Growth and puberty
The infantile phase
Growth during infancy to around 18 months of age is also largely dependent on adequate nutrition. Good health and normal thyroid function are also necessary. This phase is characterised by a rapid but decelerating growth rate, and accounts for about 15% of eventual height. By the end of this phase, children have changed from their fetal length, largely determined by the uterine environment, to their genetically determined height. An inadequate rate of weight gain during this period is called ‘failure to thrive’ (see Ch. 12).
Measurement
• Weight – readily and accurately determined with electronic scales but must be performed on a naked infant or a child dressed only in underclothing as an entire month’s or year’s weight gain can be represented by a wet nappy or heavy jeans, respectively.
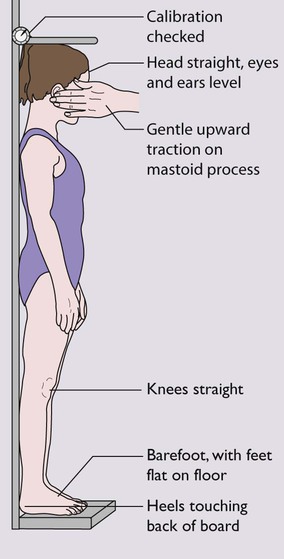
• Height – the equipment must be regularly calibrated and maintained. In children over 2 years of age, the standing height is measured as illustrated in Figure 11.2. In children under 2 years, length is measured lying horizontally (Fig. 11.3), using the mother to assist. Accurate length measurement in infants can be difficult to obtain, as the legs need to be held straight and infants often dislike being held still. For this reason, routine measurement of length in infancy is often omitted from child surveillance, but it should always be performed whenever there is doubt about an infant’s growth.
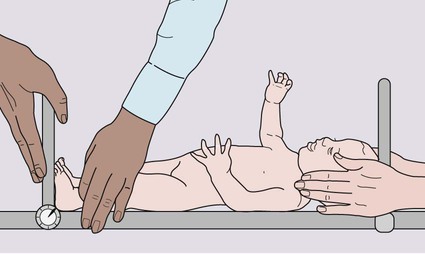
• Head circumference – the occipitofrontal circumference is a measure of head and hence brain growth. The maximum of three measurements is used. It is of particular importance in developmental delay or suspected hydrocephalus
These measurements should be plotted as a simple dot on an appropriate growth centile chart. Standards for a population should be constructed and updated every generation to allow for the trend towards earlier puberty and taller adult stature from improved childhood nutrition. In 2009, the UK adopted the World Health Organization (WHO) new global Child Growth Standards for infants and children 0–4 years old (See Appendix Fig. A1). The new charts are based on the optimal growth of healthy children totally breast-fed up to the age of 6 months. These charts allow for the lower weight of totally breast-fed infants and are therefore less likely to identify some breast-fed babies as underweight and may also allow early identification of bottle-fed babies gaining weight too rapidly.
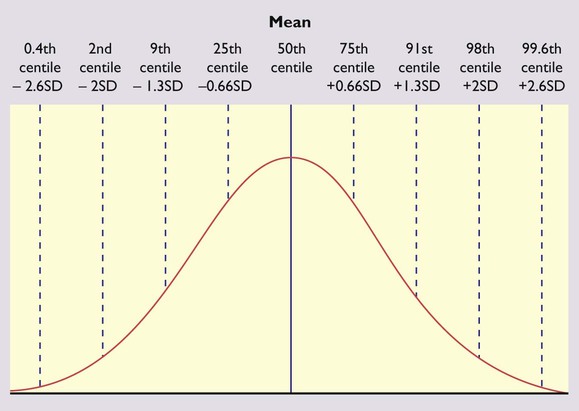
Height in a population is normally distributed and the deviation from the mean can be measured as a centile or standard deviation (Fig. 11.4). The bands on the growth reference charts have been chosen to be two-thirds of a standard deviation apart and correspond approximately to the 25th, 9th, 2nd and 0.4th centiles below the mean, and the 75th, 91st, 98th and 99.6th centiles above the mean. The further these centiles lie from the mean, the more likely it is that a child has a pathological cause for his short or tall stature. For instance, values below the 0.4th or above the 99.6th centile will occur by chance in only 4 per 1000 children and can be used as a criterion for referral from primary to specialist care. A single growth parameter should not be assessed in isolation from the other growth parameters: e.g. a child’s low weight may be in proportion to the height if short, but abnormal if tall. Serial measurements are used to show the pattern and determine the rate of growth. This is helpful in diagnosing or monitoring many paediatric conditions. The WHO charts include an adult height predictor and a BMI centile ready-reckoner.
