Chapter 208 Hansen Disease (Mycobacterium leprae)
Clinical Manifestations
The manifestations of Hansen disease reflect the host response to the infection.
Tuberculoid Leprosy
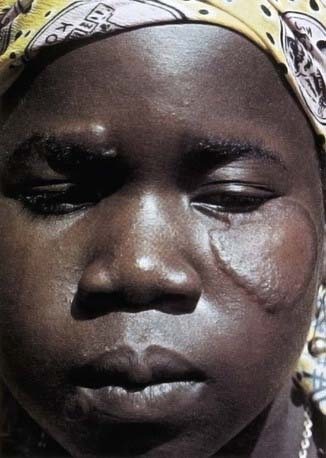
In tuberculoid leprosy, there is usually a single, large (often >10 cm in diameter) lesion with a well-demarcated, elevated erythematous rim (Fig. 208-1). The interior of the lesion is flat, atrophic, hypopigmented, occasionally scaly, and anesthetic. Rarely, there are as many as 4 lesions. The closest superficial nerve is often impressively thickened. The ulnar, posterior tibial, and great auricular nerves are most commonly affected. Periodic examination of all leprosy patients and their contacts should include palpation of these nerves. Without therapy, the skin lesion tends to enlarge slowly, but documented instances of spontaneous resolution exist. The coloration of the rim slowly fades with therapy and the induration resolves, resulting in a flat lesion with central hypopigmentation and a ring of postinflammatory hyperpigmentation. Loss of hair follicles, sweat glands, cutaneous nerve receptors, and sensation in the central portion of the lesion is irreversible. Marked improvement should be apparent within 1-2 mo after initiating therapy, but complete resolution can take up to 8-12 mo.
Borderline Leprosy
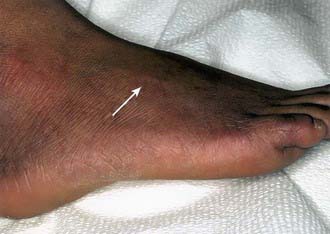
In the borderline tuberculoid (BT) pattern, the lesions are greater in number but smaller in size than in TT. There may be small satellite lesions around older lesions, and the margins of the BT lesions are less distinct. There is usually thickening of 2 or more superficial nerves (Fig. 208-2).
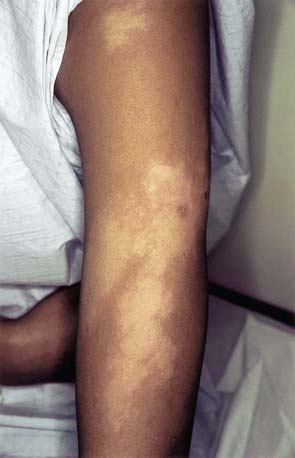
In the borderline (BB) pattern, the lesions are more numerous and more heterogeneous in appearance. They can become confluent, and plaques may be present. The borders are poorly defined, and the erythematous rim fades into the surrounding skin (Fig. 208-3). There may be anesthesia, but hypesthesia is more common. Mild to moderate nerve thickening is common, but severe muscle wasting and neuropathy are unusual.
Treatment
Physicians in the USA considering the diagnosis or treatment of leprosy should obtain consultation and assistance in patient management from the National Hansen’s Disease Programs (800-642-2477; www.hrsa.gov/hansens/), which maintains an active physician referral list of physicians in all parts of the USA.
Boggild AK, Keystone JS, Kain KC. Leprosy: a primer for Canadian physicians. CMAJ. 2004;170:71-78.
Britton WJ, Lockwood DNJ. Leprosy. Lancet. 2004;363:1209-1219.
Cambau E, Bonnafous P, Perani E, et al. Molecular detection of rifampin and ofloxacin resistance for patients who experience relapse of multibacillary leprosy. Clin Infect Dis. 2002;34:39-45.
Hartzell JD, Zapor M, Peng S, et al. Leprosy: a case series and review. South Med J. 2004;97:1252-1256.
Lawn SD, Lockwood DNJ. Leprosy after starting antiretroviral treatment. BMJ. 2007;334:217-218.
Moet FJ, Pahan D, Schurng RP, et al. Physical distance, genetic relationship, age, and leprosy classification are independent risk factors for leprosy in contacts of patients with leprosy. J Infect Dis. 2006;193:346-353.
Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
Ooi WW, Moschella SL. Update on leprosy in immigrants in the United States: status in the year 2000. Clin Infect Dis. 2001;32:930-937.
Ooi WW, Srinivasan J. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve. 2004;30:393-409.
World Health Organization. Global leprosy situation. Weekly Epidemiol Record. 2008;33:293-300.
Zhang FR, Huang W, Chen SM, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609-2618.



