[level-membership-for-pediatrics-category]
Chapter 10 Haematology
Long Cases
Haemophilia
The World Health Organization (WHO), the World Federation of Hemophilia (WFH) and various national haemophilia foundations (in Australia, the USA, Canada and many European countries) uniformly recommend that prophylaxis with an intravenous factor replacement for at least 46 weeks per year through adulthood is the standard of care. In children, the first prospective randomised controlled trial in the USA assessing the progression of arthropathy in children (under 30 months) treated (until 6 years old) with prophylaxis (25 IU/kg every other day) versus on-demand treatment (40 IU/kg initially, then 20 IU/kg at 24 and 72 hours post joint bleed) showed an 83% reduction in risk for joint damage on MRI. The evolution of a network of specialised haemophilia treatment centres in various developed countries has decreased the morbidity and mortality of haemophilia. There is no international consensus as yet regarding the optimal age to commence prophylaxis, but several studies have shown that children with no or few joint bleeds who start prophylaxis early (mean age 3 years) have a better musculoskeletal outcome. In developing countries, the high cost has precluded primary prophylaxis being adopted, and the average life expectancy for a child with severe haemophilia remains around 11 years, whereas in developed countries the life expectancy for someone with severe haemophilia is around 63 years.
Background information
Definitions
The F-VIII gene is at the telomeric end of the long arm of the X chromosome, at band Xq28. It is a large gene, 186 kilobases (kb) long, and it has 26 exons. Over 1200 mutations (missense, nonsense, splicing, and small or large deletions and insertions) have been described in the F-VIII gene. Mutations occur throughout the gene, with some concentration around exon 14. The most prevalent gene defect seen in severe haemophila A is an intron 22 inversion (int22), which accounts for 40–45% of all mutations. An inversion affecting exon 1 is present in around 5% of patients with severe haemophilia A. Around 200 smaller deletions have been described, which generally involve reading frame shifts, and non-functional gene products. Large deletions comprise 15% of haemophilia A; these result in truncated transcripts that are non-functional. Children with larger deletions and nonsense mutations are at higher risk of developing F-VIII inhibitory antibodies, and are less likely to respond to immune tolerance therapy. Most of F-VIII is synthesised in the liver endothelial cells and is immediately linked to von Willebrand’s factor (vWF) on entering the circulation; this prevents enzymatic degradation of factor F-VIII until it is required for coagulation.
Disease manifestations
• Severe haemophilia corresponds to <1% F-VIII or F-IX clotting activity: this accounts for approximately 70% of type A and 50% of type B cases. These children are predisposed to having spontaneous bleeding into joints, muscles and deep organs, including central nervous system bleeds. Without preventative treatment, these children have two to five spontaneous bleeding episodes each month. The usual age of diagnosis is within the first year of life.
• Moderately severe haemophilia means 1–5% F-VIII or F-IX clotting activity: these children rarely have spontaneous haemorrhages, but may have significant haemorrhage with mild or moderate trauma. The usual age at diagnosis is before 5 years.
• Mild disease, infers > 5% (6–35%) of F-VIII or F-IX clotting activity: these children may have bleeding with trauma or surgery. Some carrier females, who have low levels of these factors, may present clinically with gynaecological or obstetric haemorrhage.
• Around 10% of carriers have F-VIII or F-IX clotting activity lower than 35%.
• Individuals with more than 30% F-VIII or F-IX clotting activity usually do not spontaneously bleed and may not need supplemental clotting factor in the setting of minor surgery.
History
Past history
1. Initial presenting symptoms, diagnosis (when, where, how), subsequent management, progress of disease, hospitalisation details.
2. Complications of the disease (e.g. neurological deficits, joint disease) or its treatment (e.g. inhibitor formation).
3. Previous elective surgery or dental procedures (their management and outcome).
4. Outpatient clinics attended (where, how often).
5. Past treatments used (e.g. F-VIII, desmopressin [DDAVP], prothrombin complex concentrate). The majority of young children should have only received recombinant clotting factor concentrate.
6. Age when parents started administering F-VIII.
7. Age of self-administration.
8. Age of venous access port placement.
Current status
1. Average number of bleeds per year, common sites involved (e.g. knee, elbow), ‘target joint’ (most patients with significant joint disease will typically develop one joint that is more affected by recurrent bleeds), any common precipitants (e.g. sport), treatment required (type of concentrate), usual outcome, where usually managed (home or hospital), who gives infusions (patient or parent), prophylaxis regimen, details of venous access port use.
2. Ongoing symptoms of joint disease (e.g. pain, stiffness), neurological disease (e.g. weakness from peripheral nerve compression, hemiplegia from intracranial bleed).
3. Management of bleeds away from home (school, on holidays, overseas).
Social history
1. Disease impact on patient (e.g. avoidance of participation in sports such as rugby and football), self-image, schooling (attendance, performance, teacher awareness of, and attitudes towards, the disease and its treatment, peer interactions). Most patients with haemophilia are encouraged to participate in sports; there is some evidence that participation in sports reduces the incidences of bleeding episodes in boys on prophylaxis.
2. Disease impact on parents (e.g. marriage stability, fears for future, financial considerations [medical treatment, awareness of benefits available], modification of holiday plans).
3. Disease impact on siblings (e.g. sibling rivalry, hostility, genetic implications for girls).
4. Social supports (e.g. social worker, extended family). Patients with haemophilia are eligible for the carers payment.
5. Coping; for example, who attends with the patient, confidence with management, degree of understanding of the disease, expectations for the future, understanding of the prognosis.
6. Access to hospital, local doctor, paediatrician, haematologist, orthopaedic surgeon, rheumatologist.
Examination
The salient findings to be sought in the haemophilia long case are as follows.
General inspection
1. Position patient standing, undressed to underpants.
2. Parameters: weight, height, head circumference (subdural bleed).
3. Visually scan skin for bruises (number, size, age), joints for swelling and posture for evidence of neurological sequelae (e.g. hemiparesis [intracranial bleed] or foot drop [lateral popliteal palsy]).
4. Unwell (e.g. severe bleed) or well.
5. Pallor (anaemia from large bleed, e.g. retroperitoneal).
7. Vital signs: respiratory rate (e.g. pulse [anaemia], blood pressure [hypotension from bleeding], urinalysis [blood]).
Directed examination for disease extent and complications
1. Full skin examination, for distribution of bruises, including mucous membranes (mouth, tongue).
2. Full joint examination for evidence of arthropathy, focusing on the range of movement of affected joints, associated muscle wasting, any supportive devices (wheelchairs, splints, orthotic devices), gait.
3. Full neurological examination for any evidence of intracerebral or intravertebral haemorrhage, or peripheral nerve lesions. Best commenced with gait examination, followed by examination of the motor system.
4. Abdominal examination for liver and spleen size (liver disease), or tenderness (gastrointestinal or retroperitoneal bleed).
Available treatment modalities
Desmopressin (1-deamino 8-D arginine vasopressin: DDAVP)
This is a synthetic analogue of vasopressin that raises the F-VIII level by up to five times in normal subjects and seven times in some mild haemophiliacs, but has no effect in patients with severe disease. The mechanism is not fully understood. It can be given intravenously (0.3 mcg/kg in 50 mL saline over 20 minutes, peaks at 30–60 minutes) or by intranasal spray (150–300 mcg, peaks at 60–90 minutes). Tachyphylaxis may occur after several doses. Complications include facial flushing, hyponatraemia (particularly in infants—contraindicated in children under 2 years) and thrombosis (rare). Fluid restriction and urine output monitoring are important considerations. It is best for use in controlled situations such as elective minor surgery.
Management
Treatment of acute haemorrhage
1. Control of specific bleeding problems
F-VIII replacement guide
1. Minimal bleeds: first aid, antifibrinolytics if indicated. The need for factor VIII replacement should be assessed recognising the occasional difficulties with intravenous cannulation, particularly in young children.
2. Moderate bleed (e.g. joint, muscle, small oral mucosal or tongue laceration or bleed, epistaxis, gastrointestinal or genitourinary): 20–40 units/kg, given 12–24-hourly, usually for 3 days. Frequent repeated doses may be required if bleeding does not settle.
3. Severe bleed (life-threatening haemorrhage, e.g. major trauma, retropharyngeal, retroperitoneal, intracranial, large oral mucosal bleeds): 50–75 units/kg initially, followed by repeat doses of 25–40 units/kg every 12 hours until bleeding has ceased, or continuous infusion (especially intracranial bleeds, trauma).
4. Generally, haemarthroses and soft tissue bleeds require 1–3 infusions, whereas serious bleeds, such as areas with peripheral nerves at risk (e.g. psoas), retropharyngeal or retroperitoneal bleeding, may require prolonged treatment, including continuous infusions of factor VIII.