Puberty
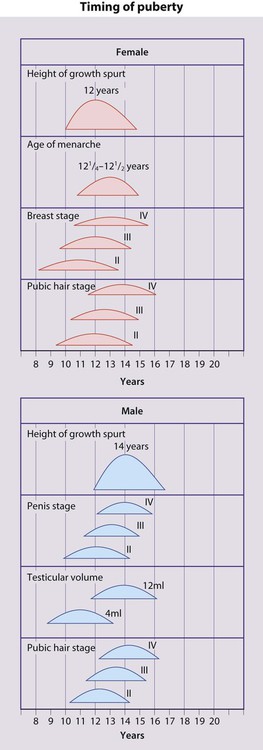
Puberty follows a well-defined sequence of changes that may be assigned stages, as shown in Figures 11.5 and 11.6. Over the last 20 years, the mean age at which puberty starts in girls has lowered. However, the age at which menarche occurs has remained stable. Therefore, females now remain in puberty for longer.
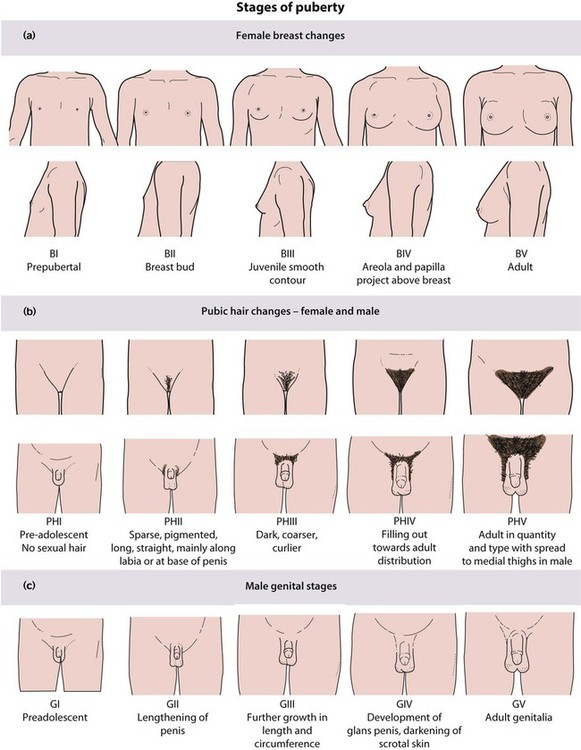
In females the features of puberty are:
• Breast development – the first sign, usually starting between 8.5 and 12.5 years
• Pubic hair growth and a rapid height spurt – occur almost immediately after breast development
• Menarche – occurs on average 2.5 years after the start of puberty and signals that growth is coming to an end, with only around 5 cm height gain remaining.
• Testicular enlargement to >4 ml volume measured using an orchidometer (Fig. 11.7) – the first sign of puberty

• Pubic hair growth – follows testicular enlargement, usually between 10 and 14 years of age
• Height spurt – when the testicular volume is 12–15 ml, after a delay of around 18 months.
In both sexes, there will be development of acne, axillary hair, body odour and mood changes.
