[level-membership-for-pediatrics-category]
Chapter 608 Guillain-Barré Syndrome
Clinical Manifestations
Weakness usually begins in the lower extremities and progressively involves the trunk, the upper limbs, and finally the bulbar muscles, a pattern known as Landry ascending paralysis. Proximal and distal muscles are involved relatively symmetrically, but asymmetry is found in 9% of patients. The onset is gradual and progresses over days or weeks. Particularly in cases with an abrupt onset, tenderness on palpation and pain in muscles is common in the initial stages. Affected children are irritable. Weakness can progress to inability or refusal to walk and later to flaccid tetraplegia. Paresthesias occur in some cases. The differential diagnosis of acute weakness is noted in Table 599-3 and of Guillain Barré syndrome in Table 608-1.
Table 608-1 DIFFERENTIAL DIAGNOSIS OF CHILDHOOD GUILLAIN-BARRÉ SYNDROME
SPINAL CORD LESIONS
PERIPHERAL NEUROPATHIES
NEUROMUSCULAR JUNCTION DISORDERS
From Agrawal S, Peake D, Whitehouse WP: Management of children with Guillain Barré syndrome, Arch Dis Child Edu Pract Ed 92:161–168, 2007.
Table 608-2 CLASSIFICATION OF GUILLAIN-BARRÉ SYNDROME AND RELATED DISORDERS AND TYPICAL ANTIGANGLIOSIDE ANTIBODIES BY PATHOLOGY
| DISORDER | ANTIBODIES |
|---|---|
| Acute inflammatory demyelinating polyradiculoneuropathy | Unknown |
| Acute motor and sensory axonal neuropathy | GM1, GM1b, GD1a |
| Acute motor axonal neuropathy | GM1, GM1b, GD1a, GalNac-GD1a |
| Acute sensory neuronopathy | GD1b |
| ACUTE PANDYSAUTONOMIA | |
| Regional Variants | |
| Fisher syndrome | GQ1b, GT1a |
| Oropharyngeal | GT1a |
| Overlap | |
| Fisher/Guillain-Barré overlap syndrome | GQ1b, GM1, GM1b, GD1a, GalNac-GD1a |
From Hughes RAC: Treatment of Guillain-Barré syndrome with corticosteroids: lack of benefit? Lancet 363:181–182, 2004.
Laboratory Findings and Diagnosis
CSF studies are essential for diagnosis. The CSF protein is elevated to more than twice the upper limit of normal, glucose level is normal, and there is no pleocytosis. Fewer than 10 white blood cells/mm3 are found. The results of bacterial cultures are negative, and viral cultures rarely isolate specific viruses. The dissociation between high CSF protein and a lack of cellular response in a patient with an acute or subacute polyneuropathy is diagnostic of Guillain-Barré syndrome. MRI of the spinal cord may be indicated to rule out disorders in Table 608-1. MRI findings include thickening of the cauda equina and intrathecal nerve roots with gadolinium enhancement. These finds are fairly sensitive and are present in >90% of patients (Fig. 608-1). Imaging in CIDP is similar but demonstrates greater enhancement of spinal nerve roots (Fig. 608-2).
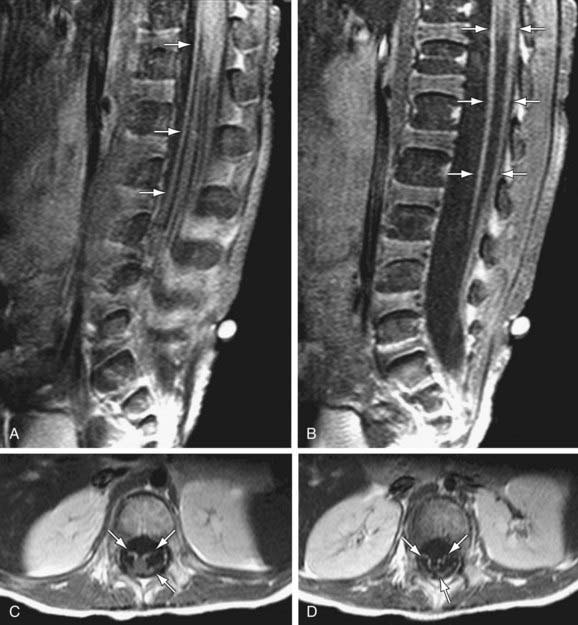
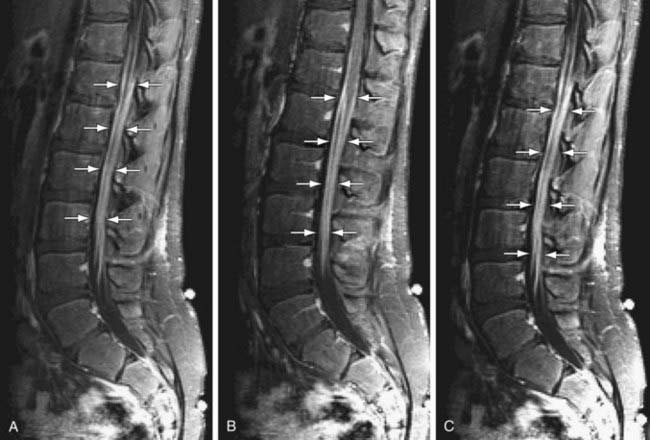
Motor NCVs are greatly reduced, and sensory nerve conduction time is often slow. Electromyography (EMG) shows evidence of acute denervation of muscle. Serum creatine kinase (CK) level may be mildly elevated or normal. Antiganglioside antibodies, mainly against GM1 and GD1, are sometimes elevated in the serum in Guillain-Barré syndrome, particularly in cases with primarily axonal rather than demyelinating neuropathy, and suggest that they might play a role in disease propagation and/or recovery in some cases (Table 608-1). Muscle biopsy is not usually required for diagnosis; specimens appear normal in early stages and show evidence of denervation atrophy in chronic stages. Sural nerve biopsy tissue shows segmental demyelination, focal inflammation, and wallerian degeneration but also is usually not required for diagnosis.
