AND NONSUSTAINED VT For PVC and nonsustained VT in the absence of structural heart disease or a genetic sudden death syndrome, no specific therapy is needed unless the patient has significant symptoms or evidence that frequent PVCs are depressing ventricular function (see below). Reassurance that the arrhythmia is benign is often sufficient to allow the patient to cope with the symptoms, which will often wax and wane in frequency over years. Avoiding stimulants, such as caffeine, is helpful in some patients. If symptoms require treatment, β-adrenergic blockers and nondihydropyridine calcium channel blockers (verapamil and diltiazem) are sometimes helpful (see Table 276-3). If these fail, more potent antiarrhythmic drugs or catheter ablation can be considered. The antiarrhythmic agents flecainide, propafenone, mexiletine, and amiodarone can be effective, but the potential for side effects warrants careful consideration. Catheter ablation can be effective if the arrhythmia occurs with sufficient frequency or is readily provoked such that its origin can be identified for ablation in a similar manner to that for idiopathic monomorphic VT as discussed below. Benefit must be carefully weighed against the procedure-related risks (see below).
PVCS AND NONSUSTAINED VT ASSOCIATED WITH ACUTE CORONARY SYNDROMES During and early after acute myocardial infarction (MI), PVCs and nonsustained VT are common and can be an early manifestation of ischemia and a harbinger of subsequent VF. Treatment with β-adrenergic blockers and correction of hypokalemia and hypomagnesemia reduce the risk of VF. Routine administration of the antiarrhythmic drugs such as lidocaine has not been shown to reduce mortality and is not indicated for suppression of PVCs or asymptomatic nonsustained VT.
Following recovery from acute MI, frequent PVCs (typically >10 PVCs per hour), repetitive PVCs with couplets, and nonsustained VT are markers for depressed ventricular function and increased mortality, but routine antiarrhythmic drug therapy to suppress these arrhythmias is not warranted. Treatment with the sodium channel blocker flecainide increased mortality. Amiodarone therapy reduces sudden death, but does not improve total mortality. Therefore, amiodarone is an option for treatment of symptomatic arrhythmias in this population when the potential benefit outweighs its potential toxicities. β-Adrenergic blockers reduce sudden death but have limited effect on spontaneous arrhythmias.
For survivors of an acute MI, an ICD reduces mortality in certain high-risk groups: patients who have survived >40 days after the acute MI and have a left ventricular (LV) ejection fraction of ≤0.30 or who have an ejection fraction <0.35 and have symptomatic heart failure (functional class II or III); and patients >5 days after MI who have a reduced LV ejection fraction, nonsustained VT, and inducible sustained VT or VF on electrophysiologic testing. ICDs do not reduce mortality when routinely implanted soon after MI or in patients after recent coronary artery revascularization surgery.
PVCS AND NONSUSTAINED VT ASSOCIATED WITH DEPRESSED VENTRICULAR FUNCTION AND HEART FAILURE PVCs and nonsustained VT are common in patients with depressed ventricular function and heart failure and are markers for disease severity and increased mortality, but antiarrhythmic drug therapy to suppress these arrhythmias has not been shown to improve survival. Antiarrhythmic drugs whose major action is blockade of the cardiac sodium channel (flecainide, propafenone, mexiletine, quinidine, and disopyramide) are avoided in patients with structural heart disease because of a risk of proarrhythmia, negative inotropic effects, and increased mortality. Therapy with the potassium channel blockers, e.g., dofetilide, does not reduce mortality. Amiodarone suppresses ventricular ectopy and reduces sudden death but does not improve overall survival. ICDs are the major therapy to protect against sudden death in patients at high risk and are recommended for those with LV ejection fraction <0.35 and New York Heart Association class II and III heart failure, in whom they reduce mortality by 20%, from 36% to 29%, over 5 years.
OTHER CARDIAC DISEASES Ventricular ectopy is associated with increased mortality in patients with hypertrophic cardiomyopathy (Chap. 287) or with congenital heart disease (Chap. 282) associated with right ventricular or LV dysfunction. In these patients, management is similar to that for patients with ventricular dysfunction. Pharmacologic suppression of the arrhythmia has not been shown to improve mortality. ICDs are indicated for patients considered at high risk for sudden cardiac death.
PVC-INDUCED VENTRICULAR DYSFUNCTION Very frequent ventricular ectopy and repetitive nonsustained VT (Fig. 277-2) can depress ventricular function, possibly through an effect similar to chronic tachycardia or by inducing ventricular dyssynchrony. Depression of ventricular function rarely occurs unless PVCs account for more than 10–20% of total beats over a 24-h period. Often the PVCs are idiopathic and unifocal, most commonly originating from the LV papillary muscles or outflow tract regions where they can be targeted for ablation. The distinction between PVC-induced ventricular dysfunction as compared to a cardiomyopathic process causing ventricular dysfunction and arrhythmia is difficult and in some cases can be made only retrospectively by observing an improvement in ventricular function after the arrhythmia is suppressed with an antiarrhythmic drug, such as amiodarone, or by catheter ablation.
Idioventricular Rhythms Three or more ventricular beats at a rate slower than 100 beats/min are termed idioventricular rhythm (Fig. 277-1C). Automaticity is the likely mechanism. Idioventricular rhythms are common during acute MI (Chap. 295) and may emerge during sinus bradycardia. Atropine may be administered to increase the sinus rates if the loss of atrioventricular synchrony leads to hemodynamic compromise. This rhythm is also common in patients with cardiomyopathies or sleep apnea. It can also be idiopathic, often emerging when the sinus rate slows during sleep. Therapy should target any underlying cause and correction of bradycardia. Specific therapy for asymptomatic idioventricular rhythm is not necessary.
Sustained Monomorphic VT Sustained monomorphic VT presents as a wide QRS tachycardia that has the same QRS configuration from beat to beat, indicating an identical sequence of ventricular depolarization for each beat (Fig. 277-3A). VT originates from a stable focus or reentry circuit. In structural heart disease, the substrate is often an area of patchy replacement fibrosis due to infarction, inflammation, or prior cardiac surgery that creates anatomical or functional reentry pathways (Fig. 277-5). Less commonly, VT is related to reentry or automaticity in a diseased Purkinje system. In the absence of structural heart disease, idiopathic VT can present as sustained monomorphic VTs that are due to focal automaticity or reentry involving a portion of the Purkinje system.
The clinical presentation can vary depending on the rate of the arrhythmia, underlying cardiac function, and autonomic adaptation in response to the arrhythmia. Whereas patients with normal cardiac function might tolerate rapid VTs, those with severe LV dysfunction often experience symptoms of hypotension, even if VT is not particularly fast. Monomorphic VT may deteriorate to VF, which may be the initial cardiac rhythm recorded at the time of resuscitation.
DIAGNOSIS Sustained monomorphic VT has to be distinguished from other causes of uniform wide QRS tachycardia. These include supraventricular tachycardia with left or right bundle branch block aberrant conduction, supraventricular tachycardias conducted to the ventricles over an accessory pathway (Chap. 276), and rapid cardiac pacing in a patient with a pacemaker or defibrillator. In the presence of known heart disease, VT is the most likely diagnosis of a wide QRS tachycardia. Hemodynamic stability during the arrhythmia does not exclude VT. A number of ECG criteria have been evaluated. The presence of AV dissociation is usually a reliable marker for VT (Fig. 277-7), but P waves can be difficult to define. A P wave following each QRS does not exclude VT because 1:1 conduction from ventricle to atrium can occur. A monophasic R wave or Rs complex in AVR or concordance from V1 to V6 of monophasic R or S waves is also relatively specific for VT (Fig. 277-7). Other QRS morphology criteria have also been described, but all have limitations and are not very reliable in patients with severe heart disease. In patients with known bundle branch block, the same QRS morphology during tachycardia as during sinus rhythm suggests supraventricular tachycardia rather than VT, but is not absolutely reliable. An electrophysiologic study is sometimes required for definitive diagnosis. Rarely, noise and movement artifacts on telemetry recordings can simulate VT; prompt recognition can avoid unnecessary tests and interventions.
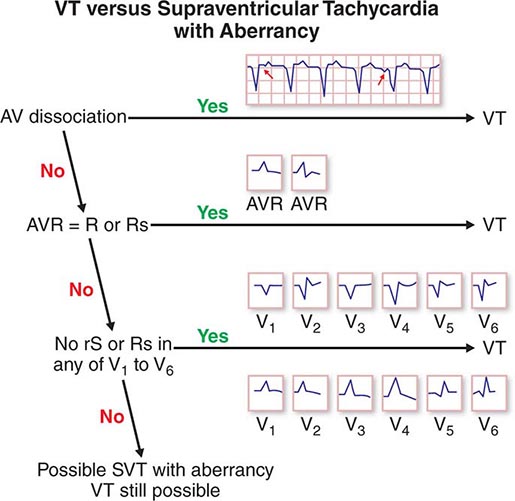
FIGURE 277-7 Algorithm for differentiation of ventricular tachycardia (VT) from supraventricular tachycardia (SVT) with aberration. AV, atrioventricular.
When LV function is depressed or there is evidence of structural myocardial disease, scar-related reentry is the most likely diagnosis. Scars are suggested by pathologic Q waves on the ECG, segmental left or right ventricular wall motion abnormalities on echocardiogram or nuclear imaging, and areas of delayed gadolinium enhancement during MRI (Fig. 277-5).
TREATMENT AND PROGNOSIS Initial management follows Advanced Cardiac Life Support (ACLS) guidelines. If hypotension, impaired consciousness, or pulmonary edema is present, QRS synchronous electrical cardioversion should be performed, ideally after sedation if the patient is conscious. For stable tachycardia, a trial of adenosine is reasonable, as this may clarify a supraventricular tachycardia with aberrancy (Chap. 276). Intravenous amiodarone is the drug of choice if heart disease is present. Following restoration of sinus rhythm, hospitalization and evaluation to define underlying heart disease are required. Assessment of cardiac biomarkers for evidence of MI is appropriate, but acute MI is rarely a cause of sustained monomorphic VT, and elevations in troponin or creatine kinase (CK)-MB are more likely to indicate myocardial damage that is secondary to hypotension and ischemia from the VT. Subsequent management is determined by the underlying heart disease and frequency of VT. If VT recurs frequently or is incessant, administration of antiarrhythmic medications or catheter ablation may be required to restore stability. More commonly, sustained monomorphic VT occurs as an isolated episode, but with a risk of recurrence. ICDs are usually considered for VT associated with structural heart disease.
Sustained Monomorphic VT in Specific Diseases • CORONARY ARTERY DISEASE Patients who present with sustained VT associated with coronary artery disease typically have a history of prior large MI and present years after the acute infarct with a remodeled ventricle and markedly depressed LV function. Even when there is biomarker evidence of acute MI, a preexisting scar from previous MI should be suspected as the cause of the VT. Infarct scars provide a durable substrate for sustained VT, and up to 70% of patients have a recurrence of the arrhythmia within 2 years. Scar-related reentry is not dependent on recurrent acute myocardial ischemia, so coronary revascularization cannot be anticipated to prevent recurrent VT, even when it may be appropriate for other indications. Depressed ventricular function, which is a risk factor for sudden death, is usually present. Implantation of an ICD is warranted for most patients provided that there is a reasonable expectation of survival with acceptable functional status for the next year after recovery from the VT episode. ICDs reduce annual mortality from 12.3% to 8.8% and lower arrhythmic deaths by 50% in patients with hemodynamically significant sustained VT or a history of cardiac arrest compared with pharmacologic therapy. Chronic amiodarone therapy may be considered for patients who are not candidates for or who decline ICD placement.
Following ICD implantation, patients remain at risk for heart failure, recurrent ischemic events, and recurrent VT, with a 5-year mortality that exceeds 30%. Attention to therapies with survival benefit, including β-adrenergic blocking agents, angiotensin-converting enzyme inhibitors, and statins, is important. Patients with frequent symptomatic recurrences of VT require antiarrhythmic drug therapy or catheter ablation.
NONISCHEMIC DILATED CARDIOMYOPATHY Sustained monomorphic VT associated with nonischemic cardiomyopathy is usually due to scar-related reentry. The etiology of scar is often unclear, but progressive replacement fibrosis is the likely cause. On cardiac MRI, scars are detectable as areas of delayed gadolinium enhancement and are more often intramural or subepicardial in location as compared with patients with prior MI. Scars that cause VT are often located adjacent to a valve annulus and can occur in either ventricle. Any cardiomyopathic process can cause scars and VT, but cardiac sarcoidosis (Chap. 390) and Chagas’ disease (Chap. 252) are particularly associated with monomorphic VT (Table 277-2). An ICD is usually indicated with additional drugs or ablation for control of recurrent VT.
|
VENTRICULAR ARRHYTHMIAS ASSOCIATED WITH DIFFERENT FORMS OF HEART DISEASE |
Abbreviation: RV, right ventricle. See text for other abbreviations.
MONOMORPHIC VT IN ARVC ARVC (Chap. 287) is a rare genetic disorder most commonly due to mutations in genes encoding for cardiac desmosomal proteins. Approximately 50% have a familial transmission with autosomal dominant inheritance. A less common, autosomal recessive form is associated with cardiocutaneous syndromes that include Naxos disease and Carvajal syndrome. Patients typically present between the second and fifth decade with palpitations, syncope, or cardiac arrest owing to sustained monomorphic VT, although polymorphic VT can also occur. Fibrosis and fibro-fatty replacement most commonly involve the right ventricular myocardium and provide the substrate for reentrant VT that usually has a left bundle branch block–like configuration, consistent with the right ventricular origin. The sinus rhythm ECG suggests the disease in more than 85% of patients, most often showing T-wave inversions in V1–V3 (Fig. 277-6). Delayed activation of the right ventricle may cause a widened QRS (≥110 ms) in the right precordial leads and a prolonged S-wave upstroke in those leads, and occasionally a deflection at the end of the QRS known as an epsilon wave (Fig. 277-6). Cardiac imaging may show right ventricular enlargement or areas of abnormal motion or reveal areas of scar on CMR imaging with gadolinium. The monomorphic VT of early ARVC can sometimes be difficult to differentiate from idiopathic right ventricular outflow tract VT.
