[level-membership-for-emergency-medicine-category]
Head injuries
Introduction
There are no reliable up-to-date figures for the total denominator of attendees who attend Emergency Departments (EDs) with a head injury (National Institute for Health and Clinical Excellence 2007). The role of ED staff is to diagnose and appropriately treat a large number of patients presenting following a head injury. Neurological damage occurs both at the time of the injury (primary insult) and evolves over the following minutes, hours and days (secondary insult). Patient outcomes improve where secondary insults are treated early, and where successful responses result in limiting and preventing further injury caused by secondary insult. Wyatt et al. (2008) argue that emergency nurses are ‘fundamental to keeping morbidity to a minimum by being vigilant and prevent secondary brain injury.’
While 90 % of traumatic brain injuries (TBIs) are considered mild (Vos et al. 2012), it is the world’s leading cause of morbidity and mortality in individuals under the age of 45 years old (Wilson 2011). It is estimated that the ED attendance rate in the UK for patients with head injuries is close to 700 000 patients (National Institute for Clinical Excellence 2007) and that 20 % of these are admitted to hospital. Half of those who die from TBI do so within the two hours of the injury. Approximately 30 % of admitted patients with a Glasgow Coma Score (GCS) of <13 will die, and if the GCS is <8, this increases to 50% (Wilson 2011).
The most common causes of a minor head injury are falls (22–43 % of injuries), assaults (30–50 % of injuries), and road traffic accidents (25 % of injuries) (Department of Health 2001, National Institute for Health and Clinical Excellence 2007). In the UK, 70–88 % of individuals that sustain a head injury are male, 10–19 % are aged 65 years or greater, and 40–50 % are children. Alcohol may be involved in up to 65 % of adult head injuries and road traffic accidents account for a large proportion of moderate to severe head injuries. Traumatic injury in which severe head injury plays a major role in over 50 % of cases, is a leading cause of death in those aged 25 years or less (Maartens & Lethbridge 2005); 5–10 % of patients who suffer a severe head injury also suffer a cervical spine injury.
A structured approach to the care of head-injured patients should be initiated based on current valid evidence. The Brain Trauma Foundation has developed recommendations for the management of moderate and severe head injuries (Bratton et al. 2007) in collaboration with the Advanced Trauma Life Support (ATLS) system (American College of Surgeons 2008), providing a mechanism for assessment and immediate management and minimizes the risk of secondary brain injury in adults. For children the Advanced Paediatric Life Support (APLS) system (Advanced Life Support Group 2011) should be employed (National Institute for Health and Clinical Excellence 2007). These guidelines are based on class II (studies based on prospectively collected data and the retrospective analysis of reliable data) and III (studies based on retrospectively collected data) evidence. There is a lack of class I (prospective randomized controlled trials) evidence to support practice.
Anatomy and physiology
The cranium is one of the strongest structures in the body and provides the bony protection for the brain. It is composed of the parietal (2), occipital, frontal, temporal (2), sphenoid, and ethmoid bones. Figure 5.1 shows an exploded view of the cranial skull; however, the bones are fused along main sutures, the sagittal, coronal, lambdoidal, and squamosal. The facial bones form the framework for the nasal and oral cavities and include the zygomatic bones (2), palatine bones (2), mandible, maxilla (2), lacrimal bones (2), nasal bones (2), vomer and the inferior nasal concha (2) (Fig. 5.2).
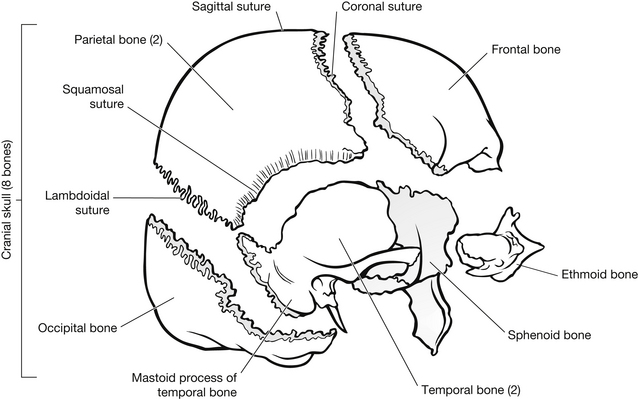
Figure 5.1 Exploded view of the cranial skull.
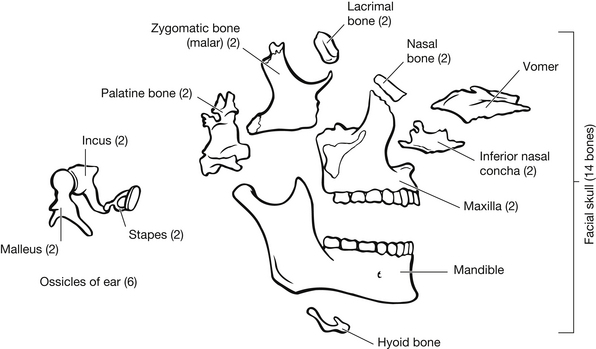
Figure 5.2 Exploded view of the facial skull.
Figure 5.3 shows the irregular internal surfaces of the skull. These irregular surfaces/bony protrusions account for injury to the brain as it moves within the skull under acceleration/deceleration forces.
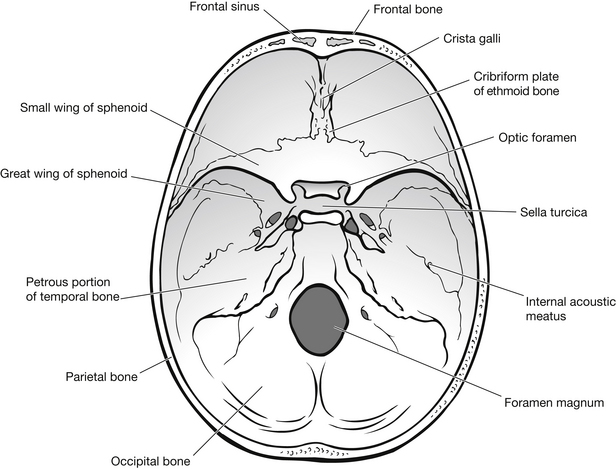
Figure 5.3 View of the base of the skull from above.
The meninges
The brain and the spinal cord are encased by three layers of membrane – the dura mater, the arachnoid mater, and the pia mater, known collectively as the meninges (Fig. 5.4).
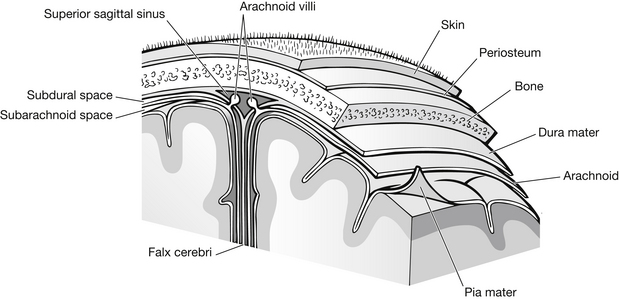
Figure 5.4 The cranial meninges.
Dura mater
The dura mater consists of two layers: the outer layer is the periosteal layer of the skull, which terminates at the foramen magnum, and the inner layer is a strong, thick membrane that is continuous with the spinal dura mater. There is a potential space between the two dura, except at the falx cerebri, which divides the left and right hemispheres of the cerebrum; the tentorium cerebelli, which divides the cerebrum and cerebellum; the falx cerebelli, which divides the lateral lobes of the cerebellum; and the diaphragm sellae. The dura creates a roof for the sella turcica (which houses the pituitary gland). These compartments provide support and protection for the brain and form the sinuses, which drain venous blood from the brain (Crossman & Neary 2000, Lindsey et al. 2004).
Arachnoid mater
The arachnoid mater is a fine serous membrane that loosely covers the brain. There is a potential space between this and the inner dura mater, known as the subdural space. Between the arachnoid mater and the pia mater is an actual space, known as the subarachnoid space, which contains the arachnoid villi, cerebrospinal fluid (CSF), and small blood vessels.
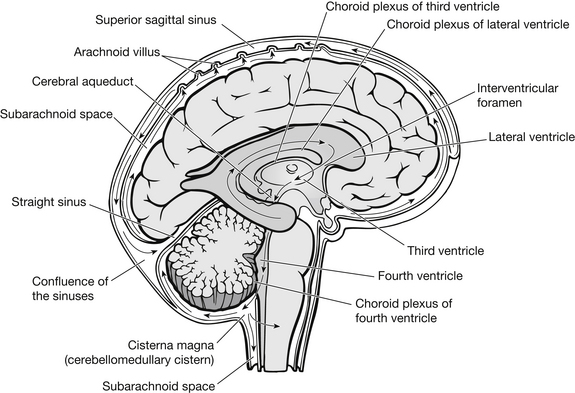
The ventricles and cerebrospinal fluid
Within the brain there are four connected cavities called ventricles, which contain CSF. These are the left and right lateral ventricles, the third ventricle and the fourth ventricle. The lateral ventricles lie in the cerebral hemispheres, the third in the diencephalon, and the fourth in the brain stem. The lateral ventricles are connected to the third ventricle by the interventricular foramen, sometimes known as the foramen of Munro, and the third ventricle is connected to the fourth by the cerebral aqueduct, sometimes known as the aqueduct of Sylvius (Fig. 5.5).
The brain
The brain consists of three main areas:
The major structures within the brain are summarized in Box 5.1.
Cerebrum
The surface area of the cerebral cortex (grey matter) on the surface of the brain is much increased by the presence of gyri and sulci (Fig. 5.6), resulting in a 3:1 proportion of grey to white matter. Below the cortex lies the white matter. The cerebral hemispheres are composed of four lobes, the frontal, parietal, temporal and occipital lobes. Box 5.2 summarizes the main functions of these lobes.
Cerebellum
Brain stem
The brain stem is the connection between the brain and the spinal cord and is continuous with the diencephalon above and the spinal cord below. Within the brain stem are ascending and descending pathways between the spinal cord and parts of the brain. All cranial nerves except the olfactory (1) and the optic (2) nerves emerge from the brain stem (Fig. 5.7). The brain stem is formed from three main structures:
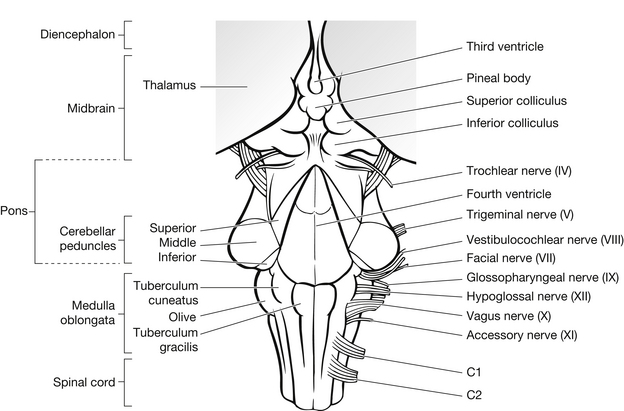
Figure 5.7 Brain stem (dorsal view).
The midbrain connects the pons and the cerebellum to the cerebrum. It is involved with visual reflexes, the movement of the eyes, focusing and the dilatation of the pupils. Contained within the midbrain and upper pons is the reticular activating system, which is responsible for the ‘awake’ state.
Cerebral circulation
The brain is supplied with blood by four major arteries: two internal carotid arteries, which supply most of the cerebrum and both eyes; and two vertebral arteries, which supply the cerebellum, brain stem and the posterior part of the cerebrum. Before the blood enters the cerebrum it passes through the circle of Willis, which is a circular shunt at the base of the brain consisting of the posterior cerebral, the posterior communicating, the internal carotids, the anterior cerebral and the anterior communicating arteries (Figs 5.8 and 5.9). These vessels are frequently anomalous; however, they allow for an adequate blood supply to all the brain, even if one or more is ineffective.
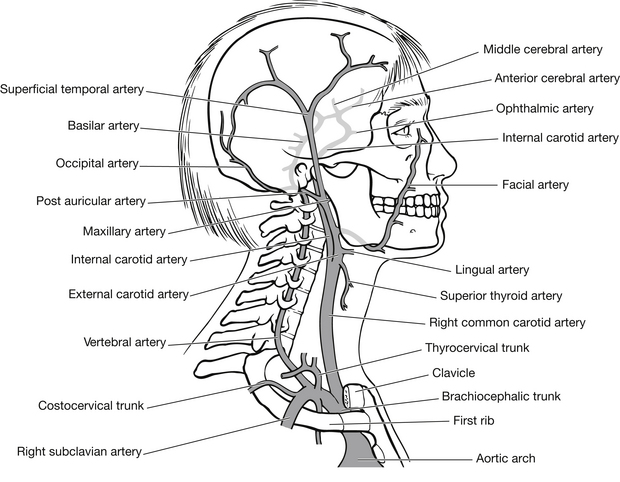
Figure 5.8 Major arteries of the head and neck.
