[level-membership-for-critical-care-medicine-category]66
Head Injury
Introduction
It also has been demonstrated that the management of serious brain trauma requires specialty teams consisting of experienced neurosurgeons, traumatologists, and intensivists (specifically, neurointensivists) working together in a dedicated unit equipped and staffed to optimally care for these patients. In the absence of such facilities, outcomes suffer.1–3
Incidence
The Centers for Disease Control and Prevention report the number of patients sustaining a traumatic brain injury (TBI) to be at least 1.7 million per year in the United States. Approximately 80% of patients are released from the emergency department, 275,000 are admitted, and 52,000 die. Nearly a third of all traumatic deaths in the U.S. involve a head injury. The ages most likely to sustain a TBI are children up to 4 years old and adults over 64, with adults greater than 74 years old having the highest rates of hospitalization and death associated with head injury.4 Teenagers and young adults continue to be at high risk for sustaining TBI, but the range has decreased over the past 20 years from between 15 and 24 to between 15 and 194,5. The incidence of head injury has been shown to be inversely proportional to economic status,6 and has remained up to three times more common in males in every age group over the past several decades.4,7,8 In the United States, falls are the most common cause of TBI and are responsible for half of the head-injured children up to 14 years of age and 60% of the adults over the age of 64.4 Although the frequency of head injury related to motor vehicle crashes has decreased over the years to a little less than 20%, these crashes are the second leading cause of head injury in all age groups and result in the largest percentage of TBI-related deaths.4,9 The incidence of TBI worldwide is difficult to quantify but is estimated to be at least 10 million people a year, with traffic accidents accounting for more than half, and falls being the second leading cause;10 this is the opposite distribution seen in the United States4. Violence is estimated to cause about 10% of head injuries throughout the world,10 as well as in the U.S.4, but there is significant variability in the specific mechanisms. For example, between 1997 and 2007 in the U.S., firearms were related to just over a third of all lethal TBIs, compared to a single hospital in South Africa that reported its experience with more than 300 stab wounds to the brain.11
Diagnostic Approach
The presenting neurologic condition of patients with head injuries is the primary factor in determining the initial management and prognosis. When a detailed history is unavailable, it is important to keep in mind that loss of consciousness may have preceded and in fact caused the traumatic event, such as aneurysmal subarachnoid hemorrhage (SAH), hypoglycemia, intoxication, or syncope. Level of consciousness is one of the most important neurologic considerations in managing a head-injured patient. Neurosurgical patients generally have an alteration in level of consciousness from either brainstem or bilateral cerebral hemispheric involvement. This may be secondary to poor perfusion due to high ICP or brainstem compression, both of which require neurosurgical intervention. Therefore, an accurate tool to measure level of consciousness is essential. Many clinical assessment tools are available for use in the critical care setting.12–24
Glasgow Coma Scale
Ideally, using the GCS, a nurse, medical student, paramedic, physician assistant, intensivist, trauma surgeon, emergency medicine physician, neurologist, and neurosurgeon all should obtain the same score when assessing a patient. The GCS system is not perfect, but it has proved itself to be, overall, a practical, straightforward grading system that can be used by health professionals in various fields and at different levels to produce reliable results.25–30 The GCS has become the most widely used assessment tool and is considered the gold standard for the evaluation of patients with head injuries.12
The GCS is a measure of level of consciousness and does not take into account focal deficits. It is based on eye opening (1-4), verbal response (1-5), and motor response (1-6) (Table 66.1). A patient with a normal level of consciousness should have the highest possible score of 15. The lowest score possible is 3, not 0 as might be expected. An intubated patient technically gets a 1 for verbal response and is assigned 1T (for Tube) for the verbal score. It is important to identify the score for each observed variable tested. For example, a typical score for an intubated patient with decorticate posturing probably would be E1 + V1T + M3 = 5T (where E denotes eye opening, V is verbal response, and M is motor response). It also is important to realize the shortcoming of this system in evaluating patients with dementia or aphasia. A point to keep in mind is that the GCS attempts to put a numeric value on level of consciousness, with 15 being representative of normal. If a demented patient from a nursing home presents with a normal level of consciousness after a fall but doesn’t know what day it is, the GCS would be calculated as E4 + V4 + M6 = 14. A GCS score of 14 describes a patient with an abnormal level of consciousness but fails to accurately communicate this particular patient’s condition. Likewise, clinicians must use caution when applying the GCS to patients with aphasia. Conversely, patients with profound focal deficits and a normal level of consciousness should have a GCS score of 15. Another problematic case is that of a quadriplegic patient with a normal level of consciousness who is able to blink the eyes or stick out the tongue upon command, thereby giving a top score of 6 on the motor assessment.
Table 66.1
| Component | Score |
| Eye Opening | |
| Spontaneously | 4 |
| To voice | 3 |
| To pain | 2 |
| No response | 1 |
| Verbal | |
| Oriented | 5 |
| Disoriented | 4 |
| Inappropriate | 3 |
| Incomprehensible sounds | 2 |
| No response | 1 |
| Motor | |
| Following commands | 6 |
| Localizing to pain | 5 |
| Withdrawing to pain | 4 |
| Abnormal flexion | 3 |
| Abnormal extension | 2 |
| No response | 1 |
The GCS has been used to predict outcome.31 The motor score itself has predictive value as well.32 The GCS score should be calculated after hemodynamic and pulmonary resuscitation and without sedatives or muscle relaxants.33
Aggressive implementation of early sedation and intubation in severely head-injured patients compromises the ability to determine an accurate GCS score.34
Computed Tomography Classification: Marshall Classification
A useful classification of brain injuries by findings on computed tomography (CT) has been devised by Marshall and colleagues. This CT classification, presented in Table 66.2, can serve as a guide in describing scans and has strong predictive power.34,35
Table 66.2
Marshall Computed Tomography Classification
| Category | Definition |
| Diffuse injury I (no visible pathology) | No visible intracranial pathology seen on computed tomography scan |
| Diffuse injury II | Cisterns are present with midline shift of 0-5 mm or lesion densities present no high- or mixed-density lesion > 25 cm3 may include bone fragments and foreign bodies |
| Diffuse injury III (swelling) | Cisterns compressed or absent with midline shift of 0-5 mm; no high- or mixed-density lesion > 25 cm3 |
| Diffuse injury IV (shift) | Midline shift > 5 mm; no high or mixed-density lesion > 25 cm3 |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High- or mixed-density lesion > 25 cm3; not surgically evacuated |
From Marshall LF, Marshall SB, Klauber MR, et al: A new classification of head injury based on computerized tomography. J Neurosurg 1991;75:S14-S20.
Maas and associates have proposed a scoring system with better predictive power (Table 66.3), with a sum total adjusted to be consistent with the GCS.34 Table 66.4 presents a mortality prediction chart based on this classification.
Table 66.3
| Predictor | Score |
| Basal Cisterns | |
| Normal | 0 |
| Compressed | 1 |
| Absent | 2 |
| Midline Shift | |
| No shift or shift < 5 mm | 0 |
| Shift > 5 mm | 1 |
| Epidural Mass Lesion | |
| Present | 0 |
| Absent | 1 |
| Intraventricular Blood or tSAH | |
| Absent | 0 |
| Present | 1 |
| Sum Score* | +1 |
*The sum score can be used to obtain the predicted probability of mortality (see Table 66.4). The authors chose to add plus 1 to make the grading numerically consistent with the grading of the motor score of the GCS and with the Marshall CT classification. tSAH, traumatic subarachnoid hemorrhage.

This scoring system is best used as one piece of data in the overall clinical assessment in considering a patient’s prognosis.36
Prediction of Outcomes
Severe brain injury outcomes can be accurately predicted using four variables: age, GCS score on admission (especially motor score), CT characteristics, and presence of ischemic and hemodynamic secondary insults. Using these four predictors, numerous investigators have used statistical modeling techniques to predict up to 80% or more of outcomes.37
Head injuries generally are classified as mild (GCS score of 13 to 15), moderate (GCS score of 9 to 12), or severe,3–8 although the evidence may be sufficient to include a GCS score of 13 in the moderate category.38
CT scanning is the diagnostic imaging study of choice in trauma. Plain x-ray films of the skull are rarely indicated as a screening study for head-injured patients. Outcomes with the strategy of obtaining CT scans in all head-injured patients are superior to those with other management strategies. The incidence of missed surgical lesions in mildly head-injured patients by CT scanning is 0.028%.39–42
Primary Head Injury
Primary head injury can be defined as the damage that occurs at the moment of impact and can take the form of skull fractures, surface contusions and lacerations, diffuse axonal injury, or diffuse vascular injury.42 Some authors include the contusions and hematomas that form as a direct and immediate effect of the impact as part of the primary injury as well.43 These hemorrhages may manifest with significant mass effect requiring surgical intervention.
Skull Fractures
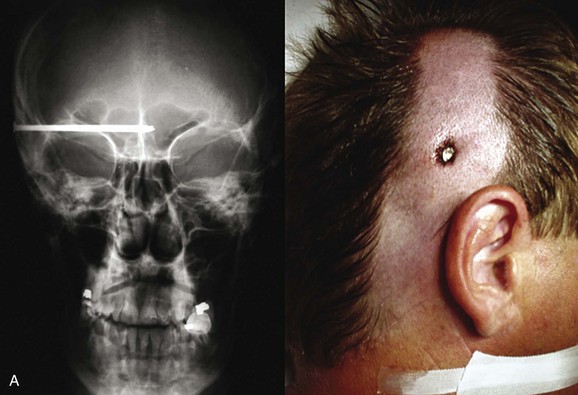
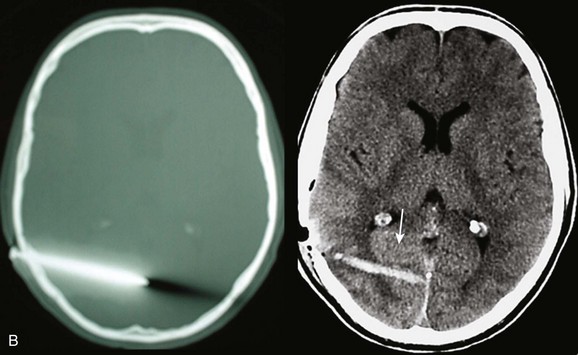
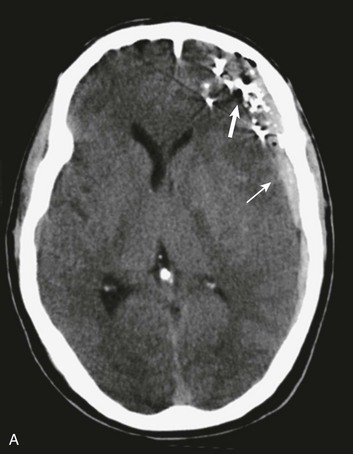
Basilar skull fractures typically are linear and involve the anterior cranial base and the petrous part of the temporal bone. These fractures have the potential for an associated dural laceration adjacent to potentially contaminated paranasal sinuses, or the external ear canal if the tympanic membrane is disrupted. This allows the potential for a cerebrospinal fluid (CSF) fistula to develop, as well as meningitis. Clinical signs of a fracture of the petrous portion of the temporal bone include hemotympanum with or without tympanic membrane disruption, hearing loss, CSF otorrhea, and Battle sign. The cranial nerves that course through the temporal bone include the facial, acoustic, and vestibular nerves; therefore, associated vestibular dysfunction or facial weakness may be noted (Fig. 66.1). Anterior cranial base fractures may be associated with “raccoon eyes,” anosmia, and CSF rhinorrhea. It is important to remember that when a fracture of the floor of the anterior cranial base is suspected, a nasogastric tube should not be inserted, because of the risk of intracranial penetration (Fig. 66.2).
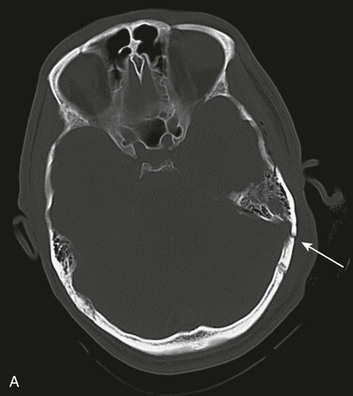
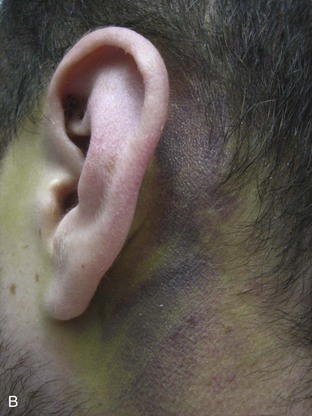
The use of prophylactic antibiotics in basilar skull fractures has been debated but generally is not recommended. When CSF leak does occur, the break in the tissue usually will heal with conservative treatment over the course of a week, unless the defect is very large or a bony spicule is identified. Keeping the patient’s head elevated and possibly using external CSF diversion after a few days are reasonable nonoperative methods; however, if the leak persists, surgical repair is indicated.44
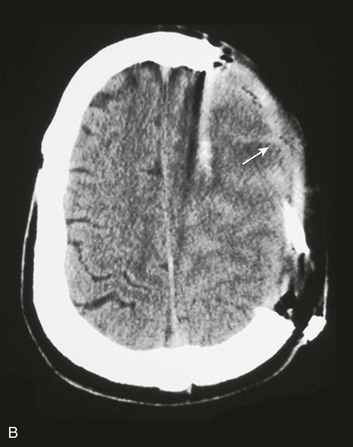
Most depressed skull fractures occur in men younger than 30 years of age.45 Despite very little literature to support any particular management strategy, closed depressed skull fractures generally are operated on if the extent of depression is greater than the full thickness of the adjacent skull, with the theoretical benefits of improved cosmetic result, a decrease in late-onset posttraumatic epilepsy, and a reduction in the incidence of persistent neurologic deficit.46 Most depressed skull fractures, however, are open45 (Fig. 66.3), meaning that a scalp laceration with galeal disruption overlying the fracture is present.
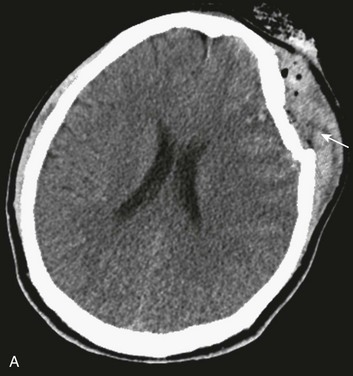
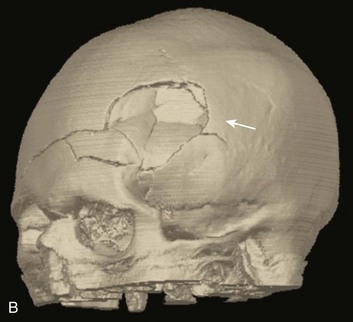
Open depressed skull fractures may be associated with significant morbidity and mortality.45,47 Infection rates are reported to be between 1.9% and 10.6%,46–50 with neurologic morbidity and mortality rates of approximately 11% each46 and an incidence of late epilepsy up to 15%.51
By convention, open depressed cranial vault fractures are treated surgically, with debridement and elevation, primarily to attempt to decrease the incidence of infection. Open depressed cranial fractures may be treated nonoperatively if clinical and radiographic examination reveals no evidence of dural penetration, significant intracranial hematoma, depression greater than 1 cm, frontal sinus involvement, gross cosmetic deformity, wound infection, pneumocephalus, or gross wound contamination.46
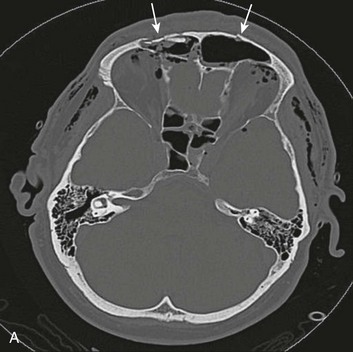
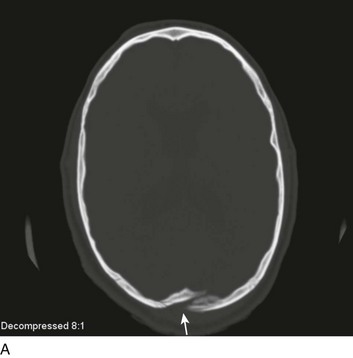
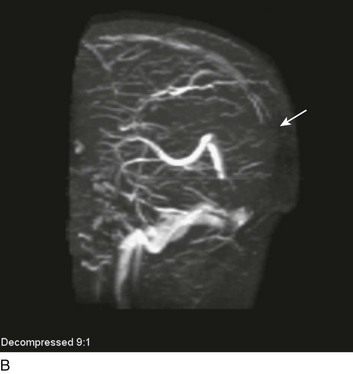
A special type of depressed fracture is fracture of the frontal sinus. Depression of the fracture may require surgery to prevent CNS infection and CSF leak (Fig. 66.4). Another special type of depressed fracture involves a major dural venous sinus (see Fig. 66.4). Under these circumstances, the risks associated with surgery are increased, and elevation of the fracture fragment generally is reserved for significant compromise of venous drainage.44
Epidural Hematoma
The next anatomic layer after the skull is the dura, which is the periosteum of the inner table of the skull and, as such, is tightly adherent to the bone. The potential space between the inner table of the skull and the dura is the epidural space. Epidural hematomas (EDHs) form in this potential space between the skull and the dura. The most common mechanism for the development of an EDH is a motor vehicle crash (in 53% of the cases), followed by falls (in 30%) and assault (in 8%).52–60 Typically, bleeding results from damage to the middle meningeal artery, but an EDH also can occur from injury to the middle meningeal vein, diploic veins, or the venous sinuses.60
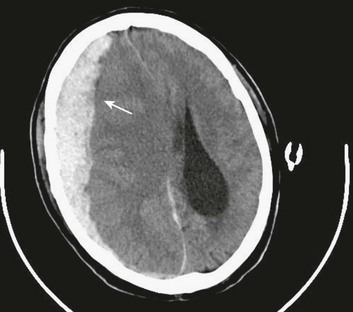
EDHs generally occur in the temporal and temporoparietal regions (Fig. 66.5).52,57,61–64 A useful way to describe the development of an EDH is to follow the events that take place when a patient presents with an initial lucid interval following brief loss of consciousness after head trauma. A lucid interval occurs when a patient initially is rendered unconscious from a concussive head injury that causes a linear skull fracture involving the middle meningeal artery or one of its branches. The middle meningeal artery is a dural vessel that runs half in the dura and half through the groove in the inner table of the skull. When a fracture extends across the bony groove of the artery, it will tear and bleed. Because the dura is tightly adherent to the inner table of the skull, significant force is needed to push the dura off the inner table. An arterial hemorrhage generally has enough pressure to strip the dura off the bone, converting the potential epidural space into a mass. The bony attachment of the dura becomes progressively stronger with age; therefore, the dura of a younger patient requires less force to push it off the bone than would be required in an older patient. Not surprisingly, the mean age of patients with EDH is between 20 and 30 years of age,53–55,63–70 and EDHs are unusual in patients older than 50.60
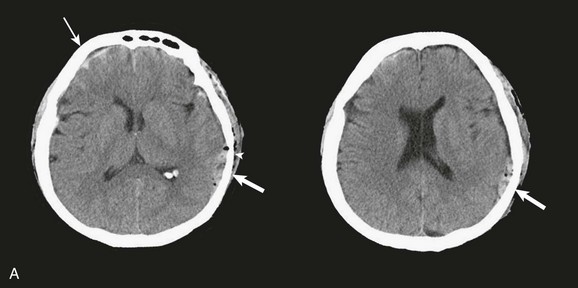
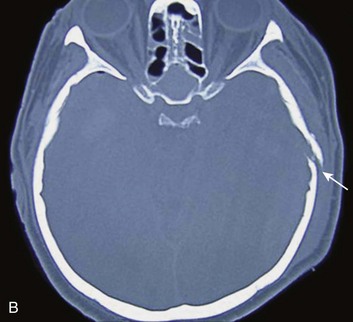
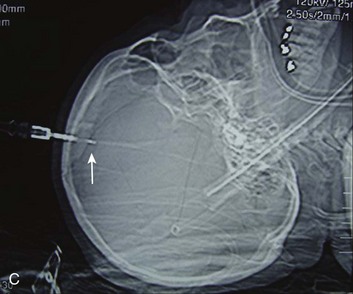
Taking a general view, the patient goes from unconscious at the time of impact from a concussive injury, to awake, to unconscious again secondary to brainstem compression—hence the term lucid interval. The lucid interval is observed in close to half of the patients undergoing surgery for EDH.*
Pupillary abnormalities occur in approximately 20% to 30% of patients with surgical EDH.52,54,59,60,67 Cranial fractures are present in between 70% and 95% of the cases.54,60,68,70,74,75 Associated intracranial lesions are found in 30% to 50% of adults with surgical EDHs,† and subdural or parenchymal lesions in association with EDH lower the chance of a good outcome.60
The overall mortality rate (for all ages and GCS scores) is approximately 10%.‡ The time lapse between the onset of pupillary abnormalities and surgery determines outcome,61,66,80 but the single most important predictor of outcome in patients operated on for EDH is the GCS score on admission and before surgery.52,53,55,61,81–83
Not all EDHs, however, require surgery. No prospective randomized trials have been conducted to compare surgical treatment with nonoperative management, nor should there be. Available data describe nonsurgical management in selected cases. In one study, approximately 10% of the total number of EDHs were treated nonoperatively. All were conscious with a GCS score greater than 11 and a midline shift on CT scan of less than 10 mm. Not one of these nonoperative hematomas was in the temporal region.84
Another study reported findings in a group of 57 selected patients treated nonoperatively with an initial GCS score of 10 or higher, with maximum hematoma thickness less than 13 mm, with five clots located in the temporal region, but only one patient had a midline shift on CT.53
An interesting approach for dealing with thin, acute EDHs in an early stage has been reported in a small, isolated series of patients using endovascular techniques to occlude the middle meningeal artery.85 Surgical decision making for an acute EDH is based on GCS score, pupillary findings, comorbid conditions, CT findings, age, and, in delayed decisions, ICP. Guidelines for the surgical management of acute EDH published in 2006 recommend surgical evacuation of an EDH less than 30 mL in volume. Smaller hematomas in patients with a GCS score greater than 8 and without focal deficits, clot thickness less than 15 mm, and less than 5 mm of midline shift may be considered for nonoperative management. Close neurologic monitoring and serial CT scanning are essential. Of importance, a temporal location for an EDH is associated with failure of nonoperative management and should lower the threshold for surgery.60
Because time between neurologic deterioration and surgery is critical, the question of whether a patient with an acute EDH should receive treatment at the nearest hospital or should be transferred to a trauma center is important. The issue is whether or not a non-neurosurgeon should operate on a patient deteriorating from an acute EDH as a true emergency. One suboptimally controlled study reported worse outcomes in a small group of patients who underwent emergency operations by non-neurosurgeons and attributed this mainly to the technical inadequacy of the operation.59,60 Other studies have documented worse outcomes in patients transferred from outlying hospitals for surgery.52,73 The take-home message is that EDHs are extra-axial hemorrhages located adjacent to the brainstem that can represent a true neurosurgical emergency, and that rapid, competent decompression makes a difference.
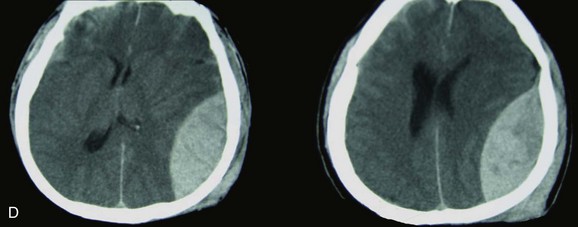
Subdural Hematoma
Proceeding deep to the skull, the next space is the subdural space. Unlike the potential epidural space, the subdural space is a real space. It is a compartment that follows the contour of the brain, which is how it appears on a CT scan (Fig. 66.6). Anatomically, bridging veins course through the subdural space from the cerebral hemispheres to the superior sagittal sinus. As the brain accelerates within the skull after impact, these veins can stretch and tear.86
The source of bleeding also can be from a cortical artery87,88 or vein. These hematomas typically form over the surface or between the cerebral hemispheres, along the tentorium, or between the temporal lobe and base of the skull.