Chronic problems
Specific discussion areas
Prophylaxis
Primary prophylaxis
Primary prophylaxis is the ongoing regular infusion of F-VIII from early childhood, before significant joint bleeding is established, to prevent most bleeding episodes. This is usually started after the first significant joint bleed (often at around 12 months of age) and is typically given through a venous access port (see below). In neonates who have intracranial bleeding, prophylaxis is best started as soon as possible (i.e. without waiting until after a first major joint bleed), implanting a venous access port during the hospital admission for the presenting intracranial bleed (in one of the author’s patients, this included a [successful] burr hole at age 4 days for a subdural haematoma with midline shift, a ‘blown’ pupil and incipient coning). Recombinant F-VIII is given three times a week, at a dose of 25–40 units/kg. Prophylaxis has led to a marked decrease in the number of bleeds suffered by these children. The age at which prophylaxis is started is an independent predictor for the development of arthropathy; the earlier it is started, the less joint disease will occur, irrespective of the variables of dose and infusion intervals used at the start of treatment before the age of 3 years. The increased cost of prophylaxis may be offset by the decrease in later interventions such as synovectomy and the avoidance of significant arthritis in the adult years.
Elective surgery and continuous infusion of replacement factors
Before any elective surgical procedures, all patients with haemophilia should be assessed for inhibitors (see below) and to establish the optimum intervals of factor infusion. This comprises evaluation of increase in F-VIII or F-IX level per unit of F-VIII or F-IX per kilogram and the biological half-life of the factor in that child. It must be ensured that there are sufficient quantities of F-VIII or F-IX available on the day of surgery and for the week after. In general, for all surgical procedures, a dose to bring the patient’s level to 100% can be given at induction for the procedure.
Inhibitors and immune tolerance therapy (ITT)
Immune tolerance therapy (ITT), also called tolerisation, refers to eradicating inhibitors by manipulating the immune system through recurrent exposure to regular infusions of F-VIII or F-IX. There is controversy over the ideal dosing, interval and product choice. Many patients can be tolerised on a regimen of plasma-derived F-VIII, in a dose of about 40 units/kg three times a week. An international study is under way to compare high-dose 200 units/kg/day infusions of F-VIII with 50 units/kg, three times a week. The advantages of ITT include better control of bleeds and reduced use of expensive products. The disadvantages include increased use of F-VIII in some cases, and a success rate of up to 80%. Inhibitors to F-IX are less common; around a third of patients achieve successful immune tolerance. In response to exposure to F-IX replacement, severe allergic reactions have been described, including anaphylaxis; also, nephrotic syndrome can develop. Hence ITT must be considered very carefully in these patients. Rituximab, a monoclonal antibody directed against CD-20 positive cells, is being evaluated for those who fail to respond to ITT. Occasionally, immune modulation with steroids, or cyclophosphamide (to inhibit antibodies), IV immunoglobulin, plasmapheresis or protein A adsorption (to remove antibodies) is needed.
Sickle cell disease (SCD)
The last decade has seen several advances in the management of sickle cell anaemia (SCA), which occurs in around 1 in 500 African Americans, and in 1 in 1000–1400 Hispanic Americans. It has become clear that blood transfusion therapy has widened clinical applications, that hydroxyurea treatment effectively decreases painful crises and the requirement for transfusions, and that a cure can be obtained through haematopoietic stem cell transplantation (HSCT), for which there are now clear indications. The two main pathophysiological processes are haemolytic anaemia and vaso-occlusion. These are secondary to deoxygenation of the haemoglobin S (HbS) molecule, which aggregates into a polymer, which then causes a distortion of the red blood cell to a ‘sickle’ shape. Sickle cells block the microvasculature; the consequent deleterious effects of SCA can involve most organ systems. The rate of sickling is related to the concentration of deoxy-HbS: it takes only seconds—and if the cell is rammed through the capillaries, it becomes reoxygenated and the polymers of HbS depolymerise. The cell shape lags behind, and repeated hypoxic stress will alter the cytoskeleton of the cell and cause an irreversibly sickled cell. Organ damage in SCA can develop throughout childhood, starting with splenic and renal changes in infancy, and continuing through to pulmonary and neurological involvement with vasculopathy in older children and adolescents.
Background information
Definitions
The term sickle cell anaemia (SCA) refers only to homozygosity for the sickle cell gene. The term sickle cell disease (SCD) is a more general one that can also include compound heterozygous states such as sickle cell/haemoglobin C disease (HbSC), sickle cell/β-thalassaemia (HbS β-thalassaemia) and other sickling disorders.
There are two groups of sickle cell syndromes:
1. Sickle states, which are relatively benign (e.g. sickle cell trait, HbAS).
2. Sickle cell diseases (SCDs), which present a variety of problems. These include: sickle cell anaemia (SCA), HbSS; sickle β0-thalassaemia (β-thalassaemia without production of any β globin); sickle haemoglobin C disease (HbSC); and sickle β+-thalassaemia (β-thalassaemia with decreased [not absent] production of β-globin). Patients with sickle β0-thalassaemia and sickle β+-thalassaemia have clinical features that are more like SCD than thalassaemia, because the sickle β-globin predominates as a result of inadequate production of normal β-globin.
Effects of α-thalassaemia
HbSS patients with α-thalassaemia have a larger number of painful events (in extremities/back/abdomen/head) but fewer acute anaemic events (this does not include splenic sequestration).
Major complications
SICKLE CELL is a mnemonic for vaso-occlusive complications:
S. Sequestration (spleen and liver)
C. Cerebrovascular accidents (CVAs)
C. Crises (painful, infarctive)
• Limb effects (bone infarcts, marrow necrosis, osteomyelitis and aseptic necrosis)
The most dangerous complications are splenic sequestration, sepsis and CVAs.
Splenic sequestration crisis
1. This is the most severe crisis in those under 5 years of age. It occurs in 10–30% of children with HbSS, most commonly between 6 months and 3 years.
2. It results from acute entrapment of a large volume of blood in the spleen—often a large fraction of the circulating blood volume.
3. These children get rapid splenic enlargement (at least 2 cm increase in spleen size from baseline) with an acute fall in haemoglobin of greater than 2 g/dL, and with a raised reticulocyte count. They present with sudden collapse, shock, profound anaemia and abdominal fullness due to the massive splenomegaly. This often occurs during an acute infection.
4. It is life-threatening: it can be fatal within 30 minutes. Shock is treated with plasma expanders and whole blood.
5. If the child is older (e.g. over 2 years), consider splenectomy in susceptible patients. If under 2 years of age, a chronic transfusion program may be needed.
6. There is a tendency for recurrence: up to 50% of children have a second episode, usually within 2 years. Elective splenectomy is generally recommended in patients presenting with the first episode of splenic sequestration.
Infection: overwhelming sepsis
1. This particularly affects children under 3 years of age, as a result of poor development of immune response to polysaccharide antigens, complicated by early loss of splenic function (this ‘autosplenectomy’ occurs in 60% by 2 years, and 90% by 5 years).
2. Pathogens are most commonly Streptococcus pneumoniae (pneumococcus)—various serotypes—and occasionally Haemophilus influenzae type B. Other pathogens include meningococcus, and other Streptococci, Salmonella and fastidious gram-negative organisms, such as DF-2 (Capnocytophaga canimorsus) after a dog bite.
3. Prevention is by immunisation against pneumococci, Haemophilus and meningococcus. Prophylactic penicillin is recommended until the age of 5 years, as it decreases the incidence of pneumococcal bacteraemia by 84%. Many units continue penicillin prophylaxis throughout childhood.
4. If children with HbSS or sickle β0-thalassaemia present febrile, after clinical assessment it is prudent to take blood for a full blood count, blood cultures, plus type and cross-match if pale, the spleen is enlarged, or there are neurological or respiratory symptoms. Then treat with ceftriaxone or cefotaxime (or vancomycin if in an area with a high prevalence of resistant pneumococci). Other investigations for sepsis (e.g. other cultures, CXR) and coexisting complications depend on presentation.
Cerebral infarction (cerebrovascular accident, CVA)
1. Central nervous system involvement is seen in up to 8% of patients; the median age is between 5 and 8 years.
2. CVA particularly affects internal carotid (ICA), anterior cerebral (ACA) and middle cerebral (MCA) arteries. Stroke involves arterial occlusion, although SCA’s haemoglobin deoxygenation and polymerisation occur in the microcirculation and venous system. Hence the aetiology of CVAs in SCA is unclear; it is possibly HbSS membrane procoagulant. Permanent sequelae can result (e.g. hemiplegia).
3. Without treatment, CVA recurs in 50–90% within 3 years; it can be progressive.
4. MRI and MR arteriography are useful in evaluating the extent of the infarction.
5. Haemorrhage into infarcted areas (from bleeding from delicate vessels from neovascularisation), or ruptured aneurysm (in the contralateral circulation from compensatory increased blood flow) with subarachnoid haemorrhage, can occur as early as 4 years of age and cause further neurological deficits.