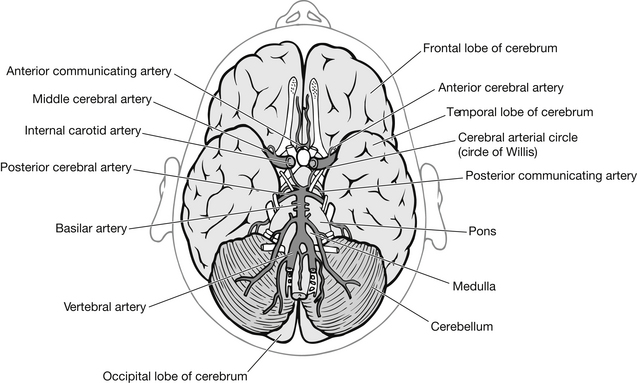
Figure 5.9 Cerebral circulation.
The venous drainage from the brain does not follow a similar pathway (Fig. 5.10). Cerebral veins empty into large venous sinuses located in the folds of the dura mater. Bridging veins connect the brain and the dural sinuses and are often the cause of subdural haematomas. These sinuses empty into the internal jugular veins, which sit on either side of the neck and return the blood to the heart via the brachiocephalic veins.
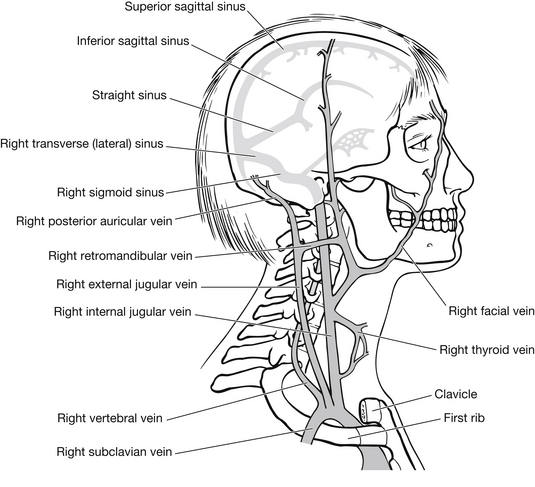
Figure 5.10 Major veins of the head and neck.
Physiology of raised intracranial pressure
Intracranial pressure (ICP) represents the pressure exerted by the CSF within the ventricles of the brain (Hickey 2009). The exact pressure varies in different areas of the brain. The normal range is 0–15 mmHg in adults, 3–7 mmHg in children, and 1.5–6 mmHg in term babies when measured from the foramen of Munro.
ICP is fundamental in maintaining adequate brain function. The brain lies in the skull, a rigid compartment. The contents of the skull are non-compressible, i.e., brain tissue (80 %), intravascular blood (10 %) and cerebrospinal fluid (10 %). Normally these components maintain a fairly constant volume, therefore creating dynamic equilibrium therein. Should one or more components increase for whatever reason, the Monro-Kellie hypothesis states that another component must decrease in quantity in order to maintain the dynamic equilibrium and thus maintain adequate cerebral blood flow (CBF). If this does not occur, ICP rises, leading to brain injury (Hickey 2009). Dynamic equilibrium is maintained by a number of compensatory mechanisms, these include:
In severe traumatic brain injury the compensatory mechanisms are rapidly exhausted (Deitch & Dayal 2006). They fail in the healthy adult brain when the ICP reaches 20 mmHg. Once exhausted, small increases in brain mass, blood, or CSF volume have a profound effect on ICP. Chestnut et al. (1993) demonstrated a clear correlation between the length of time patient’s ICP remains greater than 20 mmHg and an increased mortality and morbidity rate.
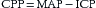
CPP is used as an indicator of CBF, and therefore oxygen delivery to the brain. Current recommendations suggest that in adults the CPP should lie between 50–70 mmHg, although adults with intact pressure autoregulation may tolerate higher CPP values (Bratton et al. 2007). When the patient’s MAP falls and/or the patient’s ICP increases, there is a risk that the cerebral perfusion pressure will fall to too low a value to maintain adequate CBF. This results in cerebral hypoxia and secondary brain injury in the form of cerebral ischaemia and potentially infarction. Autoregulation fails when CPP falls below 50 mmHg or rises above 150 mmHg, resulting in CPP and CBF become dependent on the systemic blood pressure alone.
Chemo autoregulation is triggered by changes in extracellular pH and metabolic by-products. Changes in PCO2 or a dramatic reduction in PO2 (Fig. 5.11) may trigger this. Hypercapnia >45 mmHg (or 6 kPa) (Albano 2005) is a potent vasodilator that induces hyperaemia and cerebral blood volume increases, without an adequate decrease in CSF; as a result of the compensatory response the ICP will rise. Hypocapnia <45–>30 mmHg (or 6–4 kPa) considered a critical low value (Garner & Amin 2007) is a potent vasoconstrictor that reduces brain mass and induces hypoaemia; as a result cerebral blood flow and volume decrease leading to a risk of secondary ischaemia and potentially an infarction if not treated (Albano 2005).
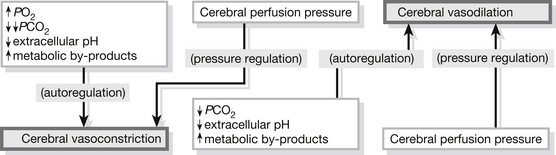
Figure 5.11 Autoregulation of the brain.
Classification of head injuries
Head injuries can be classified under three anatomical sites:
Scalp injuries
There are four types of injury to the scalp:
• abrasion – minor injury that may cause a small amount of bleeding. Treatment may not be required, but ice applied to the area may reduce any haematoma formation (Hickey 2009)
• contusion – no break in the skin, but bruising to the scalp may cause blood to leak into the subcutaneous layer
• laceration – a cut or tear of the skin and subcutaneous fascia that tends to bleed profusely. Bleeding from the scalp alone is unlikely to cause shock in the adult. In small children, a scalp laceration may be sufficient to cause hypovolaemia. Scalp lesions should be explored under local anaesthetic for foreign bodies and/or skull fracture with a skull X-ray if there is any doubt about the diagnosis. Lesion(s) should be sutured or glued according to their depth and position
• subgaleal haematoma – a haematoma below the galea, a tough layer of tissue under the subcutaneous fascia and before the skull. The veins here empty into the venous sinus, and thus any infection can spread easily to the brain, despite the skull remaining intact. There is controversy surrounding the treatment of subgaleal haematomas, due to the risks of infection; therefore some doctors argue that it is best to evacuate the haematoma, while others suggest that it is best to let it reabsorb.
If the scalp injuries are only part of other injuries, it is important they are documented to allow further investigation at a more appropriate time. They may need to be cleaned and dressed or temporarily sutured.
Skull injuries
• linear – these are the most common types of injury. They usually result from low-velocity direct force. They are usually diagnosed from skull X-ray and need no specific treatment
• depressed – usually evident clinically, but a skull X-ray to discover the full extent of the potential brain damage is usually necessary. Management is dependent on the severity of the fracture and whether there are any accompanying injuries. If there are no other injuries requiring surgical management, they may not be surgically elevated, due to the risks of infection. However, surgical intervention will normally be necessary if there are bone fragments embedded in the brain so as to elevate the bone fragments and manage the brain trauma
• open – usually evident clinically. Usually managed according to the severity of the injury. If debris is dispersed in the brain tissue then surgery will be required and there is a heightened risk of infection
• comminuted – these are detected on skull X-ray. These patients should be closely observed and any neurological deficits managed appropriately. Surgical intervention is usually required. If there are bone fragments imbedded in the brain tissue then surgery will be required to elevate the bone fragments and manage the brain trauma
• basal – these are diagnosed clinically as they are difficult to detect on X-ray. Signs include CSF leakage from the nose (rhinorrhoea) or the ear(s) (otorrhoea). Rhinorrhoea or otorrhoea indicates that a skull base fracture has breached the dura and formed a communication between the intracranial contents and an air sinus. This places the patient at risk of meningitis while the CSF leak continues. If CSF leakage is suspected, the fluid should be tested for glucose and the ‘halo test’ performed, where a small amount of fluid is placed on blotting paper; if CSF is present it will separate from blood and form a yellow ring around the outside of the blood. Patients with a base of skull fracture may also have retroauricular bruising (Battle’s signs) and periorbital bruising (‘panda eyes’ or ‘raccoon eyes’): 80–90 % of cases seal within two weeks and neurosurgical intervention is usually not considered until this time has elapsed. An exception is a fracture of the posterior wall of the frontal sinus, visualized on CT scan, where anterior fossa repair may be undertaken early.
Brain injuries
Severe brain injury is uncommon because the skull and scalp absorb the majority of the impact of the assault. The amount of brain damage suffered is relative to the force/energy of the assault. A high-energy head injury results when: a pedestrian is struck by a motor vehicle, an occupant is ejected from a motor vehicle, a person falls from a height of greater than 1 metre or more than five stairs (a lower threshold for the height of falls should be used when dealing with infants and young children under 5 years old), following a diving accident, following a high-speed motor vehicle collision, following a rollover motor accident, or a bicycle collision (National Institute for Health and Clinical Excellence 2007).
Damage to the brain as a result of trauma includes both the immediate (primary) injury caused at the moment of the impact and the secondary injury that develops during the first few minutes, hours or days after the impact (Box 5.3). These secondary injuries may have extracranial or intracranial causes.
There are no interventions that can prevent the primary brain injury. Secondary brain injuries, which further exacerbate the primary neuronal injury and lead to a worsening outcome depending upon their duration and severity, are largely preventable. The two main causes of secondary injury are delayed diagnosis and treatment of intracranial haematomas, and failure to correct systemic hypoxaemia and hypotension (Fig. 5.12). Brain injuries are usually categorized as either focal or diffuse injuries.
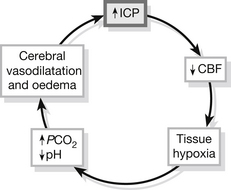
Figure 5.12 Cycle of progressive brain swelling.
Focal injuries
Haematoma: Extradural haematoma (EDH) is an accumulation of blood in the extradural space between the periosteum on the inner side of the skull and the dura mater (Fig. 5.13). Most are associated with skull fracture and are commonly caused by a laceration to the middle meningeal artery or vein, or less commonly to the dural venous sinus, following an insult to the temporal-parietal region. Consequentially, the parietal and parieto-temporal areas of the brain are affected. In 85 % of patients the EDH will be accompanied by a skull fracture (Hudak & Gallo 1994).
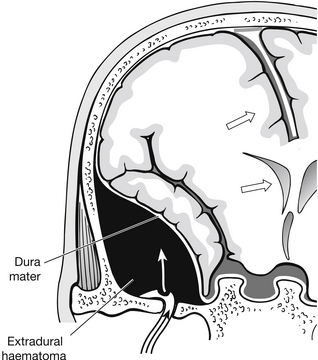
Figure 5.13 Extradural haematoma.
Patients with skull fractures may be neurologically intact on admission and later deteriorate as the EDH develops. Most often the primary brain injury causes some disturbance of consciousness and the developing haematoma results in rapid neurological deterioration (Kwiatkowski 1996). Patients with EDHs most commonly present with a history of transient loss of consciousness, followed by lucidity for a period (hours to days) dependent on the rate of the bleed, irritation and headache. Patients then rapidly lose consciousness and deteriorate very quickly. Late signs are seizures, ipsilateral pupil dilatation, unconsciousness, and contralateral hemiplegia. Surgical treatment is required to evacuate the haematoma and ligate the damaged blood vessel.
Subdural haematoma (SDH) is an accumulation of blood between the dura mater and arachnoid mater. SDHs are caused by the rupture of bridging veins from the cortical surfaces to the venous sinuses (cortical veins) (Fig. 5.14).
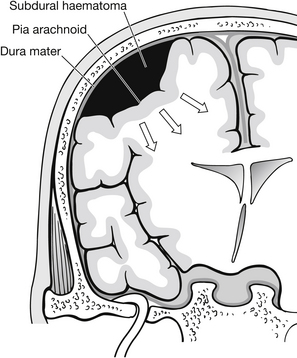
Figure 5.14 Subdural haematoma.
SDHs can be seen in isolation, but more commonly are associated with accompanying brain injury, i.e., cerebral contusions and/or intracerebral haematomas. They are the most common intracranial mass result from head trauma (Maartens & Lethbridge 2005). In most cases a large contusion is found at the frontal or temporal surface of the brain. SDHs are predisposed with increasing age and alcoholism. Both groups can suffer regular falls and have a degree of cerebral atrophy, which puts strain on the bridging veins and coagulopathy. Subdural haematomas are classified as acute, subacute and chronic:
• acute (ASDH) refers to symptoms which manifest before 72 hours post-injury. Most patients harbouring an acute SDH are unconscious immediately following major cerebral trauma. The expanding haematoma then causes additional deterioration (Duffy 2001)
• subacute refers to symptoms which manifest between 72 hours and 3 weeks post-injury
• chronic refers to symptoms which manifest after 3 weeks post-injury. The injury may have been considered as minor and the patient often does not remember a particular predisposing injury.