An acute subdural hematoma (SDH), as defined by Bullock and colleagues, appears within 14 days of injury,60 although some authors consider an SDH to be subacute when signs and symptoms develop between 3 and 20 days after trauma.89
Acute SDHs consist of clotted blood, which is fibrous and often adherent to adjacent tissue. The blood remains clotted for several days. After this time, the clot gradually and progressively lyses, resulting in a mixture of clot and fluid. After several weeks, the clot is liquefied and becomes a chronic SDH. Chronic SDHs may manifest weeks or months after what may have been mild or insignificant trauma. Chronic SDHs occur more often in elderly patients and individuals who have more intracranial space because of cortical atrophy. A membrane often surrounds these hematomas, and the collection of fluid may slowly grow in size because of repeated small bleeds or accumulation of fluid transudate from the membrane.86
The incidence of acute SDHs is approximately 20% in severe traumatic brain injury,60,90–93 and the mean age is between 31 and 47 years, with most of the patients being men.60,94–97 The mechanism of injury differs between age groups, with patients older than 65 more commonly presenting after a fall, and younger patients being involved more often in a motor vehicle crash,60,98–100 which, among comatose patients with acute SDH, is the most common mechanism of injury.60,91,93,101
Between 37% and 80% of patients with acute SDH present with a GCS score of 8 or lower,60,65,71,94,97,102 and the overall mortality rate is between 40% and 60%.* Mortality rates among comatose patients requiring surgery are somewhat higher, with reported rates between 57% and 68%.† Acute subdural hematomas frequently are associated with other brain injuries,97,102 which is one of the reasons why the mortality rate is relatively high.
A simple acute SDH is a subdural, extra-axial hematoma without any other associated brain injury (Fig. 66.7). A complex SDH is associated with parenchymal injury (Fig. 66.8).107,108 Fewer than half of acute SDHs requiring surgery are isolated, simple lesions.60,97,102
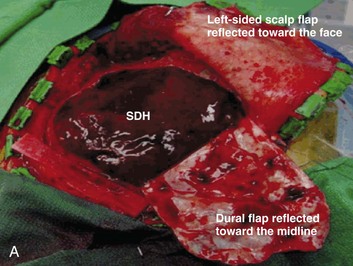
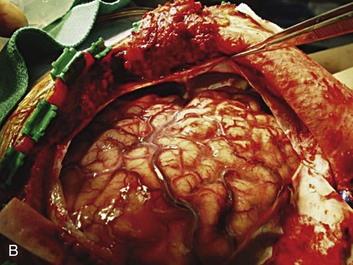
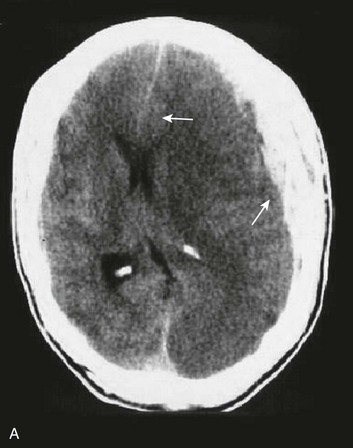
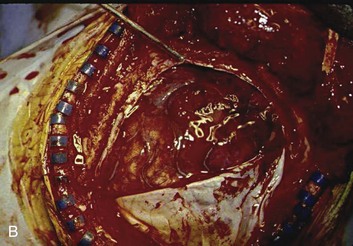
Figure 66.8 A, Computed tomography scan of acute, complex left-sided subdural hematoma (SDH) (lower arrow) with mass effect (upper arrow) out of proportion to the size of the clot, indicating more than just the effect of an extra-axial compressive mass—that is, intrinsic, parenchymal brain injury with edema. B, Intraoperative photograph of decompressed brain reveals hemorrhagic, inflamed, and edematous brain swelling out through the cranial defect. Compare with the intraoperative photograph of the simple SDH in Figure 66.7.
One useful method to identify the presence of associated injury is to measure the thickness of the subdural clot relative to the amount of brain shift on the CT scan. If the amount of shift is directly proportional to the thickness of the extra-axial clot, the injury is likely to be simple and only the brain compressed. However, if the amount of shift is more than expected as indicated by the size of the hematoma, then additional parenchymal brain injury probably is present under the clot, causing additive mass effect. Not surprisingly, the mortality for complex injuries is higher than that for simple SDHs.107,108
In addition to the obvious compressive effects that an SDH generates, the available evidence points to a direct toxic effect of the blood itself on the underlying cortex, thereby compounding the problem.109,110
Age is an important factor in acute SDHs. There is a significant increase in poor outcome among patients older than 60 years of age with severe head injury in general.93,98–101,111,112 Older patients with an acute SDH and a low GCS score do especially poorly.98–101,111,112
The decision for immediate surgery is dependent on GCS score, age, pupillary examination, comorbidities, CT findings, and salvageability with respect to the patient’s level of injury. In addition to all of these factors, decision making for delayed surgery is dependent on clinical course and ICP.60
On the basis of a contemporary search of the literature, Bullock and colleagues found that CT parameters of a midline shift greater than 5 mm and a clot thickness greater than 10 mm were independent factors requiring surgery in salvageable patients. They also identified a select group of comatose patients with smaller SDHs who could be managed nonoperatively if the patients remained neurologically stable with normal pupils and ICP of 20 mm or less.60
Traumatic Subarachnoid Hemorrhage
The subarachnoid space is a CSF-filled compartment within which are the major cerebral blood vessels. The CSF within the subarachnoid space fills the basal cisterns and interdigitates into the cortical sulci. Traumatic SAH can be caused by bleeding of cortical arteries, veins, or brain surface cerebral contusions.113
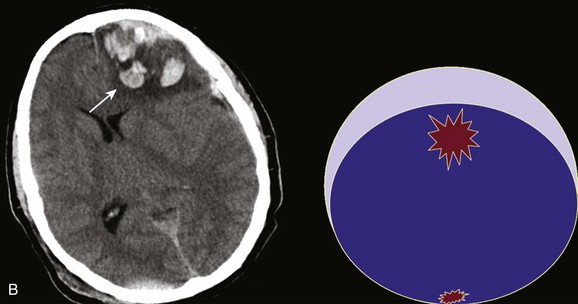
In contrast with hemorrhages in other locations, SAH is not a discrete surgical clot that requires evacuation. Trauma is the most common cause of SAH; a ruptured aneurysm is the most common cause of a spontaneous SAH. Aneurysmal SAH generally involves the suprasellar cistern, where the circle of Willis lies. Traumatic SAH can be found as small-volume hemorrhages in the sylvian fissures and especially in the interpeduncular cistern. Traumatic SAH also commonly involves the cerebral convexity and can fill cortical sulci, which can sometimes mimic sulcal effacement114 (Fig. 66.9A).
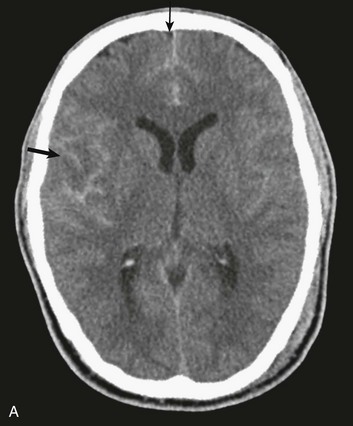
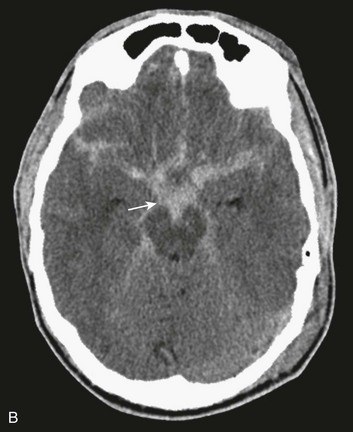
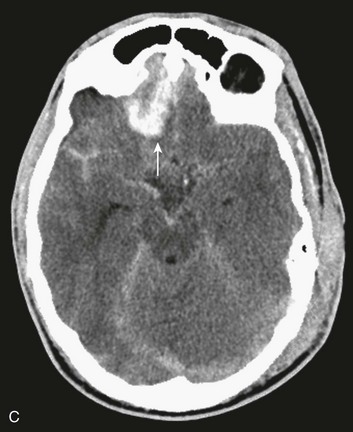
Occasionally, the distribution of the hemorrhage may be hard to differentiate from an aneurysmal hemorrhage. Under this circumstance, a vascular study should be obtained (Fig. 66.9B). Cerebral vasospasm following aneurysmal SAH is a common, well-described problem with an unclear pathogenesis. Increasing evidence suggests that SAH from trauma also may cause clinically significant cerebral vasospasm, which may be responsible for ischemia and infarction.115,116
Clearly the incidence of clinically relevant cerebral vasospasm is less than what is seen in aneurysmal SAH; however, posttraumatic SAH may cause ischemia during the acute as well as the delayed phase.115,117
In severe nonpenetrating head injury, the degree of SAH can be predictive of outcome,118,119 and although traumatic SAH can be an isolated finding, it is commonly associated with other intracranial injuries.120
Traumatic SAH can be a marker of severe primary injury and the amount of blood can also be an independent predictor of the development and progression of intraparenchymal contusions113 (Fig. 66.9C).
Intraparenchymal Contusions and Hematomas
Traumatic parenchymal mass lesions are common and are reported in 13% to 35% of severe traumatic brain injury.121–127 Contusions consist of heterogeneous areas of necrosis, pulping, infarction, hemorrhage, and edema.89,128,129 Hemorrhagic contusions are mixtures of blood and edematous cerebral parenchyma that also have a heterogeneous appearance on CT.89 Contusions commonly occur in the frontal and temporal lobes both at the poles and on the inferior surfaces as a result of contact with the rough, bony skull base130 (Fig. 66.10A).
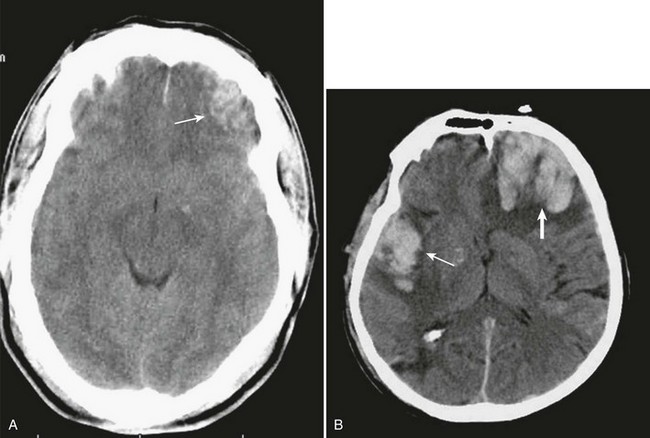
Contusions typically involve the crests of the gyri but in more severe injury may extend into the substance of the white matter. If the pia-arachnoid is torn, contusions are classified as cortical lacerations.130–132
Contusions represent one end of a spectrum of injury on which hematomas, which are well-defined, homogeneous collections of blood, are the other end (Fig. 66.10B). The amount of energy delivered may have only been enough to cause failure of small vessels, resulting in contusions. If more energy is delivered, failure of larger vessels may occur, resulting in hematomas. Alternatively, the hemorrhagic component of a contusion may continue to bleed and coalesce into a more discrete hematoma. In general, the most common traumatic parenchymal lesions are contusions,89 and they tend to evolve.122,126,127,133,134
Risk factors for the progression of intraparenchymal hemorrhage include the presence of SAH (see Fig. 66.9C), and subdural hematoma, as well as the size of the clot on the initial CT scan. Enlargement of contusions occurs approximately 40% of the time, which justifies early follow-up CT scanning.135 In addition to the problem of progressive enlargement of existing contusions is a phenomenon of delayed appearance of traumatic intracerebral contusions (DITCH). DITCH is defined as a new contusion identified on a CT scan in an area of brain that was normal on the admission CT scan. These delayed hemorrhages are reported to occur in up to 7% of patients with severe head injuries.90
Contusions can be subdivided into two groups. Coup contusions occur in the brain tissue under the impact site and usually are associated with an acceleration injury. Contrecoup contusions are located away from the point of impact and usually are associated with a deceleration injury.130,131,136–138
Understanding the biomechanics of how a contrecoup contusion develops is useful. In general, biologic tissues tolerate strain better if they are deformed slowly rather than quickly. For example, a 150-mL hematoma with accumulation of blood over minutes is likely to be lethal, whereas a 150-mL slow-growing meningioma may have no appreciable effect on the patient’s level of consciousness. In trauma, the cause of tissue damage may be any of three types of induced strains: compression, tension, and shear. The skull, brain, and blood vessels will tolerate compression better than tension, and tension better than shear. These strains are induced by contact or inertia (relative to acceleration-deceleration), or both. Contact injuries are a result of impact, which may cause inward deformation of the skull with local effects, and of shock waves, which can produce remote effects. As a consequence of contact the head is set in motion, which leads to inertial injury. Inertial injuries may cause damage by differential acceleration of the skull and brain. In addition, acceleration-deceleration can independently produce strains in the brain itself. The two clinically relevant types of acceleration are translational (when the brain moves in a straight line) and angular. Most injuries are a combination of both.139
These effects are best illustrated by a case example of a 37-year-old man who falls from a height onto the back of his head and presents with a deteriorating level of consciousness. His CT scan (Fig. 66.11) reveals soft tissue swelling at the point of impact in the right occipital region with a small underlying hemorrhage, as well as a large, hemorrhagic contusion in the left frontal lobe. The mechanism involved is primarily translational (linear) deceleration. The skull stops suddenly as it hits the ground, but the brain continues to move toward the impact site, where compression is induced (Fig. 66.12).
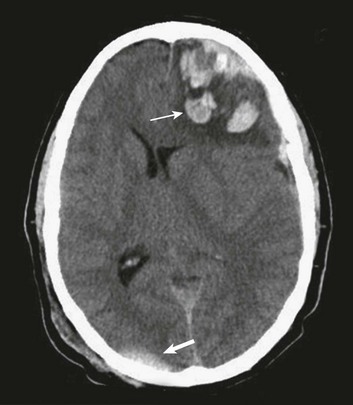
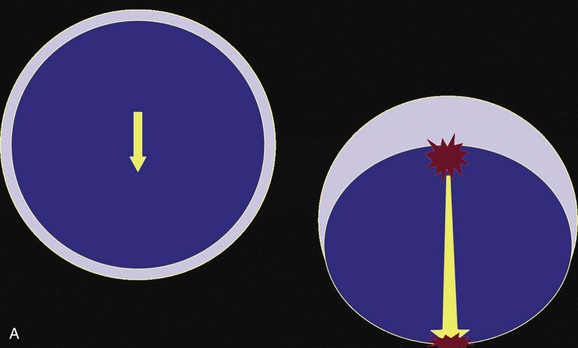
As the brain is moving toward the inner table of the skull at the impact site, it also is moving away from the skull on the opposite side, creating regions of low pressure and tensile strains. Contributing to the extensive tissue damage in the left frontal lobe also may be movement of the brain across the rough surface of the anterior cranial base.89,139
The same amount of energy is delivered to the brain in nearly a straight line, with the compressive injury at the point of impact resulting in much less damage than the contrecoup injury caused by tension. In view of the dramatic differential susceptibilities of the brain and blood vessels to compressive and tensile strains induced in trauma, it is understandable that diffuse injuries can be caused by seemingly less violent trauma when the head undergoes an injury with a large component of angular acceleration-deceleration. This is the motion that can induce shearing strains, which the tissues tolerate poorly (see “Diffuse Axonal Injury” later on).
In 2006, guidelines were published after a thorough review of the relevant but scientifically weak literature. The reviewers reported that patients with parenchymal mass lesions and signs of progressive neurologic deterioration referable to the lesion, medically refractory intracranial hypertension, or signs of mass effect on CT scan should be treated operatively. Patients with GCS scores of 6 to 8 with frontal or temporal contusions greater than 20 cc in volume with midline shift of at least 5 mm or cisternal compression on CT scan and patients with any lesion greater than 50 cc in volume should receive operative treatment. Patients with parenchymal mass lesions who do not show evidence of neurologic compromise, whose ICP is controlled, and who demonstrate no significant signs of mass effect on CT scan may be managed nonoperatively with intensive monitoring and serial imaging. For patients with refractory intracranial hypertension and diffuse parenchymal injury with clinical and radiographic evidence for impending transtentorial herniation, the guidelines also recommended, as treatment options, subtemporal decompression, temporal lobectomy, or hemispheric decompressive craniectomy (Fig. 66.13). With regard to timing of surgery, the literature supports a bifrontal decompressive craniectomy within 48 hours of injury as a treatment option for patients with diffuse, medically refractory posttraumatic cerebral edema and resultant intracranial hypertension.127
Hypothalamic-Pituitary Injury
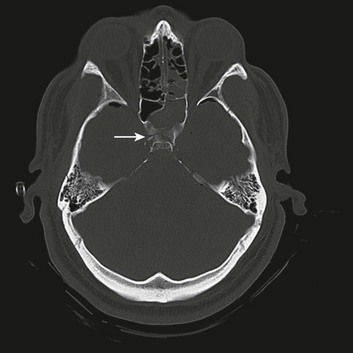
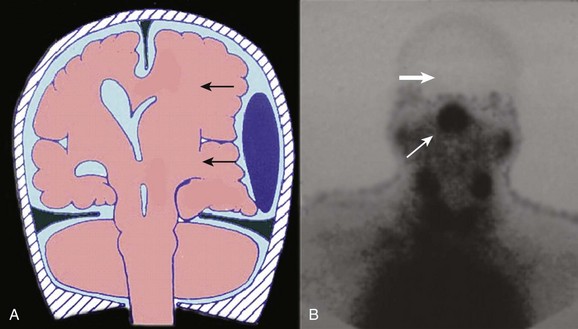
Injury to the pituitary and hypothalamus can complicate head injury. A prolonged loss of consciousness often is reported in patients with such injuries. Up to 80% of cases are associated with a fracture through the skull base. Injuries include (in decreasing order of frequency) pericapsular hemorrhage, hypothalamic infarction, posterior pituitary hemorrhage, anterior pituitary infarction, and rupture of the infundibulum.140–142 Clinically measurable decreases in pituitary hormone production are not seen until at least 75% of the gland is destroyed. Complete loss of production requires destruction of at least 90% of the gland.
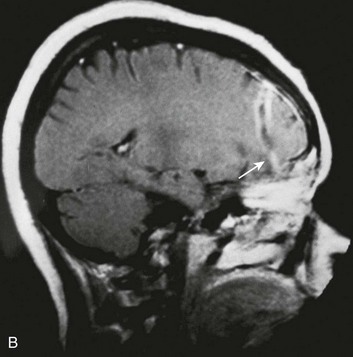
Derangements in pituitary function can result in decreased production of any of the pituitary hormones. Of particular clinical significance is impairment of adrenocorticotropic hormone release that can result in secondary adrenal insufficiency (Addison’s disease). Physiologic stressors such as trauma, surgery, or infection can result in addisonian crisis, with potentially life-threatening results. Diabetes insipidus has been reported in a significant number of severe head injuries, with a high percentage of cases remaining permanent.143 Injuries to the pituitary often are not suspected in the acute period, and the diagnosis of hypopituitarism is therefore often delayed (Fig. 66.14).
Diffuse Axonal Injury
The term diffuse axonal injury (DAI) describes brain damage in a group of patients who become immediately unconscious or go into coma at the time of the head trauma.81 Depending on the severity of the injury, patients may have mild, moderate, or severe DAI. A large number of patients with severe head injury will have DAI. Because of the gradient acceleration difference of certain brain areas during primary impact, shearing forces at the gray-white junction, corpus callosum, or brainstem may occur.131,144 The result of these forces will be diffuse tearing of axons and small blood vessels. These lesions initially may be hemorrhagic, but as they become chronic, shrinking, softening, and scarring ensue, often with cyst formation.42,145 The lesions usually are microscopic but can be large and macroscopic. Although small, focal, petechial hemorrhages may be visualized, the initial CT scan often is normal.146 Most of the clinical and experimental studies support that the diffuse injury occurs at the time of initial impact and is not the result of other adverse factors, such as decreased brain oxygenation, increased ICP, or brain swelling,145,147 although these factors tend to accentuate the amount of damage.
The term gliding contusions originally was introduced by Lindenberg and Freytag148 to describe hemorrhagic lesions in parasagittal white matter; this brain injury concept has been reanalyzed.149 Gliding contusions frequently are found in association with DAI and acute subdural hematomas. Two mechanisms have been considered in relation to the formation of these contusions. First, during angular acceleration, more movement and displacement of the cortical gray matter occur than of the deep white matter; accordingly, most of the tissue injury occurs at the gray-white junction because shearing strains predominate. The second mechanism involves excessive displacement of the bridging veins during brain acceleration. Patients with severe DAI who survive may be profoundly disabled, but some patients with mild or moderate DAI may recover with mild or no disability.81 The importance of recognizing DAI in the initial evaluation cannot be overstressed; this will affect the management and outcome. Clinically, these patients will have impaired consciousness secondary to bicortical damage, possibly in association with unremarkable imaging findings and normal ICP.
Penetrating Brain Injury
It is estimated that between 6000 to 7000 people die each year in the United States from gunshot wounds to the brain.130–139,150–152
Classification
Low-velocity penetrating brain injuries include wounds from nail guns, arrows, and knives and some types of gunshot wounds (Fig. 66.15). Gunshot wounds are divided into low-velocity injuries, such as from civilian handguns and shrapnel, and high-velocity injuries, from rifles and military weapons152 (see Table 66.1).
An Israeli report described an intermediate type of injury from spherical bolts whereby the ball bearings used by suicide bombers, having unique ballistics, may cause a type of “stab wound” injury to the brain.152
Ballistics
The biomechanics of penetrating brain injury (PBI) are important and require an understanding of the dynamics of projectiles—that is, ballistics. Ballistics can be divided into three phases. The first phase, internal ballistics, deals with the source of the projectile and its intrinsic dynamics (e.g., rifle, bomb). External ballistics, the second phase, focuses on the flight of the projectile itself and the deviation of its longitudinal axis relative to the line of flight, referred to as yaw motion. Collision of the projectile with any object during this external phase will change the speed, angle, and yaw. The third phase is referred to as terminal ballistics, which is the physical interaction between the projectile and the body.152
As the projectile directly crushes tissue, it forms a permanent cavity, and as surrounding tissue is compressed, a temporary cavity also is formed. As the missile breaks through the skull, secondary projectiles of bone fragments may be generated, causing independent, secondary permanent and temporary cavities.152–156
Terminal ballistics are influenced by many factors, such as size, shape, and stability of the penetrating object; however, most authors (but not all) believe that entrance velocity is the most important factor in determining the degree of tissue damage.152,153,155,157–159 Experimental evidence has demonstrated an immediate increase in ICP with a pressure wave transmitted throughout the cranial cavity, which probably accounts for the transient respiratory arrest observed in PBI.152,160 High-velocity injuries (velocity greater than 320 meters per second) cause shock waves that emanate from the front of the missile. These shock waves can reflect off the inside of the skull and summate, producing significant pressure gradients with remote effects.
Initial Resuscitation
Initial resuscitation in the trauma bay should be in accordance with current recommendations for head-injured patients in general. During this initial resuscitation, it is important to recognize that hypotension in PBI either is a terminal event or is related to other injuries. Once resuscitation has been accomplished, a CT scan is essential. Although skull films seldom add to the information obtained from a CT scan,152 our group has found plain films to be very helpful in PBI, particularly in industrial accidents, in which the shape of the penetrating object may be unknown.