If puberty is abnormally early or late, it can be further assessed:
Short stature
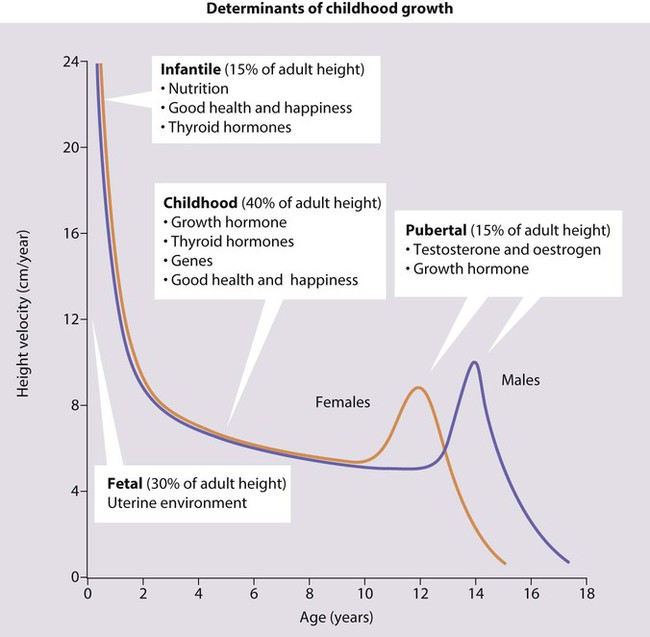
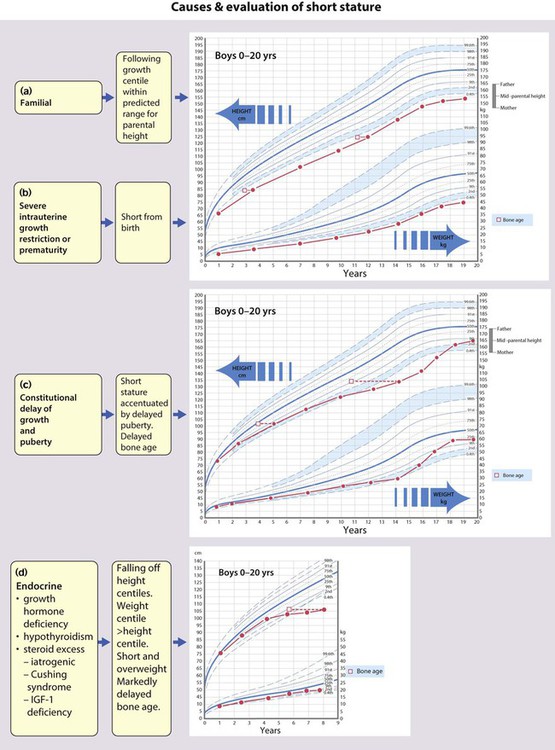
Short stature is usually defined as a height below the second centile (i.e. two standard deviations (SD) below the mean) or 0.4th centile (−2.6 SD). Only 1 in 50 children will be shorter than the 2nd centile and 1 in 250 (4 in 1000) shorter than the 0.4th centile. Most of these children will be normal, though short, with short parents, but the further the child is below these centiles, the more likely it is that there will be a pathological cause. However, the rate of growth (measured as height velocity; Fig. 11.1) may be abnormal long before a child’s height falls below these values. This growth failure can be identified from the child’s height falling across centile lines plotted on a height velocity chart (Fig. 11.1). This allows growth failure to be identified earlier, even though the child’s height is still above the 2nd centile.
Measuring height velocity is a sensitive indicator of growth failure. Two accurate measurements at least 6 months but preferably a year apart allow calculation of height velocity in cm/year (Fig. 11.1). This is plotted at the midpoint in time on a height/velocity chart. A height velocity persistently below the 25th centile is abnormal and that child will eventually become short. A disadvantage of using height velocity calculations is that they are highly dependent on the accuracy of the height measurements and so tend not to be used outside specialist growth units.
The height centile of a child must be compared with the weight centile and an estimate of their genetic target centile and range calculated from the height of their parents. This is calculated as the mean of the father’s and mother’s height with 7 cm added for the mid-parental target height of a boy, and 7 cm subtracted for a girl. The 9th–91st centile range of this estimate is given by ±10 cm in a boy and ±8.5 cm in a girl (see examples in Fig. 11.9).
Endocrine
Hypothyroidism
This is usually caused by autoimmune thyroiditis during childhood (see Ch. 25). This produces growth failure, usually with excess weight gain. It may go undiagnosed for many years and lead to short stature. When treated, catch-up growth rapidly occurs but often with a rapid entry into puberty that can limit final height. Congenital hypothyroidism is diagnosed soon after birth by screening and so does not result in any abnormality of growth.