6. The recommended treatment after cerebral infarction is long-term transfusion therapy maintaining HbS levels below 30%; this lowers the risk of recurrence to 10%.
7. Some studies show that subsequent stroke can occur after cessation of transfusion therapy. Many would recommend continuing transfusion indefinitely.
8. Prevention: some units suggest chronic transfusion of at-risk children. They may be detected by transcranial Doppler (TCD) studies that have shown that high blood flow velocity through the ICA (over 200 cm/min) clearly increases the probability of arterial occlusive stroke. TCD studies are used in the USA, but were not available in the majority of Australian centres in 2006.
Kidney involvement
1. Hyposthenuria (the inability to concentrate urine) is due to chronic sickling in the region of the loop of Henle (microvascular ischaemia induced by sickle cell, with obliteration of the vasa recta of the kidney medulla).
2. This leads to an obligatory urine output of up to 2 L/m2/day in infants, up to to 4 L/m2/day in adults, and associated nocturia, and puts patients at risk of rapid dehydration.
3. Other renal sequelae include renal tubular acidosis, impaired potassium excretion, microscopic haematuria, proteinuria and nephrotic syndrome.
4. Eventually, in adulthood, hypertension, ineffective erythropoiesis, renal osteodystrophy and end-stage renal disease (ESRD) can supervene, requiring top-up transfusion, recombinant human erythopoietin or renal transplantation.
Lung disease: acute chest syndrome (ACS)
1. Acute pulmonary disease with new respiratory symptoms (fever, cough, sputum production, dyspnoea, hypoxia), and new infiltrates on CXR.
2. May be due to pulmonary infarction or infection: for example, bacterial (Haemophilus influenzae, Staphylococcus aureus, Klebsiella pneumoniae, Mycoplasma pneumoniae, pneumococcus, Chlamydia pneumoniae, Legionella pneumophila, TB, Cryptococcus) or viral (respiratory syncytial virus, parvovirus, adenovirus, influenza, cyto-megalovirus), or both; it can be difficult to distinguish between them. Definite aetiology is not established in 65–70% of cases with pneumonia. ACS can also be caused by rib or sternal infarction.
3. Pulmonary fat embolism (PFE) is the second most common cause of ACS. Marrow infarcts during a vaso-occlusive crisis (VOC) can generate fat emboli, which cause a marked inflammatory response in the lung. Secretory phospholipase A2 (sPLA2) is an inflammatory mediator that liberates free fatty acids and causes the acute lung injury with PFE. In children with VOC and elevated sPLA2, blood transfusion may prevent the development of ACS. ACS can also be caused by rib or sternal infarction.
4. ACS is most common in the 2–4 year age group and declines with increasing age. HbF seems to protect those under 2 years. Incidence is related to genotype: it is more common in HbSS and HBS β0-thalassaemia than in HbSC or HbS β+-thalassaemia. Lower haematocrit also reduces incidence.
5. Repeated episodes of ACS are associated with the development, in adulthood, of chronic restrictive lung disease, pulmonary hypertension, cor pulmonale, hypoxia, osteonecrosis at multiple sites and myocardial infarction.
6. Treatment: avoid hypoxia; judicious intravenous rehydration, analgesia, prevention of atelectasis (with incentive spirometry if possible), antibiotics (for all with fever, and most without), transfusion with packed cells (to increase the oxygen carrying capacity), exchange transfusion for clinical deterioration or hypoxia, or those unresponsive to other therapies. Transfusion risks include viral disease transmission, acute hyperviscosity and alloimmunisation. In adults, hydroxyurea treatment leads to a significant reduction in ACS.
7. It is the single largest contributor to mortality in children under 2 years.
Dactylitis (hand–foot syndrome)
1. Infants as young as 3 months may present with painful swollen hands and feet due to symmetric infarction of red marrow and associated periosteal inflammation involving metacarpals, metatarsals and phalanges. Patients may be misdiagnosed with rheumatological disorders.
2. Dactylitis may be the first sign of diagnosis. Initial X-rays show swelling only; but after a few days, periosteal elevation and areas of osteoporosis and sclerosis may be seen.
3. It is rare after 3 years of age, as the haematopoietic tissue in hands and feet is later replaced by fatty tissue.
Abdominal involvement
1. Infarction within the spleen, mesenteric lymph nodes or liver.
2. It may present with signs of acute abdomen, with generalised abdominal pain and distension, with vomiting and diminished bowel sounds. X-ray is consistent with ileus. Management is conservative, with IV fluids and nasogastric aspiration.
3. Hepatic sequestration crisis can occur, with sickled cells held up in the liver.
Erectile problems: priapism
1. Painful failure of penile detumescence. ‘Stuttering’ priapism often occurs: the development of erections lasting 1–2 hours. Priapism lasting for over 3 hours is a medical emergency. It is caused by obstruction between the corpora cavernosa and the spongiosa, followed by sickling within the corpora cavernosa, particularly in adolescents.
2. If the child cannot urinate, this indicates involvement of the corpora spongiosa.
3. Priapism may resolve within a few hours or last more than 24 hours, with extreme pain.
4. Management is medical initially, with analgesia, IV hydration, oxygen, sedation, warmth and exchange transfusion. Non-acute cases can be treated by conjugated oestrogens or vasodilators.
5. If medical treatment is unsuccessful after 12 hours, surgical intervention is required (e.g. cavernosa aspiration and irrigation—the earlier performed, the better).
6. Priapism may cause a fibrotic corpora cavernosa, penile atrophy and impotence.
Limb (bone and joint) involvement
1. Painful episodes usually occur after the age of 3 years.
2. Pathology includes infarction of bone marrow, cortical bone and periarticular tissues.
3. It may present with limitation of movement and swelling.
4. Osteomyelitis may occur, particularly under 5 years of age, with Salmonella as the pathogen in over half the cases.
5. Differentiating between infarction and infection can be hard; bone scans may help.
6. Infarcts are the most common in the long bones, spine, sternum and ribs.
7. Vaso-occlusion of the vessels supplying the head of the femur can lead to avascular necrosis of the femoral head
Chronic haemolysis and anaemia
Sequelae include poor growth, delayed puberty and pigmented gallstones.
Haemolytic crisis
This is not common and is associated with a rise in the reticulocyte count (which differentiates this from aplasia) and the bilirubin level.
Aplastic crisis
1. This is due to compromise of the compensatory response (increased red cell production) to ongoing haemolysis.
2. In SCA, red cell survival is only 10–20 days. If infection develops, patients may have red cell aplasia for 10–14 days. Causes include parvovirus B19, pneumococci, streptococci, Salmonella species and EB virus.
3. Cases present with pallor, tachypnoea, tachycardia and weakness.
4. White cell and platelet counts are normal; reticulocytes are decreased or absent.
5. Management is with transfusion (packed cells or exchange).
History
Past history
1. Age and mode of presentation (e.g. screening as a newborn, dactylitis as an infant, sequestration as a toddler, painful crisis as a schoolchild).
2. The progress of the disease, hospitalisation details, complications experienced (e.g. aplasia, sepsis, stroke), outpatient clinics attended.
3. Past treatment used (e.g. intravenous rehydration, analgesia, bicarbonate).
Social history
1. Disease impact on patient: effects of hospitalisations, behaviour, school attendance, self-image (e.g. short stature, leg ulcers, delayed puberty), peer group anxieties, narcotic abuse in older children.
2. Disease impact on parents: for example, financial considerations (medical treatment, awareness of benefits available), modification of holiday plans.
3. Disease impact on siblings: for example, sibling rivalry, genetic implications (screening for occult disease).
4. Social supports: for example, social worker, extended family.
5. Coping: for example, level of understanding of the disease by the parent (awareness of risks of sepsis, ability to palpate spleen to detect sequestration) and the patient (understanding of precautions to take with risks of hypoxia, high altitude, exposure to cold, and of the necessity for penicillin prophylaxis); any problems with analgesic abuse.
Examination
General inspection
1. Position patient: standing, fully undressed and then lying down.
2. Parameters: height, weight, head circumference, percentiles.
4. Racial origin (e.g. African).
5. Well or unwell (e.g. sepsis, painful crisis).
6. Skin: pallor (aplastic crisis, haemolytic crisis), jaundice, scratch marks (haemolysis).
7. Joint swelling: periarticular infarction.
8. Posture: for example, hemiparesis (cerebral sickling), restricted movement (bony infarction).
9. Dysphasia (cerebral sickling).
10. Abdominal distension (sequestration crisis).
11. Vital signs: respiratory rate (e.g. pneumonia), pulse (anaemia), blood pressure (hypotension from bleeding), urinalysis (low specific gravity, blood, white cells).