LV involvement can occur and occasionally precede manifest right ventricular disease. Heart failure is rare except in late stages, and survival to advanced age can be anticipated provided that VT can be controlled. An ICD is recommended. When VT is exercise-induced, it may respond to β-adrenergic blockers and limiting exercise. Sotalol, amiodarone, and catheter ablation have been used to reduce recurrences. Ablation targets are often located in the subepicardium of the RV.
TETRALOGY OF FALLOT VT occurs in 3–14% of patients late after repair of tetralogy of Fallot (Chap. 282) and contributes to a 2% per decade risk of sudden death. Monomorphic VT is due to reentry around areas of surgically created scar in the RV (Table 277-2). Factors associated with VT risk include age >5 years at the time of repair, high-grade ventricular ectopy, inducible VT on an electrophysiologic study, abnormal right ventricular hemodynamics, and sinus rhythm QRS duration >180 ms. An ICD is usually warranted for patients who have a spontaneous episode of VT, but criteria for a prophylactic ICD in other patients have not been established. Catheter ablation is used to control recurrent episodes.
BUNDLE BRANCH REENTRY VT Reentry through the Purkinje system occurs in approximately 5% of patients with monomorphic VT in the presence of structural heart disease. The reentry circuit typically revolves retrograde via the left bundle and anterograde down the right bundle, thereby producing VT that has a left bundle branch block configuration. Catheter ablation of the right bundle branch abolishes this VT. Bundle branch reentry is usually associated with severe underlying heart disease. Other scar-related VTs are often present and often require additional therapy or ICD implantation.
IDIOPATHIC MONOMORPHIC VT Idiopathic VT in patients without structural heart disease usually presents with palpitations, lightheadedness, and occasionally syncope, often provoked by sympathetic stimulation during exercise or emotional upset. The QRS morphology of the arrhythmia suggests the diagnosis (see below). The sinus rhythm ECG is normal. Cardiac imaging shows normal ventricular function and no evidence of ventricular scar. Occasionally a patient with structural heart disease is found to have concomitant idiopathic VT, unrelated to the structural disease. Sudden death is rare.
Outflow tract VTs originate from a focus, usually with features consistent with triggered automaticity. The arrhythmia may present with sustained VT, nonsustained VT, or PVCs often provoked by exercise or emotional upset. Repeated bursts of nonsustained VTs, which may occur incessantly, are known as repetitive monomorphic VTs and can cause tachycardia-induced cardiomyopathy with depressed ventricular function that recovers after suppression of the arrhythmia (Fig. 277-2). Most originate in the right ventricular outflow tract, which gives rise to VT that has a left bundle branch block configuration in V1 and an axis that is directed inferiorly, with tall R waves in leads II, III, and AVF (Fig. 277-2). Idiopathic VT can also arise in the LV outflow tract or in sleeves of myocardium that extend along the aortic root. LV origin is suspected when lead V1 or V2 has prominent R waves. Although this typical outflow tract QRS morphology favors idiopathic VT, some cardiomyopathies, notably ARVC, can cause PVCs or VT from this region. Excluding these diseases is an initial focus of evaluation.
LV intrafascicular VT presents with sustained VT that has a right bundle branch block–like configuration. It is often exercise-induced and occurs more often in men than women. The mechanism is reentry in or near the septal ramifications of the LV Purkinje system. This VT can be terminated by intravenous administration of verapamil.
MANAGEMENT OF IDIOPATHIC VT Treatment is required for symptoms or when frequent or incessant arrhythmias depress ventricular function. β-Adrenergic blockers are first-line therapy. Nondihydropyridine calcium channel blockers (diltiazem and verapamil) are sometimes effective. Catheter ablation is warranted for severe symptoms or when beta blockers or calcium channel blockers are not effective or not desired. Efficacy and risks of catheter ablation vary with the specific site of origin of the VT, being most favorable for arrhythmias originating in the right ventricular outflow tract.
LV fascicular VT can be terminated by intravenous administration of verapamil, although chronic therapy with oral verapamil is not always effective. Catheter ablation is recommended if β-adrenergic blockers or calcium channel blockers are ineffective or not desired.
Polymorphic VT Sustained polymorphic VT can be seen with any form of structural heart disease (Table 277-2). However, unlike sustained monomorphic VT, polymorphic VT does not always indicate a structural abnormality or focus of automaticity. Reentry with continually changing reentrant paths, spiral wave reentry, and multiple automatic foci are potential mechanisms (Chap. 278e). Sustained polymorphic VT usually degenerates into VF. Polymorphic VT is typically seen in association with acute MI or myocardial ischemia, ventricular hypertrophy, and a number of genetic mutations that affect cardiac ion channels (Table 277-3).
|
CAUSES OF QT PROLONGATION AND TORSADE DE POINTES VENTRICULAR TACHYCARDIA |

POLYMORPHIC VT ASSOCIATED WITH ACUTE MI/MYOCARDIAL ISCHEMIA Acute MI or ischemia is a common cause of polymorphic VT and should be the initial consideration in management. Approximately 10% of patients with acute MI develop VT that degenerates to VF, related to reentry through the infarct border zone. The risk is greatest in the first hour of acute MI. Following resuscitation as per the ACLS guidelines, management is as for acute MI (Chap. 295). β-Adrenergic blockers, correction of electrolyte abnormalities, and prompt myocardial reperfusion are required. Repeated episodes of polymorphic VT suggest ongoing myocardial ischemia and warrant assessment of adequacy of myocardial reperfusion. Polymorphic VT and VF that occur within the first 48 h of acute MI are associated with greater in-hospital mortality, but those who survive past hospital discharge are not at increased risk for arrhythmic sudden death. Long-term therapy for postinfarct ventricular arrhythmia is determined by residual LV function, with an ICD being indicated for persistent severe LV dysfunction (LV ejection fraction <0.35).
REPOLARIZATION ABNORMALITIES AND GENETIC ARRHYTHMIA SYNDROMES • Acquired long QT Abnormal prolongation of the QT interval is associated with the polymorphic VT Torsade de Pointes (Fig. 277-8). The VT often has a characteristic initiation sequence of a premature ventricular beat that induces a pause, followed by a sinus beat that has a longer QT interval and interruption of the T wave by the PVC that is the first beat of the polymorphic VT. This characteristic initiation is termed “pause-dependent” (Fig. 277-8). Causes of QT prolongation include electrolyte abnormalities, bradycardia, and a number of medications that block repolarizing potassium currents, notably the antiarrhythmic drugs sotalol, dofetilide, and ibutilide, but also a number of other medications used for noncardiac diseases, including erythromycin, pentamidine, haloperidol, phenothiazines, and methadone (Table 277-3). Individual susceptibility may be related to genetic polymorphisms or mutations that influence repolarization.
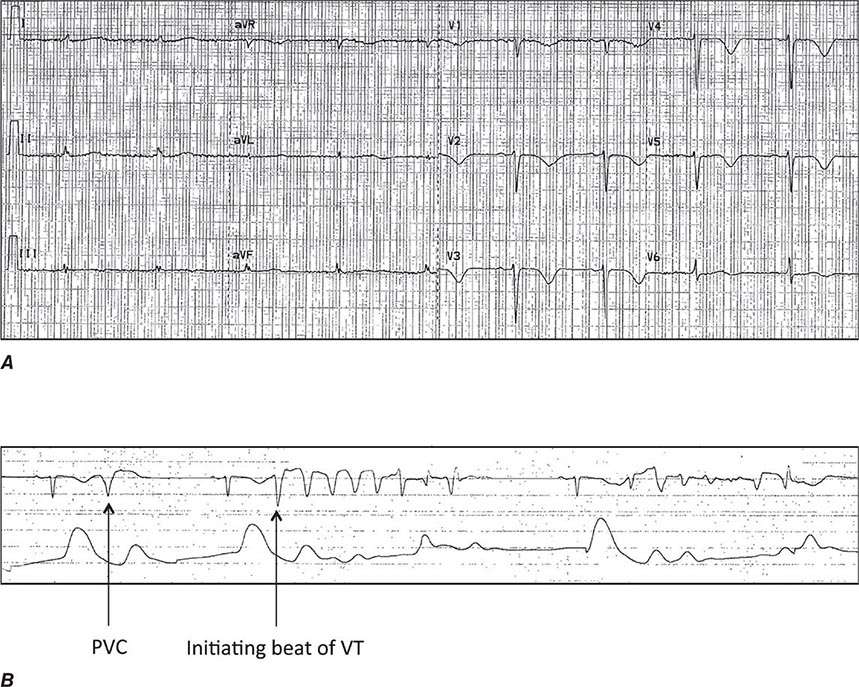
FIGURE 277-8 Electrocardiogram (ECG) of a patient with prolonged QT and episodes of torsade de pointes ventricular tachycardia (VT). A. Twelve-lead ECG showing a heart rate of 54, anterior wall T inversion, and QT interval of 600 ms. The corrected QT interval (QTc) is 585 ms. B. Telemetry ECG tracing with digital pulse waveform demonstrating bursts of torsade de pointes VT. The initiating sequence of the VT is characteristic, with a PVC inducing a pause followed by a sinus beat that had a longer QT and interruption of the T wave by a PVC that is the first beat of VT. The VT is self-terminating in this case.
Patients typically present with near-syncope, syncope, or cardiac arrest. Sustained episodes degenerate to VF requiring defibrillation. PVCs and nonsustained VT often precede episodes of sustained VT. Intravenous administration of 1–2 g of magnesium sulphate usually suppresses recurrent episodes. If magnesium alone is ineffective, increasing heart rate with isoproterenol infusion or pacing, to a rate of 100–120 beats/min as required to suppress PVCs, usually suppresses VT recurrences. These maneuvers allow time for correction of associated electrolyte disturbance (hypokalemia and hypocalcemia) and bradycardia and removal of any causative drugs (Table 277-3). Drug interactions that elevate levels of the offending agent are often a precipitating factor. Patients who experience a polymorphic VT induced by QT prolongation should be considered to have a susceptibility to the arrhythmia and should avoid all future exposure to medications known to prolong the QT interval.
Congenital long QT syndrome The congenital long QT syndrome (LQTS) is caused by mutations in genes coding for cardiac ion channels responsible for ventricular repolarization. The corrected QT (QTc) is typically prolonged to greater than 440 ms in men and 460 ms in women. Symptoms are due to Torsade de Pointes VT (Fig. 277-8). Several forms of congenital LQTS have been identified, but three groups of mutations that lead to LQTS type 1 (LQTS-1), LQTS type 2 (LQTS-2), or LQTS type 3 (LQTS-3) account for 90% of cases. The most frequently encountered mutations, LQTS1 and LQTS2, are due to abnormalities of potassium channels, but mutations affecting the sodium channel (LQTS3) and calcium channels have also been described (Table 277-3).
Patients often present with syncope or cardiac arrest, usually during childhood. In LQTS-1, episodes tend to occur during exertion, particularly swimming. In LQTS-2, sudden auditory stimuli or emotional upset predispose to events. In LQTS-3, sudden death during sleep is a notable feature. Asymptomatic patients may be discovered in the course of family screening or on a routine ECG. Genotyping can be helpful for family screening and to provide reassurance regarding the diagnosis. Correlations of genotype with risk and response to therapy are beginning to emerge. In most patients with LQTS-1 or LQTS-2, adequate doses of beta blocker therapy (the nonselective agents nadolol or propranolol) are sufficient protection from arrhythmia episodes. Markers of increased risk include QTc interval exceeding 0.5 s, female gender, and a history of syncope or cardiac arrest. Recurrent syncope despite beta blocker therapy or a high-risk profile merits consideration of an ICD. Avoidance of QT-prolonging drugs is critical for all patients with LQTS, including those who are genotype positive but have normal QT intervals.
Short QT syndrome Short QT syndrome is very rare compared to LQTS. The QTc is shorter than 0.36 s, and usually less than 0.3 s. The genetic abnormality causes a gain of function of the potassium channel (I Kr) or reduced inward depolarizing currents. The abnormality is associated with atrial fibrillation, polymorphic VT, and sudden death.
Brugada syndrome Brugada syndrome is a rare syndrome characterized by >0.2 mV of ST-segment elevation with a coved ST segment and negative T wave in more than one anterior precordial lead (V1–V3) (Fig. 277-6) and episodes of syncope or cardiac arrest due to polymorphic VT in the absence of structural heart disease. Cardiac arrest may occur during sleep or be provoked by febrile illness. Males are more commonly affected than females. Mutations involving cardiac sodium channels are identified in approximately 25% of cases. Distinction from patients with similar ST elevation owing to LV hypertrophy, pericarditis, myocardial ischemia or MI hyperkalemia, hypothermia, right bundle branch block, and ARVC is often difficult. Furthermore, the characteristic ST-segment elevation can wax and wane over time and may become pronounced during acute illness and fever. Administration of the sodium channel blocking drug flecainide, ajmaline, or procainamide can augment or unmask ST elevation in affected individuals. An ICD is indicated for individuals who have had unexplained syncope or been resuscitated from cardiac arrest. Quinidine has been used successfully to suppress frequent episodes of VT.
Early repolarization syndrome Patients resuscitated from VF who have no structural heart disease or other identified abnormality have a higher prevalence of J-point elevation with notching in the terminal QRS. A family history of sudden death is present in some patients, suggesting a potential genetic basis. J-point elevation is also seen in some patients with the Brugada syndrome and associated with a higher risk of arrhythmias. An ICD is recommended for those who have had prior cardiac arrest. It should be noted that J-point elevation is commonly seen as a normal variant, and in the absence of specific symptoms, the clinical relevance is not known.
Catecholaminergic polymorphic VT This rare familial syndrome is due to mutations in the cardiac ryanodine receptor and, less commonly, the sarcoplasmic calcium binding protein, calsequestin 2. These mutations result in abnormal sarcoplasmic calcium handling and polymorphic ventricular arrhythmias that resemble those seen with digitalis toxicity. The VT is polymorphic or has a characteristic alternating QRS morphology termed bidirectional VT. Patients usually present during childhood with exercise- or emotion-induced palpitations, syncope, or cardiac arrest. β-Adrenergic blockers (e.g., nadolol and propranolol) and an ICD are recommended. Verapamil, flecainide, or surgical left cardiac sympathetic denervation reduces or prevents recurrent VT in some patients.