Corticosteroid excess, Cushing syndrome
This is usually iatrogenic, as corticosteroid therapy is a potent growth suppressor. This effect is greatly reduced by alternate day therapy, but some growth suppression may be seen even with relatively low doses of inhaled or topical steroids in susceptible individuals. Non-iatrogenic Cushing syndrome is very unusual in childhood and may be caused by pituitary or adrenal pathology. Growth failure may be very severe, usually with excess weight gain, although normalisation of body shape and height occurs on withdrawal of treatment or treatment of the underlying steroid excess. Cushing syndrome during puberty can result in permanent loss of height (see Ch. 25).
Chromosomal disorder/syndromes
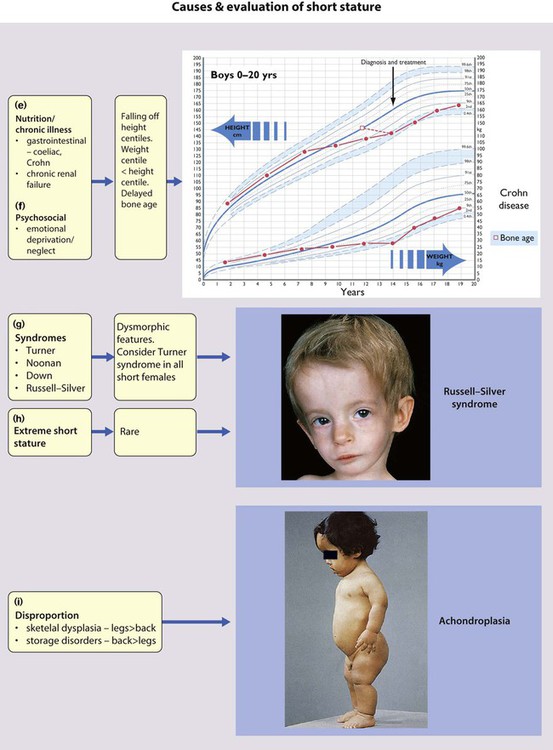
Many chromosomal disorders and syndromes are associated with short stature. Down syndrome is usually diagnosed at birth, but Turner (Fig. 8.6 and see Ch. 8), Noonan (Fig. 8.17) and Russell–Silver syndromes may present with short stature. Turner syndrome may be particularly difficult to diagnose clinically and should be considered in all short females.
Examination and investigation
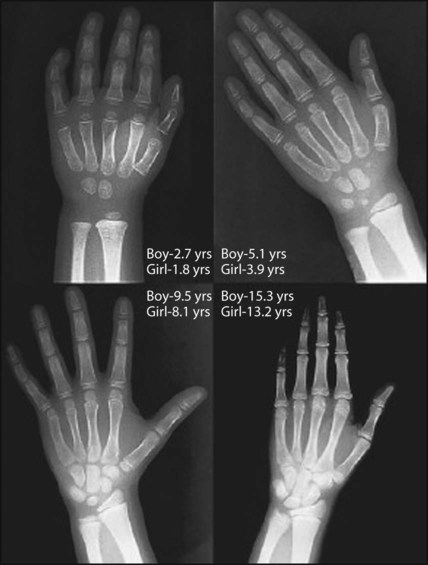
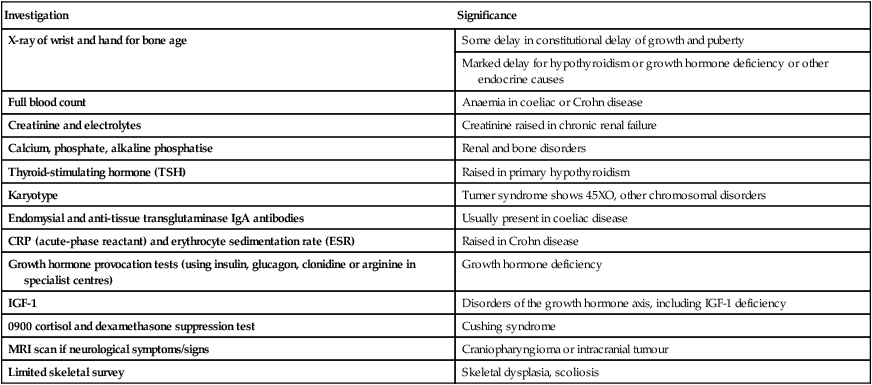
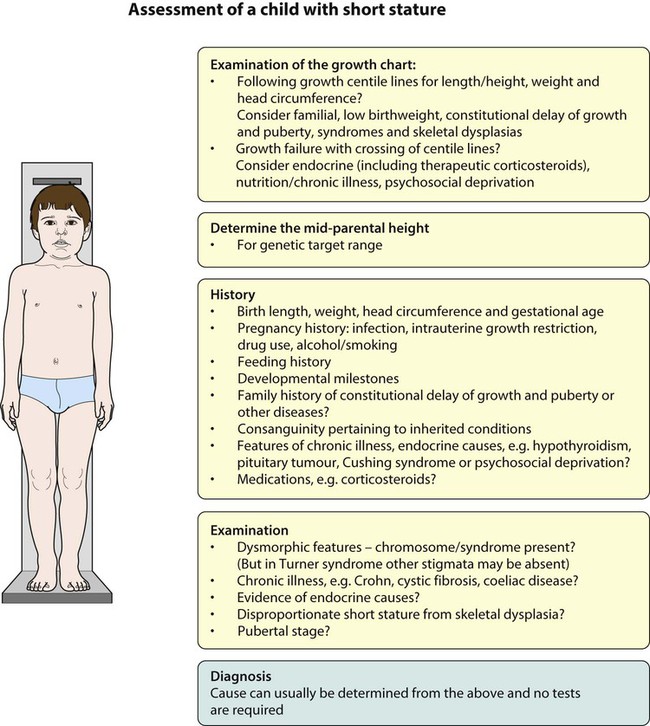
Plotting present and previous heights and weights on appropriate growth charts, together with the clinical features, usually allows the cause to be identified without any investigations (Fig. 11.9a-i). Previous height and weight measurements should be available from the parent-held personal child health record. The bone age may be helpful, as it is markedly delayed in some endocrine disorders, e.g. hypothyroidism and growth hormone deficiency, and is used to estimate adult height potential. Investigations that may be indicated are shown in Table 11.1.
Table 11.1
Investigations considered for short stature
| Investigation | Significance |
| X-ray of wrist and hand for bone age | Some delay in constitutional delay of growth and puberty |
| Marked delay for hypothyroidism or growth hormone deficiency or other endocrine causes | |
