17 Headaches
 Clinical cases for thought
Clinical cases for thoughtIntroduction
Headaches can be due to a variety of causes but are generally categorised into primary and secondary headaches (Box 17.1). The primary headaches, the main focus of this chapter, are headaches that are not caused by another disease or condition, unlike secondary headaches. The International Headache Society (IHS) maintains a list of conditions that can produce headache symptoms secondary to the disorder (International Headache Society 2004). This list is extensive and the clinician managing the headache patient needs to be familiar with these conditions and include or exclude them as appropriate. Such causes may include intracranial mass, vascular lesions, systemic disorders or infection. In particular, the clinician needs to be aware of the following danger signs that may indicate the need for aggressive investigation.
• Sudden onset of headache, severe in nature, that peaks quickly (minutes to a few hours) or that gets progressively worse. For example, subarachnoid haemorrhage may be of sudden onset, or headache due to infection such as abscess or meningitis may worsen over hours to days.
• Any patient that presents with the ‘worst headache of my life’.
• Focal neurological signs, particularly on the first occurrence of the headache, including changes in personality, mental status, and level of consciousness.
• Headache precipitated or made worse by bending, sneezing, coughing, or exertion. This may indicate raised intracranial pressure. Vomiting with headache may also be associated with raised intracranial pressure.
• Changes in the headache pattern of an existing headache sufferer.
• Association of headache with fever, rash, nucal rigidity, lymphadenopathy, and particularly if for the first time, photophobia.
• New headaches in patients under 5 or over 50 years of age.
 Box 17.1
Box 17.1Adapted from IHS 2004, with permission.
Migraine
Migraine is a common disabling condition that is ranked nineteenth by the World Health Organization amongst worldwide diseases causing disability (IHS 2004). Approximately 15–20% of the general population suffers from migraine headaches (Bolay et al. 2002). It has considerable social and economic impact; hence effective treatment strategies have the ability to make significant improvements in the quality of life of the sufferer, and have positive social benefit as well. In this chapter we will discuss the two most common forms of migraine, migraine with aura and migraine without aura, along with the childhood syndromes that are often the precursor of migraine.
Migraine without aura (MWOA)
Migraine without aura was previously known as common migraine.
The International Headache Society (2004) describes MWOA as: ‘Recurrent headache disorder manifesting in attacks lasting 4–72 hours. Typical characteristics of the headache are unilateral location, pulsating quality, moderate or severe intensity, aggravation by routine physical activity and association with nausea and/or photophobia and phonophobia’.
To be diagnosed with MWOA the individual must have had at least five attacks lasting 4–72 hours with at least two of the following characteristics: unilateral pain location, pulsating quality, moderate to severe pain intensity and/or aggravated by routine activity. It must also be accompanied by either nausea and/or vomiting or phono- and photophobia (Box 17.2) (IHS 2004).
 Box 17.2
Box 17.2
Diagnostic criteria for migraine without aura
Diagnostic criteria
A. At least five attacks fulfilling criteria B–D
B. Headache attacks lasting 4–72 hours (untreated or unsuccessfully treated)
C. Headache has at least two of the following characteristics:
From IHS 2004, with permission.
Peripheral sensitisation in migraine
Migraine is a complex phenomenon that likely has multiple causes. However, current thought on the pathophysiology of migraine is that an initial activation of meningeal nociceptors causes a release of substance P and salcitonin gene related peptide (CRGP) from the nociceptive nerve endings, which in turn causes vasodilation and protein extravasation (Dalkara et al. 2006). This process is known as neurogenic inflammation and/or peripheral sensitisation. In peripheral sensitisation the increased sensitivity of the nociceptors to activation may develop to a degree that, in combination with central sensitisation, they are susceptible to activation by the arterial pulse and head movements (Dalkara et al. 2006). In this way, normal physiological activities may become sources of pain. The causes of the initial activation are still not fully described but several possible mechanisms are discussed below.