The most common symptom of a SDH is a headache, which progressively intensifies and is eventually accompanied by vomiting, cognitive impairment(s), a depressed level of consciousness, and a focal deficit, which will vary depending on severity of the injury. Even in the absence of focal deficit, increasing ICP may lead to cognitive impairment and eventually a depressed level of consciousness (Watkins 2000). SDHs are often associated with other injuries, and therefore the symptoms can become confused within a general head injury picture. Small SDHs may be treated conservatively, as they will reabsorb over time. Larger SDHs will require evacuation, due to the secondary damage they cause.
Intracerebral haematoma (ICH) is caused by bleeding within the substance of the brain (Fig. 5.15). ICH usually affects the white matter and the basal ganglia found deep within the brain parenchyma. ICHs are related to contusions as a result of a major impact, and are usually found in the frontal, temporal, and parietal lobes. Other causes include penetrating and missile injuries and shearing of blood vessels deep within the brain following an acceleration/deceleration injury. Symptoms include headache, contralateral hemiplegia, ipsilateral dilated/fixed pupil and deteriorating level of consciousness, progressing to deep coma (GCS < 8). Treatment tends to be conservative, due to the difficulties of evacuating haematomas situated so deeply within the brain. Mortality is high within this group of patients.
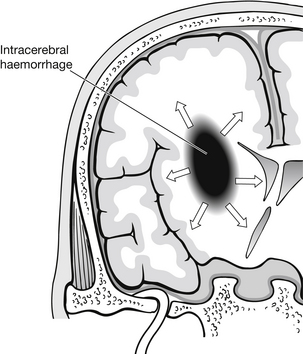
Figure 5.15 Intracerebral haematoma.
Subarachnoid haemorrhage (SAH) is seen in 30–40 % of patients following severe traumatic brain injury. The mortality and morbidity rates are double in these patients compared with those with similar injury without the SAH component (Dearden 1998). The patient either suffered the SAH prior to the insult and thus the SAH is possibly the cause of the incident (Sakas et al. 1995), or the vessels in the subarachnoid space are damaged by the shearing forces at the time of the insult.
Diffuse injuries
Diffuse injuries occur throughout the brain rather than in a specific area of the brain. They result in generalized dysfunction. Diffuse injuries range from concussion with no residual damage, to diffuse axonal injury and persistent vegetative state. Diffuse injury occurs in 50–60 % of patients with severe head trauma and is the commonest cause of unconsciousness, the vegetative state and subsequent disability (Graham et al 1995).
Concussion is a transient form of diffuse injury that occurs following blunt trauma. It causes a temporary neuronal dysfunction because of transient ischaemia or neuronal depolarization. This manifests as a headache, dizziness, inability to concentrate, disorientation, irritability, and nausea. Concussion can occur with or without memory loss. Concussion is graded in line with the severity of symptoms (Box 5.4).
Approximately one-third of patients with head injuries who are discharged from emergency departments have persistent post-concussion-type symptoms, such as headache, fatigue, inability to concentrate, irritability, and anxiety, persisting for several months due to mild diffuse axonal injury (Jackson 1995). The majority of these patients will have been knocked out for a short time and may have other mild neurological signs. As there is no treatment for mild diffuse axonal injury, and recovery is usually spontaneous, reassurance and psychological support are vital to the patient’s recovery (Box 5.5).
Acute axonal injury is usually the result of an acute rotation/deceleration injury, typically following a road traffic accident (Fig. 5.16). The patient usually becomes unconscious rapidly after injury, due to the shearing injury to the brain. Mortality is high in this patient group, and those who do survive usually suffer severe neurological dysfunction.
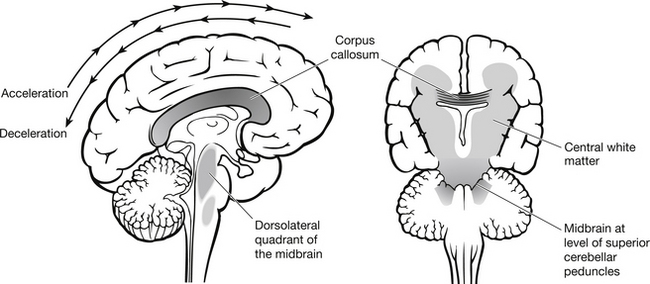
Figure 5.16 Diffuse axonal injury.
Cerebral oedema – This is a consistent reaction of the brain to an insult and it usually develops during the first 3–5 days following the insult causing an increase in ICP. Cerebral oedema following severe traumatic brain injury affects almost all patients to a greater or lesser degree. The opening of the blood–brain barrier is a central prerequisite to the development of cerebral oedema (Fernandes & Landolt 1996). Four pathophysiological mechanisms of cerebral oedema have been proposed (Table 5.1).
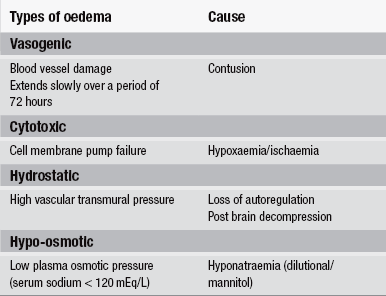
Cerebral ischaemia occurs whenever the delivery of oxygen and substrates to the brain falls below its metabolic needs, as a result of hypoxia (cardiac arrest, obstructive airway, cervical spinal injury and prolonged epileptic-type seizures), hypotension and/or intracranial hypertension (raised ICP).
Cerebral ischaemia may be global or focal, complete or incomplete. Incomplete ischaemia differs from complete ischaemia in that there is a continuing supply of glucose to the brain tissue despite tissue hypoxia. The glucose sustains anaerobic metabolism, which increases the brain lactic acid level. Neuronal damage occurs above a certain threshold. This effect is the basis for concern that increased peri-ischaemia glucose levels may increase and/or hasten the ischaemia tissue damage. Ischaemia leads instantly to cerebral oedema, which in turn worsens ischaemia (Farnsworth & Sperry 1996).
As the PaCO2 increases, unaffected normal blood vessels dilate, however blood is shunted away from the abnormal areas of the brain that do not respond to CO2 and is known as the ‘steal phenomenon’. An inverse steal (Robin Hood phenomenon) occurs when the PaCO2 is reduced and the unaffected, normal blood vessels vasocontrict, shunting blood to the abnormal areas of the brain that do not constrict (Darby et al. 1988).
The concept of ischaemia penumbra states that the area of the brain around an ischaemic brain, where blood flow provides sufficient oxygenation for the cells to survive but insufficient oxygenation for the cells to maintain normal neuronal function, can re-establish normal function if blood flow and oxygenation to this area is rapidly improved. Cerebral ischaemia is the single most important factor in determining the outcome in severe traumatic brain injury: ischaemia lesions are found in 90 % of patients at post-mortem (Dearden 1998).
Management
Patients who have sustained a head injury should be assessed and managed according to clear principles and standard practice as embodied in the ATLS system (American College of Surgeons 2008) and for children the APLS system (Advanced Life Support Group 2011) should be employed (Bavetta & Benjamin 2002, National Institute for Health and Clinical Excellence 2007). The main focus of the assessment should be the risk of clinically important brain and cervical spine injuries. Due attention should also be paid to co-existing injuries and other concerns the healthcare team may have, e.g., non-accidental injury.
External referrals
• GCS less than 15 at any time since the injury
• any loss of consciousness as a result of the injury
• any focal neurological deficit since the injury, e.g., problems understanding, speaking, reading or writing, loss of sensation in a part of the body, problems balancing, general weakness, problems walking, and any changes in eyesight
• any seizure since the injury
• any suspicion of a skull fracture or penetrating head injury, e.g., CSF leakage from the nose (rhinorrhoea) or the ear(s) (otorrhoea), black eye(s) with no associated damage around the eye(s), bleeding from one or both ears, new deafness in one or both ears, bruising behind one or both ears, penetrating injury signs, or visible trauma to the scalp or skull
• the injured person or their carer is incapable of transporting the injured person safely to the hospital emergency department without the use of ambulance services, providing any other risk factors indicating emergency department referral are present (National Institute for Health and Clinical Excellence 2007).
Telephone advice services e.g., NHS Direct should refer people who have sustained a head injury to a hospital emergency department if the related history indicates any of the following risk factors:
• amnesia for events before or after the injury: the assessment of amnesia will not be possible in preverbal children and is unlikely to be possible in any child under 5 years old
• persistent headache since the injury
• any loss of consciousness as a result of the injury from which the injured person has now recovered
• any vomiting episode since the injury: clinical judgement should be used regarding the cause of vomiting in those aged less than or equal to 12 years and whether referral is necessary
• any previous cranial neurosurgical intervention
• history of bleeding or clotting disorders
• current anticoagulant therapy such as warfarin
• current drug or alcohol intoxication
• age greater than or equal to 65 years
• suspicion of non-accidental injury
• irritability or altered behaviour particularly in infants and young children
• continuing concern by the Helpline’s personnel about the diagnosis (National Institute for Health and Clinical Excellence 2007).
In the absence of the factors listed above the telephone advice services should advise the injured person to seek medical advice from community health services, e.g., general practice and NHS walk-in centres if any of the following factors are present:
History
Accurate history-taking gives vital clues to the type and potential severity of the head injury (Shah 1999) (Box 5.6). This may have to be obtained from a witness or paramedic. If the history is obtained from the patient, it should be corroborated by a witness/relative if possible.
Assessment
Management of head injury in the emergency department revolves largely around the assessment of the risks of, and the prevention or limiting of, secondary brain injury (the causation of secondary brain injury is shown in Box 5.7) and of injury to the cervical spine, whilst the patient awaits definitive treatment such as surgery to evacuate haematoma. Due attention should also be paid to co-existing injuries.
Patients presenting to the emergency department with a GCS less than or equal to 8 should be assessed early by an anaesthetist or critical-care physician to provide appropriate airway management and to assist with resuscitation (National Institute for Health and Clinical Excellence 2007). The recommended primary investigation of choice for the detection of acute clinically important brain injuries is CT imaging (National Institute for Health and Clinical Excellence 2007). CT scanning was generally reserved for patients with moderate or severe head injuries (GCS less than 13) and should be undertaken within 30 minutes of admission to the ED (National Institute for Health and Clinical Excellence 2007). MRI for safety, logistic and resource reasons is not currently indicated as the primary investigation, although additional information of importance to the patient’s prognosis can sometimes be detected using MRI, care must be taken to ensure that the patient does not harbour an incompatible device, implant, or foreign body. Skull X-ray(s) are the recommended primary investigation of choice for skull fractures (Box 5.8). If CT scanning is not available, then skull X-rays along with high-quality patient observations have a vital role. Early imaging, rather than admission and observation for neurological deterioration, reduces the time needed to detect life-threatening complications and is associated with a better outcome (National Institute for Health and Clinical Excellence 2007). Indications for CT scanning are listed in Box 5.9.
Neurological assessment
Full neurological assessment forms part of the secondary survey, as should a thorough examination of the scalp for lacerations, haematoma, or evidence of a depressed skull fracture. The assessment and classification of patients who have suffered a head injury should be guided primarily by the adult (16 years or older) and paediatric versions of the GCS and its derivative the Glasgow Coma Score (National Institute for Health and Clinical Excellence 2007). The paediatric version should include a ‘grimace’ alternative to the verbal score to facilitate assessment in the pre-verbal or intubated patients.
The GCS, developed by Teasdale & Jennett (1974) continues to be the gold standard for assessing consciousness (Addison & Crawford 1999, Stern 2011) (Table 5.2 and Fig. 5.17). The GCS measures arousal, awareness, and activity by assessing eye opening (E), verbal response (V), and motor response (M). Each activity is allocated a score, therefore enabling objectivity, ease of recording, and comparison between recordings. It also provides useful information for patient outcome prediction. The score should be based on the sum of 15 and to avoid confusion this denominator should be specified, e.g., 13/15. When recording the GCS it is important to record the three separate response scores as well as the total GCS score, i.e., E2, V3, and M4, GCS = 9 (National Institute for Health and Clinical Excellence 2007).