Directed examination for complications
Head and neck
1. Check eyes for visual field defects (cerebral infarction), conjunctival pallor, scleral icterus and proliferative retinopathy.
2. Examine the motor cranial nerves, especially noting whether there is an upper or lower motor neurone lesion of the seventh nerve (for localisation of damage in hemiplegia).
Management
Common management issues are as follows:
1. Management of acute complications.
2. Complications of medical treatment.
Management of acute complications
Chronic transfusion therapy—prevention of primary manifestations
Hydroxyurea—the prevention of primary manifestations
Avoiding known precipitants
1. Dehydration, to which they are prone because of their fixed hyposthenuria. Encourage the patient to drink plenty of fluids, especially during viral illnesses or in hot weather. During any infection, dehydration is anticipated and early hydration is instituted.
2. Infection with encapsulated bacteria. Prophylactic penicillin may be given, as well as immunisation against pneumococcus, Haemophilus influenzae type b and meningococcus.
3. Vascular stasis. Tight clothing (e.g. fashion jeans, cycling pants) and tourniquets when in hospital must be avoided.
4. Cold temperatures, either environmental (e.g. getting caught in the rain, swimming in rivers, cold baths, washing the car or doing the laundry [adolescents]) or related to ice-cold food or drinks.
Chronic problems
Related to chronic haemolytic anaemia
Chronic ankle ulceration is common in adolescent and adult patients, and is due to inadequate healing (a result of anaemia) of minor traumatic lesions. Management may include complete bed rest (immobilisation), debridement with proteolytic enzymes (or crushed papaya, as used in Jamaica), clean dressings, antibiotics, oral zinc sulphate or pinch skin grafting.
Many units prescribe folic acid to prevent superimposed folate deficiency.
Specific discussion areas
Neonatal screening and prevention of early mortality
Neonatal screening for early detection of HbSS (and related haemoglobinopathies), coupled with comprehensive medical management (immunisations, prophylactic penicillin), has decreased the mortality from sickle haemoglobinopathies.
Haematopoietic stem cell transplantation (HSCT)—potential cure
The indications for HSCT relate to patients 16 years old or younger with SCD, with an HLA-identical sibling bone marrow donor with one or more of the following (mnemonic STAR IS BORN):
S. Stroke, central nervous system (CNS) haemorrhage or a neurological event lasting longer than 24 hours, or an abnormal cerebral magnetic resonance imaging (MRI) scan, cerebral arteriogram or MRI angiographic study and impaired neuropsychological testing
T. Transfusion requirement, but with RBC alloimmunisation of more than two antibodies during long-term transfusion therapy.
A. ACS, with a history of recurrent hospitalisations or exchange transfusions
R. Recurrent vaso-occlusive pain—three or more episodes per year for three years or more—or recurrent priapism
I. Impaired neuropsychological function and abnormal cerebral MRI scan
S. Stage I or II sickle lung disease
B. Bilateral proliferative retinopathy and major visual impairment in at least one eye
O. Osteonecrosis of multiple joints, with documented destructive changes

Thalassaemia: β-thalassaemia major
Background information
Basic defect (β-thalassaemia major)
1. Decreased synthesis of the β-globin chain of haemoglobin.
2. Normal synthesis of the α chain of haemoglobin, leading to the accumulation of unstable aggregates within the red blood cells, resulting in ineffective erythropoiesis and haemolysis. In patients with concomitant α-thalassaemia, the symptoms of haemolysis may be ameliorated.
3. Hypochromic microcytic anaemia (note: the mean cell volume may be in the normal range for children, but abnormal for that child). The number of reticulocytes is less than expected for the degree of the anaemia. Diagnostic blood film features include target cells, anisocytosis, poikilocytosis, schistocytosis, basophilic stippling and erythroblastosis.
Genetics
1. Autosomal recessive; β-globin gene on the short arm of chromosome 11, in a region that contains the embryonic- and fetal-globin genes. The expression of these globin genes is controlled by the LCR (locus control region), a major regulatory region containing a series of hypersensitive sites that interact with a variety of transcription factors.
2. The frequency of the gene in Greek Cypriots is 0.2; in Italians and Lebanese, 0.04.
3. By 2002, over 200 different molecular defects were known, mostly involving single nucleotide substitutions or oligonucleotide insertions/deletions, which inactivate the β gene expression by various mechanisms. Some mutations silence the β-globin gene (from mRNA modification at the splicing or cleavage steps; or from RNA mutations, causing abnormal translation of the gene to a globin chain product, mostly caused by premature termination codon), resulting in β0-thalassaemia; others reduce β-globin output, resulting in β+-thalassaemia (either by DNA transcriptional mutations at the promoter site, or from mRNA modification at the splicing or cleavage steps). Depending on the residual β-globin production, β+-thalassaemia may be silent, mild or severe. β0-Thalassaemia mutations are usually severe. If a single β-globin gene is affected, then the resulting phenotype depends on whether there is partial or absent gene expression. If partial, then this is silent carrier status; if absent, then this is the β-thalassaemia trait. Should both β-globin genes be affected, then the phenotype is more severe, depending on the degree of gene expression, and on the relative imbalance of the globin chains. It is termed thalassaemia intermedia if the genotype is β+/β+, but thalassaemia major if the genotype is β0/β0. The resultant imbalance between the excessive α chains and the diminished β chains leads to unpaired globin chains, which can precipitate and cause premature death (apoptosis) of red cell precursors in the bone marrow, this being called ‘ineffective erythropoiesis’.
Diagnosis
1. Usually not clinically evident until the child is 6–12 months old.
2. Definitive diagnosis is by haemoglobin electrophoresis: HbA is absent or minimal, HbF is elevated and HbA2 may be raised. The percentage of fetal haemoglobin is the most important laboratory parameter, which influences clinical severity in SCD.
Major complications
Excess erythropoiesis (causing bone marrow expansion)
1. Bony changes to face: maxillary overgrowth, protrusion of teeth, separation of orbits, frontal bossing, chronic sinusitis and impaired hearing.
2. Bones (general): cortical thinning and risk of fractures. Most adult patients get osteopenia and osteoporosis, with backache, scoliosis, fractures and other spinal deformities. Males who are diabetic and have pubertal delay are most at risk for osteoporosis. DEXA (dual X-ray absorptiometry) assesses for bone density.
3. Spinal cord compression (vertebral expansion).
Iron overload (causing parenchymal organ toxicity)
This is transfusional or from increased iron absorption. A packed red blood cell unit of 250 mL has around 175 mg of iron. Free iron is toxic to cells, so iron is normally complexed to proteins; in plasma, iron is bound to transferrin, which transports it to the cells. Iron not bound to transferrin is toxic to the endocrine organs, the liver and the heart, where myocyte damage can lead to arrhythmias and cardiac failure. Cardiac toxicity is the main cause of death related to iron overload in thalassaemia. The goal of chelation is to prevent these organ toxicities. Chelation can reverse some already developed toxicities, such as cardiac complications, but the endocrinopathies are generally irreversible.
Endocrine failure (in order of frequency)
1. Short stature: growth is usually normal until about 12 years, but no pubertal growth spurt occurs: two thirds of these children are below the 10th height percentile at 21 years. A possible pituitary or hypothalamic defect. The growth hormone level may be normal or raised.
2. Delayed puberty: gonadotropin levels are normal until puberty, but then no increase occurs. Hypogonadotrophic hypogonadism can be treated with hormonal therapy and aggressive iron chelation, to preserve fertility.
Cardiac involvement
1. Cardiomyopathy from myocardial iron deposition, hypertrophy, dilatation, degeneration of myocardial fibres; unbound iron generates toxic oxygen metabolites; higher risk for myocarditis; occasional pulmonary hypertension.
2. Pericarditis: first attack usually after 10 years of age.
3. Arrhythmias, both ventricular and atrial, can occur. The risk is much increased after 150–200 units of blood.
4. Congestive cardiac failure: once this develops, the mortality is 90% within 12–18 months. Cardiac function can be reversed by aggressive chelation. Treatment may include angiotensin-converting enzyme (ACE) inhibitors, digoxin, diuretics and a low-salt diet, in addition to chelation therapy. If refractory to medical treatment, heart transplantation is an appropriate consideration.
5. Measurement of liver iron correlates best with total body iron and predicts the threshold for risk for cardiac disease and early death (levels over 15 mg/g dry weight). The gold standard is liver biopsy. There are two non-invasive procedures that give results that correlate well with biopsy: liver iron concentration by MRI, and liver iron concentration by the superconducting quantum interference device (SQUID) technique—the latter is very expensive and has very limited availability. Ferritin over 2500 micrograms/L can help predict cardiac disease.
6. Measurement of cardiac iron loading by MRI can predict risk for cardiac disease, but cannot be validated with biopsy specimens, as can occur with the liver.
7. Heart disease is the main cause of death in transfusion-induced iron-overload patients. Cardiac iron removal can be achieved with chelation with deferasirox (DFS).