Central sensitisation
Accompanying peripheral sensitisation is the phenomenon of central sensitisation. Activation of trigeminovascular nociceptors leads to an increase in activity in the second-order neurons in the trigeminocervical nucleus (Bolay et al. 2002). Furthermore, activation in the trigeminocervical nucleus occurs with stimulation of both meningeal afferents and those from the greater occipital nerve (a branch of the C2 dorsal root), and that stimulation for just 5 minutes can lead to an increase in response in the nucleus for over an hour to other nociceptive stimuli, including if that stimulation is received from the C2 innervation (Goadsby 2005). This is particularly relevant for manual therapy practitioners with their focus on removal of pain generators from the cervical spine. It is likely that this sensitisation involves the activation of NMDA receptors in the nucleus as in vitro application of sumatriptan, an antimigraine drug, is effective in blocking NMDA receptor activation (Buzzi & Moskowitz 2005).
The cutaneous allodynia is often associated with migraine and is thought to be another symptom of this central sensitisation of the trigeminocervical nucleus. The allodynia usually occurs on the ipsilateral forehead to the headache, but has been documented to occur over the ipsi- and contralateral hands as well (Dalkara, Zervas et al. 2006).
Brainstem dysfunction and disinhibition
Brainstem dysfunction has also been implicated in migraine, both as a primary migraine generator, and through failure of brainstem pain inhibition mechanisms. Goadsby (2005) reports activation of the locus ceruleus and periaqueductal grey (PAG) in migraine without aura, and that these correspond to areas that have generated migraine-like symptoms in patients when stimulated electrically. Similarly, Goadsby reports excess iron has been found in the PAG of episodic and chronic migraineurs, and lesions in this area have been known to cause migraine. The notion of brainstem dysfunction is further supported by the presence of measured balance deficits, vestibular changes (Akdal et al. 2007; Akdal et al. 2009; Asai et al. 2009), and subclinical cerebellar deficits in migraineurs (Sandor et al. 2001).
The PAG is involved in descending pain control mechanisms, along with the locus ceruleus and raphae nuclei. In 2001, Knight and Goadsby (2001) published research that suggests that it is dysfunction in the PAG that leads to disinhibition of the trigeminocervical nucleus and hence pain.
Mechanisms leading to initial trigeminovascular activation
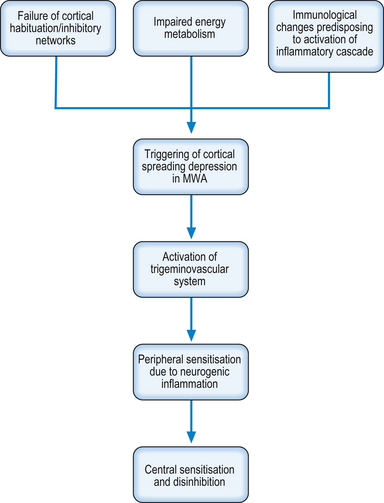
The brain of the migraineur displays different neurophysiological behaviours to that of the normal individual. Evoked potential and transcranial magnetic stimulation (TMS) studies have shown that the cerebral cortex of the migraine sufferer shows an impaired ability to habituate to sensory stimulation across all sensory modalities (Schoenen 2006; Brighina et al. 2009). This is thought to be due to impaired functioning of inhibitory networks in the cortex, and it has been suggested that this dysfunction occurs in a manner similar to the GABA circuit down-regulation that occurs in sensory deafferentation (Brighina et al. 2009). Interestingly, Brighina et al. (2009) report that repeated trains of TMS have been shown to improve the functioning of the inhibitory networks that could have long-term effects. This is consistent with the functional neurology paradigm that it is possible to improve neuronal functioning by repeated stimulation within the tolerance of fatigue, thereby promoting gene expression, protein synthesis, and thus building plasticity.