Hypertrophic cardiomyopathy (HCM) HCM is the most common genetic cardiovascular disorder, occurring in 1 in 500 individuals, and is a prominent cause of sudden death before the age of 35 years (Chap. 287). Sudden death can be due to polymorphic VT/VF. Rarely, sustained monomorphic VT occurs related to areas of ventricular scar. Risk factors include young age, nonsustained VT, failure of blood pressure to increase during exercise, recent (within 6 months) syncope, ventricular wall thickness >3 cm, and possibly the severity of LV outflow obstruction. An ICD is generally indicated for high-risk patients, but the specific risk profile warranting an ICD continues to be debated. Surgical myectomy, performed to relieve outflow obstruction, has been associated with a sudden death rate of less than 1% per year. The reported annual rate of sustained VT or sudden death after transcoronary ethanol septal ablation done to relieve outflow obstruction has been reported to range between 1 and 5%.
Genetic dilated cardiomyopathies Genetic dilated cardiomyopathies account for 30–40% of cases of nonischemic dilated cardiomyopathies. Some are associated with muscular dystrophy. Autosomal dominant, recessive, X-linked, and mitochondrial inheritance patterns are recognized. Mutations in genes coding for structural proteins of the nuclear lamina (lamin A and C) and the SCN5A gene are particularly associated with conduction system disease and ventricular arrhythmias. Patients can experience polymorphic VT and cardiac arrest or develop areas of scar causing sustained monomorphic VT. ICDs are recommended for those who have had a sustained VT or are at high risk due to significantly depressed ventricular function (LV ejection fraction of ≤0.35 and associated with heart failure) or a malignant family history of sudden death.
Ventricular Fibrillation VF is characterized by disordered electrical ventricular activation without identifiable QRS complexes (Fig. 277-3E). Spiral wave reentry and multiple circulating reentry wavefronts are possible mechanisms. Sustained polymorphic or monomorphic VT that degenerates to VF is a common cause of out-of-hospital cardiac arrest. Treatment follows ACLS guidelines with defibrillation to restore sinus rhythm. If resuscitation is successful, further evaluation is performed to identify and treat underlying heart disease and potential causes of the arrhythmia, including the possibility that monomorphic or polymorphic VT could have initiated VF. If a transient reversible cause such as acute MI is not identified, therapy to reduce the risk of sudden death with an ICD is often warranted. Chronic amiodarone therapy may be considered for individuals who are not ICD candidates.
Incessant VT and Electrical Storm VT is incessant when it continues to recur shortly after electrical, pharmacologic, or spontaneous conversion to sinus rhythm. “VT storm” or “electrical storm” refers to three or more separate episodes of VT within 24 h, most commonly encountered in patients with ICDs. Slow incessant VT is sometimes asymptomatic, but can cause heart failure or tachycardia-induced cardiomyopathy. More commonly, these presentations are life-threatening and require emergent therapy. Measures to reduce sympathetic tone, including β-adrenergic blockade, sedation, and general anesthesia, have been used effectively. Intravenous administration of amiodarone and lidocaine can be effective for suppression. Urgent catheter ablation can be lifesaving.
|
TREATMENT |
VENTRICULAR ARRHYTHMIAS |
ANTIARRHYTHMIC DRUGS
Use of antiarrhythmic drugs is based on consideration of the risks and potential benefit for the individual patient. The potential to increase the frequency of VT or cause a new VT, an undesirable effect known as “proarrhythmia,” is a potential risk. Many drugs have multiple effects, often blocking more than one channel. Drug doses, metabolism, and adverse effects are summarized in Chap. 277.
β-Adrenergic Blockers Many ventricular arrhythmias are sensitive to sympathetic stimulation, and β-adrenergic stimulation also diminishes the electrophysiologic effects of many antiarrhythmic drugs. The safety of β-blocking agents makes them the first choice of therapy for most ventricular arrhythmias. They are particularly useful for exercise-induced arrhythmias and idiopathic arrhythmias, but have limited efficacy for most arrhythmias associated with heart disease. Bradyarrhythmias are the major cardiac toxicity.
Calcium Channel Blockers The nondihydropyridine calcium channel blockers diltiazem and verapamil can be effective for some idiopathic VTs. The risk of proarrhythmia is low, but they have negative inotropic and vasodilatory effects that can aggravate hypotension.
Sodium Channel-Blocking Agents Drugs whose major effect is mediated through sodium channel blockade include mexiletine, quinidine, disopyramide, flecainide, and propafenone, which are available for chronic oral therapy (Table 277-3). Lidocaine, quinidine, and procainamide are available as intravenous formulations. Quinidine, disopyramide, and procainamide also have potassium channel-blocking effects that prolong the QT interval. These agents have potential proarrhythmic effects and, with the possible exception of quinidine, also have negative inotropic effects that may contribute to increased mortality observed in patients with prior MI. Long-term therapy is generally avoided in patients with structural heart disease but may be used to reduce symptomatic arrhythmias in patients with ICDs.
Potassium Channel-Blocking Agents Sotalol and dofetilide block the delayed rectifier potassium channel IKr, thereby prolonging the QT interval. Sotalol also has nonselective β-adrenergic blocking activity. It has a modest effect on reducing ICD shocks due to ventricular and atrial arrhythmias. Proarrhythmia with Torsade de Pointes due to QT prolongation occurs in 3–5% of patients. Both sotalol and dofetilide are excreted via the kidneys, necessitating dose adjustment or avoidance in renal insufficiency. These drugs must be avoided in patients with other risk factors for Torsade de Pointes, including QT prolongation, hypokalemia, and significant bradycardia.
Amiodarone and Dronedarone Amiodarone, which blocks multiple cardiac ionic currents and has sympatholytic activity, suppresses a variety of ventricular arrhythmias. It is administered intravenously for life-threatening arrhythmias. During chronic oral therapy, electrophysiologic effects develop over several days. It is more effective than sotalol in reducing ICD shocks and is the preferred drug for ventricular arrhythmias in patients with heart disease who are not candidates for an ICD. Bradyarrhythmias are the major cardiac adverse effect. Ventricular proarrhythmia can occur, but Torsade de Pointes VT is rare. Noncardiac toxicities are a major problem and contribute to drug discontinuation in approximately a third of patients during long-term therapy. Pneumonitis or pulmonary fibrosis occurs in approximately 1% of patients. Photosensitivity is common, and neuropathy and ocular toxicity can occur. Systematic monitoring is recommended during chronic therapy, including assessment for thyroid and liver toxicity every 6 months and lung toxicity with a chest radiograph and/or determination of lung diffusing capacity annually. Dronedarone has structural similarities to amiodarone but without the iodine moiety. Efficacy for ventricular arrhythmias is poor, and it increases mortality in patients with heart failure.
IMPLANTABLE CARDIOVERTER-DEFIBRILLATORS
ICDs are highly effective for termination of VT and VF and also provide bradycardia pacing. ICDs decrease mortality in patients at risk for sudden death due to structural heart diseases. In all cases, ICDs are recommended only if there is also expectation for survival of at least a year with acceptable functional capacity. The exception is in cases of patients with end-stage heart disease who are awaiting cardiac transplantation outside the hospital, or who have left bundle branch block QRS prolongation such that they are likely to have improvement in ventricular function with cardiac resynchronization therapy from a biventricular ICD.
ICDs can often terminate monomorphic VT by a burst of rapid pacing faster than the VT, known as antitachycardia pacing (ATP) (Fig. 277-9A). If ATP fails or is not a programmed treatment, as is often the case for rapid VT or VF, a shock is delivered (Fig. 277-9B). Shocks are painful if the patient is conscious. The most common ICD complication is the delivery of unnecessary therapy (either ATP or shocks) in response to a rapid supraventricular tachycardia or electrical noise as a result of an ICD lead fracture. Interrogation of the ICD, which can be performed remotely and communicated via Internet, is critical after an ICD shock to determine the arrhythmia diagnosis and exclude an unnecessary therapy. Device infection occurs in approximately 1% of patients.
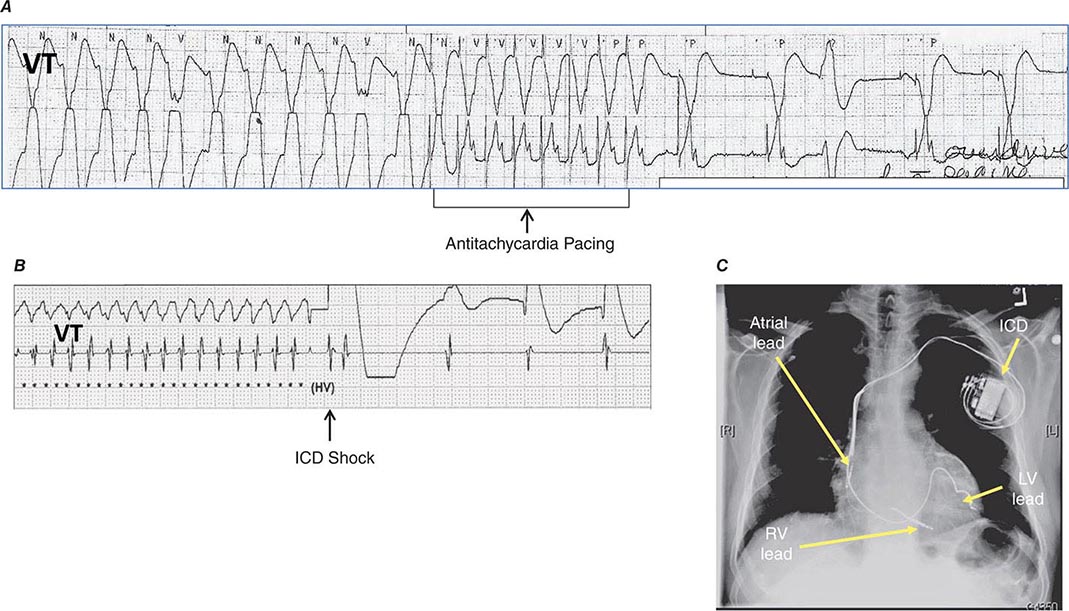
FIGURE 277-9 Implantable cardioverter-defibrillator (ICD) and therapies for ventricular arrhythmias. A. A monomorphic ventricular tachycardia (VT) is terminated by a burst of pacing impulses at a rate faster than VT (antitachycardia pacing). B. A rapid VT is converted with a high-voltage shock (arrow). The chest x-ray in panel C shows the components of an ICD capable of biventricular pacing. ICD generator in the subcutaneous tissue of the left upper chest, pacing leads in the right atrium and the left ventricular (LV) branch of the coronary sinus (LV lead), and a pacing/defibrillating lead in the right ventricle (RV lead) are shown.
Despite prompt termination of VT or VF by an ICD, the occurrence of these arrhythmias predicts subsequent increased mortality and risk of heart failure. Occurrence of VT or VF should therefore prompt assessment for potential causes including worsening heart failure, electrolyte abnormalities, and ischemia. Repeated shocks, even if appropriate, often induce posttraumatic stress disorder. Antiarrhythmic drugs mostly in the form of amiodarone or catheter ablation are often required for suppression of recurrent arrhythmias. Antiarrhythmic drug therapy can alter the VT rate and the energy required for defibrillation, thereby necessitating programming changes in the ICD’s algorithms for detection and therapy.
CATHETER ABLATION FOR VT
Catheter ablation is performed by guiding an electrode catheter to the arrhythmia origin and producing a thermal injury with radiofrequency current. The size and location of the arrhythmia substrate determine the ease and likely effectiveness of the procedure, as well as potential complications. The most common complications, which occur in <5% of patients, are related to vascular access, including bleeding, femoral hematomas, arteriovenous fistulae, and pseudoaneurysms.
Catheter ablation is a reasonable first-line therapy for many patients with symptomatic idiopathic VTs. Success rates for those originating from a focus in the right ventricular outflow tract are in the range of 80–90% but lower for idiopathic VTs arising in less common locations such as from the LV outflow tract or aortic root, along the atrioventricular valve annuli, and from the papillary muscles. Failure of ablation is often due to inability to induce the arrhythmia for precise localization or because the origin of the VT is from a site that is inaccessible or in close proximity to a coronary artery. Complications are infrequent but can include perforation with cardiac tamponade, atrioventricular block due to injury to the conduction system, and coronary artery injury for foci in proximity to a coronary vessel.
In patients with scar-related VT due to prior infarction or cardiomyopathy, ablation targets abnormal regions in the scar. Because these scars often contain multiple reentry circuits over relatively large regions, extensive areas of ablation are required, and these areas are often identified as regions of low voltage displayed on anatomic reconstructions of the ventricle (Fig. 277-5). If the circuits are not confined to the subendocardial scar, epicardial mapping and ablation can be performed via a subxiphoid pericardial puncture, similar to a pericardiocentesis. Epicardial mapping and ablation are often required for VTs due to nonischemic cardiomyopathy, but also have potentially greater risks of bleeding, coronary injury, and postprocedure pericarditis, which is usually transient. For drug-refractory VT due to prior MI, ablation abolishes VT in approximately half of patients and reduces the frequency of VT in an additional 20%. More than one procedure is necessary in up to 30% of patients. Ablation can be lifesaving for patients with very frequent or incessant VT. Procedure-related mortality is in the range of 3%, with most mortality due to continued uncontrollable VT when the procedure fails. In nonischemic heart disease, the arrhythmia substrate locations are more variable and outcomes are less well defined.
Catheter ablation can also be lifesaving for rare patients with recurrent polymorphic VT and VF that is repeatedly initiated by a uniform PVC. The initiating ectopic beat often originates from the Purkinje system or the right ventricular outflow tract and can be targeted for ablation.
When antiarrhythmic drug therapy and catheter ablation fail or are not options, surgical cryoablation, often combined with aneurysmectomy, can be effective therapy for recurrent VT due to prior MI and has also been used successfully in a few patients with nonischemic heart disease. Few centers now maintain the expertise for this therapy. Injection of absolute ethanol into the coronary arterial blood supply of the arrhythmia substrate has also been used for ablation in a small number of patients who have failed catheter ablation and drugs.