Hepatic involvement
1. Cirrhosis and hepatic fibrosis: the risk for this is increased after 7 years of treatment.
2. Liver function tests may be abnormal, but it is rare to see symptoms of cirrhosis. If hepatic enzyme levels are increased four- to fivefold, then there is a possibility of hepatitis and liver biopsy may be needed.
3. Common cause of morbidity and early death; worsened by coexistent hepatitis C.
4. If hepatitis C (HCV) is suspected, liver biopsy is needed and the HCV RNA polymerase chain reaction (PCR) should be checked. Image to exclude hepatocellular carcinoma. Treatment with alpha interferon and ribavirin can improve liver disease, but the side effects can include haemolysis, which increases transfusion requirements.
History
Past history
2. Diagnosis (age, where, how).
3. Age at first transfusion and at commencement of desferrioxamine (DFO).
4. Number of transfusions per year.
5. Previous hospitalisations (other than routine transfusions), previous surgery (e.g. splenectomy), medications given (e.g. penicillin with splenectomy, vitamin C).
Specific complications
The current status of the following complications (mnemonic THALASSAEMIA):
T. Tanner stage (pubertal delay)
H. Heart (cardiomyopathy)/Haematopoiesis (extramedullary)/Hypercoagulable
• Liver/Long tracts (neurological involvement)/Leg ulcers
A. Appearance (thalassaemic facies, pigmentation)
M. Metabolic (hypocalcaemia)/Malocclusion
I. Iron overload/Icterus/Infection/Iatrogenic (DFO, DFS side effects)
Social history
1. Disease impact on patient: for example, effects of hospitalisations, behaviour, school performance, amount of school missed, self-image, pubertal anxieties, peer group anxieties, stigma of ‘bad blood’, adolescent denial, poor compliance, anxiety, ‘live for today’ approach, concerns about developing relationships, wanting a family.
2. Disease impact on parents: for example, marriage stability, financial considerations (family income, cost of DFO and pump), dealing with non-compliance and teenage rebellion.
3. Disease impact on siblings: for example, sibling rivalry.
4. Social supports: for example, extended family, Australian Thalassaemia Association and state thalassaemia societies, social worker, financial benefits obtained (e.g. Children’s Disability Allowance).
5. Coping: for example, compliance, degree of self-management, perception of the disease and its rate of progression, future plans, career thoughts.
Standard management principles
Blood transfusion
1. Regular transfusions every 4–6 weeks.
2. Timing of first transfusion usually at haemoglobin <60 g/L.
3. Maintain haemoglobin above 100 g/L (range 100–150 g/L, e.g. pretransfusion Hb about 95 g/L, post-transfusion 140 g/L, with a mean of about 120 g/L). This level is enough to suppress endogenous erythropoiesis and compensatory marrow hyperplasia, while avoiding unnecessary iron overload.
4. Use packed cells, or filtered red blood cells, negative for hepatitis B, C and HIV (phenotyped filtered red cells).
5. Watch for reactions at the time of transfusions.
6. Pre-transfusion investigations: group and cross-match (genotype); full blood count (for haemoglobin level and reticulocyte count, and to assess for hypersplenism); initial viral serology for hepatitis B, C (± G: research units), cytomegalovirus (CMV), toxoplasmosis, Ebstein–Barr virus, HIV; assessment for alloantibodies. The red cell genotype determines all the important red cell groups, including Duffy and Kell. Antibody screening at the time of cross-match is to detect any irregular alloantibodies.
Chelation with desferrioxamine (DFO)
1. Start when ferritin level is around 1000–2000 mcg/L (before 3–4 years) or at preschool age. DFO was previously standard therapy. It has high molecular weight (MW), is poorly absorbed from the gut and so is given parenterally, aiming for negative iron balance.
2. The DFO molecule has six binding sites and wraps itself around the iron nucleus.
3. Dosage for DFO is 20-30 mg/kg for young children, increasing to 40 mg/kg after age 6 years, subcutaneously over 8–10 hours, 6 days per week.
4. Side effects of DFO include local irritation and hypotension if given too quickly intravenously. More severe effects were originally described when doses in the range 100 to 200 mg/kg/day were used. These included problems with vision (cataracts, night blindness, reduction of visual fields, decreased visual acuity, and pigmentary retinopathy) and hearing (sensorineural deafness). Other side effects described have included bone abnormalities (pseudorickets, metaphyseal changes, flat vertebral bodies) and altered renal function. At doses less than 50 mg/kg/day, these effects are not often seen.
5. Pre-chelation evaluation may include clinical photography, bone X-rays, ferritin level, full blood count, liver function tests, thyroid function tests, fasting blood glucose level, tests of the hypothalamic–pituitary axis, calcium, phosphate and magnesium, audiovisual assessment (audiometry; slit-lamp examination), baseline electrocardiogram.
6. Avoidance of red meat and cereals should be recommended.
7. Patients who are compliant with iron chelation therapy should have a life expectancy of more than 50 years.
8. DFO treatment is suspended if the patient has sepsis, as DFO promotes Yersinia enterocolitica gastroenteritis.
Chelation with deferasirox (DFS)
1. Deferasirox (DFS) is the first oral agent approved for use in the USA. It is a tridentate chelator; two molecules of DFS are needed to bind one atom of iron.
2. It is supplied as a dispersible tablet to be dissolved in water or juice. It has a long half-life (up to 16 hours) and so can be given once daily. The recommended initial daily dose is 20 mg/kg. For patients who are already well controlled and receiving DFO, the starting dose of DFS is half that of DFO; hence DFS 20 mg/kg is equivalent to DFO 40 mg/kg.
3. The response to DFS depends on the transfusion requirements; for patients with a lower transfusional iron intake, it is effective in decreasing liver iron stores, but it is ineffective in some patients with higher transfusional iron intake.
4. DFS is effective in removing myocardial iron, which is associated with a concomitant decrease in total body iron. Iron levels can be quantified in a non-invasive manner using cardiac MRI to measure myocardial T2∗; T2∗ is a measure of magnetic relaxation, and it shortens when the particulate stored iron interrupts the magnetic microenvironment, so that as T2∗ falls, the risk of left ventricular dysfunction increases.
5. The side effects of DFS include gastrointestinal problems (nausea, vomiting, abdominal pain), skin rashes, elevated transaminases, cataracts and audiotoxicity.
6. As with DFO, pre-chelation evaluation may include clinical photography, bone X-rays, ferritin level, full blood count, liver function tests, thyroid function tests, fasting blood glucose level, tests of the hypothalamic–pituitary axis, calcium, phosphate and magnesium, audiovisual assessment (audiometry; slit-lamp examination) and baseline electrocardiogram.
7. Avoidance of red meat and cereals should be recommended, as above.
8. Patients who are compliant with iron chelation therapy should have a life expectancy of more than 50 years, as above.
Splenectomy
1. The main indication is hypersplenism, usually evidenced by increased transfusion requirements. Another indication is a ‘large spleen’, causing discomfort.
2. Most authorities recommend the use of pneumococcal and meningococcal vaccines (given before splenectomy) and prophylactic penicillin.
3. Consideration should also be given to gall bladder ultrasound for calculi, with a view to the possible need for cholecystectomy, which can be performed at the same time as splenectomy. Appendicectomy can be performed at the same time.
4. If a patient with a splenectomy is febrile, blood cultures should be taken and treatment instituted with intravenous antibiotics in hospital or oral antibiotics at home.
5. Some units recommend splenectomy only if transfusion requirements exceed 180–200 mL/kg/year.
Common management issues
When to start transfusions?
This is really a clinical decision. There is no specific haemoglobin level, but if the level is below 50 g/L, there needs to be a good reason not to transfuse. Careful assessment in the first 2 months after diagnosis is needed, and treatment with folate. If there is any failure to thrive, significant anaemia or (especially) cardiac compromise related to anaemia, then transfusion should probably be instituted.
Curative therapies: haematopoietic stem cell transplantation (HSCT)
The enthusiasm for this mode of treatment varies between countries. Some countries prefer to wait for breakthroughs in genetic engineering or a HbF switch mechanism; whereas in Italy, BMTx is quite popular but has a 10% mortality rate related to infection or acute graft-versus-host reaction. The donor must be fully compatible (usually a sibling who is normal or heterozygous). It is not justified to offer any mismatched or matched unrelated donor transplantation. This treatment modality should be discussed with the parents of any child with thalassaemia, so that they are aware of this option in the future.