It is logical to assume that this inability to habituate to sensory stimulation could predispose the cortex to excitotoxic events, and evidence suggests that this is the case (Dalkara et al. 2006). This is further supported by evidence that there is impaired energy metabolism in migraineurs (Lodi et al. 2006), which will predispose to anaerobic metabolism. Anaerobic metabolism is toxic to neurons and the metabolic by-products associated with it such as lactic acid are able to activate nociceptive fibres. Functional neurological interventions to promote aerobic metabolism and to protect from anaerobic metabolism should be considered by the clinician. Interventions that may be considered include:
• Supplementation with co-enzyme Q10 due to its antioxidant capabilities and involvement in oxidative phosphorylation (Lodi et al. 2006);
• Supplementation with other antioxidants;
• Supplementation with magnesium to reduce the likelihood of activation of NMDA receptors on nociceptive fibres (Lodi et al. 2006);
• Reduction of circulating adrenaline levels as adrenaline reduces the threshold to activation of C fibres via the alpha 1 receptors. Examples may be stress reduction or the reduction of caffeine intake;
• Ensuring good respiratory mechanisms through preservation of the lumbar lordosis and normal rib mechanics;
• Management of autonomic function to ensure optimum vascular supply to the brain; and
• Lifestyle advice to ensure the patient only stimulates their nervous system within the tolerance of fatigue.
It is also thought that the migraineurs have immunological changes that predispose them to the activation of the inflammatory cascade, which in turn leads to the migraine attack. These triggers may be infections, foods, or other environmental factors (Longoni & Ferrarese 2006). Any management plan for the migraineur should include the identification and, as far as possible, the removal of the triggers. Headache diaries including diet and environmental exposures can be useful for identifying the triggers.
Migraine with aura (MWA)
Migraine with aura was previously known as classic migraine.
The International Headache Society (2004) describes MWA as: ‘Recurrent disorder manifesting in attacks of reversible focal neurological symptoms that usually develop gradually over 5–20 minutes and last for less than 60 minutes. Headache with the features of migraine without aura usually follows the aura symptoms. Less commonly, headache lacks migrainous features or is completely absent’ (Box 17.3).
 Box 17.3
Box 17.3
Diagnostic criteria for migraine with aura
Diagnostic criteria
A. At least 2 attacks fulfilling criterion B
B. Migraine aura fulfilling criteria 1 and 2 for one of the subforms:
Adapted from IHS 2004, with permission.
The signs and symptoms of the aura vary depending on the area of the brain involved. Almost all of individuals experience visual symptoms (99%), with sensory (31%), aphasic (18%), and motor (6%) symptoms being next most common (Russell & Olesen 1996). This is consistent with the discovery that the occipital cortex more excitable in migraineurs (Aurora et al. 1999). It is thought the aura corresponds to a phenomenon known as cortical spreading depression.
Cortical spreading depression
The aura of MWA is thought to be due to the phenomenon known as cortical spreading depression (CSD) (Hadjikhani et al. 2001). In CSD there is a slow travelling wave of neuronal and glial depolarisation in the cortex, which is followed by a long-lasting suppression of neural activity (Buzzi & Moskowitz 2005). It is accompanied by increases in extracellular ions, neurotransmitter such as glutamate, and a temporary increase in cortical blood flow, which is followed by a longer period of decreased flow. This is also associated with an increase in glucose utilisation in the cortex (Shinohara et al. 1979). This paradoxical finding of decreased neuronal activity in the presence of increased glucose utilisation could, in the author’s opinion, be explained by a shift to anaerobic metabolism in the affected neurons.
The blood flow changes can spread across multiple brain regions independent of vascular territories (Hoskins et al. 2006). These changes in blood flow are due to activation of the trigeminovascular system (the meningeal nociceptive system) by CSD, which is sufficient to cause headache and vasodilation (Bolay et al. 2002), triggering the sensitisation described above.
So what causes CSD to occur? Causes include high concentrations of excitatory amino acids and K+ ions, as well as energy failure (Somjen 2001). As mentioned earlier, there is impaired energy functioning in migraineurs, which under stress may fail, triggering CSD. In addition, the aforementioned failure to habituate to repeated sensory stimuli may cause sufficient elevation of excitatory amino acids and K+ to initiate CSD (Dalkara et al. 2006). In turn, the CSD causes the activation of the trigeminovascular system leading to the pain and other symptoms of the migraine headache (Fig. 17.1).