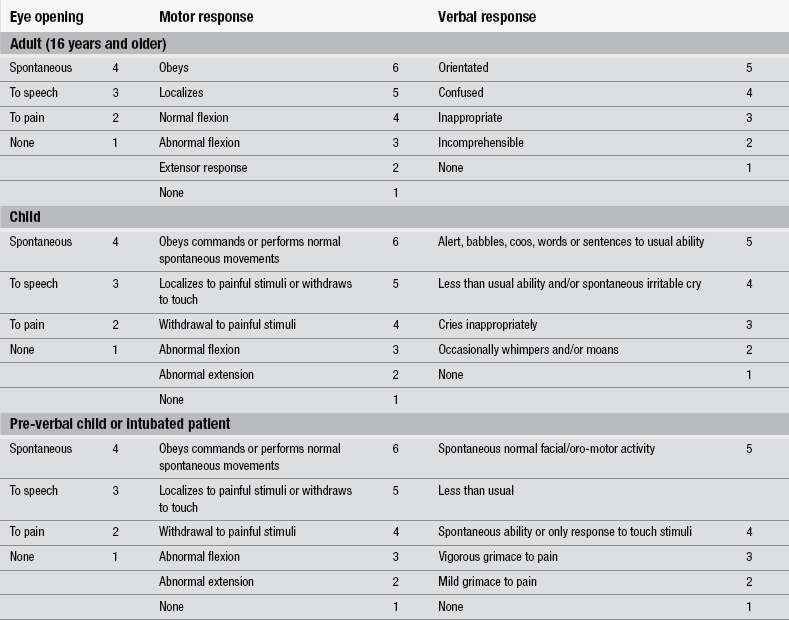
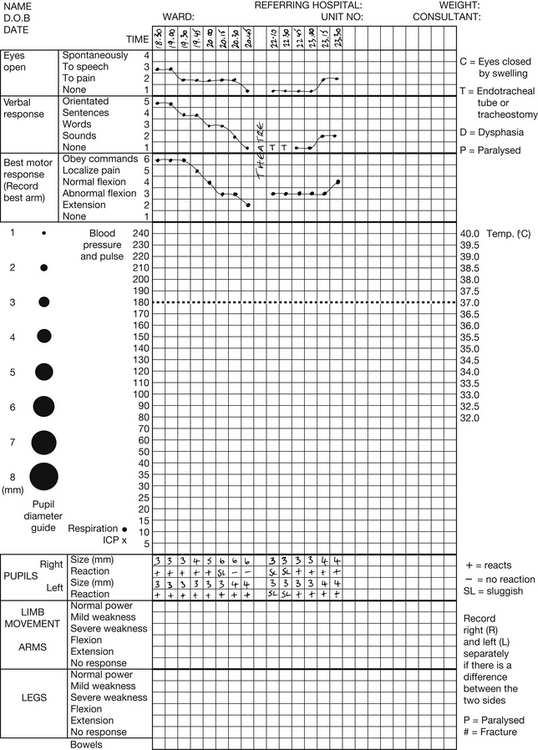
Figure 5.17 Glasgow Coma Scale.
When applying verbal stimulus, it is good practice to commence with normal voice and then increase volume to elicit a response. It is important to ascertain whether the patient is deaf, wears a hearing aid, and whether English is the patient’s spoken language. When applying a painful stimulus, it is good practice to commence with light pressure and then increase to elicit a response. When assessing motor function, always record the response from the best arm. There is no need to record left and right differences, as the GCS does not aim to measure focal deficit. It is not appropriate to measure leg response unless unavoidable, i.e., injury to both arms, as a spinal reflex rather than a brain-initiated response might be initiated (Teasdale & Jennett 1974).
The GCS may be misleading in patients who have a high cervical injury or brain stem lesion, and in those who are hypoxic, suffering haemodynamic shock, or suffer from epileptic seizures. These patients may be unable to move their limbs or show no responses to stimuli. It is important to attempt to assess the spinal patient using facial movements, being aware of the possibility of a combined head and neck injury. Patients who show no response should be re-evaluated following correction of any shock or hypoxia (National Institute for Health and Clinical Excellence 2007). Post-resuscitation, the GCS with specific emphasis on the motor response score is a powerful predictor of outcome (Hunningher & Smith 2006).
As well as providing a baseline for assessing the patient’s progress, vital signs give important information about potential secondary brain injury, e.g., respiration rate and cerebral hypoxia. When assessing vital signs in conjunction with GCS, it is important to remember the following:
• hypotension is only of neurological origin in end-stage brain injury or spinal shock; other causes of hypotension, such as hypovolaemia, should be investigated
• Cushing’s triad (hypertension, bradycardia and bradypnoea) indicates a life-threatening rise in ICP
• pyrexia with hypertension may indicate autonomic dysfunction.
Limb movement is useful to assess for focal damage. However, although it is usual for a hemiparesis or hemiplegia to occur on the contralateral side to the lesion, it may occur on the ipsilateral side. This is due to indentation of the contralateral cerebral peduncle and is known as a false localizing. Spontaneous movements are observed for equality. If there is little or no spontaneous movement, then painful stimuli must be applied to each limb in turn, comparing the result. As already stated it is most appropriate to complete this while assessing the motor component of the GCS.
Pupils are assessed for their reaction to light, size and shape, i.e., cranial nerves II (optic) and III (oculomotor) activity. Each pupil needs to be assessed and recorded individually. Pupils are measured in millimetres, the normal range being 2–6 mm in diameter. They are normally round in shape and abnormalities are described as ovoid, keyhole, or irregular (Hickey 2009). A bright light is shone into the side of each eye to assess the pupils’ reaction to light. This should produce a brisk constriction in both pupils, the consensual light reaction. Herniation of the medial temporal lobe through the tentorium directly damages the oculomotor (CNIII) nerve, resulting in dilation of the pupil and an impaired reaction to light. The pupil dilates on the side of the lesion.
Patients with head injuries can be classified into three groups depending on their GCS:
• a score of 13–15 is indicative of a minor head injury. In some patients, a one-point drop in their GCS can be alcohol- or drug-induced. This necessitates extra vigilance from nursing staff as alcohol and/or drugs may mask subtle changes in the patient’s cognition or conscious level
• a GCS of 9–12 suggests a moderate head injury, or a more serious injury evolving. Any changes in the patient’s condition should be closely monitored
• a severe head injury is classed by a GCS of 8 or less. These patients are potentially at risk of secondary brain injury, and their GCS and vital signs should be monitored at frequent intervals.
Admission to hospital
Patients should be admitted if:
• they have suffered a new surgically significant abnormality on imaging
• the patient has not returned to a GCS equal to 15 after imaging, regardless of the imaging results
• they fulfil the criteria for CT scanning but this cannot be done within the appropriate period, either because CT is not available or because the patient is not sufficiently co-operative to allow scanning
• the patient has continuing worrying signs of concern to the clinicians, e.g., persistent vomiting and severe headache
• the patient has received sedation or general anaesthetic during CT imaging
• the patient has other sources of concern to the clinicians, e.g., drug and/or alcohol intoxication, other injuries, shock, suspected non-accidental injury, and cerebrospinal fluid leak (National Institute for Health and Clinical Excellence 2007).
Patients should undergo urgent reassessment by the clinician in charge of the case if any of the following responses occur:
• development of agitation or abnormal behaviour
• a sustained (at least 30 minutes) drop of one point in GCS level (greater weight should be given to a drop of one point in the motor score of the GCS)
• any drop of greater than two points in GCS level regardless of duration or GCS sub-scale
• development of severe or increasing headache or persisting vomiting
• new or evolving neurological symptoms or signs such as pupil inequality or asymmetry of limb or facial movement (National Institute for Health and Clinical Excellence 2007).
Management of minor traumatic head injury
The majority of patients treated in emergency departments with head injury will have a ‘minor’ head injury and the majority will be discharged home. A thorough assessment of the patient’s condition should be performed, which should include pulse, respiration, blood pressure, pupil size and reaction, and the patient’s GCS (Caton-Richards 2010). Although only 1 % of head-injured patients have skull fractures (Ramrakha & Moore 1997), there are certain circumstances where a skull X-ray is appropriate (Box 5.8).
The key to managing minor head injury is giving adequate information and advice to the patient and his carer. The patient should be advised to rest quietly, avoid stressful situations, should be discouraged from taking part in strenuous activities and from undertaking long periods of visual display unit work or watching television which will exacerbate any headache. The patient should not stay at home alone for the first 48 hours after leaving hospital. Simple analgesia, such as paracetamol, that should be sufficient to alleviate headaches without masking other signs of deterioration should be suggested. The patient should be discouraged from taking alcohol or non-prescribed drugs until symptoms have subsided. The patient should not play any contact sport for at least three weeks after the injury without talking to their doctor or other appropriately qualified clinician first. Written advice should always be given to the patient/carer to reinforce any verbal information (Box 5.10). (See also Chapter 34 – Health Promotion.)
When discussing the outcomes of a minor head injury, it is important that the emergency nurse explains post-concussion-type symptoms to the patient. Head-injured patients should be discharged into the care of a responsible adult. Not all patients with a minor head injury are appropriate for discharge (Box 5.11). Particular care is needed with patients who are intoxicated and/or have taken drugs where neurological assessment is unreliable.
Management of severe traumatic head injury
Despite the fact that patients who have suffered severe injury to the brain may recover completely if they are treated quickly and appropriately, it is also possible that the patient may suffer serious disability or even death (National Institute for Health and Clinical Excellence 2007). Indeed, head injury accounts for the majority of trauma deaths in young adults in Europe (Tagliaferri et al. 2006). Age is a significant factor in determining outcome, especially in patients who become deeply unconscious. The mortality rate is 19 % in patients aged 20, but 71 % in those aged 60 and over (Hickey 2009).
There is a developing base of evidence to guide the management of patients with traumatic brain injury. A study by Patel et al. (2002) looked at the effect of neurocritical care, delivered by specialist staff and based on protocol-driven therapy, they found improved outcomes. It is universally accepted that initial management of severe traumatic brain injury requires urgent cardiopulmonary resuscitation, emergency CT scan, transfer to a neurosurgical centre, and appropriate surgical intervention, and guidelines covering this initial care are generally well established in practice (Intensive Care Society 1997, McNaughton & Harwood 2002,). In the US, the practice of ensuring that pre-hospital care, triage, and admission to designated trauma centres are coordinated within regional trauma systems has been seen to improve the outcomes for individuals with traumatic brain injury (Ghajar 2000). There is little evidence in the UK as to whether patients transported directly to a neurosciences centre compared to those who are taken to their nearest district hospital experience a better outcome (National Institute for Health and Clinical Excellence 2007). Level III evidence from Patel et al. (2002) concluded that specialist neurocritical care is associated with a significant improvement in outcome for patients with TBI.
Trauma services in the UK are presently being organized into a formal system similar to that in the US. In London four trauma networks have been established (Healthcare for London 2009). Extracranial complications occur frequently in severe traumatic brain injury and studies suggest that some complications are highly influential in determining patient outcome. Management should follow the sequence laid down by ATLS (National Institute for Health and Clinical Excellence 2007, American College of Surgeons 2008).
Assessment and management
Airway management is paramount in preventing hypoxia (PO2 <60 mmHg or 8 kPa). Hypoxia is the second most influential cause of secondary brain injury after hypotension and leads to a worse outcome (Chesnut et al. 1993). Prevention of such insults during initial resuscitation at the scene of the injury, during transfer and during hospitalization may have a major impact on outcome (Royal College of Paediatrics and Child Health 2001). Oedema and/or debris following injury, loss of the gag reflex and/or vomiting may threaten airway patency. If a clear airway cannot be maintained with simple aids, such as a Guedel or oropharyngeal airway, then intubation should be performed. Cervical spine immobilization should be maintained until a full risk assessment and imaging (if deemed necessary) has been undertaken and the possibility of a neck injury has been excluded. If urgent intubation is indicated, it should be assumed that the patient has a full stomach, and cricoid pressure should be applied to prevent vomiting or gastric regurgitation. In adults, this should be maintained until the cuff of the endotracheal tube is inflated, creating a secure airway. Short-acting sedatives and muscle relaxants should always be used for intubation, to minimize the risk of raised ICP due to noxious stimulation and coughing. Suctioning should be kept to a minimum as it also raises ICP.
Breathing
In patients with isolated head injury, acute lung injury (ALI) is common. Studies show ALI occurrence in 20 % of patients with a post-resuscitative GCS of 8 or less. ALI is an additional marker of the severity of brain injury and is associated with an increased risk of morbidity and mortality (Bratton & Davis 1997). Neurogenic pulmonary oedema (NPE) represents the most severe form of ALI and is typically reported in cases of fatal or near-fatal head injuries (Bratton & Davis 1997). The development of neurogenic pulmonary oedema may be remarkably rapid and is usually associated with an acute and significant rise in ICP. The exact mechanism responsible for this acute condition seen after severe traumatic brain injury and after abrupt elevations in ICP is unclear. It is generally accepted that there is a neurological pathway following central nervous system injury and that it is the result of massive sympathetic outflow, possibly mediated by the hypothalamus. The classic form appears early, within minutes to a few hours after injury, while the delayed form progresses slowly over a period of 12 to 72 hours (Durieux 1996).
Some studies have shown a link between CPP management and acute respiratory distress syndrome (ARDS) (Contant et al. 2001, Robertson 2001). Induced hypertension to raise CPP can cause increased pulmonary hydrostatic pressures and thereby increase the amount of water accumulating within the lungs. The results of a randomized trial comparing two head injury management strategies, one ICP targeted and the other CPP targeted, showed a fivefold increase in the incidence of ARDS in the CPP targeted group where CPP was maintained >70 mmHg (Robertson 2001).
The pathophysiological changes result in the rapid development of interstitial oedema and subsequent increased pulmonary shunt, decreased compliance, and loss of alveoli surface area. Signs and symptoms include dyspnoea, cyanosis, pallor, sweating, a weak rapid pulse, and the production of pink frothy sputum. Primary treatment consists of employing therapeutic interventions aimed at reducing ICP and to normal limits providing appropriate ventilation support and management, controlling carbon dioxide levels, and maximizing oxygenation with minimal effect on cardiac output. This complicates the management of severe traumatic brain injury, many of the therapies used to protect the lungs e.g., reduced tidal volume, permissive hypoxia and hypercarbia, increased levels of positive tracheal end pressure, and prone lying, causes a rise in ICP or decreased CPP (Robertson et al. 1999).