Prenatal diagnosis (PND)
There are over 200 different molecular defects of the haemoglobin subunit β (HBB) gene known for β-thalassaemia. Within given communities, there are a dozen or so common molecular defects. Targeted mutation analysis uses a number of PCR-based procedures, such as reverse dot blot analysis or primer specific amplification using a set of probes or primers complementary to the most common mutations in that particular community, to detect common mutations. If this fails to identify the mutation, then mutation scanning or sequence analysis can be used. Prenatal diagnosis and preimplantation genetic diagnosis (PGD; see below) can be performed only in at-risk pregnancies where the mutation is known. If the parents already have an affected child, then prenatal identification of HLA compatibility with an unaffected fetus enables collection of placental blood at delivery and future cure with cord blood transfusion. If the fetus is affected but there is an older sibling who is unaffected and HLA compatible, then the parents may continue the pregnancy with a plan for a later BMTx.
Follow-up
1. A check of the ferritin level (the degree of iron overload) every 3–6 months. Once receiving chelation, if ferritin levels are 2000 mcg/L, this suggests non-compliance. Levels over 3000 mcg/L may warrant liver MRI (for iron content; MRI images darken at a rate proportional to the iron concentration) and hospital admission for continuous intravenous DFO once or twice a month.
2. Blood tests every 6 months for calcium, magnesium, phosphate (hypoparathyroidism), liver function (hepatitis or hepatic fibrosis), thyroid function (hypothyroidism), urea, electrolytes and creatinine (renal dysfunction from DFO). If ALT is elevated, repeat in one month: if still up, check for hepatitis A, B, C or G, or CMV or EBV. If ALT is elevated for 3 months, consider liver biopsy and hepatitis viral titres by RNA analysis. HIV serology should be checked as well.
3. Yearly assessments for DFO or DFS toxicity (audiometry and slit-lamp examination) and of growth and pubertal status (bone age and, over the age of 14, tests of the hypothalamic pituitary axis). By the age of puberty, essentially all of these patients attend endocrine outpatient clinics. The boys usually require supplementation with testosterone preparations, and the girls low-dose oestrogens and progestogens. Screening for the development of diabetes mellitus includes testing for glycosuria and performing glucose tolerance tests. Approximately 50% of patients will have an abnormal glucose tolerance test by 10 years of age.
4. Yearly cardiac assessment, with a gated blood pool scan in children over 10 years to assess the left ventricular ejection fraction (at rest and during exercise), has been a preferred cardiac investigation. Some units recommend annual ECG, Holter monitoring and cardiac stress testing. Myocardial siderosis can be assessed using T2∗ MRI, as above. Cardiac iron overload has been the cause of most deaths in β-thalassaemia major.
5. Annual dental examination is useful. Dental procedure antibiotic prophylaxis in splenectomised patients is important. Reminding these patients to take their antibiotics is another important point in follow-up.
6. Other points regarding clinic follow-up include the discussion of any emotional or social problems.
Prognosis
A favourite question is: ‘What do you tell the parents of a child newly diagnosed with thalassaemia major?’ The answer should include telling the parents the following:
• The child is very likely to need transfusions.
• A wait-and-see approach regarding the need for, and frequency of, transfusions should be adopted.
• A cure by HCT, or genetic engineering, may be possible (after discussing the more standard treatment options).
• Current treatment modalities allow patients to reach young adulthood, by which time better treatments and probably a cure may be available.
• Regular erythrocyte transfusions plus adequate iron chelation has improved the prognosis of β-thalassaemia dramatically over the last two decades.
Short Cases
The haematological system
The usual introduction involves assessment for anaemia or bruising.
Assess whether the child looks sick or well. Patients with acute lymphoblastic leukaemia (ALL), CKD, haemophilia complicated by HIV or purpura due to meningococcaemia look unwell. Patients with SCA may be in pain if a recent crisis has occurred. Infants with NAI may be wary of any attempts to get close. Note any pallor (various causes of anaemia).
Next, examine the head and neck, followed by the chest wall, spine, abdomen, gait, lower limbs and heart. This is outlined in Figure 10.1.
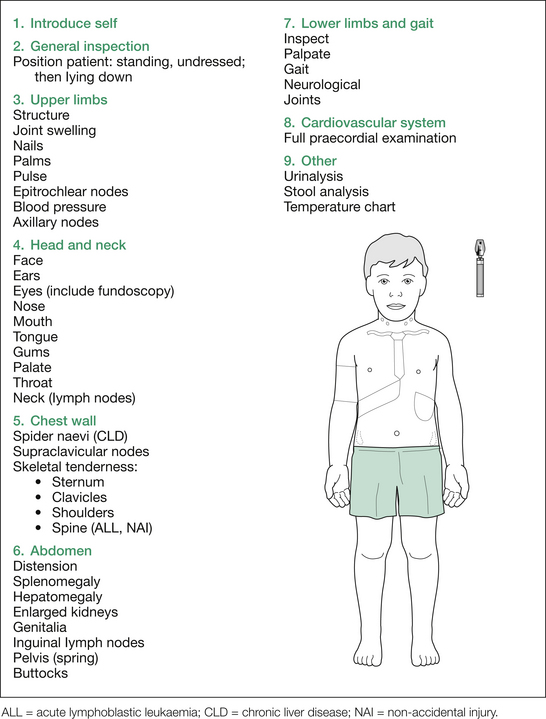
1. First, all the lymph nodes (lymphadenopathy with ALL, AML, lymphoma) and the abdomen (hepatosplenomegaly, or abdominal masses, from ALL or AML infiltrates, or extramedullary haematopoiesis).
2. Next, the musculoskeletal examination (skeletal tenderness at sternum, clavicles, ribs, spine, pelvis and tibiae [leukaemic infiltrates, SCA]; joint tenderness, swelling or decreased range of movement [ALL, AML, SCA, IBD, HSP, JIA]).
A comprehensive listing of findings not listed above is given in Table 10.1. Which of the findings listed is relevant depends on the particular case involved.
Table 10.1 Additional information: comprehensive listing of possible findings in the haematological examination
| General inspection |
| Well or unwell (DIC, ALL, meningococcaemia) |
| Sex (haemophilia, G6PD deficiency in males) |
| Race |
ALL = acute lymphoblastic leukaemia; AML = acute myeloid leukaemia; CHD = congenital heart disease; CLD = chronic liver disease; CKD = chronic kidney disease; DIC = disseminated intravascular coagulation; G6PD = glucose-6-phosphate dehydrogenase deficiency; HHT = hereditary haemorrhagic telangiectasia; HSP = Henoch–Schönlein purpura; HUS = haemolytic uraemic syndrome; IBD = inflammatory bowel disease; JIA = juvenile idiopathic arthritis; MAHA = microangiopathic haemolytic anaemia; NAI = non-accidental injury; SBE = subacute bacterial endocarditis; SCA = sickle cell anaemia; SLE = systemic lupus erythematosus; TAR = thrombocytopenia absent radius.
After presenting your findings, you may be asked by the examiners which investigations you would perform. Depending on the presenting problem, a suggested plan is given below.
Anaemia
2. Normocytic
This group can be subdivided into two groups, based on the reticulocyte count:
1. A low-reticulocyte response occurs most commonly with transient erythroblastopenia of childhood. Other causes include aplastic crises and Blackfan–Diamond anaemia.
2. A high reticulocyte count occurs with bleeding or haemolysis. Haemolysis can be classified by extrinsic or intrinsic causes.
Extrinsic causes
1. Mechanical injury to red cells (e.g. small vessel disease in HUS, or in disseminated intravascular coagulation [DIC]; or larger vessel disease, such as poorly epithelialised prosthetic cardiac valves).
2. Chronic kidney or liver disease (CKD or CLD).
3. Haemolysis mediated by antibodies, including autoimmune haemolytic anaemia (e.g. warm antibody type in Epstein–Barr virus infections, cold antibody type in mycoplasma infections) and isoimmune haemolysis (e.g. incompatible transfusions).
Bleeding
1. A full blood count and film, noting the haemoglobin level (associated anaemia, e.g. in aplastic anaemia or ALL), the white cell count (e.g. leucopenia in aplastic anaemia, blast forms in ALL), any thrombocytopenia and also the platelet size (large in Bernard–Soulier syndrome).
2. The prothrombin time, which assesses the extrinsic coagulation system.
3. The partial thromboplastin time, which assesses the intrinsic coagulation system.
In bleeding associated with vascular defects, the above tests will be normal.