Childhood periodic syndromes
These conditions are more common than is expected, and are most likely underdiagnosed, as primary care providers do not recognise the conditions as being associated with migraine (Al-Twaijri & Shevell 2002). They are generally benign, self-limiting conditions, and hence recognition of the diagnosis can avoid unnecessary testing (Al-Twaijri & Shevell 2002). However they are a common precursor of migraine in adulthood (Cuvellier & Lepine 2010). Hence early identification and treatment of any causative brain dysfunction would be expected to reduce the likelihood of progression to adult migraine.
There is generally an easily uncovered family history of migraine and more classic migraine headaches may coexist, though this may be difficult to elicit in a young child (Al-Twaijri & Shevell 2002; Cuvellier & Lepine 2010). Important clinical features of these attacks are the fact that they are episodic, reversible and stereotypical. Between attacks, the child will be healthy, but will be quite unwell during the attack. There is a large number of possible differential diagnoses that must be excluded before a diagnosis of a periodic syndrome can be made (Cuvellier & Lepine 2010). Many diagnostic possibilities exist for most of the conditions, many of which are beyond the scope of a functional neurologist; therefore, appropriate co-management is required.
Cyclic vomiting
Cyclic vomiting is a syndrome of recurrent and discrete self-limiting episodes of severe nausea and vomiting (Box 17.4). In between episodes, the patient is sign free. There are four stages to the condition. The first is the interepisodic phase. The prodrome phase last on average 1.5 hours and is when the patient first feels the sense of nausea developing. This evolves into the emetic phase when the patient vomits on average 6 times an hour, with an average phase length of 41 hours. It is accompanied by marked autonomic signs. The recovery phase lasts around 6 hours and starts when the nausea ends and ends with the return to normal function. Girls are more affected than boys. The average age at diagnosis is 6; however, it can occur at any age (Cuvellier & Lepine 2010).
 Box 17.4
Box 17.4
Diagnostic criteria for cyclical vomiting
Diagnostic criteria
From IHS 2004, with permission.
1 In particular, history and physical examination do not show signs of gastrointestinal disease.
Current evidence suggests the condition is due to dysregulation of central neural pathways involved in nausea and vomiting. Other pathogenic factors with evidence are autonomic, gastrointestinal, mitochondrial, and central neuroendocrine conditions (Cuvellier & Lepine 2010).
Abdominal migraine
Abdominal migraine has a childhood prevalence of 2.4–4.1%, and is more common in girls. The average age of onset is 7 years old, and it has peak prevalence at 10 years of age. There is usually a family history of migraine. The sufferer is healthy between attacks, but is very unwell during the attack. Weeks or months may separate the attacks (Cuvellier & Lepine 2010).
The symptoms are acute onset, recurring, midline abdominal pain, usually non-colicky, though 22% can be colicky. The attack may be preceded by an aura (Box 17.5). It usually lasts for hours and occurs with flushing, anorexia, pallor, dark shadows under the eyes, and possibly photophobia. Triggering events can include stress, exhaustion, some foods, and motion sickness (Cuvellier & Lepine 2010).
 Box 17.5
Box 17.5
Diagnostic criteria for abdominal migraine
From IHS 2004, with permission.
1 In particular, history and physical examination do not show signs of gastrointestinal or renal disease or such disease has been ruled out by appropriate investigations.
The pathophysiology of abdominal migraine has not been well studied. Proposed hypotheses include autonomic instability, hypothalamo-pituitary-adrenal disturbances, mitochondrial and ion channel disorders (Cuvellier & Lepine 2010).
Non-pharmaceutical treatment options include avoidance of triggers, including foods that the family feels initiate attacks (Cuvellier & Lepine 2010). In the few cases the author has seen, saccadic eye movements, particularly in the planes of movement facilitated by the semicircular canals, have been effective in reducing attacks. This is probably due to the increased frontal cortex and brainstem activation that results, improving the central integrative state of autonomic centres.