In patients with traumatic brain injury the effects of hypotension and hypoxia appear to be more profound than those that result when hypoxic and/or hypotensive episodes of similar magnitude occur in trauma patients without neurological involvement (Bratton et al. 2007). It is important to maintain adequate oxygenation, as a rising PCO2 level initiates autoregulation, causing cerebral vasodilatation and a rise in ICP. If unchecked, this may lead to secondary brain injury.
Intubation and ventilation should be implemented immediately in the following circumstances:
• loss of protective laryngeal reflexes
• ventilatory insufficiency as judged by arterial blood gases:
– hypoxaemia (PaO2 <13 kPa on oxygen) or hypercarbia (PaCO2 >6 kPa)
• spontaneous hyperventilation (causing PaCO2 >4 kPa)
• irregular respirations (National Institute for Health and Clinical Excellence 2007).
Intubation and ventilation should be implemented before the start of a transfer in the following circumstances in addition to the above:
• significantly deteriorating conscious level (one or more points on the motor score), even if not coma
• unstable fractures of the facial skeleton
• copious bleeding into the mouth, e.g., base of skull fracture
• seizures (National Institute for Health and Clinical Excellence 2007).
The goal is to achieve and maintain normocapnia, a PaO2 greater than 13 kPa, PaCO2 4.5–5.0 kPa, unless there is clinical or radiological evidence of raised intracranial pressure, in which case more aggressive hyperventilation is justified (National Institute for Health and Clinical Excellence 2007). Hypoxemia, hypercapnia, hypocapnia and acidosis that result from either inadequate ventilation or hypermetabolism following traumatic brain injury are all associated with poor outcomes. Induced hyperventilation should be used to reduce PCO2 levels, but this should be discussed with a neurosurgeon first (Bullock & Teasdale 1996). Hyperventilation is only recommended as a temporary measure in order to reduce elevated ICP. It should not be used in the first 24 hours after injury when CBF is reduced (Bratton et al. 2007). If hyperventilation is used, the inspired oxygen concentration should be increased (National Institute for Health and Clinical Excellence 2007).
Cardiac
High levels of sympathetic activity and of circulating catecholamines after severe traumatic brain injury can have an adverse effect on cardiac function, basal metabolic rate, and vascular and neuronal function in the central nervous system (Clifton et al. 1983). The magnitude of this hyperdynamic cardiovascular state occurring after severe head injury does not necessarily correlate with ICP, GCS or CT findings (Clifton et al. 1981). There is both clinical and experimental evidence to suggest that cerebral neurogenic factors cause arrhythmias in normal hearts, with fatal arrhythmias being reported in otherwise healthy brain-injured patients (McLeod 1982, Oppenheimer et al. 1990). A wide variety of atrial and ventricular arrhythmias, abnormalities of the QRS complex, T-wave and ST segment and QT prolongation have been documented and occur most commonly in patients with diffuse injury, oedema, and contusions. In two seminal studies, 31 % of patients admitted with head injury exhibited some form of cardiac arrhythmia (Hersch 1961) and cardiac arrhythmias were observed in 41 out of 100 patients admitted with acute subdural haematoma, with more than half of these showing ventricular arrhythmias ranging in severity from premature ventricular contractions to ventricular tachycardia and ventricular fibrillation (Van der Ark 1975). Elevations of pulse greater than 120 beats per minute have been found in one-third of patients with severe traumatic brain injury.
It is generally accepted that hypotension related to trauma is not caused by head injury, although it can be related to head injury per se in children. However, there is some evidence that episodes of hypotension following severe traumatic brain injury may be of neurogenic origin in a small proportion of patients, and that this is not simply attributable to devastating, non-survivable brain injury (Chesnut et al 1998).
Hypotension is usually a result of systemic hypovolaemia following multiple traumas. According to the Brain Trauma Foundation, hypotension is a systolic blood pressure of <90 mmHg (Bratton et al. 2007). The diagnosis and treatment of life-threatening extracranial injuries take priority over intracranial injuries according to ATLS protocols. The causes of hypotension should be identified rapidly as the provision of appropriate treatment and fluid resuscitation is imperative to prevent a drop in mean BP, CBF and CPP leading to cerebral ischaemia and secondary brain injury. Hypotension is a predominant contributing factor in secondary brain injury and has the highest correlation with morbidity and mortality (Schreiber et al. 2002). Chesnut et al. (1993) report that a single episode of hypotension during the pre-hospital phase in patients with a severe traumatic brain injury is associated with increasing morbidity and doubling mortality. Until ICP monitoring is established the aim should be to achieve a target mean arterial pressure of 90 mmHg or more or a systolic BP >120 mmHg by infusion of fluid and vasopressors as indicated. In children, blood pressure should be maintained at a level appropriate for the child’s age (Brain Trauma Foundation 2007, Maas et al. 1997, National Institute for Health and Clinical Excellence 2007). Children aged > 10 years have the same hypotension threshold definition as adults (Badjatia et al. 2007).
• clinical and laboratory assessment of volume status
• the effects of different fluids on CPP and cerebral oedema
Following traumatic brain injury the blood–brain barrier is likely to be disrupted, and different solutions have differing effects on cerebral oedema. If the serum osmolarity falls, water moves across the blood–brain barrier along the altered osmotic gradient, causing cerebral oedema, increased ICP and decreased CPP. The use of hypotonic fluids should, therefore, be avoided.
Since 0.9 % saline is isotonic, it has a negligible effect on brain water and has become the crystalloid of choice in the management of traumatic brain injury. Resuscitation with 0.9 % saline requires four times the volume of blood lost to restore haemodynamic parameters; therefore, blood loss from other injuries should be replaced with blood products (Spiekermann & Thompson 1996).
The use of hypertonic saline solutions has become the crystalloid of choice for volume resuscitation in the management of traumatic brain injury. The evidence base for its use in adults is weak; however there is some evidence of its beneficial effects in children and paediatric guidelines recommend the use of hypertonic saline as a continuous infusion (Adelson et al. 2003). Hypertonic saline is an osmotherapeutic agent that induces a shift of fluid from the intracellular to the extracellular space across the osmotic gradient it generates. It therefore reduces brain water (cerebral oedema) and potentially decreases ICP, increases blood volume, and increases plasma sodium, making it suitable for patients who are fluid depleted, e.g., following acute trauma. (Deem 2006). Studies in the human traumatic brain injury population demonstrate that hypotonic saline is not associated with the rebound intracranial hypertension often seen with mannitol (Bratton et al. 2007).
Based on data from various studies, it is widely accepted that glucose-containing solutions should not be used in the fluid management of patients following severe traumatic brain injury unless specifically indicated to correct hypoglycaemia. The mechanism by which glucose worsens neurological injury is not fully understood; however, it is believed that, in the presence of ischaemia, glucose is metabolized anaerobically leading to an accumulation of lactic acid. Increased lactic acid is thought to decrease intracellular pH, leading to vasodilatation, compromise cellular function, ischaemia and ultimately cause infarction. An alternative explanation proposes that hyperglycaemia worsens ischaemia by decreasing CBF. Other studies suggest that hyperglycaemia decreases cerebral adenosine levels, and adenosine is an inhibitor of the release of excitatory amino acids that are thought to play a major role in ischaemic cell death (Spiekermann & Thompson 1996).
Severe traumatic brain injury can be complicated by the development of a coagulopathy that can worsen blood loss and delay invasive neurosurgical treatment. Studies have reported a positive correlation between the presence and severity of disseminated intravascular coagulation (DIC) and the degree of brain injury, assessed by plasma fibrinogen degradation product levels. Although most of the acute coagulopathies associated with brain injury are not preventable, prompt treatment can be effective in reducing morbidity. Clotting studies at the time of admission to the ED can be valuable in predicting the occurrence of delayed injury, and early follow-up CT scanning is advocated in the patient with coagulopathy (Piek et al. 1992, May et al. 1997).
Disability
Uncontrolled elevation in ICP is the most common cause of mortality, morbidity and secondary brain injury after severe TBI, since it alters tissue perfusion causing cerebral ischaemia and potential infarction. It is imperative that signs of raised ICP are recognized, documented and treated promptly. Disorientation, irritation, headache, seizures, nausea and vomiting are all possible indicators of raised ICP, and later signs include deterioration in the GCS, deteriorating limb function and pupillary changes and finally alteration in the vital signs (Cushing’s triad).
Mannitol has been considered the most effective agent to reduce cerebral oedema and ICP in patients with intracranial hypertension (Bullock 1995). The Brain Trauma Foundation (2007) suggests that hypertonic saline as a bolus infusion may be used as an effective alternative to mannitol however it states that the limitations of the research conducted so far does not allow for a conclusion to be reached (Bratton et al. 2007). Mannitol reduces ICP as a result of its osmotic effects; mannitol may also reduce ICP by improving cerebral microcirculatory flow and oxygen delivery. However, mannitol opens the blood–brain barrier and this effect may become harmful after multiple doses and can exacerbate ICP by increasing brain swelling. Repetitive administration can potentially increase ICP, since mannitol accumulates within brain tissue, reversing osmotic shift and increasing cerebral oedema. The peak effect of mannitol on ICP occurs within 15 minutes of administration, but its effects on serum osmolarity last 2 to 6 hours, depending on the dose and clinical condition of the patient. Mannitol increased serum sodium levels and is excreted entirely by the kidneys, therefore there is increased risk of acute renal failure (acute tubular necrosis) if administered in large doses, particularly if serum osmolarity is >320 mOsm/L. Mannitol raises urine osmolarity and specific gravity, therefore these variables cannot be used to diagnose diabetes insipidus.
The ICP-lowering effect of bolus administration of mannitol appears to be equal over the range 0.25–1.0 g/kg, although the duration of activity may be somewhat shorter with the lower dose. Under most circumstances, the lower dose should be used in order to minimize the risk of hyperosmolality and avoid a negative fluid balance. Guidelines recommend mannitol 20 % be given as an intravenous infusion over 15 to 20 minutes, and repeated as necessary. Blood osmotic pressure must be monitored and serum osmolarity kept below 315 mOsm/L. They also recommend that if mannitol has insufficient effect then frusemide can be given additionally (Maas et al. 1997, Brain Trauma Foundation 2007).
Blood viscosity
Studies suggest that a haematocrit of 30–34 % may result in optimal oxygen delivery to brain tissue. However, if maximum vasodilatation already exists, haemodilution may decrease oxygen delivery and lead to increases in cerebral blood volume (CBV) and therefore ICP (Cesarini 1996).
Drugs
Intravenous sedation agents cause a dose-dependent reduction in cerebral metabolism, CBF and ICP while maintaining pressure autoregulation and CO2 reactivity. The use of barbiturates in traumatic brain injury is controversial and has largely been replaced by propofol, which has similar cerebrovascular effects but a more favourable pharmacological profile. Propofol has become the sedative of choice but care must be taken to avoid hypotension (Kelly 2000, Kelly et al. 1999).
Neuromuscular blocking drugs have no direct effect on ICP but may prevent rises produced by coughing and straining on the endotracheal tube. However, such agents are not associated with improved outcome and their use is the subject of much debate (Prielipp & Coursin 1995); their use should only be employed when sedation alone proves inadequate.
Patients with CSF leakage are usually prescribed prophylactic antibiotics, because of the high risk of bacterial meningitis. The available evidence, however, does not support the use of prophylactic antibiotics (Watkins 2000).
Temperature control
Hypothermia as a treatment in severe traumatic brain injury has been a major area of research during the last decade. Brain Trauma Foundation guidelines cautiously support prophylactic hypothermia as being associated with significantly higher GCS outcomes, but this is based on limited research (Bratton et al. 2007).
The interim results of a large multi-centre study showed that cooling to 32–33°C within 10 hours of injury improved neurological recovery in patients presenting with head injury and a GCS of 5–7 but not those with a lower GCS. However, this study was terminated early with no clear benefit established (Marion et al. 1997).
Some studies suggest that hypothermia provides protection against secondary injury, specifically hypotension and hypoxia, and that there may be a therapeutic window for neuroprotection in the early stages following the primary injury (Yamamoto et al. 1999). Whether this protection can be sustained over longer periods remains to be determined.
A randomized trial evaluating the use of mild hypothermia during intracranial aneurysm surgery found that, compared with normothermic patients, more patients in the hypothermic group had good outcomes and fewer of these patients had neurological deficits at the time of discharge (Hindman et al. 1999). In a further study on CBF and oxygen metabolism during mild hypothermia (33–34°C) in elective aneurysmal surgery, positron emission tomography (PET) showed luxury perfusion in almost all cases, providing the first PET evidence of decreased CBF and metabolic rate of O2 during hypothermia in humans (Kawamura et al. 2000).
There is some evidence of increased mortality in the general trauma population for patients admitted with hypothermia, and at least one study demonstrated an increased incidence of infection in head injured patients subjected to hypothermia (Sutcliffe 2001, Brain Trauma Foundation 2007).