Surgical Management
Most of what is known about the surgical management of PBI comes from the battlefield, beginning with World War I, when Harvey Cushing, the father of American neurosurgery, described his technique of en bloc craniectomy under aseptic conditions; thorough debridement of scalp, bone, brain, metal, and bone fragments; and watertight closure of the scalp. Using this management strategy a century ago, in the absence of antibiotics, Cushing reported a significant drop in postoperative mortality from 55% to 28%.161,162
Mortality decreased even further in World War II, primarily because neurosurgical personnel were present in forward military hospitals and antibiotics were introduced. The operative approach, however, was largely the same: that of radical debridement. Treatment consisted of four tiers: emergent life-saving maneuvers through hemostasis and cerebral decompression; prevention of infection through extensive debridement; preservation of nervous tissue through prevention of meningocerebral scars; and restoration of anatomic structures through accurate closure of the dura and scalp. Although untested against other management strategies, this approach became the standard of treatment for PBI by default. Indeed, in the U.S. military, thorough debridement of intracranial bone and metal fragments was official military policy through the Vietnam War.162
The overall mortality rate from PBI sustained during relatively modern wartime conditions ranges from 8% to 43% and generally is considered to be in the 20% range.162–171 The mortality rate from military gunshot wounds has been reported to be 2.5 to 4 times more than from shrapnel.162–165,170 Of interest, mortality rates from wartime PBIs in the U.S. military from World War II through the Vietnam War did not change significantly,172 which is consistent with the relatively unchanged management strategy of complete removal of intracranial bone or metal fragments, using repeated surgery as necessary.173
Aggressive debridement was intended to reduce complications such as infection, epilepsy, and cerebral edema. However, studies reveal that additional surgery to remove retained fragments results in significant morbidity and mortality.170,174,175
The use of broad-spectrum antibiotics in recent wars has resulted in data suggesting that retained bone fragments are not independently associated with an increased risk of infection. Therefore, aggressive debridement is no longer supported.162,176 The use of broad-spectrum antibiotics in PBI has now become universal.152
Multiple studies have looked at post-PBI epilepsy and reported an incidence of 22% to 53%.167,177,178 Several studies suggest that it is not necessary to remove all bone fragments in order to decrease the chance of developing epilepsy, but the value of removing metal fragments in this regard remains unclear.162,167,169,177,178 In military PBI, it appears that vigorous debridement is associated with increased morbidity and mortality and is not necessary to prevent infection, has no obvious efficacy in preventing epilepsy, and does not appear to improve survival.162
A CSF leak resulting from a PBI is the variable most highly correlated with intracranial infection.* One study found that most CSF leaks appeared within 2 weeks of the injury and less than half of them closed spontaneously. The incidence of infection was approximately 10 times higher in the group with CSF fistulas than in the group without leaks, and the mortality was greater as well.181 Because CSF leaks are the primary predictor of the development of intracranial infection,167,169,171 it is important to fix or prevent a CSF leak; therefore, a watertight closure of the scalp entry wound is necessary.181
To accomplish this objective, various surgical interventions are available. Small entrance wounds without underlying surgical pathology may be cleaned up and closed in the emergency department.182 Complex entry wounds or those with underlying surgical pathology will require more extensive surgical manipulation, which may include extending the incision to allow vigorous superficial debridement; complete excision of devitalized tissues including muscle, fascia, and periosteum; extension and debridement of the bony entry or exit site by craniectomy; and similarly generous debridement of the dural opening with watertight closure, using grafting as necessary.†
Predictive Factors
Increased mortality in penetrating head injury is associated with increasing age, suicide attempts, hypotension, coagulopathy (particularly with lower GCS scores), respiratory distress, low GCS score, bilateral fixed and dilated pupils, high ICP, and cisternal effacement on CT scan. Mortality also is increased with traumatic injuries in which a missile tract perforates the skull (through-and-through injury) or with injuries that involve both hemispheres, multiple lobes, or the ventricular system.162
In civilian gunshot wounds to the head, a specific vector analysis from Los Angeles County demonstrated a region of involvement that resulted in 100% fatality and is designated as the zona fatalis. This region comprises the midbody of the ventricle, the body of the corpus callosum, and the cingulum.184
No effect has been demonstrated for weapon caliber on outcome, independent of total kinetic energy, and no relation between midline shift and outcome has been established.162
Special Problems
Special problems that may be encountered with PBI include those associated with craniofacial entrance wounds: an increased incidence of hematoma formation, direct vascular injury, extensive contamination due to paranasal sinus involvement (see Fig. 66.15), and CSF fistulas.152
Other special problems include dural venous sinus involvement, tangential wounds, and traumatic aneurysms, with 0.4% to 0.7% of all intracranial aneurysms being caused by penetrating trauma.185
In PBI, mechanical loading of the cerebral vasculature, by either the contact forces of the projectile or the shearing forces of a pulsating temporary cavity, may cause partial or complete transection of an arterial wall. Such damage can result in SAH or formation of intracerebral and intraventricular hematomas.162,186,187
Damage to the arterial wall also may cause the development of a traumatic intracranial aneurysm (TICA). TICAs are primarily false aneurysms, which could heal or change in size over time, but they are especially vulnerable to rupture and can lead to delayed traumatic intracerebral hematoma or SAH or both.185,188–190
Patients with an increased risk of vascular injury include cases in which the trajectory passes through or near the sylvian fissure, supraclinoid carotid, cavernous sinus, or a major venous sinus. The development of substantial and otherwise unexplained SAH or delayed hematoma also should prompt consideration of a vascular injury.162
Cerebral angiography (CTA or conventional) remains the usual technique to detect intracranial aneurysms.187,189,191 Because a majority of traumatic intracranial aneurysms are not true aneurysms, excluding the aneurysm by clipping may not be possible and may require trapping between clips on the parent vessel.162
Course in the Intensive Care Unit
Not surprisingly, intracranial hypertension is common after PBI.181,192–194 Early ICP monitoring should be considered when the ICU clinician is unable to assess the patient’s neurologic status accurately; if the need to evacuate a mass lesion is unclear; or if CT scanning suggests elevated ICP.162 Another practical use for ICP monitoring is ongoing assessment of a patient with a depressed level of consciousness to look for an early sign of progressive intracranial pathology, which may require repeated imaging and surgical decompression.181,192,194
Cerebral autoregulation may be defective after PBI181,192,194; cerebral edema can appear extremely rapidly, contributing to medically refractory intracranial hypertension. Systemic hypertension may exacerbate intracranial hypertension after PBI.162
Treatment is not necessarily futile, and some patients with intracranial hypertension can be managed successfully and survive with acceptable morbidity.181,192,194,195 However, the literature does not clearly relate successful treatment of intracranial hypertension after PBI to an improved outcome. Not enough good data are available to guide management strategies for intracranial hypertension after PBI. It cannot be assumed that the intracranial pathology is the same as in nonpenetrating traumatic brain injury. Because of the paucity of data, however, treatment generally follows the methods for nonpenetrating traumatic brain injury as outlined in Guidelines for the Management of Severe Traumatic Brain Injury.162,196
Harrington and Apostolides have proposed a treatment algorithm for patients with gunshot wounds to the head. Patients who have lesions causing mass effect and a postresuscitation GCS score of 7 or greater undergo surgery, and patients with a GCS score of 3 or 4 with bilaterally fixed pupils after resuscitation receive no treatment beyond wound closure. In patients with GCS scores of 5 or 6, treatment should be individualized, depending on pupillary findings and other conditions.197
Secondary Head Injury
Basic Concepts
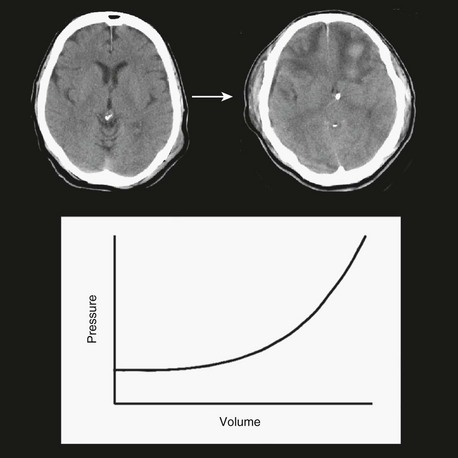
The three functional volumetric compartments in the head are blood, brain, and CSF. The volumes of these three functional compartments are approximately 1200 mL for brain, 150 mL for CSF, and roughly 150 mL for the variable blood compartment. The total volume of the intracranial compartment is relatively fixed and relatively incompressible. Therefore, if volume is added to one of these three compartments, a compensatory loss of volume from another compartment will occur; otherwise, ICP will rise. To illustrate this point, a pressure-volume curve for the intracranial compartment is shown in Figure 66.16. Initially, as volume is added to the system, no significant change in pressure occurs. With cerebral edema, for example, as the brain compartment increases in volume, venous capacitance vessels collapse, which moves blood volume out of the system; therefore, overall net volume does not change and ICP remains stable. As the brain continues to swell and increase in volume, the ventricles become smaller as CSF moves out of the system, again keeping overall net volume and ICP relatively stable. At some point the system loses this buffering capacity, and as more injury-related volume accumulates, ICP starts to rise. The rise in ICP is gradual, progressive, and nonlinear. As the system continues to get stressed with additional volume, it progressively loses compliance and, in effect, gets stiffer. The result is that increasingly smaller volumes introduced into the system result in higher changes in ICP. Using MRI methods to study cerebral edema, Marmarou and colleagues noted a rise in ICP when edema was only 1% higher than the normal water content of brain tissue. These investigators also showed that when water content increased in a contused hemisphere, ICP increased exponentially. These findings demonstrate the importance of small changes in intracranial volume, particularly when the buffering capacity of the system has been exhausted.198
Increased ICP may be associated with mass effect secondary to unbalanced forces, causing direct, mechanical compressive damage to involved structures. This condition generally necessitates surgical decompression in the operating room. Increased ICP also may result in inadequate perfusion. This condition generally requires medical management in the ICU (Fig. 66.17).
The heart delivers blood at a given mean arterial blood pressure (MABP), the systemic perfusion pressure, to the base of the skull. The blood pressure then has to overcome whatever pressure is inside the head for blood flow to get in, and whatever is left over will be the pressure that perfuses the brain—that is, cerebral perfusion pressure (CPP). Simple in concept and calculation, CPP is defined as the mean arterial blood pressure minus ICP (CPP = MABP − ICP). It is the physiologic variable that defines the pressure gradient driving cerebral blood flow (CBF) and metabolic delivery and is therefore closely related to ischemia.199,200
Technically, the MABP should be measured as the mean carotid pressure with the arterial line transducer zeroed at the level of the foramen of Monro (as opposed to the right atrium),201 but this approach is not common in clinical practice. Normal values generally are considered to be 60 to 80 mm Hg for CPP and approximately 10 mm Hg or less for ICP.
Cerebral Blood Volume
Arterial Blood Compartment
Blood Pressure
Systemic blood pressure has a direct effect on arterial cerebral blood volume. Normally, CBF will remain relatively constant over a wide range of perfusion pressures as cerebrovascular resistance (CVR) automatically adjusts. The autoregulation curve (Fig. 66.18) demonstrates the relationship among CBF, perfusion pressure, and cerebrovascular resistance. As MABP goes up, CVR increases in order to maintain a stable CBF.
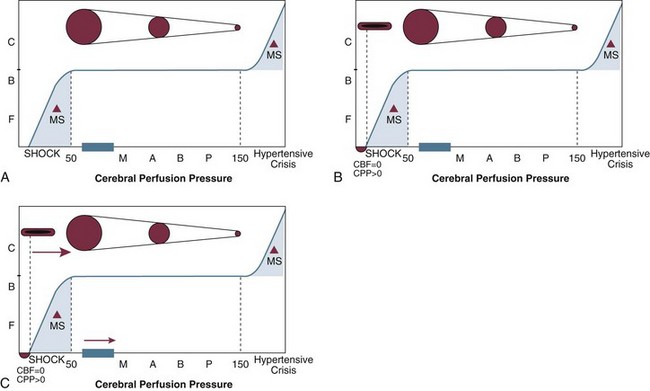
CVR often is increased by trauma. At the beginning of the autoregulation curve, CBF is zero at a perfusion pressure greater than zero. A measurable pressure in the vessel exists, yet no blood flow occurs, indicating vascular collapse (see Fig. 66.18). The cerebrovasculature in its resting state is collapsed; it takes a certain amount of energy (pressure) to open the vessels. These vessels each have critical opening and closing pressures. After a head injury, free radicals can be released, which tend to make biologic tissues less compliant. This in turn may make it more difficult for the vessels to stay open—that is, more pressure is required to keep the vessels from closing, which could shift the autoregulation curve to the right (see Fig. 66.18).202–205 This is the theoretical basis for the studies that led to the earlier recommendations to keep the minimum CPP in head-injured patients above normal values—that is, at 70 mm Hg or greater.206
Cerebral Perfusion Pressure
A low perfusion pressure jeopardizes ischemic regions of the brain. Increasing intravascular hydrostatic pressure by increasing CPP can help improve cerebral perfusion. Although CPP can be manipulated and enhancement of CPP may help to avoid both global and regional ischemia, the level at which CPP is best maintained is not entirely clear,206 and considerable interest has been directed toward attempts to determine the optimum CPP for a head-injured patient. Some studies suggest that increasing CPP to greater than 70 mm Hg leads to improved outcomes.207
A combination of studies of traumatic brain injury in which CPP was actively maintained at approximately 70 mm Hg in patients with a GCS score less than 8 reported a mean mortality rate of 21%, which was substantially less than the 40% mortality rate reported for similar patients in the Traumatic Coma Data Bank (TCDB). Good recovery and moderate disability rates were 54% in the patients whose CPP values were maintained in the 70 mm Hg range, compared with the 37% reported in the TCDB. However, it could not be concluded that the improved outcomes in these studies were a result of CPP management because none of the reported studies was a randomized, prospective trial of this treatment200,205,208–212; and in some, another treatment was actually the focus of the study.208,209
In a prospective study of 21 patients with severe traumatic brain injury in whom brain tissue PO2 (BtiO2) was monitored, ischemic episodes during the first week after injury were associated with unfavorable neurologic outcomes. Elevation of the CPP improved BtiO2, but raising CPP above 68 mm Hg did not cause a further increase in BtiO2. In the investigators’ analysis, a CPP greater than 60 mm Hg emerged as the most important factor determining a sufficient Btio2.200,213
Another prospective, controlled trial of 189 adult patients with a motor GCS score of 5 or lower within 12 hours of head injury randomized patients to an “ICP-targeted protocol” in which CPP was kept above 50 mm Hg or to a “CBF-targeted protocol” in which CPP was kept above 70 mm Hg. The investigators in this study found no significant difference in outcome. In this trial, the mean CPP for patients managed in the CBF-targeted protocol was 76 mm Hg and for those in the ICP-targeted protocol, 72 mm Hg. These differences were small but statistically significant.214
Contant and associates used data from this trial to examine risk factors for acute respiratory distress syndrome (ARDS) and consequences of its development. The risk of ARDS was five times greater among patients in the CBF-targeted group than in those in the ICP-targeted group and was associated with a more frequent use of epinephrine and a higher dose of dopamine.199,215 Patients in whom ARDS developed were two and a half times more likely to develop refractory intracranial hypertension215 and almost three times more likely to be in a vegetative state or dead at 6 months after injury.215,216 These outcomes may be a direct consequence of ARDS, and the related therapeutic limitations of treating raised ICP and low CPP in the presence of ARDS.199
Analysis of data from a study investigating the value of a neuroprotective agent in 427 patients with severe traumatic brain injury revealed that the critical CPP threshold was 60 mm Hg. Although CPPs that were persistently less than 60 mm Hg were associated with a significant decline in outcomes, no effect on neurologic outcomes was observed for patients who had CPPs that were higher, and specifically no difference was observed among those who averaged 60 mm Hg and those at 70 mm Hg or higher.199,217
In 1995 and 2000, the Brain Trauma Foundation in cooperation with the American Association of Neurological Surgeons and the Joint Section for Trauma and Critical Care published guidelines for the management of severe head injury. From the data available, based on the theoretical need to increase the minimum CPP because of a change in critical closing pressures (see Fig. 66.18), the guidelines recommended maintaining a minimum CPP of 70 mm Hg or greater—10 mm Hg above the accepted norm. In 2003, this group updated the recommendation for CPP management and published a guideline that CPP should be maintained at a minimum of 60 mm Hg. In the absence of cerebral ischemia, aggressive attempts to maintain CPP above 70 mm Hg with fluids and pressors should be avoided because of the risk of ARDS.199 In 2007, the recommendations were again modified with a level 3 recommendation to target CPP values within the range of 50 to 70 mm Hg.200 Clearly, guidelines change as new data become available and the reader is strongly advised to check for the most up-to-date recommendations.
Carbon Dioxide
Carbon dioxide is a powerful regulator of cerebrovascular resistance. At any given physiologic perfusion pressure, PaCO2 can act rapidly and severely to affect CBF in a linear fashion by vasodilating or vasoconstricting the cerebrovascular bed. For every mm Hg change in PaCO2, a 3% change in CBF can occur.218
The data on CBF in severely head-injured patients indicate that phases of CBF change over time after traumatic brain injury, with hypoperfusion occurring in the first 24 hours.218 Multiple studies have identified brain ischemia in up to 35% of serious head injuries within the first 12 hours.200,218–224 It appears that this reduction happens almost immediately. Mean CBF values measured less than 4 hours after head injury in adults were 32 mL/100 g/minute in survivors and 20 mL per 100 g per minute in nonsurvivors (normal CBF is 55 mL/100 g/minute).224 Low cerebral blood flow, to ischemic levels (less than 18 mL/100 g/minute), was observed in 31% of patients with severe head injury measured an average of 3 hours after trauma.200,225
Because of the consistent data regarding reduced CBF early after trauma, attempts at further reductions using hyperventilation have come under close scrutiny. Some evidence suggests that hyperventilation carries a particular risk of causing cerebral hypoxia when PaCO2 is 30 mm Hg or less, or within the first 24 hours226,227 because of a further reduction of an already low CBF.225 It has become clear that hyperventilation decreases BtiO2 because of decreased CBF in most patients, which negates the perceived benefit of improving intracranial and cerebral perfusion pressures.213,226,227–232
On the basis of principles reflecting a high degree of clinical certainty, the Brain Trauma Foundation (BTF) in 1995 and 2000 recommended that in the absence of increased ICP, chronic prolonged hyperventilation therapy (Pco2 of 25 mm Hg or less) should be avoided after severe traumatic brain injury.233
On the basis of principles reflecting a moderate degree of clinical certainty, this group recommended that the use of prophylactic hyperventilation (Pco2 less than 35 mm Hg) therapy during the first 24 hours after severe traumatic brain injury should be avoided because it can compromise cerebral perfusion during a time when CBF is reduced. On the basis of principles for which there is unclear clinical certainty, the group recommended that hyperventilation therapy may be necessary for brief periods during acute neurologic deterioration, or for longer periods in the presence of intracranial hypertension refractory to sedation, paralysis, CSF drainage, and osmotic diuretics.234
Oxygen
Within a normal physiologic range, oxygen causes little or no change in CBF (Fig. 66.19). Beyond a PaO2 of approximately 300 mm Hg, CBF can begin to wane. Oxygenation does become very important at PaO2 levels considered to be hypoxic. Below a PaO2 of 60 mm Hg in most head-injured patients, the cerebrovasculature can vasodilate rapidly, causing an increase in CBV with poorly oxygenated blood, with an attendant increase in intracranial volume and pressure.
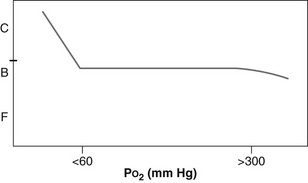
With careful clinical monitoring, our group has observed ICP spikes as a result of acute oxygen desaturation before a change in the pulse oximeter reading. With a moderate degree of clinical certainty, the BTF has recommended that hypoxia, defined as apnea or cyanosis in the field or a PaO2 less than 60 mm Hg, must be scrupulously avoided or corrected immediately. Therefore, in contrast with PaCO2, manipulation of PaO2 is not a useful tool for medical management of a head-injured patient, and hypoxia must be avoided.200
Metabolism
The metabolic state of the brain is sensitive to changes in temperature; lower temperatures lower CMRO2, and vice versa. Recognition of this relationship has led to interest in using hypothermia as a therapeutic tool in severe head injury, both as a way to lower ICP and as a neuroprotective strategy by reducing demand in the face of limited supply. Although hypothermia can be a useful adjunct to standard treatments for controlling elevated ICP it is difficult to find good data to prove a benefit in outcome. Clifton and associates found that ICP was lower in patients managed with hypothermia, but no improvement in outcome was observed, and the complication rate was higher than in a normothermic group. These investigators concluded that lowering body temperature to 33° C within 8 hours after injury was not effective in improving outcomes in patients with brain injuries.235 A Cochrane review of therapeutic hypothermia for head injury looked at 14 trials with more than 1000 participants and found no evidence that hypothermia is beneficial in the treatment of head injury, and that it increased the risk of pneumonia nearly twofold.236 A different meta-analysis using clinical trials of moderate or poor quality, identified preliminary findings suggesting a greater decrease in mortality risk when target temperatures are maintained for more than 48 hours, as well as an association with better outcomes.237,238
Although it is not clear that routine use of prophylactic hypothermia is helpful, hyperthermia is believed to be harmful to a severely head-injured patient.239 As the temperature rises, so does the CMRO2, with a concomitant increase in CBV. If the patient has lost enough compliance then small changes in intracranial volume can result in big changes in ICP (see Fig. 66.16), and CPP can fall.
Another consideration in the management of CMRO2 is drug effect. Pentobarbital is the gold standard drug for reducing cerebral metabolism and can be used in cases of refractory intracranial hypertension to induce a barbiturate coma. A condition of burst suppression is achieved, meaning that the continuous EEG recording demonstrates periods of an isoelectric tracing (straight line) interrupted by bursts of cortical electrical activity. This therapy results in vasoconstriction appropriate for the degree of reduction in metabolism, with an attendant decrease in ICP. This has the theoretical advantage of improving supply to a brain with reduced metabolic demands. However, despite the evidence for cerebral protection in experimental models, little clinical evidence is available to suggest that barbiturates improve outcome after severe head injuries in humans. Other drugs that reduce the CMRO2 and CBF are other barbiturates such as thiopental, as well as the substituted phenol propofol, the hypnotic agent etomidate, and the water-soluble short-acting benzodiazepine midazolam.239
Viscosity
When autoregulation is intact, lowering viscosity of the blood improves flow dynamics and results in an appropriate increase in CVR to maintain a constant, normal CBF.240–244
This vasoconstriction reduces intracranial volume. From a rheologic standpoint, there is an advantage to lowering viscosity by hemodilution, but this is at the expense of oxygen-carrying capacity. Reducing the hematocrit from 45% to 30% can theoretically double CBF, yet oxygen-carrying capacity would be reduced by only a third. By generalizing experimental and clinical data involving ischemia and hematocrit, it is reasonable to consider an optimum hematocrit of approximately 30% to 35%.245
A common mechanism for lowering viscosity in traumatic head injury is the use of mannitol, which causes an immediate influx of extravascular water, leading to hemodilution. The vasoconstriction resulting from this reduction in viscosity allows the same CBF and oxygen delivery to be maintained by means of smaller vessels and with a lower intracranial volume (see later section, “Hyperosmolar Therapy”). In addition, mannitol will dehydrate the red blood cells themselves by about 15%, which further improves blood flow dynamics.245
Venous Blood Compartment
During a Valsalva maneuver, intrathoracic pressure elevates and venous return to the right heart goes down. Decreased venous return means decreased venous outflow from the brain. This results in a rapid, instantaneous increase in CBV as the arterial side continues to supply the brain with blood that is not getting physiologic draining. A Valsalva maneuver also will decrease cardiac output along with a decrease in venous return, but it is a disproportionate change. A common neurosurgical practice is to have the anesthesiologist induce a controlled Valsalva maneuver toward the end of a brain operation during the final stages of hemostasis, just before beginning the closure. Intrathoracic pressure is elevated to approximately 30 mm Hg for 10 seconds as the surgeon looks for any areas of bleeding while the brain immediately physically and visibly swells as it engorges with blood. An intrathoracic pressure of 30 mm Hg will overcome a normal CVP and essentially interrupt venous outflow from the brain. The concomitant decrease in cardiac output with a pressure of 30 mm Hg will have much less of an effect than on the venous side; therefore, a relatively large volume of arterial blood will continue to fill the brain. Even a normal brain with normal compliance will immediately “feel” the effect of decreased venous outflow because it happens faster than the buffering capacity of the brain can handle (see Fig. 66.16). In fact, one mechanism the brain will use to buffer increases in volume is to collapse venous capacitance vessels, which, in the case of decreased venous outflow from the brain, becomes part of the problem, not the solution.