| Full blood count | Anaemia in coeliac or Crohn disease |
| Creatinine and electrolytes | Creatinine raised in chronic renal failure |
| Calcium, phosphate, alkaline phosphatise | Renal and bone disorders |
| Thyroid-stimulating hormone (TSH) | Raised in primary hypothyroidism |
| Karyotype | Turner syndrome shows 45XO, other chromosomal disorders |
| Endomysial and anti-tissue transglutaminase IgA antibodies | Usually present in coeliac disease |
| CRP (acute-phase reactant) and erythrocyte sedimentation rate (ESR) | Raised in Crohn disease |
| Growth hormone provocation tests (using insulin, glucagon, clonidine or arginine in specialist centres) | Growth hormone deficiency |
| IGF-1 | Disorders of the growth hormone axis, including IGF-1 deficiency |
| 0900 cortisol and dexamethasone suppression test | Cushing syndrome |
| MRI scan if neurological symptoms/signs | Craniopharyngioma or intracranial tumour |
| Limited skeletal survey | Skeletal dysplasia, scoliosis |
Growth hormone treatment of short stature
Growth hormone deficiency is treated with biosynthetic growth hormone, which is given by subcutaneous injection, usually daily. It is expensive and the management of growth hormone deficiency is undertaken at specialist centres. The best response is seen in children with the most severe hormone deficiency. Other indications include Turner syndrome, Prader–Willi syndrome (Fig. 8.19), chronic renal failure, SHOX (short stature homeobox) deficiency and intrauterine growth restriction (IUGR). In Prader–Willi syndrome (an imprinting disorder resulting in early hypotonia and feeding difficulties followed by short stature, obesity and learning difficulties), growth hormone improves muscular strength and body composition as well as modestly improving final height. Recently, recombinant IGF-1 has been used to treat children with growth hormone resistance (e.g. Laron syndrome) and IGF-1 deficiency who would have previously not responded to growth hormone treatment. Recombinant IGF-1 therapy is still very expensive and is confined to a few specialised centres.