Seizure activity
Seizure activity reflects disordered electrical discharges in the damaged brain and results in local loss of autoregulation, increased metabolic activity in the brain with concomitant increases in cerebral blood flow and tissue lactic acidosis, which may aggravate ICP. Seizures are seen in 5–15 % of patients after traumatic brain injury, especially those who have suffered a haematoma, penetrating injury, including depressed skull fracture with dural penetration, and focal neurological signs or intracranial sepsis (Dunn 1996,Watkins 2000).
In the conscious patient a single seizure requires only supportive treatment, but in the comatose patient every seizure can threaten life or neurological function and should be treated promptly. Patients having seizures should be protected from harm, if they are not in any danger they should not be handled. Extra vigilance is required, frequent GCS observations should be performed until the pre-seizure state is regained. The seizure should be observed for origin, sequence of events and time of start/finish. This information should be clearly documented in the patient’s notes. No generally accepted classification of seizures exists, but it is useful to distinguish between non-focal seizures, where loss of consciousness is the primary event, followed by convulsions affecting all four limbs, and focal or partial seizures, where activity is confined to one area of the cerebral cortex. The continuous neuronal firing that occurs during non-focal seizure activity causes a massive increase in the brain’s requirement for oxygen. If a corresponding increase in the supply of oxygen is not provided then hypoxia, ischaemia, and potential infarction will result. Anticonvulsant drugs should be used if two or more seizures occur (Watkins 2000). If seizure activity is continuous, i.e., status epilepticus or serial fitting, severe cerebral oedema may occur.
Positioning
Many studies have been undertaken to determine the influence of body position on ICP and CPP and establish the best practices for positioning of severe traumatic brain injury patients (Sullivan 2000). It has been demonstrated that head elevation decreases ICP and that head rotation and neck flexion are associated with increased ICP, decreased jugular venous return, and localized changes in cerebral blood flow. In the light of these findings, it is common practice to position head-injured patients in bed with their head elevated above the heart to varying degrees (0–30°) provided they have an adequate blood pressure and there are no contraindications, in neutral alignment with the trunk, in order to reduce ICP.
Transfer to a specialist neurosurgical unit
In an attempt to improve outcome for patients with traumatic brain injury the National Institute for Health and Clinical Excellence (2007) recommends an improvement in the rate at which such patients are transferred to the specialist neurosciences unit. Patients who have suffered an isolated severe head injury should ideally receive their treatment in a specialist neurosciences unit, regardless of the need for neurosurgery (National Institute for Health and Clinical Excellence 2007). Patients should be referred to a neurosurgeon where:
• a new surgically significant abnormality is seen on imaging
• a persisting coma (GCS less than or equal to 8) is seen after initial resuscitation
• an unexplained confusion, which persists for more than four hours is seen
• deterioration in GCS score after admission is seen. (Greatest attention should be paid to motor response deterioration)
• progressive focal neurological signs are seen
• an epileptic seizure without full recovery is seen
• a definite or a suspected penetrating injury is seen
• a cerebrospinal fluid leak is seen (National Institute for Health and Clinical Excellence 2007).
The neck should be fully examined to identify or exclude cervical injury (see also Chapter 7). Chest or abdominal injuries (Chapters 8 and 9) should be treated if potentially life-threatening and fractures splinted where appropriate (see Chapter 6). Airway management is a major cause for concern during transfer. Unconscious patients, those who are vomiting or those whose condition appears to be deteriorating (GCS less than 8) should be intubated and ventilated prior to transfer (National Institute for Health and Clinical Excellence 2007).
Any patient being transferred should be accompanied by a clinician with at least two years’ experience in an appropriate specialism, e.g., anaesthesia or a paediatrician, and one who has received supervised training in the transfer of patients with a severe head injury, along with a competent nurse escort (National Institute for Health and Clinical Excellence 2007). During transfer, the patient should be carefully observed and ECG, oxygen saturation levels, and blood pressure should be monitored continuously (Parkins 1998). If the patient is not sedated, pupil size and reaction and GCS should be recorded. All documentation and results of clinical investigations, such as CT, should accompany the patient (Jones 1993).
Sedation and neuromuscular blocking drugs are essential to optimizing the transport of patients with a traumatic head injury. In the absence of outcome-based studies the choice of sedative is left to the physician, and neuromuscular blocking agents should only be employed when sedation alone proves inadequate (Maas et al. 1997, Brain Trauma Foundation 2007).
Surgical treatment
Delay in surgical treatment of TBI has been shown to be a major preventable cause of morbidity and mortality. In a typical series of patients who had surgery for Acute Sub-Dural Haematoma (ASDH), more than 70 % had a functional recovery, i.e., good recovery or moderate disability, if the delay from injury to operation was less than two hours. Where the delay was 2–4 hours, just over 60 % made a functional recovery. Where the delay was more than four hours after the injury, fewer than 10 % made a functional recovery (Watkins 2000). The Royal College of Surgeons of England recommends that, in all circumstances, life-saving decompressive surgery must be available to all patients who require it, within four hours of the injury (Royal College of Surgeons 1999, Flannery & Buxton 2001).
Some lesions may be inaccessible to the neurosurgeon or be in such sensitive areas of the brain that the risks of surgery are too great. Alternatively, brain injury may be diffuse and surgical intervention inappropriate. Whether all haematomas must be removed remains controversial and this is particularly so in the management of intracerebral haematomas and haemorrhagic contusions. While some surgeons advocate early surgery in such cases, experience has shown that despite surgical treatment, post-operative intracranial hypertension continues to occur in virtually all patients. Guidelines outlining the specific indications for surgery are based on biological and pathophysiological principles, published evidence, and practical experience (Maas et al. 1997, Brain Trauma Foundation 2007).
Indications for decompressive surgery:
• intracranial mass lesions with >5 mm midline shift or basal cistern compression on CT scan
• a surgically significant (1 cm thick) extradural haematoma and acute subdural haematoma needs evacuation within 2–4 hours of injury to achieve optimal chance of recovery
• a conservative approach is generally adopted for small haemorrhagic contusions or other small intracerebral lesions, but operation should be considered urgent for a large intracranial haematoma (>20–30 ml) based on position, clinical condition, and ICP
• skull fracture depressed greater than the thickness of the skull or compound fractures with a torn dura (Maas et al. 1997, Brain Trauma Foundation 2007).
Decompressive craniotomy
The practice of wide bone removal for the treatment of intracranial hypertension due to cerebral oedema, refractory to medical management, has a long history and remains controversial. Polin et al. (1997) concluded that provided surgery was performed before ICP exceeded 40 mmHg for a sustained period and within 48 hours of injury, decompressive craniectomy showed a statistical advantage over medical treatment. Decompressive craniectomy, which can also increase oedema formation, is usually reserved as a last resort (Maas et al. 1997).
Brain stem death testing
The diagnosis of brainstem death is usually made in the intensive care unit.
Exclusions





Clinical tests
1. Confirmation of absent brain stem reflexes






2. Confirmation of persistent apnoea
 pre-oxygenation with 100% oxygen for 10 minutes
pre-oxygenation with 100% oxygen for 10 minutes
 allow PaCO2 to rise above 5.0 kPa before test
allow PaCO2 to rise above 5.0 kPa before test

 maintain adequate oxygenation during test
maintain adequate oxygenation during test
 allow PaCO2 to climb above 6.65 kPa
allow PaCO2 to climb above 6.65 kPa


3. Two experienced practitioners should perform clinical tests




Conclusion
Head trauma can have a devastating effect on a person’s life and some mortality and morbidity are inevitable. McMillan et al. (2011) noted that head injury is associated with increased vulnerability to death for at least 13 years after hospital admission. The emergency nurse who is equipped with the evidence-based knowledge and skills to manage patients with traumatic brain injury is fundamental to keeping morbidity and mortality to a minimum.
References
Adelson, P.D., Bratton, S.L., Carney, N.A., et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children and adolescents. Paediatric Critical Care Medicine. 2003;4(3):S1–S75.
Addison, C., Crawford, B. Not bad, just misunderstood. Nursing Times. 1999;95:52–53.
Advanced Life Support Group. Advanced Paediatric Life Support: The Practical Approach. Wiley-Blackwell: West Sussex; 2011.
Albano, C. Innovations in the management of cerebral injury. Critical Care Nurse Quarterly. 2005;28(2):135–149.
American College of Surgeons. Advanced Trauma Life Support, seventh ed. Chicago: American College of Surgeons; 2008.
Badjatia, N., Carney, N., Crocco, T.J., et al. Guidelines for prehospital management of traumatic brain injury. Prehospital Emergency Care. 2007;12(1):S1–S52. [second ed.].
Brain Trauma Foundation. American Association of Neurological Surgeons’ Joint section on neurotrauma and critical care guidelines for the management of severe head injury. Journal of Neurotrauma. 2007;13:641–734. [third ed].
Bratton, S.I., Davis, R.L. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997;40(4):707–712.
Bratton, S.L., Chesnut, R.M., Ghajar, J., et al. Guidelines for management of severe traumatic brain injury. Journal of Neurotrauma. 2007;24(Suppl. 1):S1–S106. [third ed].
Bullock, R. Mannitol and other diuretics in severe neurotrauma. New Horizons. 1995;3(3):443–452.
Bullock, R., Teasdale, G. Head injuries. In: Skinner D., Driscoll R., Earlam R., eds. ABC of Trauma. London: BMJ Publishing Group, 1996.
Caton-Richards, M. Assessing the neurological status of patients with head injuries. Emergency Nurse. 2010;17(10):28–31.
Cesarini, K.G. Pathophysiology of cerebral ischaemia. In: Palmer J.D., ed. Manual of Neurosurgery. London: Churchill Livingstone, 1996.
Chesnut, R.M., Gautille, T., Blunt, B.A., et al. Neurogenic hypotension in patients with severe head injuries. Journal of Trauma. 1998;44(6):958–963.
Chesnut, R.M., Marshall, L.F., et al. The role of secondary brain injury in determining outcome from severe head injury. Journal of Trauma Injury: Infection and Critical care. 1993;34(2):216–222.
Clifton, G.L., Ziegler, M.G., Grossman, A. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery. 1981;8:10–14.
Clifton, G.L., Robertson, C.S., Kyper, K., et al. Cardiovascular response to severe head injury. Journal of Neurosurgery. 1983;3:447–454.
Contant, C.F., Valadka, A.B., Shankar, M.D., et al. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. Journal of Neurosurgery. 2001;95:560–568.
Crossman, A.R., Neary, D. Neuroanatomy – An Illustrated Colour Text, second ed. Edinburgh: Churchill Livingstone; 2000.
Darby, J., Yonas, H., Marion, D.W., et al. Local ‘inverse steal’ induced by hyperventilation in head injury. Neurosurgery. 1998;23:84.
Dearden, N.M. Mechanism and prevention of secondary brain damage during intensive care. Clinical Neuropathology. 1998;17(47):221–228.
Deem, S. Management of acute brain injury and associated respiratory issues. Respiratory Care. 2006;51(4):357–367.
Deitch, E.A., Dayal, S.D. Intensive care unit management of the trauma patient. Critical Care Medicine. 2006;34(9):2294–2301.
Department of Health. Hospital episode statistics 2000–2001. London: Department of Health; 2001.
Duffy, C. Anaesthesia for head injury. In: Gupta A.K., Summors A., eds. Notes in Neuroanaesthesia and Critical Care. London: Greenwich Medical Media, 2001.
Dunn, L.T. Post-traumatic epilepsy. In: Palmer J.D., ed. Manual of Neurosurgery. London: Churchill Livingstone, 1996.
Durieux, M.E. Anaesthesia for head trauma. In: Stone D.J., Sperry R.J., et al, eds. The Neuroanaesthesia Handbook. St. Louis: Mosby, 1996.
Farnsworth, S.T., Sperry, R.J. Neurophysiology. In: Stone J.D., Sperry R.J., et al, eds. The Neuroanaesthesia Handbook. St Louis: Mosby, 1996.
Flannery, T., Buxton, N. Modern management of head injuries. Journal of the Royal College of Surgeons (Edinburgh). 2001;46:150–153.
Garner, A., Amin, Y. The management of raised intracranial pressure: a multidisciplinary approach. British Journal of Neurosciences Nursing. 2007;3(11):516–521.
Ghajar, J. Traumatic brain injury. Lancet. 2000;356:923–929.
Graham, D.I., Hume Adams, J., Nicoll, J.A.R., et al. The nature, distribution and causes of traumatic brain injury. Brain Pathology. 1995;5:397–406.
Healthcare for London. The shape of things to come: Developing New High-quality Major Trauma and Stroke Services for London: Compact Consultation Document. London: Healthcare for London; 2009.
Hersch, C. Electrocardiographic changes in head injuries. Circulation. 1961;23:853–860.
Hickey, J.V. The Clinical Practice of Neurological and Neurosurgical Nursing, sixth ed. Philadelphia: Lippincott Williams & Wilkins; 2009.
Hindman, B.J., Todd, M.M., Gelb, A.W., et al. Mild hypothermia as a protective therapy during intracranial aneurysm surgery: a randomized prospective pilot trial. Neurosurgery. 1999;44:23–33.
Hudak, C.M., Gallo, B.M. Critical Care Nursing, sixth ed. Philadelphia: JB Lippincott; 1994.