Brain Compartment
Another functional volumetric compartment is the brain itself. One of the consequences of traumatic brain injury is an acute increase in total brain water.246
Cerebral Edema
By definition, brain edema is an abnormal accumulation of fluid within the brain parenchyma that produces a volumetric enlargement of the brain tissue.198 The three types of cerebral edema are vasogenic, neurotoxic, and cytotoxic. Vasogenic edema occurs when the blood-brain barrier opens, resulting in movement of vascular fluid into the extracellular spaces of the brain.247 Neurotoxic edema involves astrocytic and dendritic swelling secondary to the neurotoxic effects of excitatory amino acids, particularly glutamate, seen with reperfusion.198 Cytotoxic edema results from retention of water within swollen cells, most commonly found in stroke, in which interruption of energy supply leads to pump failure and an intracellular increase in sodium and water.198
It is ischemia, associated with energy failure at the level of mitochondria, that contributes to cytotoxic brain edema and intracranial hypertension.246–254 Earlier studies led to the general acceptance that traumatic brain swelling was primarily a result of vascular engorgement, which caused increased ICP.255–259 Although opinions differ, more recent work has demonstrated that cerebral edema may be primarily responsible for brain swelling, not vascular engorgement.251,260 Marmarou and colleagues have used sophisticated MRI techniques to demonstrate that traumatic brain edema is predominantly a cellular phenomenon in both focal and diffuse brain injury.198
Although cellular edema predominates, it is likely that edema formation in head injury is complicated, with time-dependent contributions of both vasogenic and cytotoxic mechanisms. Experimental evidence points to transient compromise of the blood-brain barrier occurring immediately after injury, with resultant vasogenic edema. Studies indicate early closure of the blood-brain barrier, after which cytotoxic, cellular edema predominates.198,261–263
Hyperosmolar Therapy
Mannitol
Mannitol, an inert six-carbon alcohol of the corresponding sugar mannose, causes cellular dehydration by increasing serum osmolarity. The use of mannitol in head-injured patients to lower ICP is based on two mechanisms of action of this drug: It establishes an osmotic gradient across the blood-brain barrier to reduce water content,264,265 and it causes cerebral vasoconstriction through rheologic effects that decrease blood viscosity and enhance CBF and MABP (see earlier section, “Viscosity”).245,266,267 This vascular mechanism of action has a strong, independent effect, such that CBF can be enhanced even when ICP is not substantially reduced.268,269
With head-injured patients, mannitol is used in two clinical scenarios. The first is that of a comatose trauma patient with a surgical clot, when mannitol is given on the way to the operating room to buy time until emergent decompression is accomplished. Generally, higher doses of up to 1.2 to 1.4 g per kg can be given in this scenario.270,271
The second clinical scenario is one of cerebral edema, when mannitol is given to control ICP. Unlike with its use for emergency surgical clots, there is no consensus on the optimal dosing regimen of mannitol in the ICU to manage ICP. Doses ranging from 0.25 g per kg to 1.0 g per kg may be effective. Mannitol can be given in response to high ICP, or it can be used prophylactically regardless of ICP, using serum osmolarity as an end point of treatment,272 although some evidence indicates that it is most effective when used in the presence of high ICP or low perfusion pressure.267,273
Regardless of how mannitol is used, because of its osmotic diuresis, replacement of urinary water and electrolyte losses is critical, to avoid hypovolemia and hypotension. Mannitol typically is given as a bolus at a rate not to exceed 0.1 g per kg per minute in order to avoid hypotension.272 Caution should be exercised in patients with a propensity toward congestive heart failure, because the early effects of bolus administration cause intravascular volume expansion.272
Mannitol can extravasate into the interstitium of the brain, with breakdown of the blood-brain barrier, which may be a cause of cerebral edema. This is thought to be less of an issue with bolus dosing as opposed to continuous infusion.274 Because of this particular concern with mannitol toxicity, many clinicians prefer to use the smallest doses of mannitol necessary, and to administer it only for proven intracranial hypertension.275
Another concern is renal failure, which is a rare complication of mannitol therapy. It is generally believed that the kidneys are at risk above a serum osmolarity of 320 mOsm per L276,277; however, kidney damage may in fact be due to high serum concentrations of mannitol itself, as opposed to high serum osmolarity. It is suggested that keeping the osmolar gap (measured serum osmolarity minus calculated serum osmolarity) below 55 mOsm per kg of H2O may be better than using serum osmolarity alone to direct mannitol therapy.272,277
Furosemide, a loop diuretic, has some effectiveness for removing extracellular fluid from the brain. Although it does not work as fast as mannitol, furosemide commonly is used in conjunction with that agent because of a synergistic effect on ICP.278
Hypertonic Saline
In many ICUs, hypertonic saline is being used with increasing frequency for the treatment of raised ICP in patients with traumatic brain injury.279 It is known that patients with hypotension after severe traumatic brain injury have twice the mortality rate of normotensive patients280; therefore, aggressive resuscitation with intravenous fluids is recommended in the guidelines for the management of patients with severe traumatic brain injury.281 However, the real concern for the development of cerebral edema is reason enough to limit the amount of free water available to the injured brain during this early period. The use of hypertonic saline, both to increase mean arterial pressure and to decrease ICP, is a logical and promising approach to limit secondary injury and improve neurologic outcome.
The rapid infusion of a small volume of hypertonic solution originally was designed for the prehospital treatment of hemorrhagic shock,282–285 and a trial found a trend toward a reduction of ICP in a double-blind, randomized, controlled trial of prehospital fluid resuscitation using hypertonic saline in comatose and hypotensive head-injured patients (although no improvement in outcome was demonstrated).286
The rapid infusion of small volumes of hypertonic solution leads to an osmotic gradient that mobilizes parenchymal fluid into the vascular compartment,287,288 resulting in hemodilution, endothelial shrinkage, and improved blood flow in the microcirculation,289,290 as well as improved cardiac output.282,283,291 Hypertonic saline has been shown to reduce brain bulk and ICP292–295 and to be effective in lowering ICP both in bolus form and as a maintenance fluid.296–298
It is widely believed that the mechanism of action of both hypertonic saline and mannitol in reducing ICP is their hyperosmolar properties, although additional mechanisms are likely to be in play. Some authors have found hypertonic saline to be more effective than mannitol in selected patients.299,300
Concerns about central pontine myelinolysis and acute renal failure appear to be unfounded on the basis of clinical experience; however, a rebound effect may occur in some patients.301 A trial also reported no episodes of acute anemia or hypokalemia associated with 23.4% saline administration, nor were there any episodes of convulsions, congestive heart failure, or coagulopathy after the administration of hypertonic saline.299
In the United States, 3% sodium chloride is readily available. An initial dose of 1.5 to 3.0 mL per kg with a target for a serum sodium concentration of approximately 155 mEq per L is reasonable,295 although 23.4% saline has been shown to be safe and effective in the treatment of intracranial hypertension.299 As with hyperosmolar therapy for mannitol, it is important to maintain a normovolemic state at all times during treatment.
Cerebrospinal Fluid Compartment
In severe head injury, CSF can be displaced out of the intracranial compartment as pressure differences between the superior sagittal sinus and the intracranial compartment increase (see Fig. 66.16). The spinal CSF compartment also is an important physiologic sink, and this contribution of CSF flow to ICP is another justification for nursing traumatic brain-injured patients with the head elevated.300
A standard way of managing the CSF compartment in the trauma patient is by draining CSF through an indwelling ventricular catheter (ventriculostomy). This has proved to be a relatively safe and effective tool in lowering intracranial volume and pressure, although ventricles in younger trauma patients commonly are smaller and harder to access and have less CSF to drain. Ventricular drainage is discussed further in Chapter 17.
Specific Treatment Considerations
Craniectomy
Craniectomy is an alternative to the medical and direct surgical methods for reducing intracranial volume. This is a surgical strategy for managing a dangerous increase in intracranial volume by expanding the size of the intracranial compartment. The necessary expansion is accomplished by removing a large portion of the skull and opening the dura (see Fig. 66.13).
Intractable intracranial hypertension despite maximal medical therapy is associated with a high risk of morbidity and death.301–303 In one multicenter trial, an 86% mortality rate was found among patients whose condition failed to respond to an induced pentobarbital coma304—the last line of defense in medical management. Similarly, a significant relationship has been recognized between an ICP higher than 25 mm Hg and a poor outcome.305
Because of these ominous facts, craniectomy to increase the space available for the swollen brain has been used to lower ICP, although early outcome studies of hemicraniectomy yielded poor results.306–308 More contemporary experiences, however, have achieved better results. In 2006, Aarabi and colleagues reviewed 10 reports published since 1988, with a total of 323 patients treated with decompressive craniectomy for posttraumatic brain swelling and intractable intracranial hypertension, and calculated a collective mortality rate of 22.3%, with good outcomes in 48.3%, with the rest of the patients (29.4%) remaining severely disabled or vegetative. These results compared favorably with six previous reports in which the mortality rates ranged from 42% to 100% when ICP greater than 20 mm Hg remained refractory to medical management. Reported complications included subgaleal collections, which resolved over the course of weeks to months; delayed wound healing; bone flap resorption and infections; increased swelling and hemorrhagic contusions; and parenchymal lucencies, possibly due to ischemia.*
In addition to the demonstrated improvement in outcome, contrast-enhanced ultrasonography has been used to assess cerebral perfusion after decompressive craniectomy and found an average fivefold improvement in microvascular blood flow.319 A technical point worth emphasizing is that a decompressive craniectomy must be sufficiently large in order to be helpful.320
Management of Intracranial Hypertension
Acute, severe traumatic brain injury is a dynamic process with the potential for dangerous changes that can evolve over hours to days; therefore, all patients need to be observed closely. Ideally, ongoing assessment by neurologic examination should be at least hourly. In most patients with a GCS score of 8 or less, however, airway protection mechanisms will be impaired, necessitating intubation and, therefore, sedation, which makes an accurate neurologic assessment problematic. An ICP measuring device usually is needed at this point to adequately monitor the patient, in lieu of a reliable examination (see Chapter 16 on ICP monitoring).
The medical management of intracranial hypertension in the ICU generally occurs after surgical decompression of hematomas causing unbalanced forces, or in patients without a surgical lesion. This management strategy generally is divided into tiers of therapy. The first tier is sedation, analgesia, and intubation without hyperventilation, keeping the head elevated and the neck straight and uncompressed. The routine use of paralytics may increase the risk of pulmonary complications; therefore, muscle relaxants should be used for ICP control when sedation is inadequate.321 If ICP exceeds 20 mm Hg322 despite these maneuvers, moving to the next level of care is indicated, which includes CSF drainage if a ventriculostomy is being used, mannitol, or hypertonic saline. Beyond the first 24 hours, mild hyperventilation to keep Pco2 in the mid-30s can be used. Throughout the clinical course it also is necessary to maintain an adequate CPP of at least 60 mm Hg by ensuring normovolemia and a safe MABP. If ICP is refractory to these treatments, the next step is use of decompressive hemicraniectomies or barbiturate coma (or both). Historically, surgical decompression has been reserved as a radical, end-of-the-line treatment after the patient has failed to respond to burst suppression barbiturate coma; however, craniectomy is moving up in the treatment algorithm of most centers (Fig. 66.20).
When ICP goes up, it is important to understand why and then try to correct the underlying problem if possible. In any given head-injured patient in whom the brain has lost its buffering capacity, with resultant poor compliance, small increases in intracranial volume can cause a significant rise in pressure. When ICP rises, CPP goes down. This usually will result in a decrease in CVR and vasodilation, with its attendant increase in cerebral blood volume and increase in intracranial volume. This of course leads to a further increase in ICP, and the cycle perpetuates itself. This is the mechanism of a plateau wave.245
Anticoagulation
It is intuitive that severe coagulopathy is associated with increased mortality after head injury.323–331 Penetrating or blunt brain injury can be associated with the massive release of thromboplastin from neuronal tissue, thereby initiating the coagulation process, which evolves to an exaggerated fibrinolytic response and disseminated intra-vascular coagulopathy (DIC).324 This brain injury–induced coagulopathy may lead to significant secondary injury and delay the invasive monitoring necessary for the aggressive management of intracranial hypertension.332 The criteria for the diagnosis of DIC include abnormal clotting studies, thrombocytopenia, low fibrinogen, and, in some cases, elevation in levels of products of fibrinolysis (D-dimer or fibrin degradation products).327 The DIC caused by head trauma is relatively common and frequently is detected by a prolonged prothrombin time and elevated international normalized ratio (INR). Its presence may be an indication for more intense clinical observation and follow-up CT scanning. An important component of DIC is coagulation; however, in the setting of active bleeding, anticoagulation is not indicated. The therapeutic focus is on hemostasis and replacement of diluted or consumed blood elements and components.328–329
Preliminary data indicate that recombinant activated factor VII (rFVIIa) provides a rapid and successful correction of coagulopathy in head-injured patients.326 Originally developed for the treatment of hemophiliacs, rFVIIa also may be helpful in reversing the effects of anticoagulants and antiplatelet drugs.326,330–340 It is a vitamin K–dependent glycoprotein, similar to the human plasma-derived factor VIIa, which appears to promote hemostasis by activating the extrinsic pathway of the coagulation cascade.341 Its mechanism of action is not fully understood but includes interaction with platelets or tissue factor to augment thrombin generation and platelet activation.330,332,334,335 Factor VIIa seems to be the bottleneck, the critical factor most depleted when patients are profoundly anticoagulated.342 The use of rFVIIa to correct a coagulopathy before neurosurgical intervention was reported in 1998 in a hemophiliac patient with an EDH requiring craniotomy for evacuation.343 Although not specifically evaluated for use in DIC, rFVIIa has been administered to neurosurgical patients in whom DIC was likely to be present.329,338 Recombinant FVIIa has been used in the absence of hemophilia for neurosurgical emergencies such as EDH and SDH, SAH and intracerebral hemorrhage, tumor resection, and during placement of ICP monitors.333,336,338,340 A retrospective study found improved outcomes, without complications, when rFVIIa was used for coagulopathic patients requiring urgent neurosurgical intervention.344 The incidence of thrombosis after rFVIIa is low, but thrombosis is an extremely important potential complication334,338,339,345,346 and of particular concern in DIC.328 Precise dosing for conditions other than hemophilia has not been confirmed332; the Food and Drug Administration (FDA) has approved rFVIIa only for use in hemophilia; other uses are considered off-label.
In addition to the coagulopathy induced by a brain injury, neurosurgeons and intensivists often are faced with managing patients with traumatic intracranial hemorrhage who are pharmacologically anticoagulated. The use of these drugs is expected to increase as the population ages.329 These patients must have normal coagulation rapidly restored to prevent further bleeding and to make surgical intervention possible. One of the more common drugs encountered in clinical practice is warfarin; a typical scenario is one in which an elderly patient on warfarin sodium (Coumadin) falls and hits her head. It takes about 4 days to normalize coagulation if warfarin is stopped. With vitamin K, full reversal begins 4 to 6 hours after administration and takes about 24 hours to lower the INR. FFP contains vitamin K–dependent factors, is rapidly effective, and is given intravenously. Recombinant factor VIIa also can be used to normalize coagulation in these patients.329,336,347,348
In hospitalized patients on heparin, reversal occurs approximately 60 minutes after an intravenous infusion is stopped. If it needs to be reversed faster, protamine is recommended. Desmopressin also has been recommended to reverse the effects of heparin. Subcutaneous heparin is usually not reversed.329
With low-molecular-weight heparin (LMWH), a partial thromboplastin time (PTT) is variably altered and not a reliable measure of its effect. Reversal of LMWH effect with protamine is variable in humans.329
A common situation is the presentation of a head-injured patient on an antiplatelet agent such as aspirin or other nonsteroidal anti-inflammatory drug (NSAID). Acetylsalicylic acid (ASA) inhibits platelet aggregation for the life of the affected platelet, which is approximately 10 days. This occurs even at the lowest dose of 81 mg per day. A period of 5 to 6 days typically is needed after stopping aspirin to replace approximately half of the circulating platelets (10% per 24 hours). The effect of ASA on platelets is complete and irreversible. If treatment is necessary, platelet transfusion is given to provide unaffected platelets as long as no ASA remains in the circulation. Desmopressin (DDAVP) may secondarily improve platelet adhesion to endothelial defects and is therefore also recommended to overcome aspirin-induced platelet dysfunction. Other NSAIDs have various anticoagulant effects. If significant NSAID use has occurred before a neurosurgical emergency, the same interventional strategy may apply. Data confirming efficacy in counteracting an anticoagulant effect, however, are not available.329
Other antiplatelet agents include adenosine diphosphate (ADP) and glycoprotein receptor-blocking agents. Clopidogrel and ticlopidine irreversibly alter platelet aggregation349–352 through a mechanism that is additive to the antiplatelet effects of aspirin. The platelets are permanently affected; once the drug is stopped, recovery of normal platelet function requires about a week until new platelets are produced. Rapid correction requires platelet transfusion, and DDAVP also may have a beneficial, short-term effect.329
Abciximab, eptifibatide, and tirofiban are highly effective in generating 80% to 95% reversible inhibition in platelet aggregation.349,350,352–355 Stopping the medication allows return of normal platelet function in approximately 48 hours for abciximab and in 4 to 8 hours for the others. Platelet transfusion increases the proportion of unaffected platelets and is the primary intervention.329
Fibrinolytic and thrombolytic agents disrupt formed clots.355–357 These drugs have a prolonged half-life and frequently are used in combination with heparin or other agents to ensure continued therapeutic anticoagulation. Because circulating fibrinogen is decreased during thrombolysis, cryoprecipitate, a source of concentrated fibrinogen, is the basis for reversing treatment.329 Although these agents have well-recognized applications for cardiac358 and neurologic359,360 problems, their use generally is not seen in patients presenting with head injury.
Hypothermia also is known to cause and worsen coagulation abnormalities,332,361 the treatment for which is rewarming. Certain medical conditions also may affect coagulation, and therefore may be important as underlying factors in patients with a head injury. DDAVP increases the release of factor VIII and von Willebrand factor and is used to treat mild hemophilia A, von Willebrand disease, and chronic renal and liver disease, as well as acquired and congenital platelet disorders.361,362–365
Unintended, iatrogenic worsening of coagulopathy may result with use of hetastarch solutions, which may be administered to increase intravascular volume. These solutions may lower von Willebrand factor levels and alter platelet function. Low-molecular-weight hetastarch preparations are available that may cause less severe changes; however, if the patient requires correction of a coagulation abnormality, these solutions should be avoided if possible.365–368
Nutritional Support
Replacement of 140% of resting metabolic expenditure in nonparalyzed patients and 100% resting metabolic expenditure in paralyzed patients using enteral or parenteral formulas containing at least 15% of calories as protein by the seventh day after injury is recommended.3,369
Role of Steroid Therapy
On the basis of class 1 evidence originating from a prospective randomized trial comparing dexamethasone and placebo in 300 head-injured patients, dexamethasone was found to be ineffective in improving outcome or reducing ICP in patients with severe brain injury.3,312 Methylprednisolone, when given in high doses to moderate and severe head-injured patients, is associated with increased mortality.370 Therefore, the use of steroids is not recommended in their management.
References
1. Varelas, PN, Eastwood, D, Yun, HJ, et al. Impact of a neurointensivist on outcomes in patients with head trauma treated in a neurosciences intensive care unit. J Neurosurg. 2006; 104:713–719.
2. Smith, RF, Frateschi, L, Sloan, EP, et al. The impact of volume on outcome in seriously injured trauma patients: Two years’ experience of the Chicago Trauma System. J Trauma. 1990; 30:1066–1076.
3. Bullock, R, Chestnut, R, Clifton, G, et al, Guidelines for the management of severe traumatic brain injury A joint initiative of The Brain Trauma Foundation, The American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. 2001.
4. Faul, M, Xu, L, Wald, MM, Coronado, VG. Traumatic brain injury in the United States: Emergency Department visits, hospitalizations and deaths 2002–2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010.
5. Frankowski, RF, Anneges, JF, Whitman, S. Epidemiologic and descriptive studies, Part I: The descriptive epidemiology of head trauma in the United States. In: Becker DP, Povlishock JT, eds. Central Nervous System Trauma Status Report. Bethesda, MD: National Institute of Health (NINCDS), 1985.
6. Cooper, JD, Tabaddor, K, Hauser, WA. The epidemiology of head injury in the Bronx. Neuroepidemiology. 1983; 2:70.
7. Centers for Disease Control and Prevention. Surveillance for Traumatic Brain Injury–Related Deaths—United States, 1997–2007. MMWR. 2011; 60(No. SS-O5):1–32.
8. Sosin, DM, Sacks, JJ, Smith, SM. Head injury-associated deaths in the United States from 1979-1986. JAMA. 1989; 262:2251.
9. Kerr, JA, Kay, DW, Lassman, LP. Characteristics of patients, type of accident, and mortality in a consecutive series of head injuries admitted to a neurosurgical unit. Br J Prev Soc Med. 1971; 25:179.
10. Hyder, A, Wunderlich, C, Puvanachandra, P, Gururaj, G, Kobusingye, O. The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation. 2007; 22:341–353.
11. Du Trevor, MD, Van Dellen, JR. Penetrating stab wounds to the brain: The timing of angiography in patients with the weapon already removed. Neurosurgery. 1992; 31:905.
12. Oyesiku, NM. What is the best way to assess and classify head-injured patients? In Valadka AB, Andrews BT (eds): Neurotrauma. New York: Thieme Medical Publications; 2005.
13. Teasdale, G, Jennet, B. Assessment of coma and impaired consciousness: A practical scale. Lancet. 1974; 2:81–84.
14. Starmark, JE, Stalhammer, D, Holmgren, E, et al. A comparison of the Glasgow Coma Scale and the Reaction Level Scale (RLS85). J Neurosurg. 1988; 69:699–706.
15. Stalhammer, D, Starmark, JE, Holmgren, E, et al. Assessment of responsiveness in acute cerebral disorders: A multicentre study on the reaction level scale (RLS85). Acta Neurchir (Wien). 1988; 90:73–80.
16. Benzer, A, Mitterschiffthaler, G, Marosi, M, et al. Prediction of nonsurvival after trauma: Innsbruck Coma Scale. Lancet. 1991; 338:977–978.
17. Sugiura, K, Muraoka, K, Chishiki, T, et al. The Edinburgh-2 coma scale: A new scale for assessing impaired consciousness. Neurosurgery. 1983; 12:411–415.
18. Sugiura, K, Muraoka, K, Kanazawa, C, et al. A clinical study on a system of assessment of impaired consciousness (the second report). No To Shinkei. 1978; 30:1025–1029.
19. Sugiura, K, Kanazawa, C, Sato, S, et al. A clinical study on a system of assessment of impaired consciousness (the first report). No To Shinkei. 1977; 29:879–883.
20. Stanczak, DE, White, JG, III., Gouview, WD, et al. Assessment of level of consciousness following severe neurologic insult: A comparison of the psychometric qualities of the Glasgow Coma Scale and the Comprehensive Level of Consciousness Scale. J Neurosurg. 1984; 60:955–960.
21. Salcman, M, Schepp, RS, Ducker, TB. Calculated recovery rates in severe head trauma. Neurosurgery. 1981; 8:301–308.
22. Born, JD, Albert, A, Hans, P, et al. Relative prognostic value of best motor response and brain stem reflexes in patients with severe head injury. Neurosurgery. 1985; 16:595–601.
23. Born, JD. The Glasgow-Liege Scale: Prognostic value, and evolution of motor response and brain stem reflexes after severe head injury. Acta Neurochir (Wien). 1988; 91:1–11.
24. Yen, JK, Bourke, RS, Nelson, LR, et al. Numerical grading of clinical neurological status after serious head injury. J Neurol Neurosurg Psychiatry. 1978; 41:1125–1130.
25. Teasdale, G, Knill-Jones, R, van der Sande, J. Observer variability in assessing impaired consciousness and coma. J Neurol Neurosurg Psychiatry. 1978; 41:603–610.
26. Braakman, R, Avezaat, CJ, Maas, AI, et al. Inter observer agreement in the assessment of the motor response of the Glasgow Coma Scale. Clin Neurol Neurosurg. 1977; 80:100–106.
27. Ingersoll, GL, Leyden, DB. The Glasgow Coma Scale for patients with head injuries. Crit Care Nurse. 1987; 7:26–32.
28. Rowley, G, Fielding, K. Reliability and accuracy of the Glasgow Coma Scale with experienced and inexperienced users. Lancet. 1991; 337:535–538.
29. Fielding, K, Rowley, G. Reliability of assessments by skilled observers using the Glasgow Coma Scale. Aust J Adv Nurs. 1990; 7:13–17.
30. Jennett, B, Teasdale, G, Galbraith, S, et al. Severe head injuries in three countries. J Neurol Neurosurg Psychiatry. 1977; 40:291–298.