Thalassaemia
As mentioned in the long case, the mnemonic THALASSAEMIA helps list the important areas to examine:
T. Tanner stage (pubertal delay)
H. Heart (cardiomyopathy)/Haematopoiesis (extramedullary)/Hypercoagulable
• Liver/Long tracts (neurological involvement)/Leg ulcers
A. Appearance (thalassaemic facies, pigmentation)
M. Metabolic (hypocalcaemia)/Malocclusion
I. Iron overload/Icterus/Infection/Iatrogenic (DFO, DFS side effects)
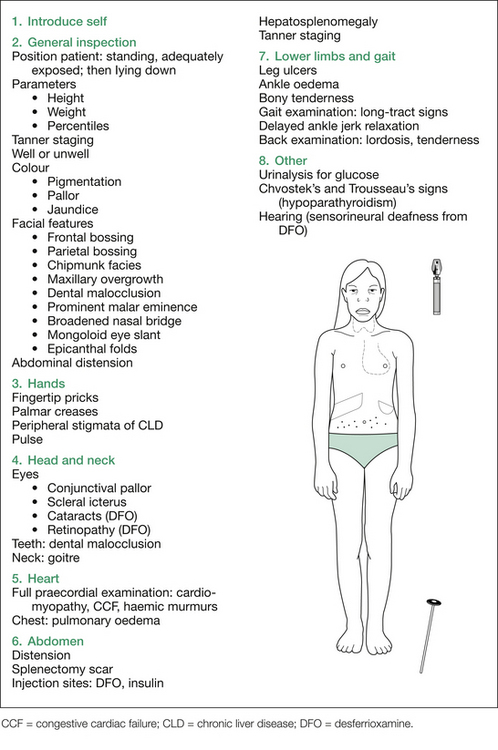
Initial inspection should include assessment of growth parameters (usually short, head circumference often increased [due to skull bossing], pubertal status delayed), colour (pigmentation from melanin and iron deposition, pallor from anaemia, jaundice from haemolysis), whether the child has any respiratory distress (e.g. anaemia or cardiomyopathy causing congestive cardiac failure [CCF], or massive splenomegaly causing marked increase in intra-abdominal pressure), and a description of any obvious bony abnormalities associated with the ‘chipmunk facies’ (see Figure 10.2). Note any obvious stigmata of CLD.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 10 Haematology
Long Cases
Haemophilia
The World Health Organization (WHO), the World Federation of Hemophilia (WFH) and various national haemophilia foundations (in Australia, the USA, Canada and many European countries) uniformly recommend that prophylaxis with an intravenous factor replacement for at least 46 weeks per year through adulthood is the standard of care. In children, the first prospective randomised controlled trial in the USA assessing the progression of arthropathy in children (under 30 months) treated (until 6 years old) with prophylaxis (25 IU/kg every other day) versus on-demand treatment (40 IU/kg initially, then 20 IU/kg at 24 and 72 hours post joint bleed) showed an 83% reduction in risk for joint damage on MRI. The evolution of a network of specialised haemophilia treatment centres in various developed countries has decreased the morbidity and mortality of haemophilia. There is no international consensus as yet regarding the optimal age to commence prophylaxis, but several studies have shown that children with no or few joint bleeds who start prophylaxis early (mean age 3 years) have a better musculoskeletal outcome. In developing countries, the high cost has precluded primary prophylaxis being adopted, and the average life expectancy for a child with severe haemophilia remains around 11 years, whereas in developed countries the life expectancy for someone with severe haemophilia is around 63 years.
Background information
Definitions
The F-VIII gene is at the telomeric end of the long arm of the X chromosome, at band Xq28. It is a large gene, 186 kilobases (kb) long, and it has 26 exons. Over 1200 mutations (missense, nonsense, splicing, and small or large deletions and insertions) have been described in the F-VIII gene. Mutations occur throughout the gene, with some concentration around exon 14. The most prevalent gene defect seen in severe haemophila A is an intron 22 inversion (int22), which accounts for 40–45% of all mutations. An inversion affecting exon 1 is present in around 5% of patients with severe haemophilia A. Around 200 smaller deletions have been described, which generally involve reading frame shifts, and non-functional gene products. Large deletions comprise 15% of haemophilia A; these result in truncated transcripts that are non-functional. Children with larger deletions and nonsense mutations are at higher risk of developing F-VIII inhibitory antibodies, and are less likely to respond to immune tolerance therapy. Most of F-VIII is synthesised in the liver endothelial cells and is immediately linked to von Willebrand’s factor (vWF) on entering the circulation; this prevents enzymatic degradation of factor F-VIII until it is required for coagulation.
Disease manifestations
• Severe haemophilia corresponds to <1% F-VIII or F-IX clotting activity: this accounts for approximately 70% of type A and 50% of type B cases. These children are predisposed to having spontaneous bleeding into joints, muscles and deep organs, including central nervous system bleeds. Without preventative treatment, these children have two to five spontaneous bleeding episodes each month. The usual age of diagnosis is within the first year of life.
• Moderately severe haemophilia means 1–5% F-VIII or F-IX clotting activity: these children rarely have spontaneous haemorrhages, but may have significant haemorrhage with mild or moderate trauma. The usual age at diagnosis is before 5 years.
• Mild disease, infers > 5% (6–35%) of F-VIII or F-IX clotting activity: these children may have bleeding with trauma or surgery. Some carrier females, who have low levels of these factors, may present clinically with gynaecological or obstetric haemorrhage.
• Around 10% of carriers have F-VIII or F-IX clotting activity lower than 35%.
• Individuals with more than 30% F-VIII or F-IX clotting activity usually do not spontaneously bleed and may not need supplemental clotting factor in the setting of minor surgery.
History
Past history
1. Initial presenting symptoms, diagnosis (when, where, how), subsequent management, progress of disease, hospitalisation details.
2. Complications of the disease (e.g. neurological deficits, joint disease) or its treatment (e.g. inhibitor formation).
3. Previous elective surgery or dental procedures (their management and outcome).
4. Outpatient clinics attended (where, how often).
5. Past treatments used (e.g. F-VIII, desmopressin [DDAVP], prothrombin complex concentrate). The majority of young children should have only received recombinant clotting factor concentrate.
6. Age when parents started administering F-VIII.
7. Age of self-administration.
8. Age of venous access port placement.
Current status
1. Average number of bleeds per year, common sites involved (e.g. knee, elbow), ‘target joint’ (most patients with significant joint disease will typically develop one joint that is more affected by recurrent bleeds), any common precipitants (e.g. sport), treatment required (type of concentrate), usual outcome, where usually managed (home or hospital), who gives infusions (patient or parent), prophylaxis regimen, details of venous access port use.
2. Ongoing symptoms of joint disease (e.g. pain, stiffness), neurological disease (e.g. weakness from peripheral nerve compression, hemiplegia from intracranial bleed).
3. Management of bleeds away from home (school, on holidays, overseas).
Social history
1. Disease impact on patient (e.g. avoidance of participation in sports such as rugby and football), self-image, schooling (attendance, performance, teacher awareness of, and attitudes towards, the disease and its treatment, peer interactions). Most patients with haemophilia are encouraged to participate in sports; there is some evidence that participation in sports reduces the incidences of bleeding episodes in boys on prophylaxis.
2. Disease impact on parents (e.g. marriage stability, fears for future, financial considerations [medical treatment, awareness of benefits available], modification of holiday plans).
3. Disease impact on siblings (e.g. sibling rivalry, hostility, genetic implications for girls).
4. Social supports (e.g. social worker, extended family). Patients with haemophilia are eligible for the carers payment.
5. Coping; for example, who attends with the patient, confidence with management, degree of understanding of the disease, expectations for the future, understanding of the prognosis.
6. Access to hospital, local doctor, paediatrician, haematologist, orthopaedic surgeon, rheumatologist.
Examination
The salient findings to be sought in the haemophilia long case are as follows.
General inspection
1. Position patient standing, undressed to underpants.
2. Parameters: weight, height, head circumference (subdural bleed).
3. Visually scan skin for bruises (number, size, age), joints for swelling and posture for evidence of neurological sequelae (e.g. hemiparesis [intracranial bleed] or foot drop [lateral popliteal palsy]).
4. Unwell (e.g. severe bleed) or well.
5. Pallor (anaemia from large bleed, e.g. retroperitoneal).
7. Vital signs: respiratory rate (e.g. pulse [anaemia], blood pressure [hypotension from bleeding], urinalysis [blood]).
Directed examination for disease extent and complications
1. Full skin examination, for distribution of bruises, including mucous membranes (mouth, tongue).
2. Full joint examination for evidence of arthropathy, focusing on the range of movement of affected joints, associated muscle wasting, any supportive devices (wheelchairs, splints, orthotic devices), gait.
3. Full neurological examination for any evidence of intracerebral or intravertebral haemorrhage, or peripheral nerve lesions. Best commenced with gait examination, followed by examination of the motor system.
4. Abdominal examination for liver and spleen size (liver disease), or tenderness (gastrointestinal or retroperitoneal bleed).
Available treatment modalities
Desmopressin (1-deamino 8-D arginine vasopressin: DDAVP)
This is a synthetic analogue of vasopressin that raises the F-VIII level by up to five times in normal subjects and seven times in some mild haemophiliacs, but has no effect in patients with severe disease. The mechanism is not fully understood. It can be given intravenously (0.3 mcg/kg in 50 mL saline over 20 minutes, peaks at 30–60 minutes) or by intranasal spray (150–300 mcg, peaks at 60–90 minutes). Tachyphylaxis may occur after several doses. Complications include facial flushing, hyponatraemia (particularly in infants—contraindicated in children under 2 years) and thrombosis (rare). Fluid restriction and urine output monitoring are important considerations. It is best for use in controlled situations such as elective minor surgery.