Benign paroxysmal vertigo of childhood
Vertigo and dizziness are relatively rare in children, and many of the common adult conditions causing it are rare in children. Posterior fossa tumours are more common in children, and may present with vertigo. Benign paroxysmal vertigo of childhood is considered to be a common cause of vertigo in children (Box 17.6). It is more common in girls and the average age of onset is 2–4 years of age (Cuvellier & Lepine 2010). Forty-three per cent have a family history of migraine, and 13% go on to develop migraine (Ralli et al. 2009).
 Box 17.6
Box 17.6
Diagnostic criteria for benign paroxysmal vertigo of childhood
From IHS 2004, with permission.
1 Often associated with nystagmus or vomiting; unilateral throbbing headache may occur in some attacks.
The vertigo lasts from a few seconds to several minutes, though rarely it may last hours, and has a sudden onset not related to position or movement. Nystagmus may be present during the attack. Audiology and EEG are normal (Ralli et al. 2009).
The pathogenesis is not known. Various authors have suggested peripheral and/or central vestibular dysfunction, but the findings of vestibular testing have not consistently borne this out (Cuvellier & Lepine 2010).
Cluster headache
Cluster headache (CH) is one of the trigeminal autonomic cephalgias, along with paroxysmal hemicrania and short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing (SUNCT). CH is characterised by unilateral attacks of head pain with ipsilateral craniofacial autonomic disturbances (Box 17.7) (Leone & Bussone 2009).
 Box 17.7
Box 17.7
Diagnostic criteria for cluster headache
Diagnostic criteria
From IHS 2004, with permission.
CH is relatively uncommon, with a lifetime incidence of 124 per 100 000 (Leone & Bussone 2009). CH can either be episodic or chronic. The episodic form is described as two or more periods lasting between 7 days and 1 year, with pain-free periods of at least 1 month. The chronic form is described as attacks that occur for more than 1 year or with pain-free periods of less than 1 month. Chronic CH may occur as a primary headache or can evolve from the episodic form (IHS 2004). CH has been found to have a genetic component, with the condition being more common in first- and second-degree relatives. DNA mutations in the mitochondria have also been suspected (Leone & Bussone 2009).
CH can be triggered by alcohol, though usually only during the cluster period, as well as by volatile substances like oil paints and solvents. Sleep and travelling across time zones can also trigger CH (Cohen et al. 2007).
Pathophysiology of cluster headache
There is debate around whether the source of CH is peripheral or central. Several features of the condition suggest a peripheral mechanism, with researchers finding raised venous level of calcitonin gene related peptide, thought to suggest peripheral inflammation. However, this is not unique to CH (Leone & Bussone 2009). Likewise, changes in blood flow due to trigeminovascular activation found in CH were found to be secondary to the pain, rather than the cause of it (Bussone 2008). CH can also be a secondary phenomenon due to intracranial lesions, and removal of the lesion usually removes the headache, and this lends support to the peripheral hypothesis (Leone & Bussone 2009).
Neuroimaging studies of CH patients have found that the ipsilateral posterior hypothalamus is activated in CH (Leone & Bussone 2009). The hypothalamus is primarily concerned with homeostasis and is involved in hormone synthesis, autonomic nervous system regulation, temperature control, behaviour, circadian rhythms, and arousal (Bussone 2008). The circadian nature of CH, the relationship with sleep, and the autonomic symptoms all point to the involvement of the hypothalamus in CH (Holland & Goadsby 2007). It is also involved in nociceptive regulation with pathways connecting both directly with the trigeminocervical nucleus (Leone & Bussone 2009) and indirectly via the periaqueductal grey, the raphae nuclei, and the locus ceruleus (Holland & Goadsby 2007).