Hunningher, A., Smith, M. Update on the management of severe head injury in adults. Care of the Critically ill. 2006;22(5):124–129.
Intensive Care Society. Guidelines for the Transport of the Critically Ill Adult. London: Intensive Care Society; 1997.
Jackson, S. Not so minor head injuries? Emergency Nurse. 1995;3(1):19–22.
Jones, H. Safe transfer. Nursing Times. 1993;89(41):31–33.
Kawamura, S., Suzuki, A., Hadeishi, H., et al. Cerebral blood flow and oxygen metabolism during mild hypothermia in patients with subarachnoid haemorrhage. Acta Neurochir (Wein). 2000;142(10):1117–1121.
Kelly, D.F. Sedation after severe head injury: beneficial to final outcome? European Journal of Anaesthesiology. 2000;17(Suppl. 18):43–54.
Kelly, D.F., Goodale, D.B., Williams, J., et al. Propofol in the treatment of moderate and severe head injury: a randomized prospective double-blind trial. Journal of Neurosurgery. 1999;90(6):1042–1052.
Kwiatkowski, S. Traumatic acute intracranial haematoma. In: Palmer J.D., ed. Manual of Neurosurgery. London: Churchill Livingstone, 1996.
Lindsey, K.W., Bone, I., Callander, R. Neurology and Neurosurgery Illustrated, fourth ed. Edinburgh: Churchill Livingstone; 2004.
Maartens, N., Lethbridge, G. Head and neck trauma. In: O’Shea R.A., ed. Principles and Practice of Trauma Nursing. Edinburgh: Elsevier Churchill Livingstone, 2005.
Maas, A.I.R., Dearden, M., Teasdale, G.M., et al. European Brain Injury Consortium Guidelines for management of severe head injury in adults. Acta Neurochir (Wien). 1997;139:286–294.
Marion, D.W., Penrod, L.E., Kelsey, S.F., et al. Treatment of traumatic brain injury with moderate hypothermia. New England Journal of Medicine. 1997;336(8):560–566.
McLeod, A.A., Neil-Dwyer, G., Meyer, C.H.A., et al. Cardiac sequelae of acute head injury. British Heart Journal. 1982;47:221–226.
McMillan, T.M., Teasdale, G.M., Weir, C.J., et al. Death after head injury: the 13 year outcome of a case control study. Journal and Neurology and Neurosurgical Psychiatry. 2011.
McNaughton, H., Harwood, M. Traumatic brain injury: assessment and management. Hospital Medicine. 2002;63:8–12.
National Institute for Health and Clinical Excellence. Head Injury Triage, Assessment, Investigation and Early Management of Head Injury in Infants, Children and Adults. London: NICE; 2007.
Oppenheimer, S.M., Cochetto, D.F., Hachinski, V.C. Cerebrogenic cardiac arrhythmias. Acta Neurology. 1990;47:513–519.
Parkins, D.R. Transportation of the injured: head injuries. Pre-hospital Immediate Care. 1998;2:25–26.
Patel, H.C., Menon, D.K., Tebbs, S., et al. Specialist neurocritical care and outcome from head injury. Intensive Care Medicine. 2002;28:547–553.
Piek, J., Chesnut, R.M., Marshall, L.F., et al. Extracranial complications of severe head injury. Journal of Neurosurgery. 1992;77:901–907.
Polin, S., Shaffrey, M.E., Bogaev, C.A., et al. Decompressive craniectomy in the treatment of severe refractory post-traumatic cerebral oedema. Neurosurgery. 1997;41:84–94.
Prielipp, R.C., Coursin, D.B. Sedative and neuromuscular blocking drug use in critically ill patients with head injuries. New Horizons. 1995;3(3):456–468.
Ramrakha, P., Moore, K. Oxford Handbook of Acute Medicine. Oxford: Oxford University Press; 1997.
Robertson, C.S. Management of cerebral perfusion pressure after traumatic brain injury. Anaesthesiology 95(6). 2001;15:13–17.
Robertson, C.S., Valadka, A.B., Hannay, H.J., et al. Prevention of secondary insults after severe head injury. Critical Care Medicine. 1999;27:2086–2095.
Royal College of Paediatrics and Child Health. Guidelines for Good Practice: Early Management of Patients with a Head Injury. London: Royal College of Paediatrics and Child Health; 2001.
Royal College of Surgeons. Report of the Working Party on the Management of Patients with Head Injury. London: Royal College of Surgeons; 1999.
Sakas, D.E., Dias, L.S., Beale, D. Subarachnoid haemorrhage presenting as a head injury. British Medical Journal. 1995;310:1186–1187.
Schreiber, M., Aoki, N., Scott, B.G., Beck, J.R. Determinants of mortality in patients with severe blunt head injury. Archives of Surgery. 2002;137:285–290.
Shah, S. Neurological assessment. Nursing Standard. 1999;13(22):49–56.
Spiekermann, B.F., Thompson, S.A. Fluid management. In: Stone D., Sperry R.J., et al, eds. The Handbook of Neuroanaesthesia. St Louis: Mosby, 1996.
Sullivan, J. Positioning of patients with severe traumatic brain injury: research based practice. Journal of Neuroscientific Nursing. 2000;32(4):204–209.
Stern, S. Observing and recording neurological dysfunction. Emergency Nurse. 2011;18(10):28–31.
Sutcliffe, A. Hypothermia (or not) for the management of head injury. Care of the Critically Ill. 2001;17(5):162–165.
Tagliaferri, F., Compagnone, C., Korsic, M., et al. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien). 2006;148:255–268.
Teasdale, G., Jennett, B. Assessment of coma and impaired consciousness: a practical scale. The Lancet. 1974;2(7872):81–84.
Van der Ark, G.D. Cardiovascular changes with acute subdural haematoma. Surgical Neurology. 1975;5(3):305–308.
Vos, P.E., Alekseenko, Y., Battistin, L., et al. Mild traumatic brain injury. European Journal of Neurology. 2012;19(2):191–198.
Watkins, L.D. Head injuries: general principles and management. Surgery. 2000;18(7):219–224.
Wilson, M. Major incidents. In: Smith J., Greaves I., Porter K., eds. Oxford Desk Reference: Major Trauma. Oxford: Oxford Medical Publications, 2011.
Wyatt, J.P., Illingworth, R.N., Graham, C.A., et al. Oxford Handbook of Emergency Medicine, third ed. Oxford: Oxford University Press; 2008.
Yamamoto, M., Marmarou, C.R., Stiefel, M.F., et al. Neuroprotective effect of hypothermia on neuronal injury in diffuse traumatic brain injury coupled with hypoxia and hypotension. Journal of Neurotrauma. 1999;16(6):487–499.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Head injuries
Introduction
There are no reliable up-to-date figures for the total denominator of attendees who attend Emergency Departments (EDs) with a head injury (National Institute for Health and Clinical Excellence 2007). The role of ED staff is to diagnose and appropriately treat a large number of patients presenting following a head injury. Neurological damage occurs both at the time of the injury (primary insult) and evolves over the following minutes, hours and days (secondary insult). Patient outcomes improve where secondary insults are treated early, and where successful responses result in limiting and preventing further injury caused by secondary insult. Wyatt et al. (2008) argue that emergency nurses are ‘fundamental to keeping morbidity to a minimum by being vigilant and prevent secondary brain injury.’
While 90 % of traumatic brain injuries (TBIs) are considered mild (Vos et al. 2012), it is the world’s leading cause of morbidity and mortality in individuals under the age of 45 years old (Wilson 2011). It is estimated that the ED attendance rate in the UK for patients with head injuries is close to 700 000 patients (National Institute for Clinical Excellence 2007) and that 20 % of these are admitted to hospital. Half of those who die from TBI do so within the two hours of the injury. Approximately 30 % of admitted patients with a Glasgow Coma Score (GCS) of <13 will die, and if the GCS is <8, this increases to 50% (Wilson 2011).
The most common causes of a minor head injury are falls (22–43 % of injuries), assaults (30–50 % of injuries), and road traffic accidents (25 % of injuries) (Department of Health 2001, National Institute for Health and Clinical Excellence 2007). In the UK, 70–88 % of individuals that sustain a head injury are male, 10–19 % are aged 65 years or greater, and 40–50 % are children. Alcohol may be involved in up to 65 % of adult head injuries and road traffic accidents account for a large proportion of moderate to severe head injuries. Traumatic injury in which severe head injury plays a major role in over 50 % of cases, is a leading cause of death in those aged 25 years or less (Maartens & Lethbridge 2005); 5–10 % of patients who suffer a severe head injury also suffer a cervical spine injury.
A structured approach to the care of head-injured patients should be initiated based on current valid evidence. The Brain Trauma Foundation has developed recommendations for the management of moderate and severe head injuries (Bratton et al. 2007) in collaboration with the Advanced Trauma Life Support (ATLS) system (American College of Surgeons 2008), providing a mechanism for assessment and immediate management and minimizes the risk of secondary brain injury in adults. For children the Advanced Paediatric Life Support (APLS) system (Advanced Life Support Group 2011) should be employed (National Institute for Health and Clinical Excellence 2007). These guidelines are based on class II (studies based on prospectively collected data and the retrospective analysis of reliable data) and III (studies based on retrospectively collected data) evidence. There is a lack of class I (prospective randomized controlled trials) evidence to support practice.
Anatomy and physiology
The cranium is one of the strongest structures in the body and provides the bony protection for the brain. It is composed of the parietal (2), occipital, frontal, temporal (2), sphenoid, and ethmoid bones. Figure 5.1 shows an exploded view of the cranial skull; however, the bones are fused along main sutures, the sagittal, coronal, lambdoidal, and squamosal. The facial bones form the framework for the nasal and oral cavities and include the zygomatic bones (2), palatine bones (2), mandible, maxilla (2), lacrimal bones (2), nasal bones (2), vomer and the inferior nasal concha (2) (Fig. 5.2).
Figure 5.1 Exploded view of the cranial skull.
Figure 5.2 Exploded view of the facial skull.
Figure 5.3 shows the irregular internal surfaces of the skull. These irregular surfaces/bony protrusions account for injury to the brain as it moves within the skull under acceleration/deceleration forces.
Figure 5.3 View of the base of the skull from above.
The meninges
The brain and the spinal cord are encased by three layers of membrane – the dura mater, the arachnoid mater, and the pia mater, known collectively as the meninges (Fig. 5.4).
Figure 5.4 The cranial meninges.
Dura mater
The dura mater consists of two layers: the outer layer is the periosteal layer of the skull, which terminates at the foramen magnum, and the inner layer is a strong, thick membrane that is continuous with the spinal dura mater. There is a potential space between the two dura, except at the falx cerebri, which divides the left and right hemispheres of the cerebrum; the tentorium cerebelli, which divides the cerebrum and cerebellum; the falx cerebelli, which divides the lateral lobes of the cerebellum; and the diaphragm sellae. The dura creates a roof for the sella turcica (which houses the pituitary gland). These compartments provide support and protection for the brain and form the sinuses, which drain venous blood from the brain (Crossman & Neary 2000, Lindsey et al. 2004).
Arachnoid mater
The arachnoid mater is a fine serous membrane that loosely covers the brain. There is a potential space between this and the inner dura mater, known as the subdural space. Between the arachnoid mater and the pia mater is an actual space, known as the subarachnoid space, which contains the arachnoid villi, cerebrospinal fluid (CSF), and small blood vessels.
The ventricles and cerebrospinal fluid
Within the brain there are four connected cavities called ventricles, which contain CSF. These are the left and right lateral ventricles, the third ventricle and the fourth ventricle. The lateral ventricles lie in the cerebral hemispheres, the third in the diencephalon, and the fourth in the brain stem. The lateral ventricles are connected to the third ventricle by the interventricular foramen, sometimes known as the foramen of Munro, and the third ventricle is connected to the fourth by the cerebral aqueduct, sometimes known as the aqueduct of Sylvius (Fig. 5.5).
The brain
The brain consists of three main areas:
The major structures within the brain are summarized in Box 5.1.
Cerebrum
The surface area of the cerebral cortex (grey matter) on the surface of the brain is much increased by the presence of gyri and sulci (Fig. 5.6), resulting in a 3:1 proportion of grey to white matter. Below the cortex lies the white matter. The cerebral hemispheres are composed of four lobes, the frontal, parietal, temporal and occipital lobes. Box 5.2 summarizes the main functions of these lobes.
Cerebellum
Brain stem
The brain stem is the connection between the brain and the spinal cord and is continuous with the diencephalon above and the spinal cord below. Within the brain stem are ascending and descending pathways between the spinal cord and parts of the brain. All cranial nerves except the olfactory (1) and the optic (2) nerves emerge from the brain stem (Fig. 5.7). The brain stem is formed from three main structures:
Figure 5.7 Brain stem (dorsal view).
The midbrain connects the pons and the cerebellum to the cerebrum. It is involved with visual reflexes, the movement of the eyes, focusing and the dilatation of the pupils. Contained within the midbrain and upper pons is the reticular activating system, which is responsible for the ‘awake’ state.