31. Teasdale, G, Jennett, B. Assessment and prognosis of coma after head injury. Acta Neurochir (Wien). 1976; 34:45–55.
32. Jagger, J, Jane, JA, Rimel, R. The Glasgow Coma Scale: To sum or not to sum? Lancet. 1983; 2:97.
33. The Brain Trauma Foundation, The American Association of Neurological Surgeons, The Joint Section on Neurotrauma and Critical Care. Management and prognosis of severe traumatic brain injury. Glascow Coma Scale score. J Neurotrauma. 2000; 17:563–571.
34. Maas, AI, Hukkelhoven, CW, Marshall, LF, Steyerberg, EW. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: A comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery. 2005; 57:1173–1182.
35. Marshall, LF, Marshall, SB, Klauber, MR, et al. A new classification of head injury based on computerized tomography. J Neurosurg. 1991; 75:S14–S20.
36. Valadka, AB. Comment. Neurosurgery. 2005; 57:1181.
37. Bullock, MR. Comment. Neurosurgery. 2005; 57:1181.
38. Stein, SC. Minor head injury: 13 is an unlucky number. J Trauma. 2001; 50:759–760.
39. Dacey, RG, Jr., Alves, WM, Rimel, RW, et al. Neurosurgical complications after apparently minor head injury: Assessment of risk in a series of 610 patients. J Neurosurg. 1986; 65:203–210.
40. Culotta, VP, Sementilli, ME, Gerold, K, et al. Clinicopathological heterogenicity in the classification of mild head injury. Neurosurgery. 1996; 38:245–250.
41. Stein, SC, Burnett, MG. When are computed tomography scans and skull x-rays indicated for patients with minor head injury? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme Medical Publications; 2005:19–24.
42. Graham, DI, Gennarelli, TA. Pathology of brain damage after head injury. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:133–153.
43. Firlik, AD, Marion, DW. Intracranial pressure physiology and pathophysiology. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:221–228.
44. Golfinos, JG, Cooper, PR. Skull fracture and post-traumatic cerebrospinal fluid fistulae. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:155–174.
45. Braakman, R. Depressed skull fracture: Data, treatment, and follow-up in 225 consecutive cases. J Neurol Neurosurg Psychiatry. 1972; 34:395–402.
46. Bullock, MR, Chestnut, R, Ghajar, J. Surgical management of depressed cranial fractures. Neurosurgery. 58(Suppl), 2006. [S2-56–S2-60].
47. Wylen, EL, Willis, BK, Nanda, A. Infection rate with replacement of bone fragment in compound depressed skull fractures. Surg Neurol. 1999; 51:452–457.
48. Jennett, B, Miller, J. Infection after depressed fracture of skull. Implications for management of nonmissile injuries. J Neurosurg. 1972; 36:333–339.
49. Mendelow, AD, Campbell, D, Tsementzis, SA, et al. Prophylactic antimicrobial management of compound depressed skull fracture. J R Coll Surg Edinb. 1983; 28:80–83.
50. van den Heever, CM, van der Merwe, DJ. Management of depressed skull fractures. Selective conservative management of nonmissile injuries. J Neurosurg. 1989; 71:186–190.
51. Jennett, B, Miller, J, Braakman, R. Epilepsy after nonmissile depressed skull fracture. J Neurosurg. 1974; 41:208–216.
52. Bricolo, A, Pasut, L. Extradural hematoma: Toward zero mortality. A prospective study. Neurosurgery. 1984; 14:8–12.
53. Cucciniello, B, Martellotta, N, Nigro, D, Citro, E. Conservative management of extradural haematomas. Acta Neurochir (Wien). 1993; 120:47–52.
54. Kuday, C, Uzan, M, Hanci, M. Statistical analysis of the factors affecting the outcome of extradural haematomas: 115 cases. Acta Neurochir (Wien). 1994; 131:203–206.
55. Lee, E, Hung, Y, Wang, L, et al. Factors influencing the functional outcome of patients with acute epidural hematomas: Analysis of 200 patients undergoing surgery. J Trauma. 1998; 45:946–952.
56. Meier, U, Heinitz, A, Kintzel, D. Surgical outcome after severe craniocerebral trauma in childhood and adulthood. A comparative study [in German]. Unfallchirurg. 1994; 97:406–409.
57. Mohanty, A, Kolluri, V, Subbakrishna, D, et al. Prognosis of extradural haematomas in children. Pediatr Neurosurg. 1995; 23:57–63.
58. Servadei, F, Faccani, G, Roccella, P, et al. Asymptomatic extradural hematomas. Results of a multicenter study of 158 cases in minor head injury. Acta Neurochir (Wien). 1989; 96:39–45.
59. Wester, K. Decompressive surgery for “pure” epidural hematomas: Does neurosurgical expertise improve the outcome? Neurosurgery. 1999; 44:495–500.
60. Bullock, RM, Chestnut, R, Ghajar, J, et al. Surgical management of acute epidural hematomas. Neurosurgery. 58(Suppl), 2006. [S2-S7–S2-15].
61. Cohen, J, Montero, A, Israel, Z. Prognosis and clinical relevance of anisocoria craniotomy latency for epidural hematoma in comatose patients. J Trauma. 1996; 41:120–122.
62. Maggi, G, Aliberti, F, Petrone, G, Ruggiero, C. Extradural hematomas in children. J Neurosurg Sci. 1998; 42:95–99.
63. Paterniti, S, Fiore, P, Macri, E, et al. Extradural haematoma. Report of 37 consecutive cases with survival. Acta Neurochir (Wien). 1994; 131:207–210.
64. Rivas, J, Lobato, R, Sarabia, R, et al. Extradural hematoma: Analysis of factors influencing the courses of 161 patients. Neurosurgery. 1988; 23:44–51.
65. Cordobes, F, Lobato, R, Rivas, J, et al. Observations on 82 patients with extradural hematoma. Comparison of results before and after the advent of computerized tomography. J Neurosurg. 1981; 54:179–186.
66. Haselsberger, K, Pucher, R, Auer, L. Prognosis after acute subdural or epidural haemorrhage. Acta Neurochir (Wien). 1988; 90:111–116.
67. Jamjoom, A. The influence of concomitant intradural pathology on the presentation and outcome of patients with acute traumatic extradural haematoma. Acta Neurochir (Wien). 1992; 115:86–89.
68. Jamjoom, A. The difference in the outcome of surgery for traumatic extradural hematoma between patients who are admitted directly to the neurosurgical unit and those referred from another hospital. Neurosurg Rev. 1997; 20:227–230.
69. Jones, N, Molloy, C, Kloeden, C, et al. Extradural haematoma: Trends in outcome over 35 years. Br J Neurosurg. 1993; 7:465–471.
70. Sullivan, T, Jarvik, J, Cohen, W. Follow-up of conservatively managed epidural hematomas: Implications for timing of repeat CT. AJNR Am J Neuroradiol. 1999; 20:107–113.
71. van den Brink, WA, Zwienenberg, M, Zandee, SM, et al. The prognostic importance of the volume of traumatic epidural and subdural haematomas revisited. Acta Neurochir (Wien). 1999; 141:509–514.
72. Otsuka, S, Nakatsu, S, Matsumoto, S, et al. Study on cases with posterior fossa epidural hematoma: Clinical features and indications for operation. Neurol Med Chir (Tokyo). 1990; 30:24–28.
73. Poon, W, Li, A. Comparison of management outcome of primary and secondary referred patients with traumatic extradural hematoma in a neurosurgical unit. Injury. 1991; 22:323–325.
74. Hunt, J, Hill, D, Besser, M, et al. Outcome of patients with neurotrauma: The effect of a regionalized trauma system. Aust N Z J Surg. 1995; 65:83–86.
75. Pillay, R, Peter, J. Extradural haematomas in children. S Afr Med J. 1995; 85:672–674.
76. Lobato, R, Cordobes, F, Rivas, J, et al. Outcome from severe head injury related to the type of intracranial lesion. A computerized tomography study. J Neurosurg. 1983; 59:762–774.
77. Seelig, J, Marshall, L, Toutant, S, et al. Traumatic acute epidural hematoma: Unrecognized high lethality in comatose patients. Neurosurgery. 1984; 15:617–620.
78. Cook, R, Dorsch, N, Fearnside, M, Chaseling, R. Outcome prediction in extradural haematomas. Acta Neurochir (Wien). 1988; 95:90–94.
79. Heinzelmann, M, Platz, A, Imhof, H. Outcome after acute extradural haematoma, influence of additional injuries and neurological complications in the ICU. Injury. 1996; 27:345–349.
80. Sakas, D, Bullock, M, Teasdale, G. One-year outcome following craniotomy or traumatic hematoma in patients with fixed dilated pupils. J Neurosurg. 1995; 82:961–965.
81. Gennarelli, T, Spielman, G, Langfitt, T, et al. Influence of the type of intracranial lesion on outcome from severe head injury. J Neurosurg. 1982; 56:26–32.
82. Lobato, R, Rivas, J, Cordobes, F, et al. Acute epidural hematoma: An analysis of factors influencing the outcome of patients undergoing surgery in coma. J Neurosurg. 1988; 68:48–57.
83. Uzan, M, Yentur, E, Hanci, M, et al. Is it possible to recover from uncal herniation? Analysis of 71 head injured cases. J Neurosurg Sci. 1998; 42:89–94.
84. Bullock, R, Smith, R, van Dellen, JR. Nonoperative management of extradural hematoma. Neurosurgery. 1985; 16:602–606.
85. Suzuki, S, Endo, M, Kurata, A, et al. Efficacy of endovascular surgery for the treatment of acute epidural hematomas. Am J Neuroradiol. 2004; 25:1177–1180.
86. Gennarelli, TA, Thibault, LE. Biomechanics of acute subdural hematoma. J Trauma. 1982; 22:680–686.
87. Drake, CG. Subdural hematoma from arterial rupture. J Neurosurg. 1961; 8:597.
88. Shenkin, HA. Acute subdural hematoma: Review of 39 consecutive cases with high incidence of cortical artery rupture. J Neurosurg. 1982; 57:254–257.
89. Cooper, PR. Post-traumatic intracranial mass lesions. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:293–348.
90. Ersahin, Y, Mutluer, S. Posterior fossa extradural hematomas in children. Pediatr Neurosurg. 1993; 19:31–33.
91. Seelig, J, Becker, D, Miller, J, et al. Traumatic acute subdural hematoma: Major mortality reduction in comatose patients treated within four hours. N Engl J Med. 1981; 304:1511–1518.
92. Servadei, F, Nasi, M, Cremonini, A, et al. Importance of a reliable admission Glasgow Coma Scale score for determining the need for evacuation of posttraumatic subdural hematomas: A prospective study of 65 patients. J Trauma. 1998; 44:868–873.
93. Wilberger, JJ, Harris, M, Diamond, D. Acute subdural hematoma: Morbidity, mortality, and operative timing. J Neurosurg. 1991; 74:212–218.
94. Dent, D, Croce, M, Menke, P, et al. Prognostic factors after acute subdural hematoma. J Trauma. 1995; 39:36–42.
95. Gabl, M, Mohsenipour, I, Benedetto, K. Acute posttraumatic subdural hematoma in advanced age [in German]. Unfallchirurgie. 1989; 15:273–278.
96. Koc, R, Akdemir, H, Oktem, I, et al. Acute subdural hematoma: Outcome and outcome prediction. Neurosurg Rev. 1997; 20:239–244.
97. Massaro, F, Lanotte, M, Faccani, G, Triolo, C. One hundred and twenty-seven cases of acute subdural haematoma operated on. Correlation between CT scan findings and outcome. Acta Neurochir (Wien). 1996; 138:185–191.
98. Howard, MA, 3rd., Gross, AS, Dacey, RJ, Jr., Winn, HR. Acute subdural hematomas: An age-dependent clinical entity. J Neurosurg. 1989; 71:858–863.
99. Cagetti, B, Cossu, M, Pau, A, et al. The outcome from acute subdural and epidural intracranial haematomas in very elderly patients. Br J Neurosurg. 1992; 6:227–231.
100. Jamjoom, A. Justification for evacuating acute subdural haematomas in patients above the age of 75 years. Injury. 1992; 23:518–520.
101. Kotwica, Z, Brzezinski, J. Acute subdural haematoma in adults: An analysis of outcome in comatose patients. Acta Neurochir (Wien). 1993; 121:95–99.
102. Servadei, F, Nasi, M, Giuliani, G, et al. CT prognostic factors in acute subdural haematomas: The value of the “worst” CT scan. Br J Neurosurg. 2000; 14:110–116.
103. Fell, D, Fitzgerald, S, Moiel, R, Caram, P. Acute subdural hematomas. Review of 144 cases. J Neurosurg. 1975; 42:37–42.
104. Hatashita, S, Koga, N, Hosaka, Y, Takagi, S. Acute subdural hematoma: Severity of injury, surgical intervention, and mortality. Neurol Med Chir (Tokyo). 1993; 33:13–18.
105. Zumkeller, M, Behrmann, R, Heissler, H, Dietz, H. Computed tomographic criteria and survival rate for patients with acute subdural hematoma. Neurosurgery. 1996; 39:708–712.
106. Domenicucci, M, Strzelecki, J, Delfini, R. Acute posttraumatic subdural hematomas: “Intradural” computed tomographic appearance as a favorable prognostic factor. Neurosurgery. 1998; 42:51–55.
107. Jamieson, KG, Yelland, JDN. Surgically treated traumatic subdural hematomas. J Neurosurg. 1972; 37:137–149.
108. Stone, JL, Rifai, MHS, Sugar, O, et al. Subdural hematomas. I. Acute subdural hematomas: Progress in definition, clinical pathology, and therapy. Surg Neurol. 1983; 19:216–231.
109. Miller, JD, Bullock, R, Graham, DJ, et al. Ischemic brain damage in a model of acute subdural hematoma. Neurosurgery. 1990; 27:433–439.
110. Yilmazlar, S, Kuday, C, Oz, B, et al. Blood degradation products play a role in cerebral ischemia caused by acute subdural hematoma. J Neurosurg Sci. 1997; 41:379–385.
111. Brain Trauma Foundation. Early indicators of prognosis in severe traumatic brain injury. J Neurotrauma. 2000; 17:535–627.
112. Kotwica, Z, Jakubowski, J. Acute head injuries in the elderly. An analysis of 136 consecutive patients. Acta Neurochir (Wien). 1992; 118:98–102.
113. Chieregato, A, Fainardi, E, Morselli-Labate, AM, et al. Factors associated with neurological outcome and lesion progression in traumatic subarachnoid hemorrhage. Neurosurgery. 2005; 56:671–680.
114. Britt, PM, Heiserman, JE. Imaging evaluation. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:361–396.
115. Chandler, JP, Batjer, HH, Kuznits, S, et al. Intracranial and cervical vascular injuries. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:361–396.
116. Sander, D, Kligelhofer, J. Cerebral vasospasm following post-traumatic subarachnoid hemorrhage evaluated by transcranial Doppler ultrasonography. J Neurol Sci. 1993; 11:1.
117. Marshall, LF, Bruce, DA, Bruno, L, et al. Vertebrobasilar spasm: A significant cause of neurologic deficit in head injury. J Neurosurg. 1978; 48:560–564.
118. Greene, KA, Jacobawitz, R, Marciano, FF, et al. Impact of traumatic subarachnoid hemorrhage on outcome in nonpenetrating head injury. Part II: Relationship to clinical course and outcome variables during acute hospitalization. J Trauma. 1996; 41:964–971.
119. Greene, KA, Marciano, FF, Johnson, BA, et al. Impact of traumatic subarachnoid hemorrhage on outcome in nonpenetrating head injury. Part I: A proposed computerized tomography grading scale. J Neurosurg. 1995; 83:445–452.
120. Kakarieka, A. Review of traumatic subarachnoid hemorrhage. Neurol Res. 1997; 19:230–232.
121. Bullock, R, Golek, J, Blake, G. Traumatic intracerebral hematoma: which patients should undergo surgical evacuation? CT scan features and ICP monitoring as a basis for decision making. Surg Neurol. 1989; 32:181–187.
122. Lobato, RD, Gomez, PA, Alday, R, et al. Sequential computerized tomography changes and related final outcome in severe head injury patients. Acta Neurochir (Wien). 1997; 139:385–391.
123. Miller, JD, Butterworth, JF, Gudeman, SK, et al. Further experience in the management of severe head injury. J Neurosurg. 1981; 54:289–299.
124. Nordstrom, C, Messeter, K, Sundbarg, G, Wahlander, S. Severe traumatic brain lesions in Sweden. Part I: Aspects of management in non-neurosurgical clinics. Brain Inj. 1989; 3:247–265.
125. Singounas, E. Severe head injury in a paediatric population. J Neurosurg Sci. 1992; 36:201–206.
126. Soloniuk, D, Pitts, LH, Lovely, M, Bartkowski, H. Traumatic intracerebral hematomas: Timing of appearance and indications for operative removal. J Trauma. 1986; 26:787–794.
127. Bullock, RM, Chestnut, R, Ghajar, J, et al. Surgical management of traumatic parenchymal lesions. Neurosurgery. 58(Suppl), 2006. [S2-25–S2-46].
128. Schonauer, M, Schisano, G, Cimino, R, et al. Space occupying contusions of cerebral lobes after closed brain injury: Considerations about 51 cases. J Neurosurg Sci. 1979; 23:279–288.
129. Evans, JP, Scheinker, IM. Histological studies of the brain following head trauama. II. Post-traumatic petechial and massive intracerebral hemorrhage. J Neurosurg. 1946; 3:101–113.
130. Graham, DI, Gennarelli, TA. Pathology of brain damage after head injury. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:134–153.
131. Adams, JH, Graham, DI, Genuarelli, TA. Contemporary neuropathological considerations regarding brain damage after head injury. In: Becker DP, Povlishock JT, eds. Central Nervous System Trauma: Status Report. Bethesda, MD: National Institute of Neurological and Communicative Disorders and Stroke, National Institutes of Health, 1985.
132. Ribas, G, Jane, JA. Traumatic contusions and intracerebral hematomas. J Neurotrauma. 1992; 9(Suppl 1):S265.
133. Servadei, F, Murray, GD, Penny, K, et al. The value of the “worst” computed tomographic scan in clinical studies of moderate and severe head injury. European Brain Injury Consortium. Neurosurgery. 2000; 46:70–75.
134. Servadei, F, Nanni, A, Nasi, M, et al. Evolving brain lesions in the first 12 hours after head injury: Analysis of 37 comatose patients. Neurosurgery. 1995; 37:899–906.
135. Chang, EF, Meeker, M, Holland, MC. Acute traumatic intraparenchymal hemorrhage: Risk factors for progression in the early post-injury period. Neurosurgery. 2006; 58:647–656.
136. Adams, JH, Graham, DI, Gennarelli, TA. Neuropathology of acceleration induced head injury in the subhuman primate. In: Grossman RG, Gildenberg PL, eds. Head Injury: Basic Clinical Aspects. New York: Raven Press, 1992.
137. Courville, CB. Coup-contrecoup mechanism of craniocerebral injuries—some observations. Arch Surg. 1942; 4:19.
138. Ommaya, AK, Grubb, RL, Nauman, RA. Coup and contrecoup injury: Observations on the mechanics of visible brain injuries in the rhesus monkey. J Neurosurg. 1971; 35:305.
139. Gennarelli, TA, Thibault, LE. Biomechanics of head injury. In: Wilkins RH, Rengachary SS, eds. Neurosurgery. New York: McGraw-Hill; 1985:1531–1536.
140. Daniel, PM, Treip, CS. The pathology of the pituitary gland in head injury. In: Gardner-Hill H, ed. Modern Trends in Endocrinology. London: Butterworth, 1961.
141. Crompton, MR. Hypothalamic and pituitary lesions. In: Vinken PJ, Bruyn GN, eds. Handbook of Clinical Neurology. Injuries of the Brain and Skull, part I. Amsterdam: North-Holland, 1975.
142. Landau, H, Adin, I, Spitz, IM. Pituitary insufficiency following head injury. Israel J Med Sci. 1978; 14:785.
143. Edwards, OM, Clark, JDA. Post-traumatic hypopituitarism. Six cases and a review of the literature. Medicine. 1986; 65:281.
144. Blumbergs, PC, Jones, NR, Nath, JB. Diffuse axonal injury in head trauma. J Neurol Neurosurg Psychiatry. 1989; 52:838.
145. Gennarelli, TA, Thibault, LE, Adams, JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982; 12:564.
146. Levi, L, Guilburd, J, Lemberger, A, et al. Diffuse axonal injury: Analysis of 100 patients with radiological signs. Neurosurgery. 1990; 27:429.
147. Gennarelli, TA. Head injury in man and experimental animals—Clinical aspects. Acta Neuroclin. 1983; 32(Suppl):1.
148. Lindenberg, R, Freytag, E. A mechanism of cerebral contusions: A pathologic anatomic study. Arch Pathol. 1960; 69:440.
149. Michaud, LJ, Rivara, FP, Grady, MS, et al. Predictors of survival and severity of disability after severe brain injury in children. Neurosurgery. 1992; 31:254.
150. Sosin, DM, Sacks, JJ, Smith, SM. Head injury associated deaths in the United States from 1979 to 1986. JAMA. 1989; 262:2251–2255.
151. Wintemute, GJ. The relationship between firearm design and firearm violence: Handguns in the 1990’s. JAMA. 1996; 275:1749–1753.
152. Harrington, T, Apostolides, P. Penetrating brain injury. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:349–359.
153. Roth, J, Mayo, A, Elran, H, Razon, N, et al. Brain injuries caused by spherical bolts. J Neurosurg. 2005; 102:864–869.
154. Allen, IV, Scott, R, Tanner, JA. Experimental high-velocity missile head injury. Injury. 1982; 14:183–193.
155. Jahrsdoerfer, RA, Johns, ME, Cantrell, RW. Penetrating wounds of the head and neck. Arch Otolaryngol. 1979; 105:721–725.
156. Marion DW, ed. Traumatic Brain Injury. New York: Thieme Medical Publishers, 1999.
157. Fackler, ML. Wound ballistics. A review of common misconceptions. JAMA. 1988; 259:2730–2736.
158. Mendelson, JA. The relationship between mechanisms of wounding and principles of treatment of missile wounds. J Trauma. 1991; 31:1181–1202.
159. Lindsey, D. The idolatry of velocity, or lies, damn lies, and ballistics. J Trauma. 1980; 20:1068–1069.
160. Carey, ME, Sarna, GS, Farrell, JB. Experimental missile wound to the brain. J Neurosurg. 1989; 71:754–764.
161. Cushing, H. A study of a series of wounds involving the brain and its enveloping structures. Br J Surg. 1918; 5:558–684.
162. Walters, BC, Aarabi, B, Chestnut, RM, et al. Methodology for development of guidelines for the management of penetrating brain injury. J Trauma. 2001; 51:S16–S25.
163. Levi, L, Borovich, B, Guilburd, JL, et al. Wartime neurosurgical experience in Lebanon, 1982-85, I: Penetrating craniocerebral injuries. Isr J Med Sci. 1990; 26:548–554.
164. Levi, L, Borovich, B, Guilburd, JL, et al. Wartime neurosurgical experience in Lebanon, 1982-85. II: Penetrating craniocerebral injuries. Isr J Med Sci. 1990; 26:555–558.
165. Aarabi, B. Surgical outcome in 435 patients who sustained missile head wounds during the Iran-Iraq war. Neurosurgery. 1990; 27:692–695.
166. Ameen, A. The management of acute craniocerebral injuries caused by missiles: Analysis of 110 consecutive penetrating wounds of the brain from Basrah. Injury. 1984; 16:88–90.
167. Brandvold, B, Levi, L, Feinsod, M, George, ED. Penetrating craniocerebral injuries in the Israeli involvement in the Lebanese conflict, 1982-85. J Neurosurg. 1990; 72:15–21.
168. Carey, ME, Young, HF, Mathis, JL. The neurosurgical treatment of craniocerebral missile wounds in Vietnam. Surg Gynecol Obstet. 1972; 135:386–390.
169. Gonul, E, Baysefer, A, Kahraman, S, et al. Causes of infections and management results in penetrating craniocerebral injuries. Neurosurg Rev. 1997; 20:177–181.
170. Hammon, WM, Kempe, LG. Analysis of 2187 consecutive penetrating wounds of the brain from Vietnam. J Neurosurg. 1971; 34:127–131.
171. Taha, JM, Haddad, FS, Brown, JA. Intracranial infection after missile injuries to the brain: Report of 30 cases from the Lebanese conflict. Neurosurgery. 1991; 29:864–868.