It has been suggested that the hypothalamus is not the trigger of the attack but that it balances the various factors that allow for the development of the attack, and that dysfunction in it may allow for the factors to be out of balance (Holland & Goadsby 2007). Hypothalamic stimulation has been shown to be effective for CH. However, it takes weeks to months to have an effect, and it has no effect in the acute period. It has been suggested that its mechanism of action may not be through the activation of the hypothalamus itself, but through the activation of its connections, namely the pain matrix areas. The long-term stimulation may raise the levels of the metabolism in the hypometabolic areas described below (Leone & Bussone 2009). This is consistent with the functional neurology model.
Neuroimaging has also shown other parts of the pain matrix including the anterior cingulate, contralateral posterior thalamus, ipsilateral basal ganglia, bilateral insulae, and cerebellar hemispheres are activated in CH attacks (Leone & Bussone 2009). A PET study found decreased glucose utilisation (hypometabolism) in the prefrontal, orbitofrontal, and perigenual anterior cingulate cortices both during the headache period and out of it, and hypometabolism in the cerebellopontine area during the headache period. It has been suggested that there is a deficiency of top-down modulation of nociceptive circuits in CH sufferers, which leads to inadequate control of trigger mechanisms, thereby promoting the headache periods (Sprenger et al. 2007).
Treatment of cluster headache
There are several pharmaceutical treatments for CH. Subcutaneous sumatriptan has a rapid effect and high success rate during an attack (Cohen et al. 2007).
For the functional neurologist, inhalation of 100% oxygen at 7–12 L/min via a nonrebreathing face mask is rapidly effective for most sufferers in the acute attack (Cohen et al. 2007). Oxygen has been shown to have a direct effect on the inhibitory projections from the brainstem to the central pain pathway (Bussone 2008).
Interestingly greater occipital nerve block has been shown to be effective in aborting a period of CH (Cohen et al. 2007). This presumably occurs due to decreased nociceptive drive into the trigeminocervical nucleus, which may suggest that manual cervical spine therapies may also be effective.
Tension-type headache
Tension-type headache (TTH) is one of the most common headache disorders, with an average lifetime prevalence of 46%. Children may be affected, but the peak incidence is 40–49 years of age. Females are slightly more affected than males, and become more so as chronicity increases (Loder & Rizzoli 2008).
The pain of TTH is usually mild to moderate, pressure or muscle type pain lasting hours to days with no neurological or constitutional symptoms. It is usually bilateral and may extend into the neck (Box 17.8). Patients often only begin to seek care as the severity and frequency of the headache increases (Loder & Rizzoli 2008).
 Box 17.8
Box 17.8
Diagnostic criteria for tension-type headache
Diagnostic criteria
A. One of the following, and fulfilling criteria B–D
B. Headache lasting from 30 minutes to 7 days
C. Headache has at least two of the following characteristics:
Adapted from IHS 2004, with permission.
1 History and physical and neurological examinations do not suggest any of the disorders listed in groups 5–12 of the classification, or history and/or physical and/or neurological examinations do suggest such disorder but it is ruled out by appropriate investigations, or such disorder is present but headache does not occur for the first time in close temporal relation to the disorder.
Pathophysiology of tension-type headache
The pathophysiology of TTH has still not been fully described but the involvement of peripheral and central mechanisms has been recognised (Speciali et al. 2008). It is thought likely that the infrequent form of TTH is primarily due to peripheral mechanisms, with central mechanisms coming to bear in the frequent and chronic forms of the condition (Bendtsen & Jensen 2009).
The main model for TTH is that tender/trigger points in peripheral muscles produce nociceptive activation, which in time will cause central sensitisation. Trigger points are hyperirritable spots within a tight band of muscle that are painful to compression, and produce referred pain in a site distant from the spot (Simons et al. 1999). Trigger points can arise from a variety of causes including psychological stress and biomechanical dysfunction, and it has been shown that trigger points can cause peripheral sensitisation of muscle nociceptors (Fernandez-de-las-Penas et al. 2007). Furthermore, trigger points and postural changes are more common in headache sufferers (Marcus et al. 1999). Hence it is likely that, at least in infrequent TTH, myofascial factors are the likely cause of pain.