SUMMARY
Patients with ventricular arrhythmias fall into three general groups. The first are those who have associated structural heart disease that must be detected. The risk of life-threatening arrhythmias causing sudden death is indicated by the nature of the arrhythmia—sustained (or causing cardiac arrest) or nonsustained, in which case the risk of life-threatening arrhythmias is assessed from the severity of the heart disease, usually the severity of ventricular dysfunction. ICDs provide the most protection from sudden arrhythmic death. The second group comprises those who do not have recognizable structural heart disease, but have a genetic syndrome associated with increased risk of sudden death. A family history of sudden death and abnormal electrocardiogram most frequently suggest the diagnosis. The third group includes individuals with benign idiopathic arrhythmias who may require therapy to control symptoms, but who are not at significant risk for life-threatening arrhythmias. The appropriate recognition of these patients is facilitated by thoughtful application of ECG and cardiac imaging. High-risk individuals benefit from specialized care for consideration of ICDs, catheter ablation, and antiarrhythmic drug therapy.
278e |
Atlas of Cardiac Arrhythmias |
The electrocardiograms in this atlas supplement those illustrated in Chaps. 274 and 276. The interpretations emphasize findings of specific teaching value.
All of the figures are adapted from cases in ECG Wave-Maven, Copyright 2003, Beth Israel Deaconess Medical Center, http://ecg.bidmc.harvard.edu.
The abbreviations used in this chapter are as follows:
AF—atrial fibrillation
AV—atrioventricular
LBBB—left bundle branch block
LV—left ventricular
LVH—left ventricular hypertrophy
MI—myocardial infarction
NSR—normal sinus rhythm
RBBB—right bundle branch block
VT—ventricular tachycardia
WPW—Wolff-Parkinson-White
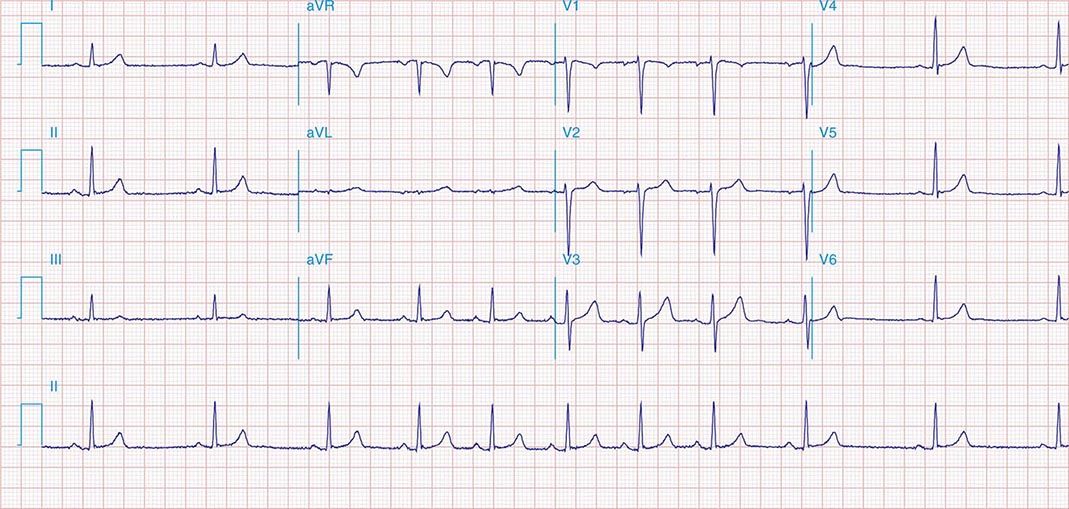
FIGURE 278e-1 Respiratory sinus arrhythmia, a physiologic finding in a healthy young adult. The rate of the sinus pacemaker is relatively slow at the beginning of the strip during expiration, then accelerates during inspiration and slows again with expiration. Changes are due to cardiac vagal tone modulation with breathing.
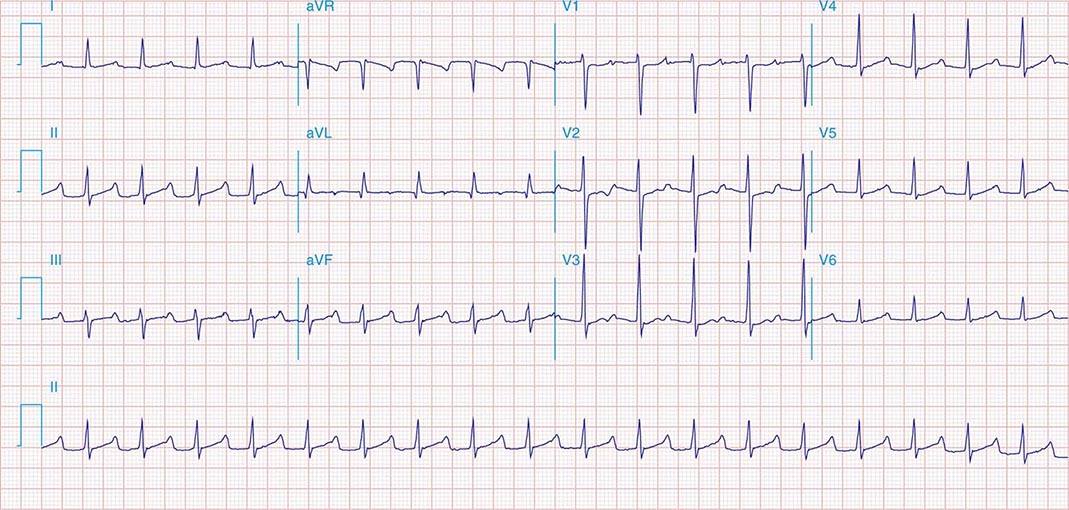
FIGURE 278e-2 Sinus tachycardia (110/min) with first-degree AV “block” (conduction delay) with PR interval = 0.28 s. The P wave is visible after the ST-T wave in V1–V3 and superimposed on the T wave in other leads. Atrial (nonsinus) tachycardias may produce a similar pattern, but the rate is usually faster.
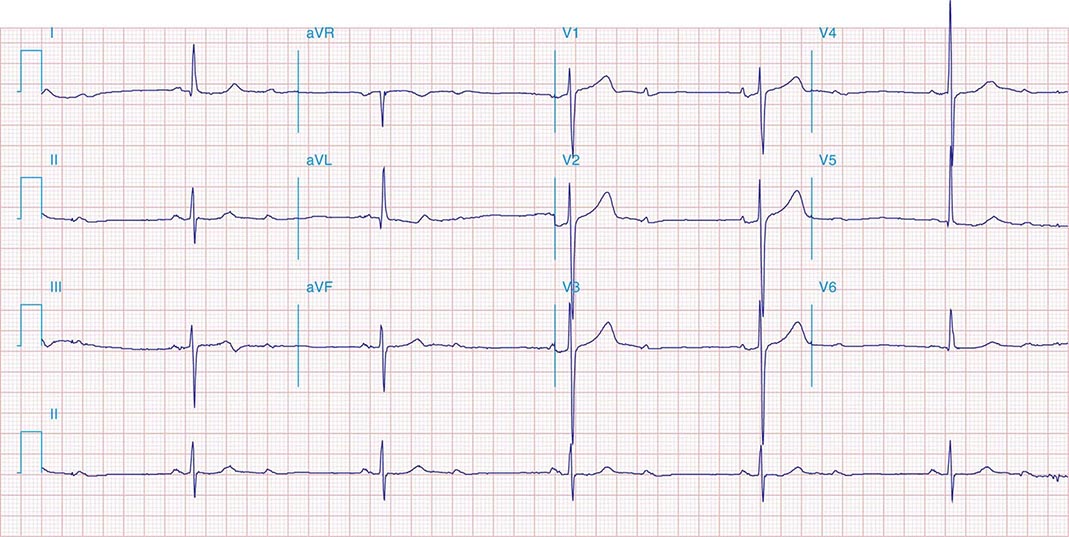
FIGURE 278e-3 Sinus rhythm (P wave rate about 60/min) with 2:1 AV (second-degree) block causing marked bradycardia (ventricular rate of about 30/min). LVH is also present, along with left atrial abnormality.
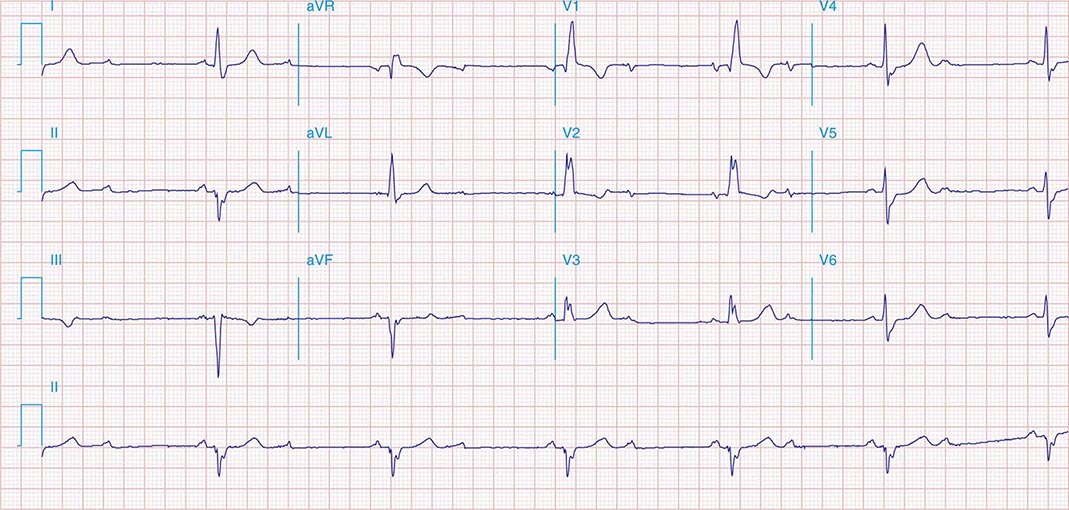
FIGURE 278e-4 Sinus rhythm (P wave rate about 60/min) with 2:1 (second-degree) AV block yielding a ventricular (pulse) rate of about 30/min. Left atrial abnormality. RBBB with left anterior fascicular block. Possible inferior MI.
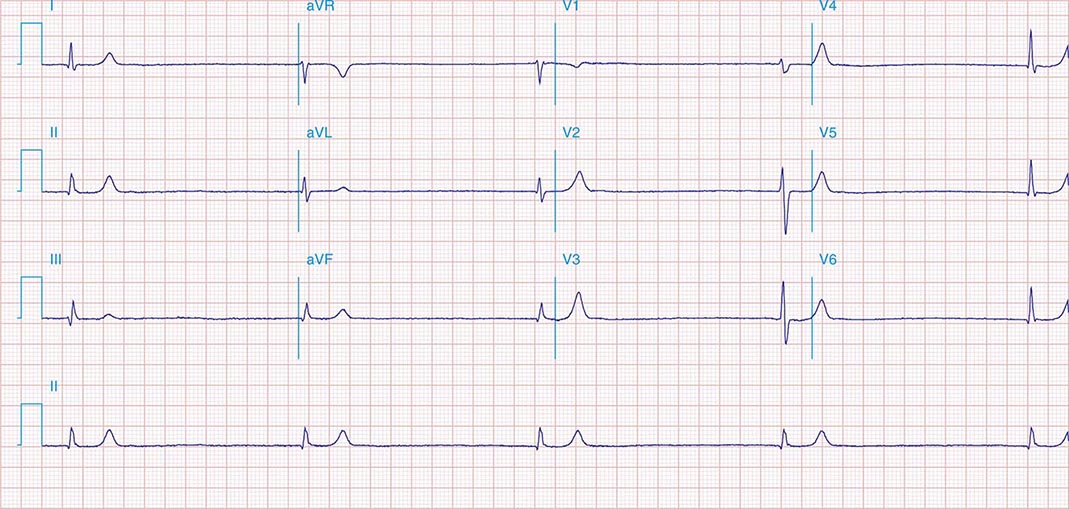
FIGURE 278e-5 Marked junctional bradycardia (25 beats/min). Rate is regular with a flat baseline between narrow QRS complexes, without evident P waves. Patient was on atenolol, with possible underlying sick sinus syndrome. The serum potassium was slightly elevated at 5.5 mEq/L.
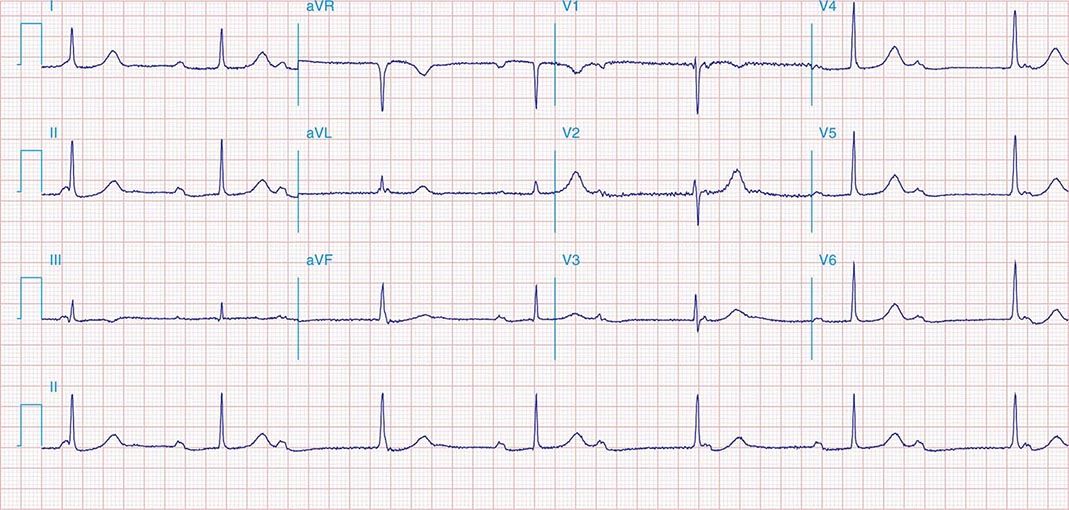
FIGURE 278e-6 Sinus rhythm at a rate of 64/min (P wave rate) with third-degree (complete) AV block yielding an effective heart (pulse) rate of 40/min. The slow, narrow QRS complexes indicate an AV junctional escape pacemaker. Left atrial abnormality.