Tall stature
This is a less common presenting complaint than short stature, as many parents are proud that their child is tall. However, some adolescents (mainly females) become concerned about excessive height during their pubertal growth spurt. The causes are shown in Table 11.2. Most tall stature is inherited from tall parents. Obesity in childhood ‘fuels’ early growth and may result in tall stature; however, because puberty is often somewhat earlier than average, it does not increase final height.
Table 11.2
Causes of excessive growth or tall stature
| Familial | Most common cause |
| Obesity | Puberty is advanced, so final height centile is less than in childhood |
| Secondary | Hyperthyroidism |
| Excess sex steroids – precocious puberty from whatever cause | |
| Excess adrenal androgen steroids – congenital adrenal hyperplasia | |
| True gigantism (excess GH secretion) | |
| Syndromes | Long-legged tall stature: |
| Marfan syndrome | |
| Homocystinuria | |
| Klinefelter syndrome (47 XXY and XXY karyotype) | |
| Proportionate tall stature at birth: | |
| Maternal diabetes | |
| Primary hyperinsulinism | |
| Beckwith syndrome | |
| Sotos syndrome – associated with large head, characteristic facial features and learning difficulties |
Abnormal head growth
Microcephaly
Microcephaly, a head circumference below the 2nd centile, may be:
• Familial – when it is present from birth and development is often normal
• An autosomal recessive condition – when it is associated with developmental delay
• Caused by a congenital infection
• Acquired after an insult to the developing brain, e.g. perinatal hypoxia, hypoglycaemia or meningitis, when it is often accompanied by cerebral palsy and seizures (see Case History 11.1).
Macrocephaly
Macrocephaly is a head circumference above the 98th centile. The causes of a large head are listed in Box 11.1. Most are normal children and often the parents have large heads. A rapidly increasing head circumference, even if the head circumference is still below the 98th centile, suggests raised intracranial pressure and may be due to hydrocephalus, subdural haematoma or brain tumour. It must be investigated promptly by intracranial ultrasound if the anterior fontanelle is still open, otherwise by CT or MRI scan.
 If an infant’s head circumference is enlarging and crossing centile lines – check for raised intracranial pressure.
If an infant’s head circumference is enlarging and crossing centile lines – check for raised intracranial pressure.Asymmetric heads
Skull asymmetry may result from an imbalance of the growth rate at the coronal, sagittal or lambdoid sutures, although the head circumference increases normally. Occipital plagiocephaly, a parallelogram-shaped head with flattening of the back of the skull, is seen with increased frequency since the advice to parents that babies should sleep lying on their back to reduce the risk of sudden infant death syndrome. It improves with time as the infant becomes more mobile. Plagiocephaly is also seen in infants with hypotonia. Preterm infants may develop long, flat heads from lying on their sides for long periods on the hard surface of incubators unless provided with a soft surface to lie on and their head position is changed frequently (see Fig. 11.11). Under these circumstances, it is not associated with abnormal development.
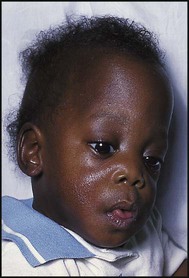
Case History
Figure 11.10 shows the head circumference chart of Tim, who was healthy and was developing normally. At 9 months of age, he was rushed to hospital as he was unrousable from profound hypoglycaemia secondary to the deliberate administration of insulin by his mother, who had diabetes. Although Tim was taken into care and had no further hypoglycaemic episodes, his head circumference shows cessation of growth. He has developed moderate learning difficulties and mild cerebral palsy.
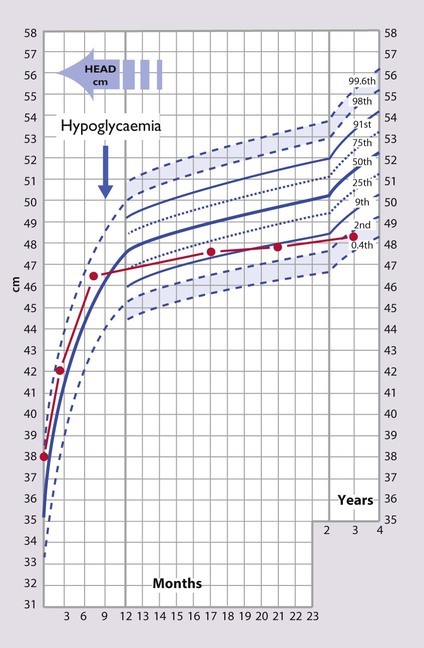
Craniosynostosis
The sutures of the skull bones start to fuse during infancy but do not finally fuse until late childhood. Premature fusion of one or more sutures (craniosynostosis) may lead to distortion of the head shape. Craniosynostosis is usually localised (Box 11.2). It most often affects the sagittal suture, when it results in a long narrow skull (Fig. 11.12). Rarely it affects the lambdoid suture to result in skull asymmetry, which needs to be differentiated from plagiocephaly, where there is asymmetric flattening of one side of the skull from positional moulding.