172. Carey, ME, Young, HF, Rish, BL, Mathis, JL. Follow-up study of 103 American soldiers who sustained a brain wound in Vietnam. J Neurosurg. 1974; 41:542–549.
173. Rosegay, H. Craniocerebral injuries. In: Ravitch MM, ed. Current Problems in Surgery: Military Surgical Practices of the United States Army in Vietnam. Chicago: Year Book Medical Publishers, 1966.
174. Chaudhri, KA, Choudhury, AR, al Moutaery, KR, Cybulski, GR. Penetrating craniocerebral shrapnel injuries during “Operation Desert Storm”: Early results of a conservative surgical treatment. Acta Neurochir (Wien). 1994; 126:120–123.
175. Martin, J, Campbell, EH. Early complications following penetrating wounds of the skull. J Neurosurg. 1946; 2:58–73.
176. Aarabi, B, Taghipour, M, Alibaii, E, Kamgarpour, A. Central nervous system infections after military missile head wounds. Neurosurgery. 1998; 42:500–509.
177. Caveness, WF, Walker, AE, Ashcroft, PB, et al. Incidence of posttraumatic epilepsy in Korean veterans as compared with those from World War I and World War II. J Neurosurg. 1962; 19:122–129.
178. Salazar, AM, Jabbari, B, Vance, SC, et al. Epilepsy after penetrating head injury, I: Clinical correlates—A report of the Vietnam Head Injury Study. Neurology. 1985; 35:1406–1414.
179. Rish, BL, Caveness, WF, Dillon, JD, et al. Analysis of brain abscess after penetrating craniocerebral injuries in Vietnam. Neurosurgery. 1981; 9:535–541.
180. Aarabi, B. Comparative study of bacteriological contamination between primary and secondary exploration of missile head wounds. Neurosurgery. 1987; 20:610–616.
181. Nagib, MG, Rockswold, GL, Sherman, RS, Lagaard, MW. Civilian gunshot wounds to the brain: Prognosis and management. Neurosurgery. 1986; 18:533–537.
182. Suddaby, L, Weir, B, Forsyth, C. The management of 22 caliber gunshot wounds of the brain: A review of 49 cases. Can J Neurol Sci. 1987; 14:268–272.
183. Byrnes, DP, Crockard, HA, Gordon, DS, Gleadhill, CA. Penetrating craniocerebral missile injuries in the civil disturbances in Northern Ireland. Br J Surg. 1974; 61:169–176.
184. Kim, KA, Wang, MY, McNatt, SA, et al. Vector analysis correlating bullet trajectory to outcome after civilian through-and-through gunshot wound to the head: Using imaging cues to predict fatal outcome. Neurosurgery. 2005; 57:737–747.
185. Aarabi, B. Management of traumatic aneurysms caused by high velocity missile head wounds. Neurosurg Clin North Am. 1995; 6:775–797.
186. Aldrich, EF, Eisenberg, HM, Saydjari, C, et al. Predictors of mortality in severely head-injured patients with civilian gunshot wound: A report from the NIH Traumatic Coma Data Bank. Surg Neurol. 1992; 38:418–423.
187. Levy, ML, Rezai, A, Masri, LS, et al. The significance of subarachnoid hemorrhage after penetrating craniocerebral injury: Correlations with angiography and outcome in civilian population. Neurosurgery. 1993; 32:532–540.
188. Aarabi, B. Traumatic aneurysms of brain due to high velocity missile head wounds. Neurosurgery. 1988; 22:1056–1063.
189. Haddad, FS, Haddad, GF, Taha, J. Traumatic intracranial aneurysms caused by missiles: Their presentation and management. Neurosurgery. 1997; 28:1–7.
190. Amirjamshidi, A, Rahmat, H, Abbassioun, K. Traumatic aneurysms and arteriovenous fistulas of intracranial vessels associated with penetrating head injuries occurring during war: Principles and pitfalls in diagnosis and management—A survey of 31 cases and review of the literature. J Neurosurg. 1996; 84:769–780.
191. Jinkins, JR, Dadsetan, MR, Sener, RN, et al. Value of acute-phase angiography in the detection of vascular injuries caused by gunshot wounds to the head: Analysis of 12 cases. AJR Am J Roentgenol. 1992; 159:365–368.
192. Crockard, HA. Early intracranial pressure studies in gunshot wounds of the brain. J Trauma. 1975; 15:339–347.
193. Lillard, PL. Five years experience with penetrating craniocerebral gunshot wounds. Surg Neurol. 1978; 9:79–83.
194. Sarnaik, AP, Kopec, J, Moylan, P, et al. Role of aggressive intracranial pressure control in management of pediatric craniocerebral gunshot wounds with unfavorable features. J Trauma. 1989; 29:1434–1437.
195. Miner, ME, Ewing-Cobbs, L, Kopaniky, DR, et al. The results of treatment of gunshot wounds to the brain in children. Neurosurgery. 1990; 26:20–25.
196. Guidelines for the management of severe traumatic brain injury. Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. J Neurotrauma. 2000; 17:451–627.
197. Harrington, T, Apostolides, P. Penetrating brain injury. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:349–359.
198. Marmarou, A, Signoretti, S, Fatouros, PP, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2000; 104:720–730.
199. Brain Trauma Foundation: Guidelines for the management of severe traumatic brain injury, 3rd edition. Chap. 1, Blood pressure and oxygenation. Journal of Neurotrauma. 2007; 24(1):S-7–S-13.
200. Brain Trauma Foundation. Guidelines for the management of severe traumatic brain injury, 3rd edition. Chap. 9, Cerebral perfusion thresholds. Journal of Neurotrauma. 2007; 24(1):S-59–S-64.
201. Rosner, MJ, Coley, IB. Cerebral perfusion pressure, intracranial pressure, and head elevation. J Neurosurg. 1986; 65:636–641.
202. Bouma, GJ, Muizelaar, JP. Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation. J Neurosurg. 1990; 73:368–374.
203. Bruce, DA, Langfitt, TW, Miller, JD, et al. Regional cerebral blood flow, intracranial pressure, and brain metabolism in comatose patients. J Neurosurg. 1973; 38:131–144.
204. Hoff, JT. Cerebral perfusion pressure, autoregulation and the PVI reflection point. In: Betz AL, ed. Intracranial Pressure VII. Berlin: Springer-Verlag; 1988:829–833.
205. Fortune, JB, Feustel, PJ, Weigle, CGM, et al. Continuous measurement of jugular venous oxygen saturation in response to transient elevations of blood pressure in head-injured patients. J Neurosurg. 1994; 80:461–468.
206. Brain Trauma Foundation: Guidelines for the management of severe traumatic brain injury (1st edition). Chap. 8, Guidelines for cerebral perfusion. 1995, pp 8-1–8-10.
207. Rosner, MJ, Rosner, SD, Johnson, AH. Cerebral perfusion pressure: Management protocol and clinical results. J Neurosurg. 1995; 83:949–962.
208. Clifton, GL, Allen, S, Barrodale, P, et al. A phase II study of moderate hypothermia in severe brain injury. J Neurotrauma. 1993; 10:263–271.
209. Marion, DW, Penrod, LE, Kelsey, SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997; 336:540–546.
210. Rosner, MJ, Daughton, S. Cerebral perfusion pressure management in head injury. J Trauma. 1990; 30:933–941.
211. Yoshida, A, Shima, T, Okada, Y, et al. Outcome of patients with severe head injury—Evaluation by cerebral perfusion pressure. In: Nakamura N, Hashimoto T, Yasue M, eds. Recent Advances in Neurotraumatology. Hong Kong: Springer-Verlag; 1993:309–312.
212. Marshall, LF, Gautille, T, Klauber, MR, et al. The outcome of severe closed head injury. J Neurosurg. 1991; 75:S28–S36.
213. Kiening, KL, Hartl, R, Unterberg, AW, et al. Brain tissue pO2-monitoring in comatose patients: Implications for therapy. Neurol Res. 1997; 19:233–240.
214. Robertson, CS, Valadka, AB, Hannay, HJ, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999; 27:2086–2095.
215. Contant, CF, Valadka, AB, Gopinath, SP, et al. Adult respiratory distress syndrome: A complication of induced hypertension after severe head injury. J Neurosurg. 2001; 95:560–568.
216. Bratton, SL, Davis, RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997; 40:707–712.
217. Juul, N, Morris, GF, Marshall, SB, et al. Intracranial hypertension and cerebral perfusion pressure: Influence on neurological deterioration and outcome in severe head injury. The Executive Committee of the International Selfotel Trial. J Neurosurg. 2000; 92:106.
218. Martin, NA, Patwardhan, RV, Alexander, MJ, et al. Characterization of cerebral hemodynamic phases following severe head trauma: Hypoperfusion, hyperemia, and vasospasm. J Neurosurg. 1997; 87:9–19.
219. Chestnut, RM, Marshall, LF, Klauber, MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993; 34:216–222.
220. Stocchetti, N, Furlan, A, Volta, F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma. 1996; 40:764–767.
221. Bouma, GJ, Muizelaar, JP, Choi, PG, et al. Cerebral circulation and metabolism after severe traumatic brain injury: The elusive role of ischemia. J Neurosurg. 1991; 75:685–693.
222. Vigue, B, Ract, C, Benayed, M, et al. Early SjvO2 monitoring in patients with severe brain trauma. Intensive Care Med. 1999; 25:445–451.
223. Eker, C, Asgeirsson, B, Grande, PO, et al. Improved outcome after severe head injury with a new therapy based on principles for brain volume regulation and preserved microcirculation. Crit Care Med. 1998; 26:1881–1886.
224. Schroder, ML, Muizelaar, JP, Kuta, AJ, et al. Thresholds for cerebral ischemia after severe head injury: Relationship with late CT findings and outcome. J Neurotrauma. 1996; 13:17–23.
225. Bouma, GJ, Muizelaar, JP, Stringer, WA, et al. Ultraearly evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J Neurosurg. 1992; 77:360–368.
226. Carmona Suazo, JA, Maas, AI, van den Brink, WA, et al. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med. 2000; 28:3268–3274.
227. Dings, J, Meixensberger, J, Amschler, J, et al. Brain tissue PO2 in relation to cerebral perfusion pressure, TCD findings and TCD-CO2 reactivity after severe head injury. Acta Neurochir (Wien). 1996; 138:425–434.
228. Kiening, KL, Sarrafzadeh, AS, Stover, JF, Unterberg, AW. Should I monitor brain tissue PO2? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme Medical Publishers; 2005:62–67.
229. Imberti, R, Bellinzona, G, Langer, M. Cerebral tissue Po2 and SjvO2 changes during moderate hyperventilation in patients with severe traumatic brain injury. J Neurosurg. 2002; 96:97–102.
230. Zhi, DS, Zhang, S, Zhou, LG. Continuous monitoring of brain tissue oxygen pressure in patients with severe head injury during moderate hypothermia. Surg Neurol. 1999; 52:393–396.
231. Dings, J, Meixensberger, J, Amschler, J, et al. Continuous monitoring of brain tissue PO2: A new tool to minimize the risk of ischemia caused by hyperventilation therapy. Zentralbl Neurochir. 1996; 57:177–183.
232. Schneider, GH, Sarrafzadeh, AS, Kiening, KL, et al. Influence of hyperventilation on brain tissue Po2, Pco2, and pH in patients with intracranial hypertension. Acta Neurochir Suppl (Wien). 1998; 71:62–65.
233. Unterberg, AW, Kiening, KL, Hartl, R, et al. Multimodal monitoring in patients with head injury: Evaluation of the effects of treatment on cerebral oxygenation. J Trauma. 1997; 42:S32–S37.
234. Muizelaar, JP, Marmarou, A, Ward, JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: A randomized clinical trial. J Neurosurg. 1991; 75:731–739.
235. Clifton, GL, Miller, ER, Choi, SC, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001; 344:556–563.
236. Alderson, P, Gadkary, C, Signorini, DF. Therapeutic hypothermia for head injury. Update of Cochrane Database Syst Rev. (1):2002.
237. Henderson, W, Dhingra, V, Chittock, D, et al. Hypothermia in the management of traumatic brain injury. A systematic review and meta-analysis. Intensive Care Med. 2003; 29:1637–1644.
238. Brain Trauma Foundation. Prophylactic hypothermia. J Neurotrauma. 2007; 24(Suppl 1):S-21–S-25.
239. Clifton, GL. Is keeping cool still hot? An update on hypothermia in brain injury. Curr Opin Crit Care. 2004; 10:116–119.
240. Guy, L, Ludbrook, GL. Sedation and anesthesia. In: Reilly P, Bullock R, eds. Head Injury. London: Chapman & Hall; 1997:364–383.
241. Kee, DB, Wood, JH. Rheology of the cerebral circulation. Neurosurgery. 1984; 15:125–131.
242. Korosue, K, Heros, RC. Mechanism of cerebral blood flow augmentation by hemodilution in rabbits. Stroke. 1992; 23:1487–1492.
243. Muizelaar, JP, Wei, EP, Kontos, HA, et al. Cerebral blood flow is regulated by changes in blood pressure and blood viscosity. Stroke. 1986; 17:44–48.
244. Muizelaar, JP, Wei, EP, Kontos, HA, Becker, DP. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg. 1983; 59:822–828.
245. Deutsch, H, Ullman, JS. What is the optimal hematocrit and hemoglobin for head injured patients? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme Medical Publishers; 2005:88–90.
246. Klatzo, I. Evolution of brain edema concepts. Acta Neurochir Suppl. 1994; 60:3–6.
247. Rosener, MJ. Pathophysiology and management of increased intracranial pressure. In: Andrews BT, ed. Neurosurgical Intensive Care. San Francisco: McGraw-Hill; 1993:57–112.
248. Aarabi, B, Hesdorffer, D, Ahn, E, et al. Outcome following decompressive cranietomy for malignant swelling due to severe head injury. J Neurosurg. 2006; 104:469–479.
249. Bergsneider, M, Wu, C, Huang, H, et al, Early abnormalities of regional brain metabolism following human traumatic brain injury: FDG and O-15 PET studies of hyperglycolysis, metabolic depression and ischemia Paper presented at the American Association of Neurological Surgeons Annual Meeting, April 16-21. 2005.
250. Cruz, J, Jaggi, JL, Hoffstad, OJ. Cerebral blood flow, vascular resistance, and oxygen metabolism in acute brain trauma: Redefining the role of cerebral perfusion pressure? Crit Care Med. 1995; 23:142–1417.
251. Marmarou, A. Pathophysiology of traumatic brain edema: Current concepts. Acta Neurochir Suppl. 2003; 86:7–10.
252. Marmarou, A, Fatouros, PP, Barzo, P, et al. Contribution of edema and cerebral blood volume to traumatic brain swelling in head injured patients. J Neurosurg. 2000; 93:183–193.
253. Robertson, CS, Gopinath, SP, Uzura, M, et al. Metabolic changes in the brain during transient ischemia measured with microdialysis. Neurol Res. 1998; 20(Suppl 1):S91–S94.
254. Verweij, BH, Muizelaar, JP, Vinas, FC, et al. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 2000; 93:815–820.
255. Zauner, A, Daugherty, WP, Bullock, MR, Warner, DS. Brain oxygenation and energy metabolism: Part 1—Biological function and pathophysiology. Neurosurgery. 2002; 51:289–302.
256. Langfitt, TW. Etiology and management of acute brain swelling. W V Med J. 1966; 62:49.
257. Langfitt, TW, Marshall, WJ, Kassell, NF, Schutta, HS. The pathophysiology of brain swelling produced by mechanical trauma and hypertension. Scand J Clin Lab Invest Suppl. 1968; 102:xiv.
258. Langfitt, TW, Tannanbaum, HM, Kassell, NF. The etiology of acute brain swelling following experimental brain injury. J Neurosurg. 1966; 24:47–56.
259. Langfitt, TW, Weinstein, JD, Kassell, NF. Cerebral vasomotor paralysis as a cause of brain swelling. Trans Am Neurol Assoc. 1964; 89:214–215.
260. Langfitt, TW, Weinstein, JD, Sklar, FH, et al. Contribution of intracranial blood volume to three forms of experimental brain swelling. Johns Hopkins Med J. 1968; 122:261–270.
261. Kita, H, Marmarou, A. The cause of acute brain swelling after the closed head injury in rats. Acta Neurochir Suppl (Wien). 1994; 60:452–455.
262. Barzo, P, Marmarou, A, Fatouros, P, et al. Magnetic resonance imaging-monitored acute blood-brain barrier changes in experimental traumatic brain injury. J Neurosurg. 1996; 85:1113–1121.
263. Barzo, P, Marmarou, A, Fatouros, P, et al. Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion weighted-imaging. J Neurosurg. 1997; 87:900–907.
264. Stroop, R, Thomale, UW, Pauser, S, et al. Magnetic resonance imaging studies with cluster algorithm for characterization of brain edema after controlled cortical impact (CCII). Acta Neurochir Suppl. 1998; 71:303–305.
265. Marshall, LF, Smith, RW, Rauscher, LA, et al. Mannitol dose requirements in brain-injured patients. J Neurosurg. 1978; 48:169–172.
266. Nath, F, Galbraith, S. The effect of mannitol on cerebral white matter water content. J Neurosurg. 1986; 65:41–43.
267. Burke, AM, Quest, DO, Chien, S, et al. The effects of mannitol on blood viscosity. J Neurosurg. 1981; 55:550–553.
268. Rosner, MJ, Coley, I. Cerebral perfusion pressure: A hemodynamic mechanism of mannitol and the postmannitol hemogram. Neurosurgery. 1987; 21:147–156.
269. Bruce, DA, Langfitt, TW, Miller, JD, et al. Regional cerebral blood flow, intracranial pressure, and brain metabolism in comatose patients. J Neurosurg. 1973; 38:131–144.
270. Muizelaar, JP, Lutz, HA, III., Becker, DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984; 61:700–706.
271. Cruz, J, Minoja, G, Okuchi, K. Improving clinical outcomes from acute subdural hematomas with the emergency preoperative administration of high doses of mannitol: A randomized trial. Neurosurgery. 2001; 49:864–871.
272. Cruz, J, Minoja, G, Okuchi, K. Major clinical and physiological benefits of early high doses of mannitol for intraparenchymal temporal lobe hemorrhages with abnormal pupillary widening: A randomized trial. Neurosurgery. 2002; 51:628–638.
273. Schrot, RJ, Muizelaar, JP. Is there a “best” way to give mannitol? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme; 2005:142–147.
274. Mendelow, AD, Teasdale, GM, Russell, T, et al. Effect of mannitol on cerebral blood flow and cerebral perfusion pressure in human head injury. J Neurosurg. 1985; 63:43–48.
275. Kaufmann, AM, Cardoso, ER. Aggravation of vasogenic cerebral edema by multiple-dose mannitol. J Neurosurg. 1992; 77:584–589.
276. Chestnut, RM. Medical management of intracranial pressure. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:229–263.
277. Becker, D, Vries, J. The alleviation of increased intracranial pressure by the chronic administration of osmotic agents. In: Brock M, Dietz H, eds. Intracranial Pressure. Berlin: Springer-Verlag; 1972:309–315.
278. Dorman, HR, Sondheimer, JH, Cadnapaphornchai, P. Mannitol induced acute renal failure. Medicine (Baltimore). 1990; 69:153–159.
279. Wilberger, JE. Emergency care and initial evaluation. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:27–40.
280. Maas, A. Effects of 23. 4% sodium chloride solution in reducing intracranial pressure in patients with traumatic brain injury: A preliminary study. Neurosurgery. 2005; 57:736.
281. Brain Trauma Foundation. Guidelines for prehospital management of traumatic brain injury, 2nd edition. Chap. 5 Treatment: Fluid resuscitation. Prehospital Emergency Care. 2007; 12(1):S32–S36.
282. Bullock, RM, Chesnut, RM, Clifton, GL, et al. Management and prognosis of severe traumatic brain injury. J Neurotrauma. 2000; 17:449–553.
283. Bitterman, H, Triolo, J, Lefer, AM. Use of hypertonic saline in the treatment of hemorrhagic shock. Circ Shock. 1987; 21:271–283.
284. de Felippe, J, Jr., Timoner, J, Velasco, IT, et al. Treatment of refractory hypovolaemic shock by 7. 5% sodium chloride injections. Lancet. 1980; 2:1002–1004.
285. Kreimeier, U, Bruckner, UB, Niemczyk, S, Messmer, K. Hyperosmotic saline dextran for resuscitation from traumatic-hemorrhagic hypotension: Effect on regional blood flow. Circ Shock. 1990; 32:83–99.
286. Nakayama, S, Sibley, L, Gunther, RA, et al. Small-volume resuscitation with hypertonic saline (2,400 mOsm/liter) during hemorrhagic shock. Circ Shock. 1984; 13:149–159.
287. Cooper, DJ, Myles, PS, McDermott, FT, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: A randomized controlled trial. JAMA. 2004; 291:1350–1357.
288. Miller, JD, Becker, DP, Ward, JD, et al. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977; 47:503–516.
289. Velasco, IT, Pontieri, V, Rocha e Silva, M, Jr., Lopes, OU. Hyperosmotic NaCl and severe hemorrhagic shock. Am J Physiol. 1980; 239:664–673.
290. Mazzoni, MC, Borgstrom, P, Arfors, KE, Intaglietta, M. Dynamic fluid redistribution in hyperosmotic resuscitation of hypovolemic hemorrhage. Am J Physiol. 1988; 255:H629–H637.
291. Messmer, K, Kreimeier, U. Microcirculatory therapy in shock. Resuscitation. 1989; 18(Suppl):51–61.
292. Maningas, PA. Resuscitation with 7. 5% NaCl in 6% dextran-70 during hemorrhagic shock in swine: Effects on organ blood flow. Crit Care Med. 1987; 15:1121–1126.
293. Gemma, M, Cozzi, S, Tommasino, C, et al. 7. 5% hypertonic saline versus 20% mannitol during elective neurosurgical supratentorial procedures. J Neurosurg Anesthesiol. 1979; 9:329–334.
294. Munar, F, Ferrer, AM, de Nadal, M, et al. Cerebral hemodynamic effects of 7. 2% hypertonic saline in patients with head injury and raised intracranial pressure. J Neurotrauma. 2000; 17:41–51.
295. De Vivo, P, Del Gaudio, A, Ciritella, P, et al. Hypertonic saline solution: A safe alternative to mannitol 18% in neurosurgery. Minerva Anestesiol. 2001; 67:603–611.
296. Prough, DS. Should I use hypertonic saline to treat high intracranial pressure? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme; 2005:148–151.
297. Fisher, B, Thomas, D, Peterson, B. Hypertonic saline lowers raised intracranial pressure in children after head trauma. J Neurosurg Anesthesiol. 1992; 4:4–10.
298. Suarez, JI, Qureshi, AI, Bhardwaj, A, et al. Treatment of refractory intracranial hypertension with 23. 4% saline. Crit Care Med. 1998; 26:1118–1122.
299. Khanna, S, Davis, D, Peterson, B, et al. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000; 28:1144–1151.
300. Ware, ML, Nemani, VM, Meeker, M, et al. Effects of 23. 4% sodium chloride solution in reducing intracranial pressure in patients with traumatic brain injury: A preliminary study. Neurosurgery. 2005; 57:727–736.
301. Vialet, R, Albanese, J, Thomachot, L, et al. Isovolume hypertonic solutes (sodium chloride or mannitol) in the treatment of refractory posttraumatic intracranial hypertension: 2 mL/kg 7. 5% saline is more effective than 2 mL/kg 20% mannitol. Crit Care Med. 2003; 31:1683–1687.
302. Vassar, MJ, Perry, CA, Holcroft, JW. Analysis of potential risks associated with 7. 5% sodium chloride resuscitation of traumatic shock. Arch Surg. 1990; 125:1309–1315.
303. (comment) Marion, DA, Poca, MA, Sahuquillo, J, et al. Posture-induced changes in intracranial pressure: A comparative study in patients with and without a cerebrospinal fluid block at the craniovertebral junction. Neurosurgery. 2006; 58:899–906.
304. Marshall, LF, Smith, RW, Shapiro, HM. The outcome of aggressive treatment in severe head injuries. Part I: the significance of intracranial pressure monitoring. J Neurosurg. 1979; 50:20–25.
305. Eisenberg, HM, Frankowski, RF, Contant, CF, et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg. 1988; 69:15–23.