Agrawal S, Peake D, Whitehouse WP. Management of children with Guillain Barré syndrome. Arch Dis Child Edu Pract Ed. 2007;92:161-168.
Bradshaw DY, Jones HR. Pseudomeningoencephalitic presentation of pediatric Guillain-Barré syndrome. J Child Neurol. 2001;16:505-508.
Centers for Disease Control and Prevention. Cluster of tick paralysis cases—Colorado, 2006. MMWR Morb Mortal Wkly Rep. 2006;55:933-936.
DeWals P, Deceuninck G, Boucher RM, Ouakki M. Risk of Guillain-Barré syndrome following serogroup C meningococcal conjugate vaccine in Québec, Canada. Clin Infect Dis. 2008;46:e75-e77.
Dornonville de la Coeur C. Jakobsen J: Residual neuropathy in long-term population-based follow-up of Guillain-Barré syndrome. Neurology. 2005;64:246-253.
Hiraga A, Kuwabara S. Early prediction of prognosis in Guillain Barré syndrome. Lancet Neurol. 2007;6:572-573.
Hiraga A, Mori M, Ogawara K, et al. Recovery patterns and long-term prognosis for axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2005;76:719-722.
Jackson AH, Barquis GD, Shah BL. Congenital Guillain-Barré syndrome. J Child Neurol. 1996;11:407-410.
Koller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. 2005;352:1343-1356.
Korinthenberg R, Schessl J, Kirschner J, et al. Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barré syndrome: A randomized trial. Pediatrics. 2005;116:8-14.
Kountouras J, Deretzi G, Zavos C, et al. Association between Helicobacter pylori infection and acute inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2005;12:139-143.
Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66:597-603.
Landaverde JM, Danovaro-Holliday MC, Trumbo SP, et al. Guillain-Barré syndrome in children aged <15 years in Latin America and the Caribbean: baseline rates in the context of the influenza A (H1N1) pandemic. J Infect Dis. 2010;201:746-750.
Latov N, Deng C, Dalakas MC, et al. Timing and course of clinical response to intravenous immunoglobulin in chronic inflammatory demyelinating polyradiculoneuropathy. Arch Neurol. 2010;67(7):802-807.
Lee DH, Linker RA, Paulus W, et al. Subcutaneous immunoglobulin infusion: a new therapeutic option in chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2008;37:406-409.
Notturno F, Pace M, De Angelis MV, et al. Susceptibility to chronic inflammatory demyelinating polyradiculoneuropathy is associated with polymorphic GA repeat in the SH2D2A gene. J Neuroimmunol. 2008;197:124-177.
Pandey CK, Raza M, Tripathhi M, et al. The comparative evaluation of gabapentin and carbamazepine for pain management in Guillain-Barré syndrome patients in the intensive care unit. Anesth Analg. 2005;101:220-225.
Schessl J, Koga M, Funakoshi K, et al. Prospective study on anti-ganglioside antibodies in childhood Guillain-Barré syndrome. Arch Dis Child. 2007;92:48-52.
Van Koningsveld R, Schmitz PIM, van der Meche FGA, et al. Effect of methylprednisone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: randomized trial. Lancet. 2004;363:192.
Van Koningsveld R, Steyerberg E, Hughes RAC, et al. A clinical prognostic scoring system for Guillain Barré syndrome. Lancet Neurol. 2007;6:589-594.
Venance SL, Cannon SC, Fialho D, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129:8-17.
Walgaard C, Lingsma HF, Ruts L, et al. Prediction of respiratory insufficiency in Guillain-Barré syndrome. Ann Neurol. 2010;67:781-787.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 608 Guillain-Barré Syndrome
Clinical Manifestations
Weakness usually begins in the lower extremities and progressively involves the trunk, the upper limbs, and finally the bulbar muscles, a pattern known as Landry ascending paralysis. Proximal and distal muscles are involved relatively symmetrically, but asymmetry is found in 9% of patients. The onset is gradual and progresses over days or weeks. Particularly in cases with an abrupt onset, tenderness on palpation and pain in muscles is common in the initial stages. Affected children are irritable. Weakness can progress to inability or refusal to walk and later to flaccid tetraplegia. Paresthesias occur in some cases. The differential diagnosis of acute weakness is noted in Table 599-3 and of Guillain Barré syndrome in Table 608-1.
Table 608-1 DIFFERENTIAL DIAGNOSIS OF CHILDHOOD GUILLAIN-BARRÉ SYNDROME
SPINAL CORD LESIONS
PERIPHERAL NEUROPATHIES
NEUROMUSCULAR JUNCTION DISORDERS
From Agrawal S, Peake D, Whitehouse WP: Management of children with Guillain Barré syndrome, Arch Dis Child Edu Pract Ed 92:161–168, 2007.
Congenital Guillain-Barré syndrome
[/not-level-membership-for-pediatrics-category]