Craniosynostosis may be generalised (Box 11.2), when it may be a feature of a syndrome (Fig. 11.13). The fused suture may be felt or seen as a palpable ridge and confirmed on skull X-ray or cranial CT scan. If necessary, the condition can be treated surgically because of raised intracranial pressure or for cosmetic reasons. Such operations are performed in specialist centres for craniofacial reconstructive surgery.
Premature sexual development
• Precocious puberty when it is accompanied by a growth spurt
Precocious puberty
Precocious puberty (PP) may be categorised according to the levels of the pituitary-derived gonadotropins, follicle-stimulating hormone (FSH) and luteinising hormone (LH), (Fig. 11.14) as:
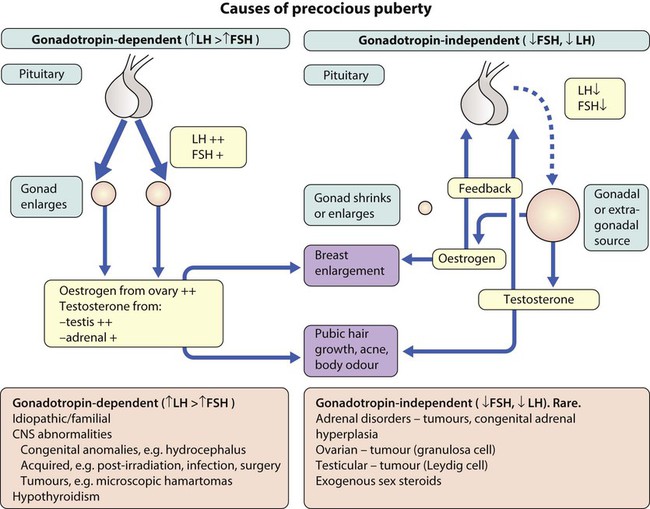
• Gonadotropin-dependent (central, ‘true’ PP) from premature activation of the hypothalamic–pituitary–gonadal axis
• Gonadotropin-independent (pseudo, ‘false’ PP) from excess sex steroids.
 Precocious puberty in females is commonly due to the premature onset of normal puberty.
Precocious puberty in females is commonly due to the premature onset of normal puberty. Central precocious puberty in males more often has an organic cause.
Central precocious puberty in males more often has an organic cause.Management
The management of precocious puberty is directed towards:
• detection and treatment of any underlying pathology, e.g. intracranial tumour in males, and reducing the rate of skeletal maturation if necessary. Skeletal maturation is assessed by bone age. An early growth spurt may result in early cessation of growth and a reduction in adult height.
• addressing psychological/behavioural difficulties associated with early progression through puberty.
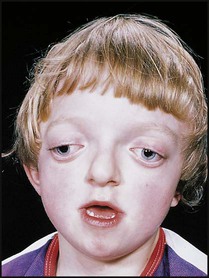
Case History
11.2 Precocious puberty in a boy
This 6-year-old boy presented with precocious puberty (Fig. 11.15a,b). He was noted to have multiple café-au-lait spots consistent with a diagnosis of neurofibromatosis type 1. An MRI scan showed a mass in the hypothalamus which proved to be an optic glioma. He was treated with radiotherapy, although full remission was not possible to achieve. The site of injection of gonadotropin super-agonist treatment to suppress his sexual development is covered by the plaster.
Premature breast development (thelarche)
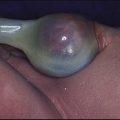
This usually affects females between 6 months and 2 years of age. The breast enlargement may be asymmetrical and rarely progresses beyond stage 3. It is differentiated from precocious puberty by the absence of axillary and pubic hair and of a growth spurt. It is non-progressive and self-limiting. Investigations are not usually required (see Case History 11.3).