Cerebral circulation
The brain is supplied with blood by four major arteries: two internal carotid arteries, which supply most of the cerebrum and both eyes; and two vertebral arteries, which supply the cerebellum, brain stem and the posterior part of the cerebrum. Before the blood enters the cerebrum it passes through the circle of Willis, which is a circular shunt at the base of the brain consisting of the posterior cerebral, the posterior communicating, the internal carotids, the anterior cerebral and the anterior communicating arteries (Figs 5.8 and 5.9). These vessels are frequently anomalous; however, they allow for an adequate blood supply to all the brain, even if one or more is ineffective.
Figure 5.8 Major arteries of the head and neck.
Figure 5.9 Cerebral circulation.
The venous drainage from the brain does not follow a similar pathway (Fig. 5.10). Cerebral veins empty into large venous sinuses located in the folds of the dura mater. Bridging veins connect the brain and the dural sinuses and are often the cause of subdural haematomas. These sinuses empty into the internal jugular veins, which sit on either side of the neck and return the blood to the heart via the brachiocephalic veins.
Figure 5.10 Major veins of the head and neck.
Physiology of raised intracranial pressure
Intracranial pressure (ICP) represents the pressure exerted by the CSF within the ventricles of the brain (Hickey 2009). The exact pressure varies in different areas of the brain. The normal range is 0–15 mmHg in adults, 3–7 mmHg in children, and 1.5–6 mmHg in term babies when measured from the foramen of Munro.
ICP is fundamental in maintaining adequate brain function. The brain lies in the skull, a rigid compartment. The contents of the skull are non-compressible, i.e., brain tissue (80 %), intravascular blood (10 %) and cerebrospinal fluid (10 %). Normally these components maintain a fairly constant volume, therefore creating dynamic equilibrium therein. Should one or more components increase for whatever reason, the Monro-Kellie hypothesis states that another component must decrease in quantity in order to maintain the dynamic equilibrium and thus maintain adequate cerebral blood flow (CBF). If this does not occur, ICP rises, leading to brain injury (Hickey 2009). Dynamic equilibrium is maintained by a number of compensatory mechanisms, these include:
In severe traumatic brain injury the compensatory mechanisms are rapidly exhausted (Deitch & Dayal 2006). They fail in the healthy adult brain when the ICP reaches 20 mmHg. Once exhausted, small increases in brain mass, blood, or CSF volume have a profound effect on ICP. Chestnut et al. (1993) demonstrated a clear correlation between the length of time patient’s ICP remains greater than 20 mmHg and an increased mortality and morbidity rate.
CPP is used as an indicator of CBF, and therefore oxygen delivery to the brain. Current recommendations suggest that in adults the CPP should lie between 50–70 mmHg, although adults with intact pressure autoregulation may tolerate higher CPP values (Bratton et al. 2007). When the patient’s MAP falls and/or the patient’s ICP increases, there is a risk that the cerebral perfusion pressure will fall to too low a value to maintain adequate CBF. This results in cerebral hypoxia and secondary brain injury in the form of cerebral ischaemia and potentially infarction. Autoregulation fails when CPP falls below 50 mmHg or rises above 150 mmHg, resulting in CPP and CBF become dependent on the systemic blood pressure alone.
Chemo autoregulation is triggered by changes in extracellular pH and metabolic by-products. Changes in PCO2 or a dramatic reduction in PO2 (Fig. 5.11) may trigger this. Hypercapnia >45 mmHg (or 6 kPa) (Albano 2005) is a potent vasodilator that induces hyperaemia and cerebral blood volume increases, without an adequate decrease in CSF; as a result of the compensatory response the ICP will rise. Hypocapnia <45–>30 mmHg (or 6–4 kPa) considered a critical low value (Garner & Amin 2007) is a potent vasoconstrictor that reduces brain mass and induces hypoaemia; as a result cerebral blood flow and volume decrease leading to a risk of secondary ischaemia and potentially an infarction if not treated (Albano 2005).
Figure 5.11 Autoregulation of the brain.
Classification of head injuries
Head injuries can be classified under three anatomical sites:
Scalp injuries
There are four types of injury to the scalp:
• abrasion – minor injury that may cause a small amount of bleeding. Treatment may not be required, but ice applied to the area may reduce any haematoma formation (Hickey 2009)
• contusion – no break in the skin, but bruising to the scalp may cause blood to leak into the subcutaneous layer
• laceration – a cut or tear of the skin and subcutaneous fascia that tends to bleed profusely. Bleeding from the scalp alone is unlikely to cause shock in the adult. In small children, a scalp laceration may be sufficient to cause hypovolaemia. Scalp lesions should be explored under local anaesthetic for foreign bodies and/or skull fracture with a skull X-ray if there is any doubt about the diagnosis. Lesion(s) should be sutured or glued according to their depth and position
• subgaleal haematoma – a haematoma below the galea, a tough layer of tissue under the subcutaneous fascia and before the skull. The veins here empty into the venous sinus, and thus any infection can spread easily to the brain, despite the skull remaining intact. There is controversy surrounding the treatment of subgaleal haematomas, due to the risks of infection; therefore some doctors argue that it is best to evacuate the haematoma, while others suggest that it is best to let it reabsorb.
If the scalp injuries are only part of other injuries, it is important they are documented to allow further investigation at a more appropriate time. They may need to be cleaned and dressed or temporarily sutured.
Skull injuries
• linear – these are the most common types of injury. They usually result from low-velocity direct force. They are usually diagnosed from skull X-ray and need no specific treatment
• depressed – usually evident clinically, but a skull X-ray to discover the full extent of the potential brain damage is usually necessary. Management is dependent on the severity of the fracture and whether there are any accompanying injuries. If there are no other injuries requiring surgical management, they may not be surgically elevated, due to the risks of infection. However, surgical intervention will normally be necessary if there are bone fragments embedded in the brain so as to elevate the bone fragments and manage the brain trauma
• open – usually evident clinically. Usually managed according to the severity of the injury. If debris is dispersed in the brain tissue then surgery will be required and there is a heightened risk of infection
• comminuted – these are detected on skull X-ray. These patients should be closely observed and any neurological deficits managed appropriately. Surgical intervention is usually required. If there are bone fragments imbedded in the brain tissue then surgery will be required to elevate the bone fragments and manage the brain trauma
• basal – these are diagnosed clinically as they are difficult to detect on X-ray. Signs include CSF leakage from the nose (rhinorrhoea) or the ear(s) (otorrhoea). Rhinorrhoea or otorrhoea indicates that a skull base fracture has breached the dura and formed a communication between the intracranial contents and an air sinus. This places the patient at risk of meningitis while the CSF leak continues. If CSF leakage is suspected, the fluid should be tested for glucose and the ‘halo test’ performed, where a small amount of fluid is placed on blotting paper; if CSF is present it will separate from blood and form a yellow ring around the outside of the blood. Patients with a base of skull fracture may also have retroauricular bruising (Battle’s signs) and periorbital bruising (‘panda eyes’ or ‘raccoon eyes’): 80–90 % of cases seal within two weeks and neurosurgical intervention is usually not considered until this time has elapsed. An exception is a fracture of the posterior wall of the frontal sinus, visualized on CT scan, where anterior fossa repair may be undertaken early.
Brain injuries
Severe brain injury is uncommon because the skull and scalp absorb the majority of the impact of the assault. The amount of brain damage suffered is relative to the force/energy of the assault. A high-energy head injury results when: a pedestrian is struck by a motor vehicle, an occupant is ejected from a motor vehicle, a person falls from a height of greater than 1 metre or more than five stairs (a lower threshold for the height of falls should be used when dealing with infants and young children under 5 years old), following a diving accident, following a high-speed motor vehicle collision, following a rollover motor accident, or a bicycle collision (National Institute for Health and Clinical Excellence 2007).
Damage to the brain as a result of trauma includes both the immediate (primary) injury caused at the moment of the impact and the secondary injury that develops during the first few minutes, hours or days after the impact (Box 5.3). These secondary injuries may have extracranial or intracranial causes.
There are no interventions that can prevent the primary brain injury. Secondary brain injuries, which further exacerbate the primary neuronal injury and lead to a worsening outcome depending upon their duration and severity, are largely preventable. The two main causes of secondary injury are delayed diagnosis and treatment of intracranial haematomas, and failure to correct systemic hypoxaemia and hypotension (Fig. 5.12). Brain injuries are usually categorized as either focal or diffuse injuries.
Figure 5.12 Cycle of progressive brain swelling.
Focal injuries
Haematoma: Extradural haematoma (EDH) is an accumulation of blood in the extradural space between the periosteum on the inner side of the skull and the dura mater (Fig. 5.13). Most are associated with skull fracture and are commonly caused by a laceration to the middle meningeal artery or vein, or less commonly to the dural venous sinus, following an insult to the temporal-parietal region. Consequentially, the parietal and parieto-temporal areas of the brain are affected. In 85 % of patients the EDH will be accompanied by a skull fracture (Hudak & Gallo 1994).
Figure 5.13 Extradural haematoma.
Patients with skull fractures may be neurologically intact on admission and later deteriorate as the EDH develops. Most often the primary brain injury causes some disturbance of consciousness and the developing haematoma results in rapid neurological deterioration (Kwiatkowski 1996). Patients with EDHs most commonly present with a history of transient loss of consciousness, followed by lucidity for a period (hours to days) dependent on the rate of the bleed, irritation and headache. Patients then rapidly lose consciousness and deteriorate very quickly. Late signs are seizures, ipsilateral pupil dilatation, unconsciousness, and contralateral hemiplegia. Surgical treatment is required to evacuate the haematoma and ligate the damaged blood vessel.
Subdural haematoma (SDH) is an accumulation of blood between the dura mater and arachnoid mater. SDHs are caused by the rupture of bridging veins from the cortical surfaces to the venous sinuses (cortical veins) (Fig. 5.14).
Figure 5.14 Subdural haematoma.
SDHs can be seen in isolation, but more commonly are associated with accompanying brain injury, i.e., cerebral contusions and/or intracerebral haematomas. They are the most common intracranial mass result from head trauma (Maartens & Lethbridge 2005). In most cases a large contusion is found at the frontal or temporal surface of the brain. SDHs are predisposed with increasing age and alcoholism. Both groups can suffer regular falls and have a degree of cerebral atrophy, which puts strain on the bridging veins and coagulopathy. Subdural haematomas are classified as acute, subacute and chronic:
• acute (ASDH) refers to symptoms which manifest before 72 hours post-injury. Most patients harbouring an acute SDH are unconscious immediately following major cerebral trauma. The expanding haematoma then causes additional deterioration (Duffy 2001)
• subacute refers to symptoms which manifest between 72 hours and 3 weeks post-injury
• chronic refers to symptoms which manifest after 3 weeks post-injury. The injury may have been considered as minor and the patient often does not remember a particular predisposing injury.
The most common symptom of a SDH is a headache, which progressively intensifies and is eventually accompanied by vomiting, cognitive impairment(s), a depressed level of consciousness, and a focal deficit, which will vary depending on severity of the injury. Even in the absence of focal deficit, increasing ICP may lead to cognitive impairment and eventually a depressed level of consciousness (Watkins 2000). SDHs are often associated with other injuries, and therefore the symptoms can become confused within a general head injury picture. Small SDHs may be treated conservatively, as they will reabsorb over time. Larger SDHs will require evacuation, due to the secondary damage they cause.
Intracerebral haematoma (ICH) is caused by bleeding within the substance of the brain (Fig. 5.15). ICH usually affects the white matter and the basal ganglia found deep within the brain parenchyma. ICHs are related to contusions as a result of a major impact, and are usually found in the frontal, temporal, and parietal lobes. Other causes include penetrating and missile injuries and shearing of blood vessels deep within the brain following an acceleration/deceleration injury. Symptoms include headache, contralateral hemiplegia, ipsilateral dilated/fixed pupil and deteriorating level of consciousness, progressing to deep coma (GCS < 8). Treatment tends to be conservative, due to the difficulties of evacuating haematomas situated so deeply within the brain. Mortality is high within this group of patients.
Figure 5.15 Intracerebral haematoma.
Subarachnoid haemorrhage (SAH) is seen in 30–40 % of patients following severe traumatic brain injury. The mortality and morbidity rates are double in these patients compared with those with similar injury without the SAH component (Dearden 1998). The patient either suffered the SAH prior to the insult and thus the SAH is possibly the cause of the incident (Sakas et al. 1995), or the vessels in the subarachnoid space are damaged by the shearing forces at the time of the insult.
Diffuse injuries
Diffuse injuries occur throughout the brain rather than in a specific area of the brain. They result in generalized dysfunction. Diffuse injuries range from concussion with no residual damage, to diffuse axonal injury and persistent vegetative state. Diffuse injury occurs in 50–60 % of patients with severe head trauma and is the commonest cause of unconsciousness, the vegetative state and subsequent disability (Graham et al 1995).
Concussion is a transient form of diffuse injury that occurs following blunt trauma. It causes a temporary neuronal dysfunction because of transient ischaemia or neuronal depolarization. This manifests as a headache, dizziness, inability to concentrate, disorientation, irritability, and nausea. Concussion can occur with or without memory loss. Concussion is graded in line with the severity of symptoms (Box 5.4).