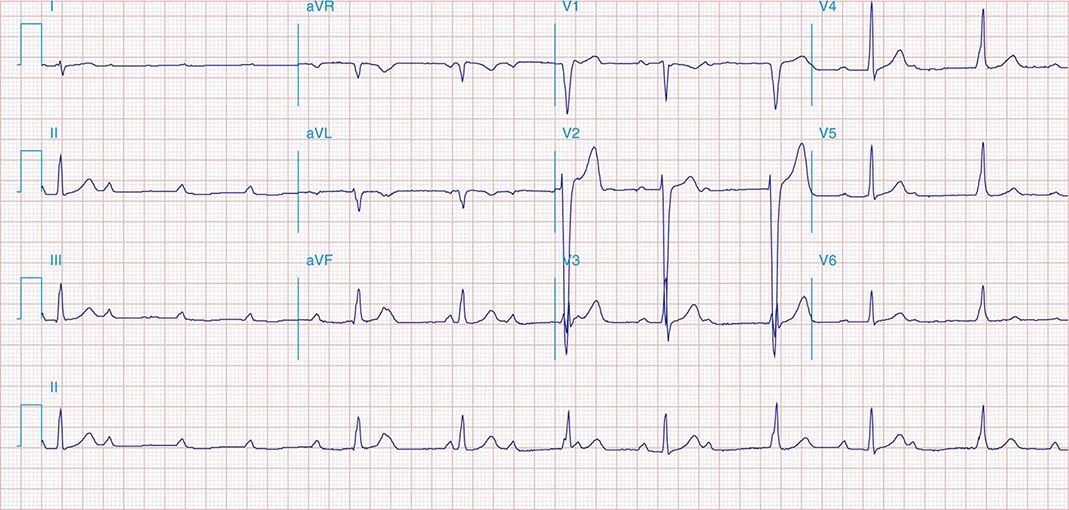
FIGURE 278e-7 Sinus rhythm at a rate of 90/min with advanced second-degree AV block and possible transient complete heart block with Lyme carditis.
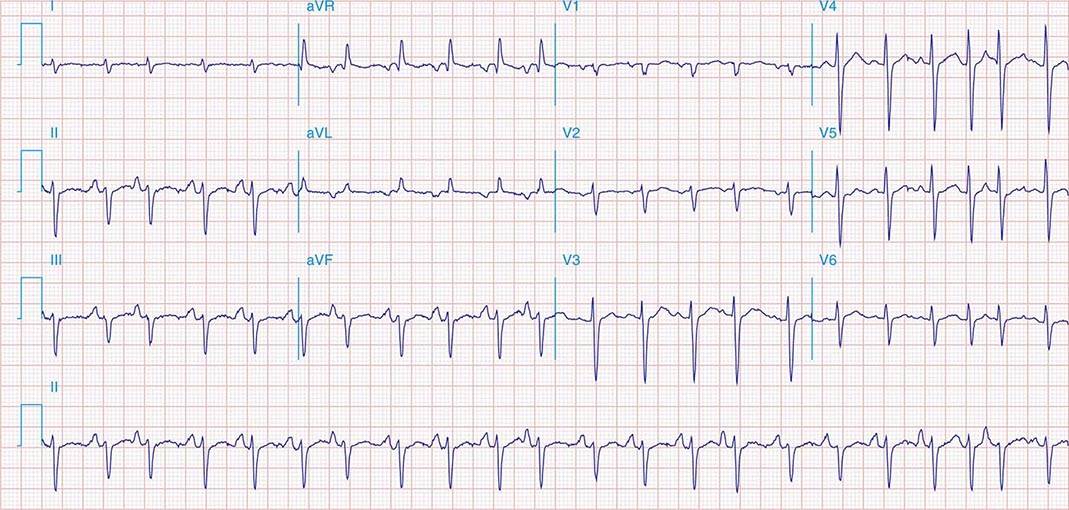
FIGURE 278e-8 Multifocal atrial tachycardia with varying P-wave morphologies and P-P intervals; right atrial overload with peaked P waves in II, III, and aVF (with vertical P-wave axis); superior QRS axis; slow R-wave progression with delayed transition in precordial leads in patient with severe chronic obstructive lung disease.
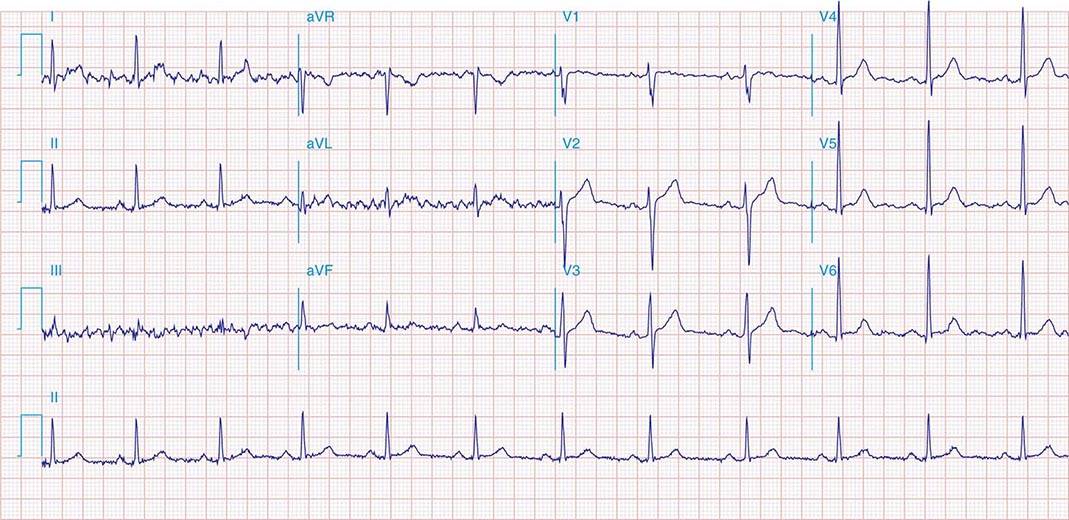
FIGURE 278e-9 NSR in a patient with Parkinson’s disease. Tremor artifact, best seen in limb leads. This tremor artifact may sometimes be confused with atrial flutter/fibrillation. Borderline voltage criteria for LVH are present.
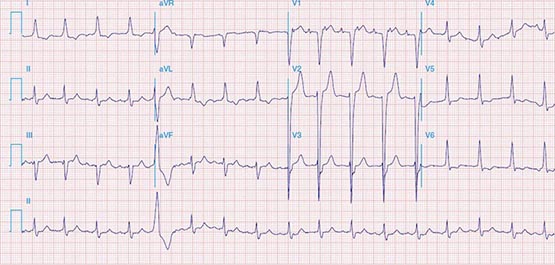
FIGURE 278e-10 Atrial tachycardia with atrial rate of about 200/min (note lead V1), 2:1 AV block (conduction), and one premature ventricular complex. Also present: LVH with intraventricular conduction delay and slow precordial R-wave progression (cannot rule out prior anterior MI).
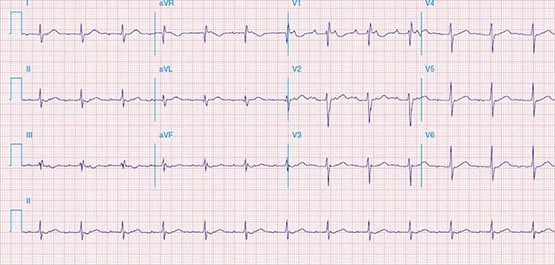
FIGURE 278e-11 Atrial tachycardia with 2:1 block. P-wave rate is about 150/min, with ventricular (QRS) rate of about 75/min. The nonconducted (“extra”) P waves just after the QRS complex are best seen in lead V1. Also, note incomplete RBBB and borderline QT prolongation.
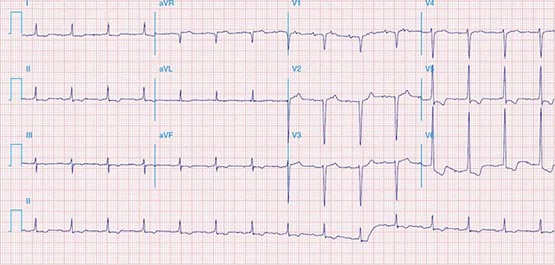
FIGURE 278e-12 Atrial tachycardia (180/min with 2:1 AV block [see lead V1]). LVH by precordial voltage and nonspecific ST-T changes. Slow R-wave progression (V1–V4) raises consideration of prior anterior MI.
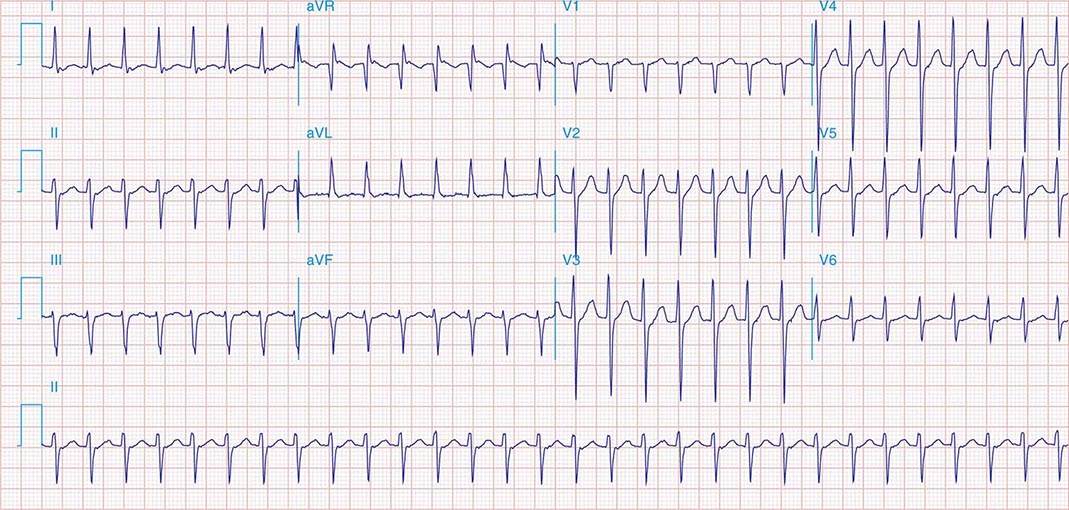
FIGURE 278e-13 AV nodal reentrant tachycardia (AVNRT) at a rate of 150/min. Note subtle “pseudo” R waves in lead aVR due to retrograde atrial activation, which usually occurs nearly simultaneous with ventricles in AVNRT. Left-axis deviation consistent with left anterior fascicular block (hemiblock) is also present.
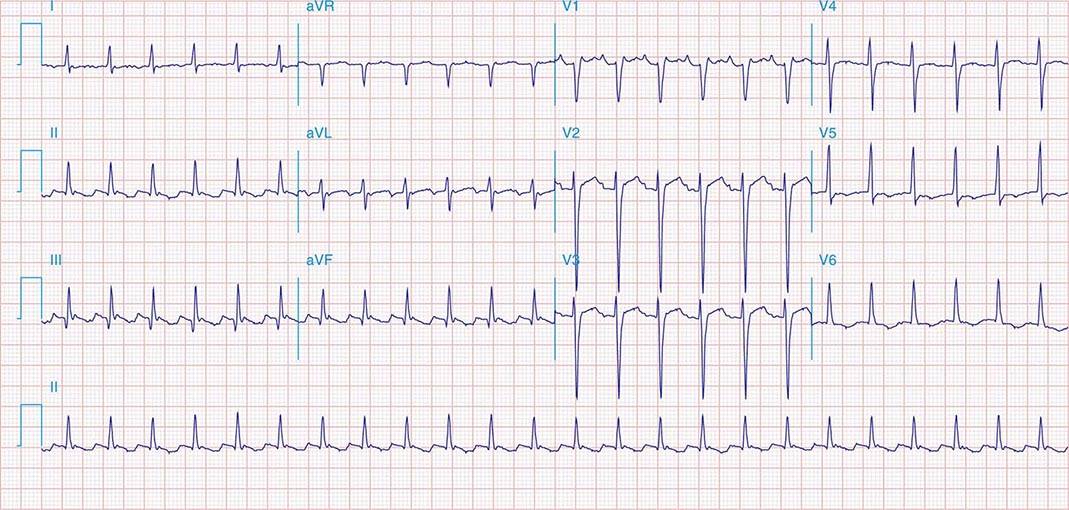
FIGURE 278e-14 Atrial flutter with 2:1 AV conduction. Note typical atrial flutter waves, partly hidden in the early ST segment, seen, for example, in leads II and V1.
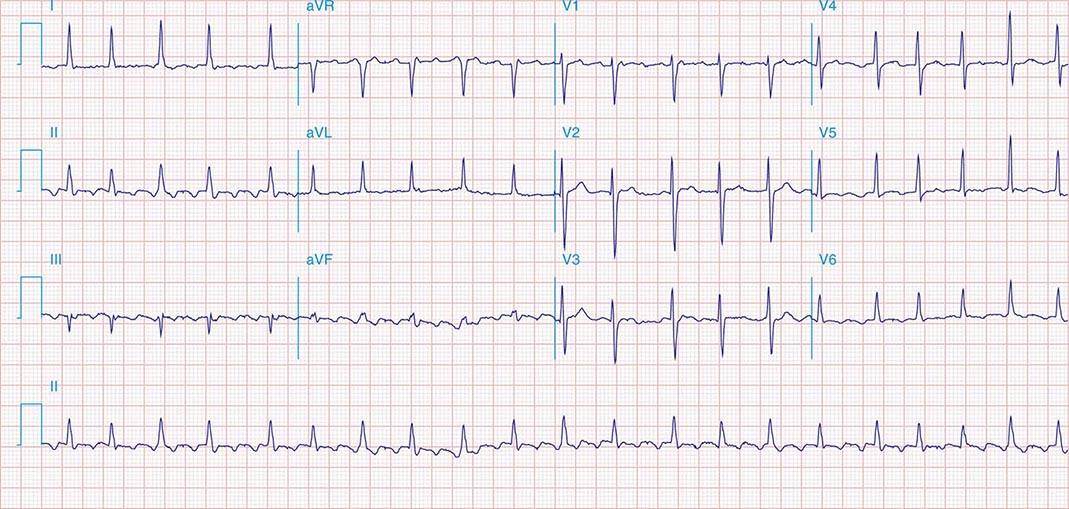
FIGURE 278e-15 Atrial flutter with atrial rate 300/min and variable (predominant 2:1 and 3:1) AV conduction. Typical flutter waves are best seen in lead II.