306. Clifton, GL, Miller, ER, Choi, SC, Levin, HS. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002; 30:739–745.
307. Clark, K, Nash, TM, Hutchison, GC. The failure of circumferential craniectomy in acute traumatic cerebral swelling. J Neurosurg. 1968; 29:367–371.
308. Cooper, PR, Rovit, RL, Ransohoff, J. Hemicraniectomy in the treatment of acute subdural hematoma: A reappraisal. Surg Neurol. 1976; 5:25–28.
309. Kjellberg, RN, Prieto, AJr. Bifrontal decompressive craniotomy for massive cerebral edema. J Neurosurg. 1971; 34:488–493.
310. Polin, RS, Shaffrey, ME, Bogaev, CA, et al. Decompressive bifrontal craniectomy in the treatment of severe refractory posttraumatic cerebral edema. Neurosurgery. 1997; 41:84–94.
311. Albanese, J, Leone, M, Alliez, JR, et al. Decompressive craniectomy for severe traumatic brain injury: Evaluation of the effects at one year. Crit Care Med. 2003; 1:2535–2538.
312. De Lucca, GP, Volpin, L, Fornezza, U, et al. The role of decompressive craniectomy in the treatment of uncontrollable post-traumatic intracranial hypertension. Acta Neurochir Suppl. 2000; 76:401–404.
313. Gaab, MR, Rittierodt, M, Lorenz, M, Heissler, HE. Traumatic brain swelling and operative decompression: A prospective investigation. Acta Neurochir Suppl. 1990; 51:326–328.
314. Gower, DJ, Lee, KS, McWhorter, JM. Role of subtemporal decompression in severe closed head injury. Neurosurgery. 1988; 23:417–422.
315. Guerra, WK, Gaab, MR, Dietz, H, et al. Surgical decompression for traumatic brain swelling: Indications and results. J Neurosurg. 1999; 90:187–196.
316. Schneider, GH, Bardt, T, Lanksch, WR, Unterberg, A. Decompressive craniectomy following traumatic brain injury: ICP, CPP, and neurological outcome. Acta Neurochir Suppl. 2002; 81:77–79.
317. Taylor, A, Butt, W, Rosenfeld, J, et al. A randomized trial of early decompressive craniectomy in children with traumatic brain injury and sustained intracranial hypertension. Childs Nerv Syst. 2001; 17:154–162.
318. Whitfield, PC, Patel, H, Hutchinson, PJ, et al. Bifrontal decompressive craniectomy in the management of posttraumatic intracranial hypertension. Br J Neurosurg. 2001; 15:500–507.
319. Marmarou, A, Anderson, RL, Ward, JD, et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991; 75(Suppl):S59–S66.
320. Heppner, P, Ellegala, DB, Durieux, M, et al. Contrast ultrasonographic assessment of cerebral perfusion in patients undergoing decompressive craniectomy for traumatic brain injury. J Neurosurg. 2006; 104:738–745.
321. Grady, MS. Decompressive craniectomy. J Neurosurg. 2006; 104:467–468.
322. Hsiang, JK, Chestnut, RM, Crisp, CB, et al. Early, routine paralysis for intracranial pressure control in severe head injury: Is it necessary? Crit Care Med. 1994; 22:1471–1476.
323. Sahuquillo, J. At what level should I start treating elevated intracranial pressure? In: Valadka AB, Andrews BT, eds. Neurotrauma. New York: Thieme; 2005:135–141.
324. Part, II. Prognosis in penetrating brain injury. J Trauma. 2001; 51(2 Suppl):S62.
325. Tien, R, Chesnut, RM. Medical management of the traumatic brain-injured patient. In: Cooper PR, Golfinos JG, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:457-482.
326. Vajkoczy, P, Schurer, L, Munch, E, Schmiedek, P. Penetrating craniocerebral injuries in a civilian population in mid-Europe. Clin Neurol Neurosurg. 1999; 101:175–181.
327. Morenski, JD, Tobias, JD, Jimenez, DF. Recombinant activated factor VII for cerebral injury-induced coagulopathy in pediatric patients. Report of three cases and review of the literature. J Neurosurg. 2003; 98:611–616.
328. Bakhtiari, K, Meijers, JC, deJonge, E, Levi, M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004; 32:2416–2421.
329. Hardy, JF, de Moerloose, P, Samama, M. Massive transfusion and coagulopathy: Pathophysiology and implication for clinical management. Can J Anesth. 2004; 51:293–310.
330. Powner, DJ, Hartwell, EA, Hoots, WK. Counteracting the effects of anticoagulants and antiplatelet agents during neurosurgical emergencies. Neurosurg. 2005; 57:823–831.
331. Allen, GA, Hoffman, M, Roberts, HR, Monroe, DM. Recombinant activated factor VII: Its mechanism of action and role in the control of hemorrhage. Can J Anesth. 2002; 49:S7–S14.
332. Fewel, ME, Park, P. The emerging role of recombinant-activated factor VII in neurocritical care. Neurocrit Care. 2004; 1:19–29.
333. Grounds, M. Recombinant factor VIIa (rFVIIa) and its use in severe bleeding in surgery and trauma: A review. Blood Rev. 2003; 17:S11–S21.
334. Karadimov, D, Binev, K, Nachkov, Y, Platikanov, V. Use of activated recombinant factor VII (NovoSeven) during neurosurgery. J Neurosurg Anesthesiol. 2003; 15:330–332.
335. Key, NS. Recombinant FVIIa for intractable hemorrhage: More questions than answers. Transfusion. 2003; 43:1649–1651.
336. Lau, HK. The interaction between platelets and factor VII/VIIa. Transfus Apheresis Sci. 2003; 28:279–283.
337. Lin, J, Hanigan, WC, Tarantino, M, Wang, J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: Preliminary findings. J Neurosurg. 2003; 98:737–740.
338. Mayer, SA. Ultra-early hemostatic therapy for intracerebral hemorrhage. Stroke. 2003; 34:224–229.
339. Park, P, Fewel, ME, Garton, HJ, et al. Recombinant activated factor VII for the rapid correction of coagulopathy in nonhemophilic neurosurgical patients. Neurosurgery. 2003; 53:34–39.
340. Roberts, HR. Recombinant factor VIIa (NovoSeven) and the safety of treatment. Semin Hematol. 2001; 38:48–50.
341. Veshchev, I, Elran, H, Salame, K. Recombinant coagulation factor VIIa for rapid preoperative correction of warfarin-related coagulopathy in patients with acute subdural hematoma. Med Sci Monit. 2002; 8:CS98–CS100.
342. Roberts, HR, Thoughts on the mechanism of action of FVIIa Presented at 2nd Symposium on New Aspects of Hemophilia Treatment, Copenhagen. 1991.
343. Penning-Van Beest, FJ, Gomez Garcia, EB, Van Der Meer, FJ, et al. Levels of vitamin K-dependent procoagulant and anticoagulant proteins in overanticoagulated patients. Blood Coagul Fibrinolysis. 2002; 13:733–739.
344. Lusher, J, Ingerslev, J, Roberts, H, Hedner, U. Clinical experience with recombinant factor VIIa: A review. Blood Coagul Fibrinolysis. 1998; 9:119–128.
345. Roitberg, B, Emechebe-Kennedy, O, Amin-Hanjani, S, et al. Human recombinant factor VII for emergency reversal of coagulopathy in neurosurgical patients: A retrospective comparative study. Neurosurgery. 2005; 57:832–836.
346. Hedner, U, Erhardtsen, E. Potential role for rFVIIa in transfusion medicine. Transfusion. 2002; 42:114–124.
347. O’Connell, NM, Perry, DJ, Hodgson, AJ, et al. Recombinant FVIIa in the management of uncontrolled hemorrhage. Transfusion. 2003; 43:1711–1716.
348. Deveras, RA, Kessler, CM. Reversal of warfarin-induced excessive anticoagulation with recombinant human factor VIIa concentrate. Ann Intern Med. 2002; 137:884–888.
349. Sorensen, B, Johansen, P, Nielsen, GL, et al. Reversal of the international normalized ratio with recombinant activated factor VII in central nervous system bleeding during warfarin thromboprophylaxis: Clinical and biochemical aspects. Blood Coagul Fibrinol. 2003; 14:469–477.
350. Kam, PC, Nethery, CM. The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anaesthesia. 2003; 58:28–35.
351. Kovesi, T, Royston, D. Is there a bleeding problem with platelet-active drugs? Br J Anaesth. 2002; 88:159–163.
352. Mannucci, PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: The first twenty years. Haemophilia. 2000; 6(Suppl 1):60–67.
353. Patrono, C, Coller, B, FitzGerald, GA, et al. Platelet-active drugs: The relationships among dose, effectiveness, and side effects. Chest. 2004; 126(3 Suppl):234S–264S.
354. Li, YF, Spencer, FA, Becker, RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J. 2001; 142:204–210.
355. Schroeder, WS, Gandhi, PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolygic agents. Curr Cardiol Report. 2003; 5:310–317.
356. Sane, DC, Califf, RM, Topol, EJ, et al. Bleeding during thrombolytic therapy for acute myocardial infarction: Mechanisms and management. Ann Intern Med. 1989; 111:1010–1022.
357. Warkentin, TE, Crowther, MA. Reversing anticoagulants both old and new. Can J Anesth. 2002; 49:S11–S25.
358. Menon, V, Harrington, RA, Hochman, J, et al. Thrombolysis and adjunctive therapy in acute myocardial infarction. Chest. 2004; 126(3 Suppl):549S–575S.
359. Albers, GW, Amarenco, P, Easton, JD, et al. Antithrombotic and thrombolytic therapy of ischemic stroke. Chest. 2004; 126(Suppl 3):483S–512S.
360. Ng, PP, Higashida, RT, Cullen, SP, et al. Intraarterial thrombolysis trials in acute ischemic stroke. J Vasc Interv Radiol. 2004; 15(1 Pt 2):S77–S85.
361. DeLoughery, TG. Coagulation defects in trauma patients: Etiology, recognition, and therapy. Crit Care Clin. 2004; 20:13–24.
362. Kaufmann, JE, Vischer, UM. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J Thromb Haemost. 2003; 1:682–689.
363. Lethagen, S. Desmopessin in mild hemophilia A: Indications, limitations, efficacy, and safety. Semin Thromb Hemost. 2003; 29:101–106.
364. Samama, CM, Bastien, O, Forestier, F, et al. for the Expert Group: Antiplatelet agents in the perioperative period: Expert recommendations of the French Society of Anesthesiology and Intensive Care (SFAR) 2001: Summary statement. Can J Anesth. 2002; 49:S26–S35.
365. de Jonge, E, Levi, M. Effects of different plasma substitutes on blood coagulation: A comparative review. Crit Care Med. 2001; 29:1261–1267.
366. de Jonge, E, Levi, M, Buller, HR, et al. Decreased circulating levels of von Willebrand factor after intravenous administration of a rapidly degradable hydroxyethyl starch (HES 2000/0. 5/6) in healthy human subjects. Intensive Care Med. 2001; 27:1825–1829.
367. Gan, TJ, Bennett-Guerrero, E, Phillips-Bute, B, et al. Hextend, a physiologically balanced plasma expander for large volume use in major surgery: A randomized phase III clinical trial. Anesth Analg. 1999; 88:992–998.
368. Neff, TA, Doelberg, M, Jungheinrich, C, et al. Repetitive large-dose infusion of the novel hydroxyethyl starch 130/0. 4 in patients with severe head injury. Anesth Analg. 2003; 96:1453–1459.
369. Rapp, RP, Young, B, Twyman, D, et al. The favorable effect of early parenteral feeding on survival in head-injured patients. J Neurosurg. 1983; 58:907–912.
370. Roberts, I, Yates, D, Sandercock, P, et al. Effect of intravenous corticosteroids on death within 14 days in 10,008 adults with clinically significant head injury (MRC CRASH trial): Randomized placebo controlled trial. Lancet. 2004; 364:1321–1328.
*See references 52, 55, 60, 65, 69, 71–73.
†See references 52, 54, 55, 57–60, 63–67, 73, 76, 77.
‡See references 54, 55, 64, 65, 68, 72, 73, 78, 79.
*See references 60, 66, 92, 95, 96, 103–106.
†See references 60, 76, 81, 91, 92, 101, 106.
*See references 162, 166, 167, 170, 179–182.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]66
Head Injury
Introduction
It also has been demonstrated that the management of serious brain trauma requires specialty teams consisting of experienced neurosurgeons, traumatologists, and intensivists (specifically, neurointensivists) working together in a dedicated unit equipped and staffed to optimally care for these patients. In the absence of such facilities, outcomes suffer.1–3
Incidence
The Centers for Disease Control and Prevention report the number of patients sustaining a traumatic brain injury (TBI) to be at least 1.7 million per year in the United States. Approximately 80% of patients are released from the emergency department, 275,000 are admitted, and 52,000 die. Nearly a third of all traumatic deaths in the U.S. involve a head injury. The ages most likely to sustain a TBI are children up to 4 years old and adults over 64, with adults greater than 74 years old having the highest rates of hospitalization and death associated with head injury.4 Teenagers and young adults continue to be at high risk for sustaining TBI, but the range has decreased over the past 20 years from between 15 and 24 to between 15 and 194,5. The incidence of head injury has been shown to be inversely proportional to economic status,6 and has remained up to three times more common in males in every age group over the past several decades.4,7,8 In the United States, falls are the most common cause of TBI and are responsible for half of the head-injured children up to 14 years of age and 60% of the adults over the age of 64.4 Although the frequency of head injury related to motor vehicle crashes has decreased over the years to a little less than 20%, these crashes are the second leading cause of head injury in all age groups and result in the largest percentage of TBI-related deaths.4,9 The incidence of TBI worldwide is difficult to quantify but is estimated to be at least 10 million people a year, with traffic accidents accounting for more than half, and falls being the second leading cause;10 this is the opposite distribution seen in the United States4. Violence is estimated to cause about 10% of head injuries throughout the world,10 as well as in the U.S.4, but there is significant variability in the specific mechanisms. For example, between 1997 and 2007 in the U.S., firearms were related to just over a third of all lethal TBIs, compared to a single hospital in South Africa that reported its experience with more than 300 stab wounds to the brain.11
Diagnostic Approach
The presenting neurologic condition of patients with head injuries is the primary factor in determining the initial management and prognosis. When a detailed history is unavailable, it is important to keep in mind that loss of consciousness may have preceded and in fact caused the traumatic event, such as aneurysmal subarachnoid hemorrhage (SAH), hypoglycemia, intoxication, or syncope. Level of consciousness is one of the most important neurologic considerations in managing a head-injured patient. Neurosurgical patients generally have an alteration in level of consciousness from either brainstem or bilateral cerebral hemispheric involvement. This may be secondary to poor perfusion due to high ICP or brainstem compression, both of which require neurosurgical intervention. Therefore, an accurate tool to measure level of consciousness is essential. Many clinical assessment tools are available for use in the critical care setting.12–24
Glasgow Coma Scale
Ideally, using the GCS, a nurse, medical student, paramedic, physician assistant, intensivist, trauma surgeon, emergency medicine physician, neurologist, and neurosurgeon all should obtain the same score when assessing a patient. The GCS system is not perfect, but it has proved itself to be, overall, a practical, straightforward grading system that can be used by health professionals in various fields and at different levels to produce reliable results.25–30 The GCS has become the most widely used assessment tool and is considered the gold standard for the evaluation of patients with head injuries.12
The GCS is a measure of level of consciousness and does not take into account focal deficits. It is based on eye opening (1-4), verbal response (1-5), and motor response (1-6) (Table 66.1). A patient with a normal level of consciousness should have the highest possible score of 15. The lowest score possible is 3, not 0 as might be expected. An intubated patient technically gets a 1 for verbal response and is assigned 1T (for Tube) for the verbal score. It is important to identify the score for each observed variable tested. For example, a typical score for an intubated patient with decorticate posturing probably would be E1 + V1T + M3 = 5T (where E denotes eye opening, V is verbal response, and M is motor response). It also is important to realize the shortcoming of this system in evaluating patients with dementia or aphasia. A point to keep in mind is that the GCS attempts to put a numeric value on level of consciousness, with 15 being representative of normal. If a demented patient from a nursing home presents with a normal level of consciousness after a fall but doesn’t know what day it is, the GCS would be calculated as E4 + V4 + M6 = 14. A GCS score of 14 describes a patient with an abnormal level of consciousness but fails to accurately communicate this particular patient’s condition. Likewise, clinicians must use caution when applying the GCS to patients with aphasia. Conversely, patients with profound focal deficits and a normal level of consciousness should have a GCS score of 15. Another problematic case is that of a quadriplegic patient with a normal level of consciousness who is able to blink the eyes or stick out the tongue upon command, thereby giving a top score of 6 on the motor assessment.
Table 66.1
| Component | Score |
| Eye Opening | |
| Spontaneously | 4 |
| To voice | 3 |
| To pain | 2 |
| No response | 1 |
| Verbal | |
| Oriented | 5 |
| Disoriented | 4 |
| Inappropriate | 3 |
| Incomprehensible sounds | 2 |
| No response | 1 |
| Motor | |
| Following commands | 6 |
| Localizing to pain | 5 |
| Withdrawing to pain | 4 |
| Abnormal flexion | 3 |
| Abnormal extension | 2 |
| No response | 1 |
The GCS has been used to predict outcome.31 The motor score itself has predictive value as well.32 The GCS score should be calculated after hemodynamic and pulmonary resuscitation and without sedatives or muscle relaxants.33
Aggressive implementation of early sedation and intubation in severely head-injured patients compromises the ability to determine an accurate GCS score.34
Computed Tomography Classification: Marshall Classification
A useful classification of brain injuries by findings on computed tomography (CT) has been devised by Marshall and colleagues. This CT classification, presented in Table 66.2, can serve as a guide in describing scans and has strong predictive power.34,35
Table 66.2
Marshall Computed Tomography Classification
| Category | Definition |
| Diffuse injury I (no visible pathology) | No visible intracranial pathology seen on computed tomography scan |
| Diffuse injury II | Cisterns are present with midline shift of 0-5 mm or lesion densities present no high- or mixed-density lesion > 25 cm3 may include bone fragments and foreign bodies |
| Diffuse injury III (swelling) | Cisterns compressed or absent with midline shift of 0-5 mm; no high- or mixed-density lesion > 25 cm3 |
| Diffuse injury IV (shift) | Midline shift > 5 mm; no high or mixed-density lesion > 25 cm3 |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High- or mixed-density lesion > 25 cm3; not surgically evacuated |
From Marshall LF, Marshall SB, Klauber MR, et al: A new classification of head injury based on computerized tomography. J Neurosurg 1991;75:S14-S20.
Maas and associates have proposed a scoring system with better predictive power (Table 66.3), with a sum total adjusted to be consistent with the GCS.34 Table 66.4 presents a mortality prediction chart based on this classification.
Table 66.3
| Predictor | Score |
| Basal Cisterns | |
| Normal | 0 |
| Compressed | 1 |
| Absent | 2 |
| Midline Shift | |
| No shift or shift < 5 mm | 0 |
| Shift > 5 mm | 1 |
| Epidural Mass Lesion | |
| Present | 0 |
| Absent | 1 |
| Intraventricular Blood or tSAH | |
| Absent | 0 |
| Present | 1 |
| Sum Score* | +1 |
*The sum score can be used to obtain the predicted probability of mortality (see Table 66.4). The authors chose to add plus 1 to make the grading numerically consistent with the grading of the motor score of the GCS and with the Marshall CT classification. tSAH, traumatic subarachnoid hemorrhage.
This scoring system is best used as one piece of data in the overall clinical assessment in considering a patient’s prognosis.36
Prediction of Outcomes
Severe brain injury outcomes can be accurately predicted using four variables: age, GCS score on admission (especially motor score), CT characteristics, and presence of ischemic and hemodynamic secondary insults. Using these four predictors, numerous investigators have used statistical modeling techniques to predict up to 80% or more of outcomes.37
Head injuries generally are classified as mild (GCS score of 13 to 15), moderate (GCS score of 9 to 12), or severe,3–8 although the evidence may be sufficient to include a GCS score of 13 in the moderate category.38
CT scanning is the diagnostic imaging study of choice in trauma. Plain x-ray films of the skull are rarely indicated as a screening study for head-injured patients. Outcomes with the strategy of obtaining CT scans in all head-injured patients are superior to those with other management strategies. The incidence of missed surgical lesions in mildly head-injured patients by CT scanning is 0.028%.39–42
Primary Head Injury
Primary head injury can be defined as the damage that occurs at the moment of impact and can take the form of skull fractures, surface contusions and lacerations, diffuse axonal injury, or diffuse vascular injury.42 Some authors include the contusions and hematomas that form as a direct and immediate effect of the impact as part of the primary injury as well.43 These hemorrhages may manifest with significant mass effect requiring surgical intervention.
Skull Fractures
Basilar skull fractures typically are linear and involve the anterior cranial base and the petrous part of the temporal bone. These fractures have the potential for an associated dural laceration adjacent to potentially contaminated paranasal sinuses, or the external ear canal if the tympanic membrane is disrupted. This allows the potential for a cerebrospinal fluid (CSF) fistula to develop, as well as meningitis. Clinical signs of a fracture of the petrous portion of the temporal bone include hemotympanum with or without tympanic membrane disruption, hearing loss, CSF otorrhea, and Battle sign. The cranial nerves that course through the temporal bone include the facial, acoustic, and vestibular nerves; therefore, associated vestibular dysfunction or facial weakness may be noted (Fig. 66.1). Anterior cranial base fractures may be associated with “raccoon eyes,” anosmia, and CSF rhinorrhea. It is important to remember that when a fracture of the floor of the anterior cranial base is suspected, a nasogastric tube should not be inserted, because of the risk of intracranial penetration (Fig. 66.2).
The use of prophylactic antibiotics in basilar skull fractures has been debated but generally is not recommended. When CSF leak does occur, the break in the tissue usually will heal with conservative treatment over the course of a week, unless the defect is very large or a bony spicule is identified. Keeping the patient’s head elevated and possibly using external CSF diversion after a few days are reasonable nonoperative methods; however, if the leak persists, surgical repair is indicated.44
Most depressed skull fractures occur in men younger than 30 years of age.45 Despite very little literature to support any particular management strategy, closed depressed skull fractures generally are operated on if the extent of depression is greater than the full thickness of the adjacent skull, with the theoretical benefits of improved cosmetic result, a decrease in late-onset posttraumatic epilepsy, and a reduction in the incidence of persistent neurologic deficit.46 Most depressed skull fractures, however, are open45 (Fig. 66.3), meaning that a scalp laceration with galeal disruption overlying the fracture is present.