Management
Treatment of acute haemorrhage
1. Control of specific bleeding problems
F-VIII replacement guide
1. Minimal bleeds: first aid, antifibrinolytics if indicated. The need for factor VIII replacement should be assessed recognising the occasional difficulties with intravenous cannulation, particularly in young children.
2. Moderate bleed (e.g. joint, muscle, small oral mucosal or tongue laceration or bleed, epistaxis, gastrointestinal or genitourinary): 20–40 units/kg, given 12–24-hourly, usually for 3 days. Frequent repeated doses may be required if bleeding does not settle.
3. Severe bleed (life-threatening haemorrhage, e.g. major trauma, retropharyngeal, retroperitoneal, intracranial, large oral mucosal bleeds): 50–75 units/kg initially, followed by repeat doses of 25–40 units/kg every 12 hours until bleeding has ceased, or continuous infusion (especially intracranial bleeds, trauma).
4. Generally, haemarthroses and soft tissue bleeds require 1–3 infusions, whereas serious bleeds, such as areas with peripheral nerves at risk (e.g. psoas), retropharyngeal or retroperitoneal bleeding, may require prolonged treatment, including continuous infusions of factor VIII.
Chronic problems
Specific discussion areas
Prophylaxis
Primary prophylaxis
Primary prophylaxis is the ongoing regular infusion of F-VIII from early childhood, before significant joint bleeding is established, to prevent most bleeding episodes. This is usually started after the first significant joint bleed (often at around 12 months of age) and is typically given through a venous access port (see below). In neonates who have intracranial bleeding, prophylaxis is best started as soon as possible (i.e. without waiting until after a first major joint bleed), implanting a venous access port during the hospital admission for the presenting intracranial bleed (in one of the author’s patients, this included a [successful] burr hole at age 4 days for a subdural haematoma with midline shift, a ‘blown’ pupil and incipient coning). Recombinant F-VIII is given three times a week, at a dose of 25–40 units/kg. Prophylaxis has led to a marked decrease in the number of bleeds suffered by these children. The age at which prophylaxis is started is an independent predictor for the development of arthropathy; the earlier it is started, the less joint disease will occur, irrespective of the variables of dose and infusion intervals used at the start of treatment before the age of 3 years. The increased cost of prophylaxis may be offset by the decrease in later interventions such as synovectomy and the avoidance of significant arthritis in the adult years.
Elective surgery and continuous infusion of replacement factors
Before any elective surgical procedures, all patients with haemophilia should be assessed for inhibitors (see below) and to establish the optimum intervals of factor infusion. This comprises evaluation of increase in F-VIII or F-IX level per unit of F-VIII or F-IX per kilogram and the biological half-life of the factor in that child. It must be ensured that there are sufficient quantities of F-VIII or F-IX available on the day of surgery and for the week after. In general, for all surgical procedures, a dose to bring the patient’s level to 100% can be given at induction for the procedure.
Inhibitors and immune tolerance therapy (ITT)
Immune tolerance therapy (ITT), also called tolerisation, refers to eradicating inhibitors by manipulating the immune system through recurrent exposure to regular infusions of F-VIII or F-IX. There is controversy over the ideal dosing, interval and product choice. Many patients can be tolerised on a regimen of plasma-derived F-VIII, in a dose of about 40 units/kg three times a week. An international study is under way to compare high-dose 200 units/kg/day infusions of F-VIII with 50 units/kg, three times a week. The advantages of ITT include better control of bleeds and reduced use of expensive products. The disadvantages include increased use of F-VIII in some cases, and a success rate of up to 80%. Inhibitors to F-IX are less common; around a third of patients achieve successful immune tolerance. In response to exposure to F-IX replacement, severe allergic reactions have been described, including anaphylaxis; also, nephrotic syndrome can develop. Hence ITT must be considered very carefully in these patients. Rituximab, a monoclonal antibody directed against CD-20 positive cells, is being evaluated for those who fail to respond to ITT. Occasionally, immune modulation with steroids, or cyclophosphamide (to inhibit antibodies), IV immunoglobulin, plasmapheresis or protein A adsorption (to remove antibodies) is needed.
Sickle cell disease (SCD)
The last decade has seen several advances in the management of sickle cell anaemia (SCA), which occurs in around 1 in 500 African Americans, and in 1 in 1000–1400 Hispanic Americans. It has become clear that blood transfusion therapy has widened clinical applications, that hydroxyurea treatment effectively decreases painful crises and the requirement for transfusions, and that a cure can be obtained through haematopoietic stem cell transplantation (HSCT), for which there are now clear indications. The two main pathophysiological processes are haemolytic anaemia and vaso-occlusion. These are secondary to deoxygenation of the haemoglobin S (HbS) molecule, which aggregates into a polymer, which then causes a distortion of the red blood cell to a ‘sickle’ shape. Sickle cells block the microvasculature; the consequent deleterious effects of SCA can involve most organ systems. The rate of sickling is related to the concentration of deoxy-HbS: it takes only seconds—and if the cell is rammed through the capillaries, it becomes reoxygenated and the polymers of HbS depolymerise. The cell shape lags behind, and repeated hypoxic stress will alter the cytoskeleton of the cell and cause an irreversibly sickled cell. Organ damage in SCA can develop throughout childhood, starting with splenic and renal changes in infancy, and continuing through to pulmonary and neurological involvement with vasculopathy in older children and adolescents.
Background information
Definitions
The term sickle cell anaemia (SCA) refers only to homozygosity for the sickle cell gene. The term sickle cell disease (SCD) is a more general one that can also include compound heterozygous states such as sickle cell/haemoglobin C disease (HbSC), sickle cell/β-thalassaemia (HbS β-thalassaemia) and other sickling disorders.
There are two groups of sickle cell syndromes:
1. Sickle states, which are relatively benign (e.g. sickle cell trait, HbAS).
2. Sickle cell diseases (SCDs), which present a variety of problems. These include: sickle cell anaemia (SCA), HbSS; sickle β0-thalassaemia (β-thalassaemia without production of any β globin); sickle haemoglobin C disease (HbSC); and sickle β+-thalassaemia (β-thalassaemia with decreased [not absent] production of β-globin). Patients with sickle β0-thalassaemia and sickle β+-thalassaemia have clinical features that are more like SCD than thalassaemia, because the sickle β-globin predominates as a result of inadequate production of normal β-globin.
Effects of α-thalassaemia
HbSS patients with α-thalassaemia have a larger number of painful events (in extremities/back/abdomen/head) but fewer acute anaemic events (this does not include splenic sequestration).
Major complications
SICKLE CELL is a mnemonic for vaso-occlusive complications:
S. Sequestration (spleen and liver)
C. Cerebrovascular accidents (CVAs)
C. Crises (painful, infarctive)
• Limb effects (bone infarcts, marrow necrosis, osteomyelitis and aseptic necrosis)
The most dangerous complications are splenic sequestration, sepsis and CVAs.
Splenic sequestration crisis
1. This is the most severe crisis in those under 5 years of age. It occurs in 10–30% of children with HbSS, most commonly between 6 months and 3 years.
2. It results from acute entrapment of a large volume of blood in the spleen—often a large fraction of the circulating blood volume.
3. These children get rapid splenic enlargement (at least 2 cm increase in spleen size from baseline) with an acute fall in haemoglobin of greater than 2 g/dL, and with a raised reticulocyte count. They present with sudden collapse, shock, profound anaemia and abdominal fullness due to the massive splenomegaly. This often occurs during an acute infection.
4. It is life-threatening: it can be fatal within 30 minutes. Shock is treated with plasma expanders and whole blood.
5. If the child is older (e.g. over 2 years), consider splenectomy in susceptible patients. If under 2 years of age, a chronic transfusion program may be needed.
6. There is a tendency for recurrence: up to 50% of children have a second episode, usually within 2 years. Elective splenectomy is generally recommended in patients presenting with the first episode of splenic sequestration.
Infection: overwhelming sepsis
1. This particularly affects children under 3 years of age, as a result of poor development of immune response to polysaccharide antigens, complicated by early loss of splenic function (this ‘autosplenectomy’ occurs in 60% by 2 years, and 90% by 5 years).
2. Pathogens are most commonly Streptococcus pneumoniae (pneumococcus)—various serotypes—and occasionally Haemophilus influenzae type B. Other pathogens include meningococcus, and other Streptococci, Salmonella and fastidious gram-negative organisms, such as DF-2 (Capnocytophaga canimorsus) after a dog bite.
[/not-level-membership-for-pediatrics-category]