In chronic forms of TTH, three aspects may come into play. Sensitisation of second-order neurons in the trigeminocervical nucleus, sensitisation of suprasegmental neurons, and decreased activity in anti-nociceptive systems could all contribute to myofascial pain and tenderness (Bendtsen 2000). It is possible that the persistent peripheral sensitisation caused by trigger points could lead to central sensitisation occurring (Fernandez-de-las-Penas et al. 2007), as research has shown that persistent peripheral nociceptive input leads to input in the trigeminocervical ganglion (Goadsby 2005).
Studies of sensitivity to stimuli (pressure, thermal, and electrical) have found that chronic TTH sufferers have lowered thresholds, not only in the cranium, but in the periphery as well. The generalised nature of these changes indicate that the sensitisation is central rather than peripheral (Bendtsen 2000).
Prolonged activation of second-order nociceptive neurons can cause the activation of NMDA receptors. Once activated, the magnesium plug is removed from the receptor, and there is a calcium influx into the cell. This initiates a process of cellular events that can lead to long-term changes and lowered threshold to activation in the neuron. As the excitability of the neuron increases, it may become sensitive to mechanical stimuli, rather than simply nociceptive, which manifests clinically as allodynia (Bendtsen 2000).
Failure of descending pain inhibitory mechanisms will also contribute to central sensitisation. Structures such as the periaqueductal grey, the raphae nuclei, and the locus ceruleus act to regulate nociceptive transmission. Decreased output from these may in turn bring the lower-order neurons closer to threshold, making subsequent excitation and plasticity easier to achieve. Studies have shown cerebellar/brainstem dysfunction and changes in TTH sufferers including deviation of subjective visual vertical (a vestibular/otolithic sign) (Asai et al. 2009), increased sway in stabilometry (Ishizaki et al. 2002), and an increased reliance on vestibular inputs in posture control (So & Bent 2009).
In addition, TTH sufferers show the same volumetric brain changes seen in many chronic pain conditions. These changes are in the regions of the brain known to be involved in pain processing, namely the cingulate cortex, insula, and the orbitofrontal cortex and parahippocampus bilaterally. These changes are also seen in conditions such as irritable bowel syndrome, chronic back pain, and fibromyalgia. The changes correspond with the duration of chronic pain, so it is likely they are the consequence, rather than the cause of the chronic pain (May 2009).
Treatment of tension-type headache
Treatment of infrequent TTH should focus on removal of pain generators in the periphery and the removal of any triggering factors such as psychological stress, postural aberrancies, etc. Pain generators may include myofascial tenderness and trigger points. Ischaemic compression-type trigger point therapy has been found to be effective in treating trigger points (Simons et al. 1999). Some studies have found cervical spine manipulation to be effective in treating TTH, but this is yet to be replicated in a randomised, controlled trial. Other therapies such as acupuncture, ultrasound, and stretching may also be effective.
In the frequent and chronic forms of TTH, it is certainly reasonable to remove the peripheral pain generators, as they may be contributing to central sensitisation. Nutritional therapies such as magnesium supplementation to decrease NMDA receptor activation, and omega 3 fatty acids to minimise peripheral inflammation should also be considered.
Conclusion
 Clinical case answers
Clinical case answersCase 17.1
References
Akdal G., Dönmez B. Balance in migraineurs. Neurology. 2007;68(12 supp 1):sA89.
Bendtsen L., Jensen R. Tension-type headache. Neurol. Clin.. 2009;27(2):525-535.
Cuvellier J.C., Lepine A. Childhood periodic syndromes. Pediatr. Neurol.. 2010;42(1):1-11.
Goadsby P.J. Migraine pathophysiology. Headache. 2005;45(Suppl 1):S14-S24.
Loder E., Rizzoli P. Tension-type headache. BMJ. 2008;336(7635):88-92.
Lodi R., Tonon C. Energy metabolism in migraine. Neurol. Sci.. 2006;27(Suppl 2):S82-S85.
Speciali J.G., Eckeli A.L. Tension-type headache. Expert Rev. Neurother.. 2008;8(5):839-853.