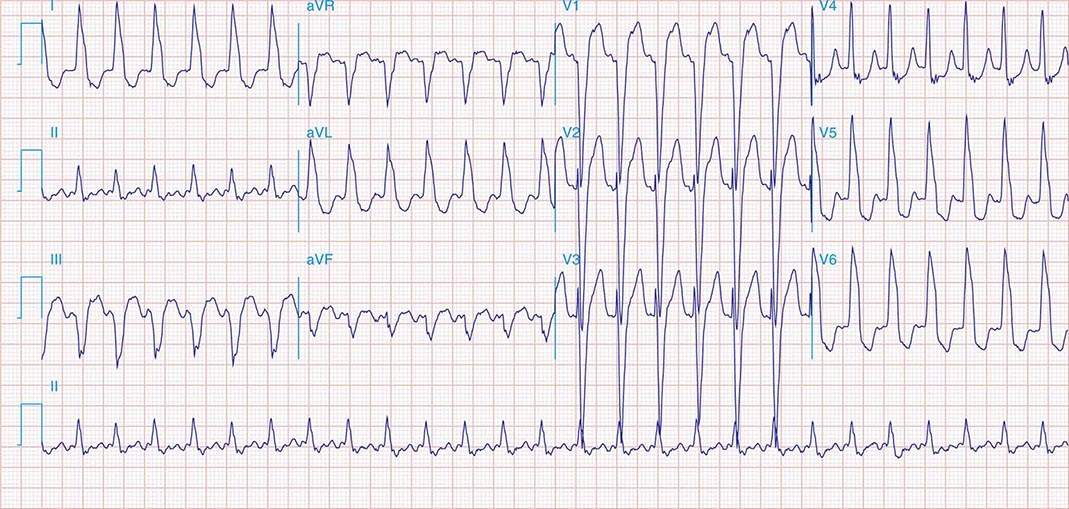
FIGURE 278e-16 Wide complex tachycardia. Atrial flutter with 2:1 AV conduction (block) and LBBB, not to be mistaken for VT. Typical atrial flutter activity is clearly present in lead II, at a cycle rate of about 320/min, yielding an effective ventricular rate of about 160/min.
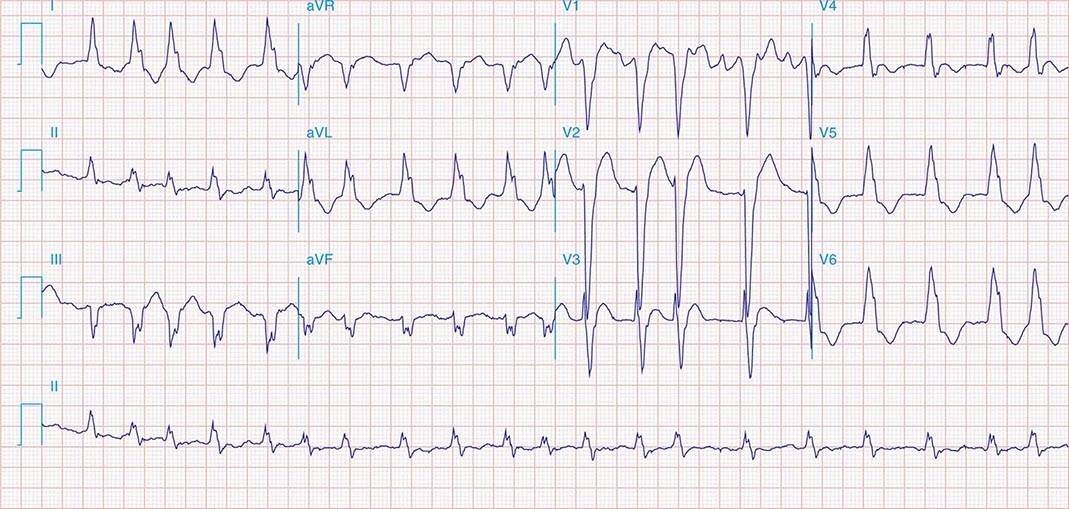
FIGURE 278e-17 AF with LBBB. The ventricular rhythm is erratically irregular. Coarse fibrillatory waves are best seen in lead V1, with a typical LBBB pattern.
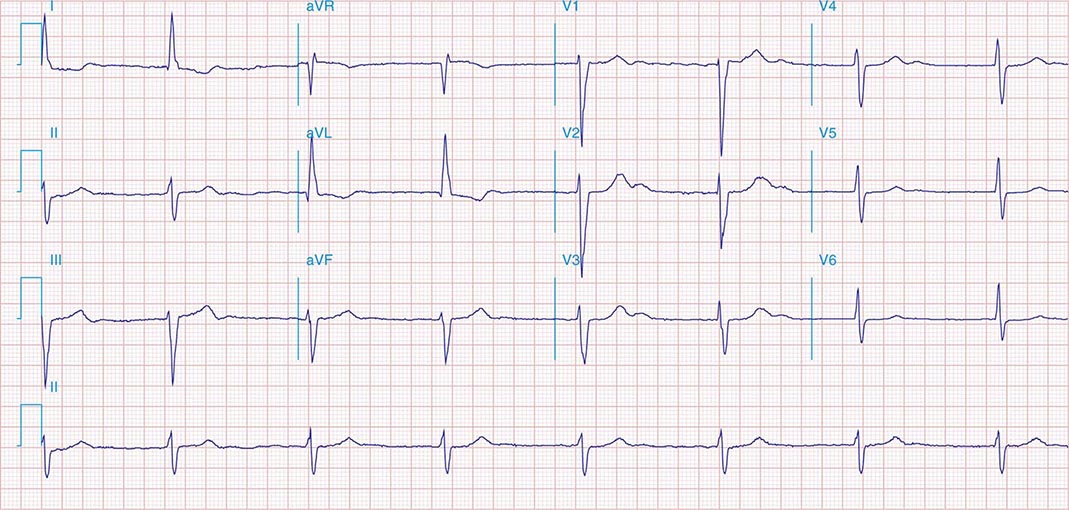
FIGURE 278e-18 AF with complete heart block and a junctional escape mechanism causing a slow regular ventricular response (45/min). The QRS complexes show an intraventricular conduction delay with left-axis deviation and LVH. Q-T (U) prolongation is also present.
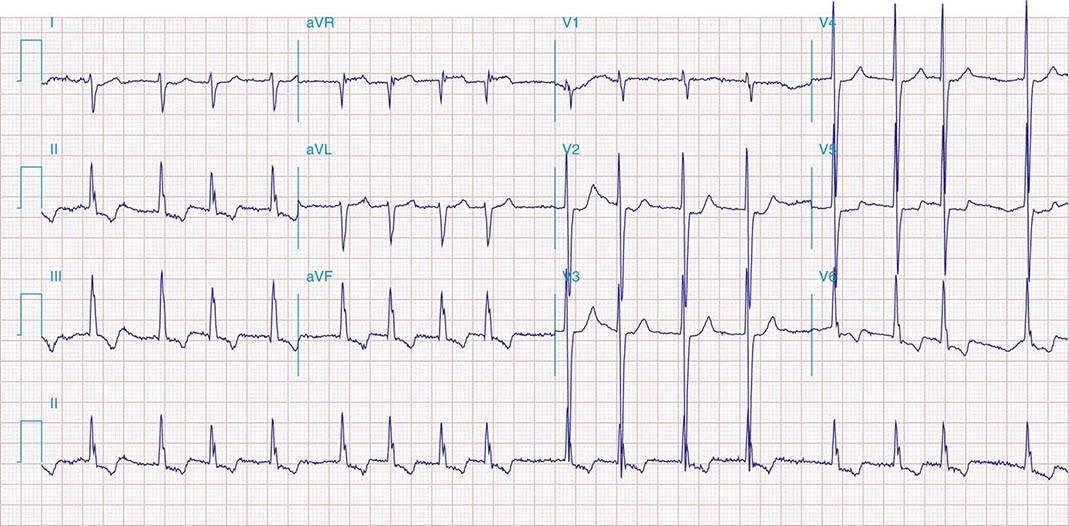
FIGURE 278e-19 AF with right-axis deviation and LVH. Tracing suggests biventricular hypertrophy in a patient with mitral stenosis and aortic valve disease.
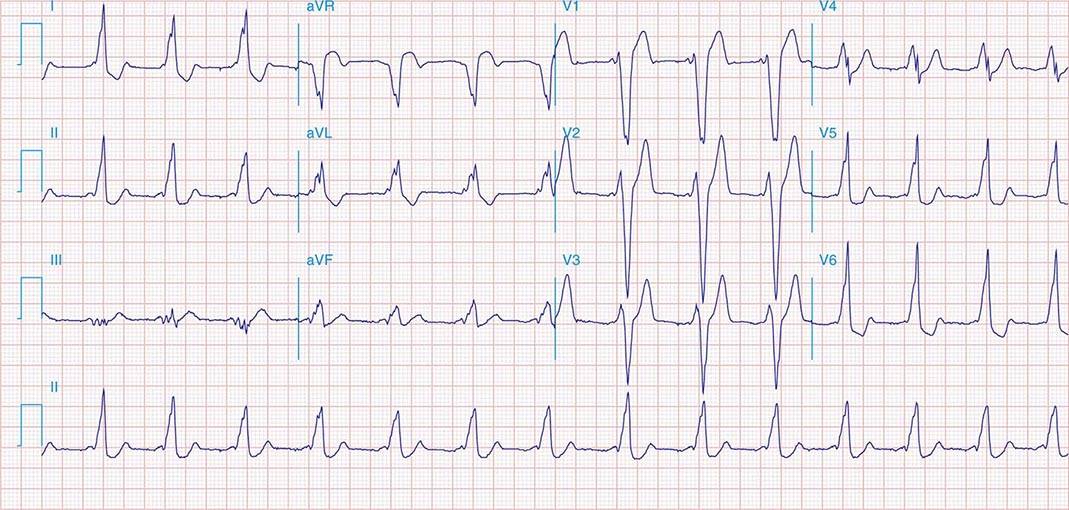
FIGURE 278e-20 WPW preexcitation pattern, with triad of short PR, wide QRS, and delta waves. Polarity of the delta waves (slightly positive in leads V1 and V2 and most positive in lead II and lateral chest leads) is consistent with a right-sided bypass tract.
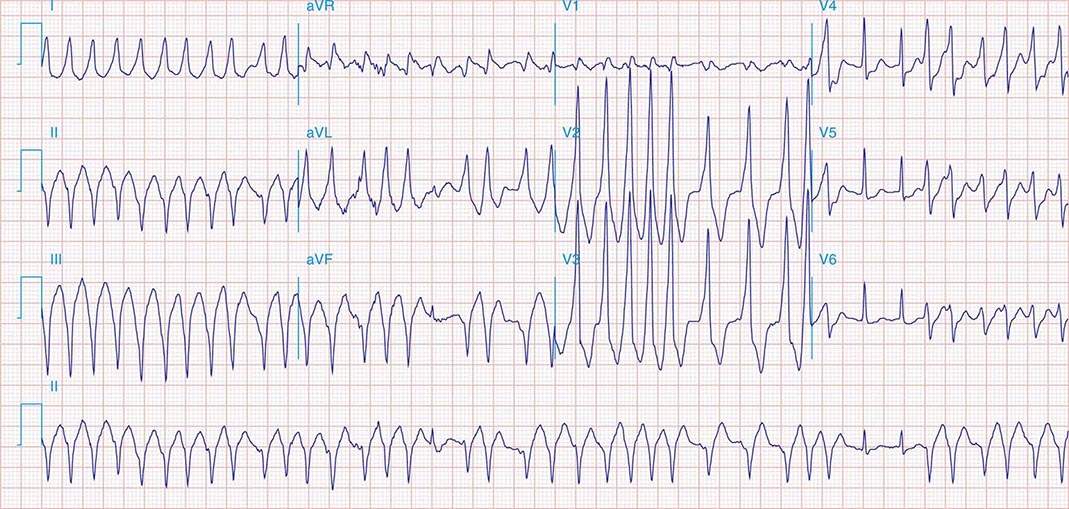
FIGURE 278e-21 AF in patient with the WPW syndrome and antegrade conduction down the bypass tract leading to a wide complex tachycardia. Rhythm is “irregularly irregular,” and rate is extremely rapid (about 230/min). Not all beats are preexcited.
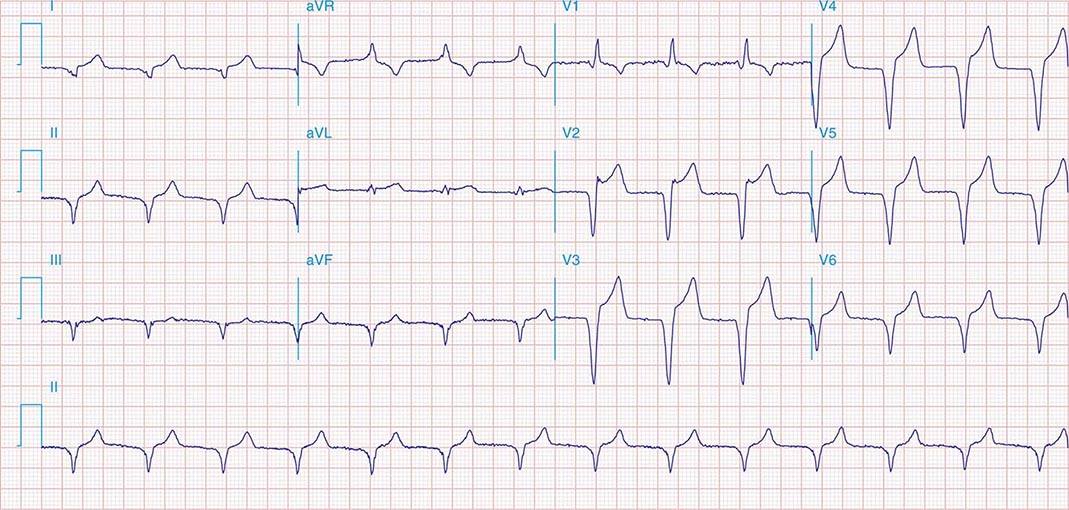
FIGURE 278e-22 Accelerated idioventricular rhythm (AIVR) originating from the LV and accounting for RBBB morphology. ST elevations in the precordial leads from underlying acute MI.