Open depressed skull fractures may be associated with significant morbidity and mortality.45,47 Infection rates are reported to be between 1.9% and 10.6%,46–50 with neurologic morbidity and mortality rates of approximately 11% each46 and an incidence of late epilepsy up to 15%.51
By convention, open depressed cranial vault fractures are treated surgically, with debridement and elevation, primarily to attempt to decrease the incidence of infection. Open depressed cranial fractures may be treated nonoperatively if clinical and radiographic examination reveals no evidence of dural penetration, significant intracranial hematoma, depression greater than 1 cm, frontal sinus involvement, gross cosmetic deformity, wound infection, pneumocephalus, or gross wound contamination.46
A special type of depressed fracture is fracture of the frontal sinus. Depression of the fracture may require surgery to prevent CNS infection and CSF leak (Fig. 66.4). Another special type of depressed fracture involves a major dural venous sinus (see Fig. 66.4). Under these circumstances, the risks associated with surgery are increased, and elevation of the fracture fragment generally is reserved for significant compromise of venous drainage.44
Epidural Hematoma
The next anatomic layer after the skull is the dura, which is the periosteum of the inner table of the skull and, as such, is tightly adherent to the bone. The potential space between the inner table of the skull and the dura is the epidural space. Epidural hematomas (EDHs) form in this potential space between the skull and the dura. The most common mechanism for the development of an EDH is a motor vehicle crash (in 53% of the cases), followed by falls (in 30%) and assault (in 8%).52–60 Typically, bleeding results from damage to the middle meningeal artery, but an EDH also can occur from injury to the middle meningeal vein, diploic veins, or the venous sinuses.60
EDHs generally occur in the temporal and temporoparietal regions (Fig. 66.5).52,57,61–64 A useful way to describe the development of an EDH is to follow the events that take place when a patient presents with an initial lucid interval following brief loss of consciousness after head trauma. A lucid interval occurs when a patient initially is rendered unconscious from a concussive head injury that causes a linear skull fracture involving the middle meningeal artery or one of its branches. The middle meningeal artery is a dural vessel that runs half in the dura and half through the groove in the inner table of the skull. When a fracture extends across the bony groove of the artery, it will tear and bleed. Because the dura is tightly adherent to the inner table of the skull, significant force is needed to push the dura off the inner table. An arterial hemorrhage generally has enough pressure to strip the dura off the bone, converting the potential epidural space into a mass. The bony attachment of the dura becomes progressively stronger with age; therefore, the dura of a younger patient requires less force to push it off the bone than would be required in an older patient. Not surprisingly, the mean age of patients with EDH is between 20 and 30 years of age,53–55,63–70 and EDHs are unusual in patients older than 50.60
Taking a general view, the patient goes from unconscious at the time of impact from a concussive injury, to awake, to unconscious again secondary to brainstem compression—hence the term lucid interval. The lucid interval is observed in close to half of the patients undergoing surgery for EDH.*
Pupillary abnormalities occur in approximately 20% to 30% of patients with surgical EDH.52,54,59,60,67 Cranial fractures are present in between 70% and 95% of the cases.54,60,68,70,74,75 Associated intracranial lesions are found in 30% to 50% of adults with surgical EDHs,† and subdural or parenchymal lesions in association with EDH lower the chance of a good outcome.60
The overall mortality rate (for all ages and GCS scores) is approximately 10%.‡ The time lapse between the onset of pupillary abnormalities and surgery determines outcome,61,66,80 but the single most important predictor of outcome in patients operated on for EDH is the GCS score on admission and before surgery.52,53,55,61,81–83
Not all EDHs, however, require surgery. No prospective randomized trials have been conducted to compare surgical treatment with nonoperative management, nor should there be. Available data describe nonsurgical management in selected cases. In one study, approximately 10% of the total number of EDHs were treated nonoperatively. All were conscious with a GCS score greater than 11 and a midline shift on CT scan of less than 10 mm. Not one of these nonoperative hematomas was in the temporal region.84
Another study reported findings in a group of 57 selected patients treated nonoperatively with an initial GCS score of 10 or higher, with maximum hematoma thickness less than 13 mm, with five clots located in the temporal region, but only one patient had a midline shift on CT.53
An interesting approach for dealing with thin, acute EDHs in an early stage has been reported in a small, isolated series of patients using endovascular techniques to occlude the middle meningeal artery.85 Surgical decision making for an acute EDH is based on GCS score, pupillary findings, comorbid conditions, CT findings, age, and, in delayed decisions, ICP. Guidelines for the surgical management of acute EDH published in 2006 recommend surgical evacuation of an EDH less than 30 mL in volume. Smaller hematomas in patients with a GCS score greater than 8 and without focal deficits, clot thickness less than 15 mm, and less than 5 mm of midline shift may be considered for nonoperative management. Close neurologic monitoring and serial CT scanning are essential. Of importance, a temporal location for an EDH is associated with failure of nonoperative management and should lower the threshold for surgery.60
Because time between neurologic deterioration and surgery is critical, the question of whether a patient with an acute EDH should receive treatment at the nearest hospital or should be transferred to a trauma center is important. The issue is whether or not a non-neurosurgeon should operate on a patient deteriorating from an acute EDH as a true emergency. One suboptimally controlled study reported worse outcomes in a small group of patients who underwent emergency operations by non-neurosurgeons and attributed this mainly to the technical inadequacy of the operation.59,60 Other studies have documented worse outcomes in patients transferred from outlying hospitals for surgery.52,73 The take-home message is that EDHs are extra-axial hemorrhages located adjacent to the brainstem that can represent a true neurosurgical emergency, and that rapid, competent decompression makes a difference.
Subdural Hematoma
Proceeding deep to the skull, the next space is the subdural space. Unlike the potential epidural space, the subdural space is a real space. It is a compartment that follows the contour of the brain, which is how it appears on a CT scan (Fig. 66.6). Anatomically, bridging veins course through the subdural space from the cerebral hemispheres to the superior sagittal sinus. As the brain accelerates within the skull after impact, these veins can stretch and tear.86
The source of bleeding also can be from a cortical artery87,88 or vein. These hematomas typically form over the surface or between the cerebral hemispheres, along the tentorium, or between the temporal lobe and base of the skull.
An acute subdural hematoma (SDH), as defined by Bullock and colleagues, appears within 14 days of injury,60 although some authors consider an SDH to be subacute when signs and symptoms develop between 3 and 20 days after trauma.89
Acute SDHs consist of clotted blood, which is fibrous and often adherent to adjacent tissue. The blood remains clotted for several days. After this time, the clot gradually and progressively lyses, resulting in a mixture of clot and fluid. After several weeks, the clot is liquefied and becomes a chronic SDH. Chronic SDHs may manifest weeks or months after what may have been mild or insignificant trauma. Chronic SDHs occur more often in elderly patients and individuals who have more intracranial space because of cortical atrophy. A membrane often surrounds these hematomas, and the collection of fluid may slowly grow in size because of repeated small bleeds or accumulation of fluid transudate from the membrane.86
The incidence of acute SDHs is approximately 20% in severe traumatic brain injury,60,90–93 and the mean age is between 31 and 47 years, with most of the patients being men.60,94–97 The mechanism of injury differs between age groups, with patients older than 65 more commonly presenting after a fall, and younger patients being involved more often in a motor vehicle crash,60,98–100 which, among comatose patients with acute SDH, is the most common mechanism of injury.60,91,93,101
Between 37% and 80% of patients with acute SDH present with a GCS score of 8 or lower,60,65,71,94,97,102 and the overall mortality rate is between 40% and 60%.* Mortality rates among comatose patients requiring surgery are somewhat higher, with reported rates between 57% and 68%.† Acute subdural hematomas frequently are associated with other brain injuries,97,102 which is one of the reasons why the mortality rate is relatively high.
A simple acute SDH is a subdural, extra-axial hematoma without any other associated brain injury (Fig. 66.7). A complex SDH is associated with parenchymal injury (Fig. 66.8).107,108 Fewer than half of acute SDHs requiring surgery are isolated, simple lesions.60,97,102
Figure 66.8 A, Computed tomography scan of acute, complex left-sided subdural hematoma (SDH) (lower arrow) with mass effect (upper arrow) out of proportion to the size of the clot, indicating more than just the effect of an extra-axial compressive mass—that is, intrinsic, parenchymal brain injury with edema. B, Intraoperative photograph of decompressed brain reveals hemorrhagic, inflamed, and edematous brain swelling out through the cranial defect. Compare with the intraoperative photograph of the simple SDH in Figure 66.7.
One useful method to identify the presence of associated injury is to measure the thickness of the subdural clot relative to the amount of brain shift on the CT scan. If the amount of shift is directly proportional to the thickness of the extra-axial clot, the injury is likely to be simple and only the brain compressed. However, if the amount of shift is more than expected as indicated by the size of the hematoma, then additional parenchymal brain injury probably is present under the clot, causing additive mass effect. Not surprisingly, the mortality for complex injuries is higher than that for simple SDHs.107,108
In addition to the obvious compressive effects that an SDH generates, the available evidence points to a direct toxic effect of the blood itself on the underlying cortex, thereby compounding the problem.109,110
Age is an important factor in acute SDHs. There is a significant increase in poor outcome among patients older than 60 years of age with severe head injury in general.93,98–101,111,112 Older patients with an acute SDH and a low GCS score do especially poorly.98–101,111,112
The decision for immediate surgery is dependent on GCS score, age, pupillary examination, comorbidities, CT findings, and salvageability with respect to the patient’s level of injury. In addition to all of these factors, decision making for delayed surgery is dependent on clinical course and ICP.60
On the basis of a contemporary search of the literature, Bullock and colleagues found that CT parameters of a midline shift greater than 5 mm and a clot thickness greater than 10 mm were independent factors requiring surgery in salvageable patients. They also identified a select group of comatose patients with smaller SDHs who could be managed nonoperatively if the patients remained neurologically stable with normal pupils and ICP of 20 mm or less.60
Traumatic Subarachnoid Hemorrhage
The subarachnoid space is a CSF-filled compartment within which are the major cerebral blood vessels. The CSF within the subarachnoid space fills the basal cisterns and interdigitates into the cortical sulci. Traumatic SAH can be caused by bleeding of cortical arteries, veins, or brain surface cerebral contusions.113
In contrast with hemorrhages in other locations, SAH is not a discrete surgical clot that requires evacuation. Trauma is the most common cause of SAH; a ruptured aneurysm is the most common cause of a spontaneous SAH. Aneurysmal SAH generally involves the suprasellar cistern, where the circle of Willis lies. Traumatic SAH can be found as small-volume hemorrhages in the sylvian fissures and especially in the interpeduncular cistern. Traumatic SAH also commonly involves the cerebral convexity and can fill cortical sulci, which can sometimes mimic sulcal effacement114 (Fig. 66.9A).
Occasionally, the distribution of the hemorrhage may be hard to differentiate from an aneurysmal hemorrhage. Under this circumstance, a vascular study should be obtained (Fig. 66.9B). Cerebral vasospasm following aneurysmal SAH is a common, well-described problem with an unclear pathogenesis. Increasing evidence suggests that SAH from trauma also may cause clinically significant cerebral vasospasm, which may be responsible for ischemia and infarction.115,116
Clearly the incidence of clinically relevant cerebral vasospasm is less than what is seen in aneurysmal SAH; however, posttraumatic SAH may cause ischemia during the acute as well as the delayed phase.115,117
In severe nonpenetrating head injury, the degree of SAH can be predictive of outcome,118,119 and although traumatic SAH can be an isolated finding, it is commonly associated with other intracranial injuries.120
Traumatic SAH can be a marker of severe primary injury and the amount of blood can also be an independent predictor of the development and progression of intraparenchymal contusions113 (Fig. 66.9C).
Intraparenchymal Contusions and Hematomas
Traumatic parenchymal mass lesions are common and are reported in 13% to 35% of severe traumatic brain injury.121–127 Contusions consist of heterogeneous areas of necrosis, pulping, infarction, hemorrhage, and edema.89,128,129 Hemorrhagic contusions are mixtures of blood and edematous cerebral parenchyma that also have a heterogeneous appearance on CT.89 Contusions commonly occur in the frontal and temporal lobes both at the poles and on the inferior surfaces as a result of contact with the rough, bony skull base130 (Fig. 66.10A).
Contusions typically involve the crests of the gyri but in more severe injury may extend into the substance of the white matter. If the pia-arachnoid is torn, contusions are classified as cortical lacerations.130–132
Contusions represent one end of a spectrum of injury on which hematomas, which are well-defined, homogeneous collections of blood, are the other end (Fig. 66.10B). The amount of energy delivered may have only been enough to cause failure of small vessels, resulting in contusions. If more energy is delivered, failure of larger vessels may occur, resulting in hematomas. Alternatively, the hemorrhagic component of a contusion may continue to bleed and coalesce into a more discrete hematoma. In general, the most common traumatic parenchymal lesions are contusions,89 and they tend to evolve.122,126,127,133,134
Risk factors for the progression of intraparenchymal hemorrhage include the presence of SAH (see Fig. 66.9C), and subdural hematoma, as well as the size of the clot on the initial CT scan. Enlargement of contusions occurs approximately 40% of the time, which justifies early follow-up CT scanning.135 In addition to the problem of progressive enlargement of existing contusions is a phenomenon of delayed appearance of traumatic intracerebral contusions (DITCH). DITCH is defined as a new contusion identified on a CT scan in an area of brain that was normal on the admission CT scan. These delayed hemorrhages are reported to occur in up to 7% of patients with severe head injuries.90
Contusions can be subdivided into two groups. Coup contusions occur in the brain tissue under the impact site and usually are associated with an acceleration injury. Contrecoup contusions are located away from the point of impact and usually are associated with a deceleration injury.130,131,136–138
Understanding the biomechanics of how a contrecoup contusion develops is useful. In general, biologic tissues tolerate strain better if they are deformed slowly rather than quickly. For example, a 150-mL hematoma with accumulation of blood over minutes is likely to be lethal, whereas a 150-mL slow-growing meningioma may have no appreciable effect on the patient’s level of consciousness. In trauma, the cause of tissue damage may be any of three types of induced strains: compression, tension, and shear. The skull, brain, and blood vessels will tolerate compression better than tension, and tension better than shear. These strains are induced by contact or inertia (relative to acceleration-deceleration), or both. Contact injuries are a result of impact, which may cause inward deformation of the skull with local effects, and of shock waves, which can produce remote effects. As a consequence of contact the head is set in motion, which leads to inertial injury. Inertial injuries may cause damage by differential acceleration of the skull and brain. In addition, acceleration-deceleration can independently produce strains in the brain itself. The two clinically relevant types of acceleration are translational (when the brain moves in a straight line) and angular. Most injuries are a combination of both.139
These effects are best illustrated by a case example of a 37-year-old man who falls from a height onto the back of his head and presents with a deteriorating level of consciousness. His CT scan (Fig. 66.11) reveals soft tissue swelling at the point of impact in the right occipital region with a small underlying hemorrhage, as well as a large, hemorrhagic contusion in the left frontal lobe. The mechanism involved is primarily translational (linear) deceleration. The skull stops suddenly as it hits the ground, but the brain continues to move toward the impact site, where compression is induced (Fig. 66.12).
As the brain is moving toward the inner table of the skull at the impact site, it also is moving away from the skull on the opposite side, creating regions of low pressure and tensile strains. Contributing to the extensive tissue damage in the left frontal lobe also may be movement of the brain across the rough surface of the anterior cranial base.89,139
The same amount of energy is delivered to the brain in nearly a straight line, with the compressive injury at the point of impact resulting in much less damage than the contrecoup injury caused by tension. In view of the dramatic differential susceptibilities of the brain and blood vessels to compressive and tensile strains induced in trauma, it is understandable that diffuse injuries can be caused by seemingly less violent trauma when the head undergoes an injury with a large component of angular acceleration-deceleration. This is the motion that can induce shearing strains, which the tissues tolerate poorly (see “Diffuse Axonal Injury” later on).
In 2006, guidelines were published after a thorough review of the relevant but scientifically weak literature. The reviewers reported that patients with parenchymal mass lesions and signs of progressive neurologic deterioration referable to the lesion, medically refractory intracranial hypertension, or signs of mass effect on CT scan should be treated operatively. Patients with GCS scores of 6 to 8 with frontal or temporal contusions greater than 20 cc in volume with midline shift of at least 5 mm or cisternal compression on CT scan and patients with any lesion greater than 50 cc in volume should receive operative treatment. Patients with parenchymal mass lesions who do not show evidence of neurologic compromise, whose ICP is controlled, and who demonstrate no significant signs of mass effect on CT scan may be managed nonoperatively with intensive monitoring and serial imaging. For patients with refractory intracranial hypertension and diffuse parenchymal injury with clinical and radiographic evidence for impending transtentorial herniation, the guidelines also recommended, as treatment options, subtemporal decompression, temporal lobectomy, or hemispheric decompressive craniectomy (Fig. 66.13). With regard to timing of surgery, the literature supports a bifrontal decompressive craniectomy within 48 hours of injury as a treatment option for patients with diffuse, medically refractory posttraumatic cerebral edema and resultant intracranial hypertension.127
Hypothalamic-Pituitary Injury
Injury to the pituitary and hypothalamus can complicate head injury. A prolonged loss of consciousness often is reported in patients with such injuries. Up to 80% of cases are associated with a fracture through the skull base. Injuries include (in decreasing order of frequency) pericapsular hemorrhage, hypothalamic infarction, posterior pituitary hemorrhage, anterior pituitary infarction, and rupture of the infundibulum.140–142 Clinically measurable decreases in pituitary hormone production are not seen until at least 75% of the gland is destroyed. Complete loss of production requires destruction of at least 90% of the gland.
Derangements in pituitary function can result in decreased production of any of the pituitary hormones. Of particular clinical significance is impairment of adrenocorticotropic hormone release that can result in secondary adrenal insufficiency (Addison’s disease). Physiologic stressors such as trauma, surgery, or infection can result in addisonian crisis, with potentially life-threatening results. Diabetes insipidus has been reported in a significant number of severe head injuries, with a high percentage of cases remaining permanent.143 Injuries to the pituitary often are not suspected in the acute period, and the diagnosis of hypopituitarism is therefore often delayed (Fig. 66.14).
Diffuse Axonal Injury
The term diffuse axonal injury (DAI) describes brain damage in a group of patients who become immediately unconscious or go into coma at the time of the head trauma.81 Depending on the severity of the injury, patients may have mild, moderate, or severe DAI. A large number of patients with severe head injury will have DAI. Because of the gradient acceleration difference of certain brain areas during primary impact, shearing forces at the gray-white junction, corpus callosum, or brainstem may occur.131,144 The result of these forces will be diffuse tearing of axons and small blood vessels. These lesions initially may be hemorrhagic, but as they become chronic, shrinking, softening, and scarring ensue, often with cyst formation.42,145 The lesions usually are microscopic but can be large and macroscopic. Although small, focal, petechial hemorrhages may be visualized, the initial CT scan often is normal.146 Most of the clinical and experimental studies support that the diffuse injury occurs at the time of initial impact and is not the result of other adverse factors, such as decreased brain oxygenation, increased ICP, or brain swelling,145,147 although these factors tend to accentuate the amount of damage.
The term gliding contusions originally was introduced by Lindenberg and Freytag148 to describe hemorrhagic lesions in parasagittal white matter; this brain injury concept has been reanalyzed.149 Gliding contusions frequently are found in association with DAI and acute subdural hematomas. Two mechanisms have been considered in relation to the formation of these contusions. First, during angular acceleration, more movement and displacement of the cortical gray matter occur than of the deep white matter; accordingly, most of the tissue injury occurs at the gray-white junction because shearing strains predominate. The second mechanism involves excessive displacement of the bridging veins during brain acceleration. Patients with severe DAI who survive may be profoundly disabled, but some patients with mild or moderate DAI may recover with mild or no disability.81 The importance of recognizing DAI in the initial evaluation cannot be overstressed; this will affect the management and outcome. Clinically, these patients will have impaired consciousness secondary to bicortical damage, possibly in association with unremarkable imaging findings and normal ICP.
Penetrating Brain Injury
It is estimated that between 6000 to 7000 people die each year in the United States from gunshot wounds to the brain.130–139,150–152
Classification
Low-velocity penetrating brain injuries include wounds from nail guns, arrows, and knives and some types of gunshot wounds (Fig. 66.15). Gunshot wounds are divided into low-velocity injuries, such as from civilian handguns and shrapnel, and high-velocity injuries, from rifles and military weapons152 (see Table 66.1).
An Israeli report described an intermediate type of injury from spherical bolts whereby the ball bearings used by suicide bombers, having unique ballistics, may cause a type of “stab wound” injury to the brain.152
Ballistics
The biomechanics of penetrating brain injury (PBI) are important and require an understanding of the dynamics of projectiles—that is, ballistics. Ballistics can be divided into three phases. The first phase, internal ballistics, deals with the source of the projectile and its intrinsic dynamics (e.g., rifle, bomb). External ballistics, the second phase, focuses on the flight of the projectile itself and the deviation of its longitudinal axis relative to the line of flight, referred to as yaw motion. Collision of the projectile with any object during this external phase will change the speed, angle, and yaw. The third phase is referred to as terminal ballistics, which is the physical interaction between the projectile and the body.152
As the projectile directly crushes tissue, it forms a permanent cavity, and as surrounding tissue is compressed, a temporary cavity also is formed. As the missile breaks through the skull, secondary projectiles of bone fragments may be generated, causing independent, secondary permanent and temporary cavities.152–156
Terminal ballistics are influenced by many factors, such as size, shape, and stability of the penetrating object; however, most authors (but not all) believe that entrance velocity is the most important factor in determining the degree of tissue damage.152,153,155,157–159 Experimental evidence has demonstrated an immediate increase in ICP with a pressure wave transmitted throughout the cranial cavity, which probably accounts for the transient respiratory arrest observed in PBI.152,160 High-velocity injuries (velocity greater than 320 meters per second) cause shock waves that emanate from the front of the missile. These shock waves can reflect off the inside of the skull and summate, producing significant pressure gradients with remote effects.
Initial Resuscitation
Initial resuscitation in the trauma bay should be in accordance with current recommendations for head-injured patients in general. During this initial resuscitation, it is important to recognize that hypotension in PBI either is a terminal event or is related to other injuries. Once resuscitation has been accomplished, a CT scan is essential. Although skull films seldom add to the information obtained from a CT scan,152 our group has found plain films to be very helpful in PBI, particularly in industrial accidents, in which the shape of the penetrating object may be unknown.
Surgical Management
Most of what is known about the surgical management of PBI comes from the battlefield, beginning with World War I, when Harvey Cushing, the father of American neurosurgery, described his technique of en bloc craniectomy under aseptic conditions; thorough debridement of scalp, bone, brain, metal, and bone fragments; and watertight closure of the scalp. Using this management strategy a century ago, in the absence of antibiotics, Cushing reported a significant drop in postoperative mortality from 55% to 28%.161,162
Mortality decreased even further in World War II, primarily because neurosurgical personnel were present in forward military hospitals and antibiotics were introduced. The operative approach, however, was largely the same: that of radical debridement. Treatment consisted of four tiers: emergent life-saving maneuvers through hemostasis and cerebral decompression; prevention of infection through extensive debridement; preservation of nervous tissue through prevention of meningocerebral scars; and restoration of anatomic structures through accurate closure of the dura and scalp. Although untested against other management strategies, this approach became the standard of treatment for PBI by default. Indeed, in the U.S. military, thorough debridement of intracranial bone and metal fragments was official military policy through the Vietnam War.162
The overall mortality rate from PBI sustained during relatively modern wartime conditions ranges from 8% to 43% and generally is considered to be in the 20% range.162–171 The mortality rate from military gunshot wounds has been reported to be 2.5 to 4 times more than from shrapnel.162–165,170 Of interest, mortality rates from wartime PBIs in the U.S. military from World War II through the Vietnam War did not change significantly,172 which is consistent with the relatively unchanged management strategy of complete removal of intracranial bone or metal fragments, using repeated surgery as necessary.173
Aggressive debridement was intended to reduce complications such as infection, epilepsy, and cerebral edema. However, studies reveal that additional surgery to remove retained fragments results in significant morbidity and mortality.170,174,175
The use of broad-spectrum antibiotics in recent wars has resulted in data suggesting that retained bone fragments are not independently associated with an increased risk of infection. Therefore, aggressive debridement is no longer supported.162,176 The use of broad-spectrum antibiotics in PBI has now become universal.152
Multiple studies have looked at post-PBI epilepsy and reported an incidence of 22% to 53%.167,177,178 Several studies suggest that it is not necessary to remove all bone fragments in order to decrease the chance of developing epilepsy, but the value of removing metal fragments in this regard remains unclear.162,167,169,177,178 In military PBI, it appears that vigorous debridement is associated with increased morbidity and mortality and is not necessary to prevent infection, has no obvious efficacy in preventing epilepsy, and does not appear to improve survival.162
A CSF leak resulting from a PBI is the variable most highly correlated with intracranial infection.* One study found that most CSF leaks appeared within 2 weeks of the injury and less than half of them closed spontaneously. The incidence of infection was approximately 10 times higher in the group with CSF fistulas than in the group without leaks, and the mortality was greater as well.181 Because CSF leaks are the primary predictor of the development of intracranial infection,167,169,171 it is important to fix or prevent a CSF leak; therefore, a watertight closure of the scalp entry wound is necessary.181
To accomplish this objective, various surgical interventions are available. Small entrance wounds without underlying surgical pathology may be cleaned up and closed in the emergency department.182 Complex entry wounds or those with underlying surgical pathology will require more extensive surgical manipulation, which may include extending the incision to allow vigorous superficial debridement; complete excision of devitalized tissues including muscle, fascia, and periosteum; extension and debridement of the bony entry or exit site by craniectomy; and similarly generous debridement of the dural opening with watertight closure, using grafting as necessary.†
Predictive Factors
Increased mortality in penetrating head injury is associated with increasing age, suicide attempts, hypotension, coagulopathy (particularly with lower GCS scores), respiratory distress, low GCS score, bilateral fixed and dilated pupils, high ICP, and cisternal effacement on CT scan. Mortality also is increased with traumatic injuries in which a missile tract perforates the skull (through-and-through injury) or with injuries that involve both hemispheres, multiple lobes, or the ventricular system.162
[/not-level-membership-for-critical-care-medicine-category]