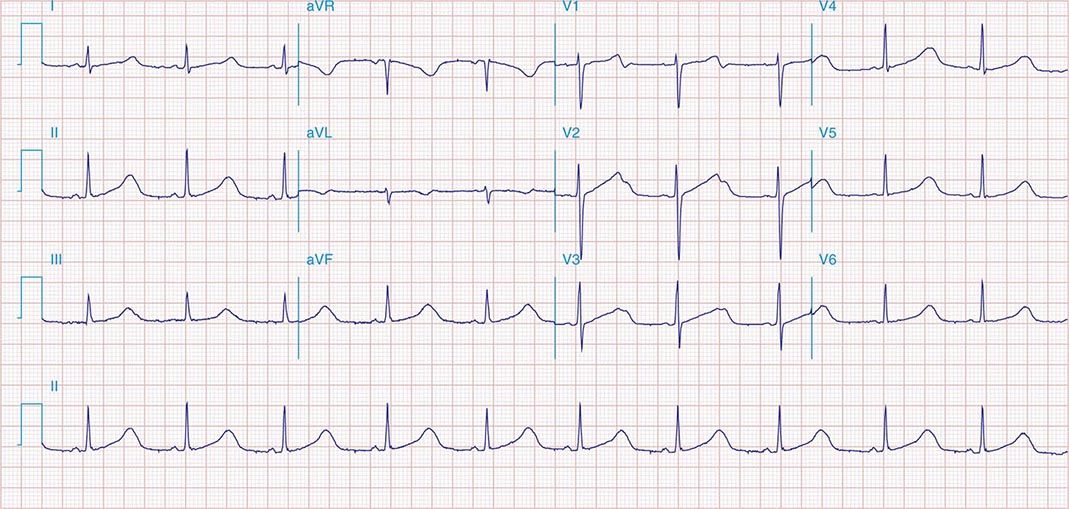
FIGURE 278e-23 Prolonged (0.60 s) QT interval in a patient with a hereditary long QT syndrome.
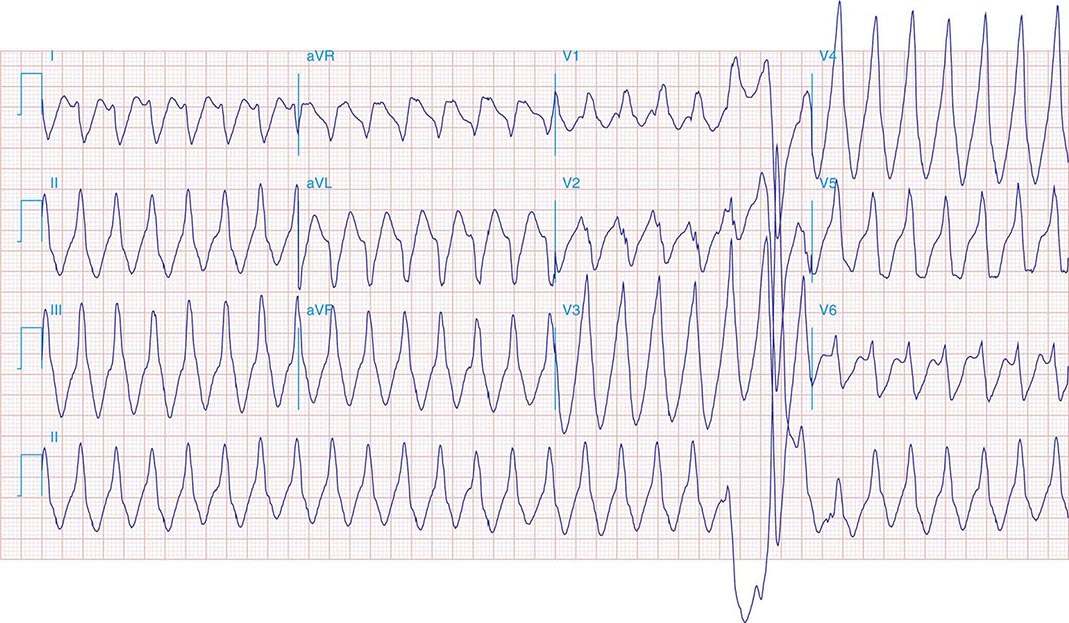
FIGURE 278e-24 Monomorphic VT at rate of 170/min. The RBBB morphology in V1 and the R:S ratio <1 in V6 are both suggestive of VT. The morphology of the VT is suggestive of origin from the left side of the heart, near the base (RBBB with inferior/rightward axis). Baseline artifact is present in leads V1–V3.
SECTION 4 |
DISORDERS OF THE HEART |
279 |
Heart Failure: Pathophysiology and Diagnosis |
HEART FAILURE
DEFINITION
Despite repeated attempts to develop a mechanistic definition that encompasses the heterogeneity and complexity of heart failure (HF), no single conceptual paradigm has withstood the test of time. The current American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) guidelines define HF as a complex clinical syndrome that results from structural or functional impairment of ventricular filling or ejection of blood, which in turn leads to the cardinal clinical symptoms of dyspnea and fatigue and signs of HF, namely edema and rales. Because many patients present without signs or symptoms of volume overload, the term “heart failure” is preferred over the older term “congestive heart failure.”
EPIDEMIOLOGY
![]() HF is a burgeoning problem worldwide, with more than 20 million people affected. The overall prevalence of HF in the adult population in developed countries is 2%. HF prevalence follows an exponential pattern, rising with age, and affects 6–10% of people over age 65. Although the relative incidence of HF is lower in women than in men, women constitute at least one-half the cases of HF because of their longer life expectancy. In North America and Europe, the lifetime risk of developing HF is approximately one in five for a 40-year-old. The overall prevalence of HF is thought to be increasing, in part because current therapies for cardiac disorders, such as myocardial infarction (MI), valvular heart disease, and arrhythmias, are allowing patients to survive longer. Very little is known about the prevalence or risk of developing HF in emerging nations because of the lack of population-based studies in those countries. HF was once thought to arise primarily in the setting of a depressed left ventricular (LV) ejection fraction (EF); however, epidemiologic studies have shown that approximately one-half of patients who develop HF have a normal or preserved EF (EF ≥50%). Accordingly, the historical terms “systolic” and “diastolic” HF have been abandoned, and HF patients are now broadly categorized into HF with a reduced EF (HFrEF; formerly systolic failure) or HF with a preserved EF (HRpEF; formerly diastolic failure).
HF is a burgeoning problem worldwide, with more than 20 million people affected. The overall prevalence of HF in the adult population in developed countries is 2%. HF prevalence follows an exponential pattern, rising with age, and affects 6–10% of people over age 65. Although the relative incidence of HF is lower in women than in men, women constitute at least one-half the cases of HF because of their longer life expectancy. In North America and Europe, the lifetime risk of developing HF is approximately one in five for a 40-year-old. The overall prevalence of HF is thought to be increasing, in part because current therapies for cardiac disorders, such as myocardial infarction (MI), valvular heart disease, and arrhythmias, are allowing patients to survive longer. Very little is known about the prevalence or risk of developing HF in emerging nations because of the lack of population-based studies in those countries. HF was once thought to arise primarily in the setting of a depressed left ventricular (LV) ejection fraction (EF); however, epidemiologic studies have shown that approximately one-half of patients who develop HF have a normal or preserved EF (EF ≥50%). Accordingly, the historical terms “systolic” and “diastolic” HF have been abandoned, and HF patients are now broadly categorized into HF with a reduced EF (HFrEF; formerly systolic failure) or HF with a preserved EF (HRpEF; formerly diastolic failure).
ETIOLOGY
As shown in Table 279-1, any condition that leads to an alteration in LV structure or function can predispose a patient to developing HF. Although the etiology of HF in patients with a preserved EF differs from that of patients with depressed EF, there is considerable overlap between the etiologies of these two conditions. In industrialized countries, coronary artery disease (CAD) has become the predominant cause in men and women and is responsible for 60–75% of cases of HF. Hypertension contributes to the development of HF in 75% of patients, including most patients with CAD. Both CAD and hypertension interact to augment the risk of HF, as does diabetes mellitus.
|
ETIOLOGIES OF HEART FAILURE |
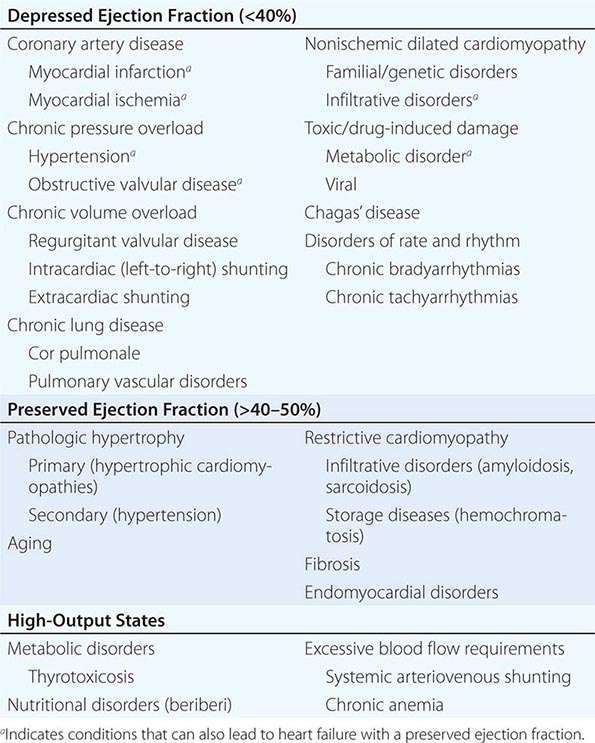
In 20–30% of the cases of HF with a depressed EF, the exact etiologic basis is not known. These patients are referred to as having nonischemic, dilated, or idiopathic cardiomyopathy if the cause is unknown (Chap. 287). Prior viral infection or toxin exposure (e.g., alcoholic or chemotherapeutic) also may lead to a dilated cardiomyopathy. Moreover, it is becoming increasingly clear that a large number of cases of dilated cardiomyopathy are secondary to specific genetic defects, most notably those in the cytoskeleton. Most forms of familial dilated cardiomyopathy are inherited in an autosomal dominant fashion. Mutations of genes that encode cytoskeletal proteins (desmin, cardiac myosin, vinculin) and nuclear membrane proteins (laminin) have been identified thus far. Dilated cardiomyopathy also is associated with Duchenne’s, Becker’s, and limb-girdle muscular dystrophies. Conditions that lead to a high cardiac output (e.g., arteriovenous fistula, anemia) are seldom responsible for the development of HF in a normal heart; however, in the presence of underlying structural heart disease, these conditions can lead to overt HF.
GLOBAL CONSIDERATIONS
![]() Rheumatic heart disease remains a major cause of HF in Africa and Asia, especially in the young. Hypertension is an important cause of HF in the African and African-American populations. Chagas’ disease is still a major cause of HF in South America. Not surprisingly, anemia is a frequent concomitant factor in HF in many developing nations. As developing nations undergo socioeconomic development, the epidemiology of HF is becoming similar to that of Western Europe and North America, with CAD emerging as the single most common cause of HF. Although the contribution of diabetes mellitus to HF is not well understood, diabetes accelerates atherosclerosis and often is associated with hypertension.
Rheumatic heart disease remains a major cause of HF in Africa and Asia, especially in the young. Hypertension is an important cause of HF in the African and African-American populations. Chagas’ disease is still a major cause of HF in South America. Not surprisingly, anemia is a frequent concomitant factor in HF in many developing nations. As developing nations undergo socioeconomic development, the epidemiology of HF is becoming similar to that of Western Europe and North America, with CAD emerging as the single most common cause of HF. Although the contribution of diabetes mellitus to HF is not well understood, diabetes accelerates atherosclerosis and often is associated with hypertension.
PROGNOSIS
Despite many recent advances in the evaluation and management of HF, the development of symptomatic HF still carries a poor prognosis. Community-based studies indicate that 30–40% of patients die within 1 year of diagnosis and 60–70% die within 5 years, mainly from worsening HF or as a sudden event (probably because of a ventricular arrhythmia). Although it is difficult to predict prognosis in an individual, patients with symptoms at rest (New York Heart Association [NYHA] class IV) have a 30–70% annual mortality rate, whereas patients with symptoms with moderate activity (NYHA class II) have an annual mortality rate of 5–10%. Thus, functional status is an important predictor of patient outcome (Table 279-2).
|
NEW YORK HEART ASSOCIATION CLASSIFICATION |
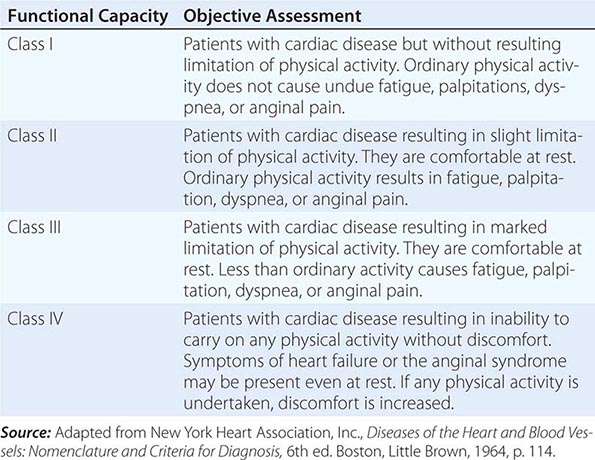
PATHOGENESIS
Figure 279-1 provides a general conceptual framework for considering the development and progression of HFrEF. As shown, HF may be viewed as a progressive disorder that is initiated after an index event either damages the heart muscle, with a resultant loss of functioning cardiac myocytes, or, alternatively, disrupts the ability of the myocardium to generate force, thereby preventing the heart from contracting normally. This index event may have an abrupt onset, as in the case of an MI; it may have a gradual or insidious onset, as in the case of hemodynamic pressure or volume overloading; or it may be hereditary, as in the case of many of the genetic cardiomyopathies. Regardless of the nature of the inciting event, the feature that is common to each of these index events is that they all in some manner produce a decline in the pumping capacity of the heart. In most instances, patients remain asymptomatic or minimally symptomatic after the initial decline in pumping capacity of the heart or develop symptoms only after the dysfunction has been present for some time.
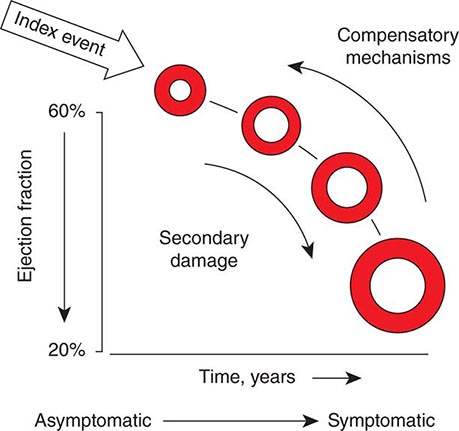
FIGURE 279-1 Pathogenesis of heart failure with a depressed ejection fraction. Heart failure begins after an index event produces an initial decline in the heart’s pumping capacity. After this initial decline in pumping capacity, a variety of compensatory mechanisms are activated, including the adrenergic nervous system, the renin-angiotensin-aldosterone system, and the cytokine system. In the short term, these systems are able to restore cardiovascular function to a normal homeostatic range with the result that the patient remains asymptomatic. However, with time, the sustained activation of these systems can lead to secondary end-organ damage within the ventricle, with worsening left ventricular remodeling and subsequent cardiac decompensation. (From D Mann: Circulation 100:999, 1999.)
Although the precise reasons why patients with LV dysfunction may remain asymptomatic is not certain, one potential explanation is that a number of compensatory mechanisms become activated in the presence of cardiac injury and/or LV dysfunction allowing patients to sustain and modulate LV function for a period of months to years. The compensatory mechanisms that have been described thus far include (1) activation of the renin-angiotensin-aldosterone (RAA) and adrenergic nervous systems, which are responsible, respectively, for maintaining cardiac output through increased retention of salt and water (Fig. 279-2), and (2) increased myocardial contractility. In addition, there is activation of a family of countervailing vasodilatory molecules, including the atrial and brain natriuretic peptides (ANP and BNP), prostaglandins (PGE2 and PGI2), and nitric oxide (NO), that offsets the excessive peripheral vascular vasoconstriction. Genetic background, sex, age, or environment may influence these compensatory mechanisms, which are able to modulate LV function within a physiologic/homeostatic range so that the functional capacity of the patient is preserved or is depressed only minimally. Thus, patients may remain asymptomatic or minimally symptomatic for a period of years; however, at some point patients become overtly symptomatic, with a resultant striking increase in morbidity and mortality rates. Although the exact mechanisms that are responsible for this transition are not known, as will be discussed below, the transition to symptomatic HF is accompanied by increasing activation of neurohormonal, adrenergic, and cytokine systems that lead to a series of adaptive changes within the myocardium collectively referred to as LV remodeling.
FIGURE 279-2 Activation of neurohormonal systems in heart failure. The decreased cardiac output in heart failure (HF) patients results in an “unloading” of high-pressure baroreceptors (circles) in the left ventricle, carotid sinus, and aortic arch. This unloading of the peripheral baroreceptors leads to a loss of inhibitory parasympathetic tone to the central nervous system (CNS), with a resultant generalized increase in efferent sympathetic tone, and nonosmotic release of arginine vasopressin (AVP) from the pituitary. AVP (or antidiuretic hormone [ADH]) is a powerful vasoconstrictor that increases the permeability of the renal collecting ducts, leading to the reabsorption of free water. These afferent signals to the CNS also activate efferent sympathetic nervous system pathways that innervate the heart, kidney, peripheral vasculature, and skeletal muscles.
Sympathetic stimulation of the kidney leads to the release of renin, with a resultant increase in the circulating levels of angiotensin II and aldosterone. The activation of the renin-angiotensin-aldosterone system promotes salt and water retention and leads to vasoconstriction of the peripheral vasculature, myocyte hypertrophy, myocyte cell death, and myocardial fibrosis. Although these neurohormonal mechanisms facilitate short-term adaptation by maintaining blood pressure, and hence perfusion to vital organs, the same neurohormonal mechanisms are believed to contribute to end-organ changes in the heart and the circulation and to the excessive salt and water retention in advanced HF. (Modified from A Nohria et al: Neurohormonal, renal and vascular adjustments, in Atlas of Heart Failure: Cardiac Function and Dysfunction, 4th ed, WS Colucci [ed]. Philadelphia, Current Medicine Group 2002, p. 104.)
In contrast to our understanding of the pathogenesis of HF with a depressed EF, our understanding of the mechanisms that contribute to the development of HF with a preserved EF is still evolving. That is, although diastolic dysfunction (see below) was thought to be the only mechanism responsible for the development of HF with a preserved EF, community-based studies suggest that additional extracardiac mechanisms may be important, such as increased vascular stiffness and impaired renal function.
BASIC MECHANISMS OF HEART FAILURE
Heart Failure with a Reduced Ejection Fraction LV remodeling develops in response to a series of complex events that occur at the cellular and molecular levels (Table 279-3). These changes include (1) myocyte hypertrophy; (2) alterations in the contractile properties of the myocyte; (3) progressive loss of myocytes through necrosis, apoptosis, and autophagic cell death; (4) β-adrenergic desensitization; (5) abnormal myocardial energetics and metabolism; and (6) reorganization of the extracellular matrix with dissolution of the organized structural collagen weave surrounding myocytes and subsequent replacement by an interstitial collagen matrix that does not provide structural support to the myocytes. The biologic stimuli for these profound changes include mechanical stretch of the myocyte, circulating neurohormones (e.g., norepinephrine, angiotensin II), inflammatory cytokines (e.g., tumor necrosis factor [TNF]), other peptides and growth factors (e.g., endothelin), and reactive oxygen species (e.g., superoxide). The sustained overexpression of these biologically active molecules is believed to contribute to the progression of HF by virtue of the deleterious effects they exert on the heart and the circulation. Indeed, this insight forms the clinical rationale for using pharmacologic agents that antagonize these systems (e.g., angiotensin-converting enzyme [ACE] inhibitors and beta blockers) in treating patients with HF (Chap. 280).
|
OVERVIEW OF LEFT VENTRICULAR REMODELING |
Source: Adapted from D. Mann: Pathophysiology of heart failure, in Braunwald’s Heart Disease, 8th ed, PL Libby et al (eds). Philadelphia, Elsevier, 2008, p. 550.
To understand how the changes that occur in the failing cardiac myocyte contribute to depressed LV systolic function in HF, it is instructive first to review the biology of the cardiac muscle cell (Chap. 265e). Sustained neurohormonal activation and mechanical overload result in transcriptional and posttranscriptional changes in the genes and proteins that regulate excitation-contraction coupling and cross-bridge interaction (see Figs. 265e-6 and 265e-7). The changes that regulate excitation-contraction include decreased function of sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA2A), resulting in decreased calcium uptake into the sarcoplasmic reticulum (SR), and hyperphosphorylation of the ryanodine receptor, leading to calcium leakage from the SR. The changes that occur in the cross-bridges include decreased expression of α-myosin heavy chain and increased expression of β-myosin heavy chain, myocytolysis, and disruption of the cytoskeletal links between the sarcomeres and the extracellular matrix. Collectively, these changes impair the ability of the myocyte to contract and therefore contribute to the depressed LV systolic function observed in patients with HF.
Myocardial relaxation is an adenosine triphosphate (ATP)-dependent process that is regulated by uptake of cytoplasmic calcium into the SR by SERCA2A and extrusion of calcium by sarcolemmal pumps (see Fig. 265e-7). Accordingly, reductions in ATP concentration, as occurs in ischemia, may interfere with these processes and lead to slowed myocardial relaxation. Alternatively, if LV filling is delayed because LV compliance is reduced (e.g., from hypertrophy or fibrosis), LV filling pressures will similarly remain elevated at end diastole (see Fig. 265e-11). An increase in heart rate disproportionately shortens the time for diastolic filling, which may lead to elevated LV filling pressures, particularly in noncompliant ventricles. Elevated LV end-diastolic filling pressures result in increases in pulmonary capillary pressures, which can contribute to the dyspnea experienced by patients with diastolic dysfunction. In addition to impaired myocardial relaxation, increased myocardial stiffness secondary to cardiac hypertrophy and increased myocardial collagen content may contribute to diastolic failure. Importantly, diastolic dysfunction can occur alone or in combination with systolic dysfunction in patients with HF.
Left Ventricular Remodeling Ventricular remodeling refers to the changes in LV mass, volume, and shape and the composition of the heart that occur after cardiac injury and/or abnormal hemodynamic loading conditions. LV remodeling may contribute independently to the progression of HF by virtue of the mechanical burdens that are engendered by the changes in the geometry of the remodeled LV. In addition to the increase in LV end-diastolic volume, LV wall thinning occurs as the left ventricle begins to dilate. The increase in wall thinning, along with the increase in afterload created by LV dilation, leads to a functional afterload mismatch that may contribute further to a decrease in stroke volume. Moreover, the high end-diastolic wall stress might be expected to lead to (1) hypoperfusion of the subendocardium, with resultant worsening of LV function; (2) increased oxidative stress, with the resultant activation of families of genes that are sensitive to free radical generation (e.g., TNF and interleukin 1β); and (3) sustained expression of stretch-activated genes (angiotensin II, endothelin, and TNF) and/or stretch activation of hypertrophic signaling pathways. Increasing LV dilation also results in tethering of the papillary muscles with resulting incompetence of the mitral valve apparatus and functional mitral regurgitation, which in turn leads to further hemodynamic overloading of the ventricle. Taken together, the mechanical burdens that are engendered by LV remodeling contribute to the progression of HF. Recent studies have shown that LV remodeling can be reversed following medical and device therapy and that reverse LV remodeling is associated with improved clinical outcomes in patients with HFrEF. Indeed, one of the goals of therapy for HF is to prevent and/or reverse LV remodeling.
CLINICAL MANIFESTATIONS
Symptoms The cardinal symptoms of HF are fatigue and shortness of breath. Although fatigue traditionally has been ascribed to the low cardiac output in HF, it is likely that skeletal-muscle abnormalities and other noncardiac comorbidities (e.g., anemia) also contribute to this symptom. In the early stages of HF, dyspnea is observed only during exertion; however, as the disease progresses, dyspnea occurs with less strenuous activity, and it ultimately may occur even at rest. The origin of dyspnea in HF is probably multifactorial (Chap. 47e). The most important mechanism is pulmonary congestion with accumulation of interstitial or intra-alveolar fluid, which activates juxtacapillary J receptors, which in turn stimulate the rapid, shallow breathing characteristic of cardiac dyspnea. Other factors that contribute to dyspnea on exertion include reductions in pulmonary compliance, increased airway resistance, respiratory muscle and/or diaphragm fatigue, and anemia. Dyspnea may become less frequent with the onset of right ventricular (RV) failure and tricuspid regurgitation.
ORTHOPNEA Orthopnea, which is defined as dyspnea occurring in the recumbent position, is usually a later manifestation of HF than is exertional dyspnea. It results from redistribution of fluid from the splanchnic circulation and lower extremities into the central circulation during recumbency, with a resultant increase in pulmonary capillary pressure. Nocturnal cough is a common manifestation of this process and a frequently overlooked symptom of HF. Orthopnea generally is relieved by sitting upright or sleeping with additional pillows. Although orthopnea is a relatively specific symptom of HF, it may occur in patients with abdominal obesity or ascites and patients with pulmonary disease whose lung mechanics favor an upright posture.
PAROXYSMAL NOCTURNAL DYSPNEA (PND) This term refers to acute episodes of severe shortness of breath and coughing that generally occur at night and awaken the patient from sleep, usually 1–3 h after the patient retires. PND may manifest as coughing or wheezing, possibly because of increased pressure in the bronchial arteries leading to airway compression, along with interstitial pulmonary edema that leads to increased airway resistance. Whereas orthopnea may be relieved by sitting upright at the side of the bed with the legs in a dependent position, patients with PND often have persistent coughing and wheezing even after they have assumed the upright position. Cardiac asthma is closely related to PND, is characterized by wheezing secondary to bronchospasm, and must be differentiated from primary asthma and pulmonary causes of wheezing.
CHEYNE-STOKES RESPIRATION Also referred to as periodic respiration or cyclic respiration, Cheyne-Stokes respiration is present in 40% of patients with advanced HF and usually is associated with low cardiac output. Cheyne-Stokes respiration is caused by an increased sensitivity of the respiratory center to arterial PCO2. There is an apneic phase, during which arterial PO2 falls and arterial PCO2 rises. These changes in the arterial blood gas content stimulate the respiratory center, resulting in hyperventilation and hypocapnia, followed by recurrence of apnea. Cheyne-Stokes respirations may be perceived by the patient or the patient’s family as severe dyspnea or as a transient cessation of breathing.
ACUTE PULMONARY EDEMA See Chap. 326
Other Symptoms Patients with HF also may present with gastrointestinal symptoms. Anorexia, nausea, and early satiety associated with abdominal pain and fullness are common complaints and may be related to edema of the bowel wall and/or a congested liver. Congestion of the liver and stretching of its capsule may lead to right upper-quadrant pain. Cerebral symptoms such as confusion, disorientation, and sleep and mood disturbances may be observed in patients with severe HF, particularly elderly patients with cerebral arteriosclerosis and reduced cerebral perfusion. Nocturia is common in HF and may contribute to insomnia.