Delayed puberty
Delayed puberty is often defined as the absence of pubertal development by 14 years of age in females and 15 years in males. The causes of delayed puberty are listed in Box 11.3.
• identify and treat any underlying pathology
• ensure normal psychological adaptation to puberty and adulthood
• accelerate growth and promote entry into puberty if necessary.
Case History
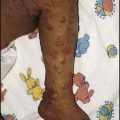
This 18-month-old female developed enlargement of both breasts (Fig. 11.16). There was no pubic hair growth, sweatiness or body odour and her height was in the mid-parental range. Her bone age was only mildly advanced (21 months) and a pelvic ultrasound showed a prepubertal uterus, small volume ovaries with two cysts in the left ovary. Her subsequent growth rate was normal. A diagnosis of premature thelarche was made.
Disorders of sexual differentiation (DSD)
The fetal gonad is initially bipotential (Fig. 11.17). In the male, a testis-determining gene on the Y chromosome (SRY) is responsible for the differentiation of the gonad into a testis. The production of testosterone and its metabolite, dihydrotestosterone, results in the development of male genitalia. In the absence of SRY, the gonads become ovaries and the genitalia female.
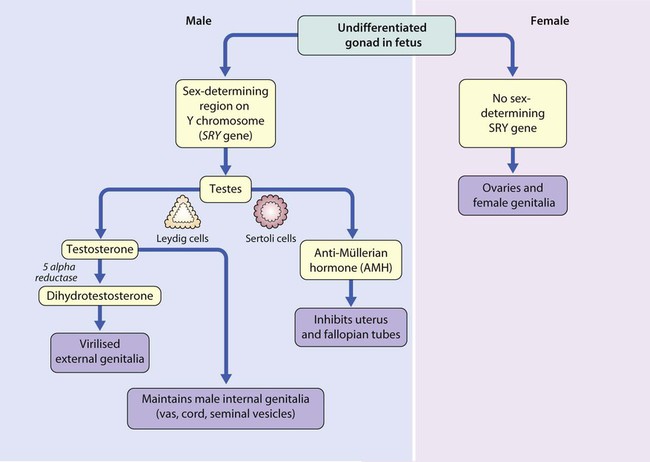
• Excessive androgens producing virilisation in a female – the commonest cause of this is congenital adrenal hyperplasia
• Inadequate androgen action, producing under-virilisation in a male – this can result from inability to respond to androgens (a receptor problem – androgen insensitivity syndrome, which may be complete or partial) or to convert testosterone to dihydrotestosterone (5α-reductase deficiency) or abnormalities of the synthesis of androgens from cholesterol
• Gonadotrophin insufficiency, also seen in several syndromes such as Prader–Willi syndrome and congenital hypopituitarism, which results in a small penis and cryptorchidism
• Ovotesticular disorder of sex development (DSD) (previously known as true hermaphroditism), caused by both XX- and Y-containing cells being present in the fetus leading to both testicular and ovarian tissue being present and a complex external phenotype; this is rare.
 If there is abnormal sexual differentiation at birth:
If there is abnormal sexual differentiation at birth:Congenital adrenal hyperplasia (CAH)
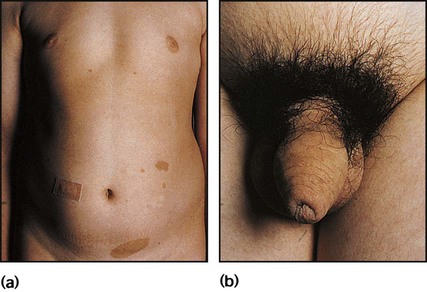
A number of autosomal recessive disorders of adrenal steroid biosynthesis result in congenital adrenal hyperplasia. Its incidence is about 1 in 5000 births, and it is commoner in the offspring of consanguineous marriages. Over 90% have a deficiency of the enzyme 21-hydroxylase, which is needed for cortisol biosynthesis. About 80% are also unable to produce aldosterone, leading to salt loss (low sodium and high potassium) (Fig. 11.18). In the fetus, the resulting cortisol deficiency stimulates the pituitary to produce adrenocorticotrophic hormone (ACTH), which drives overproduction of adrenal androgens.
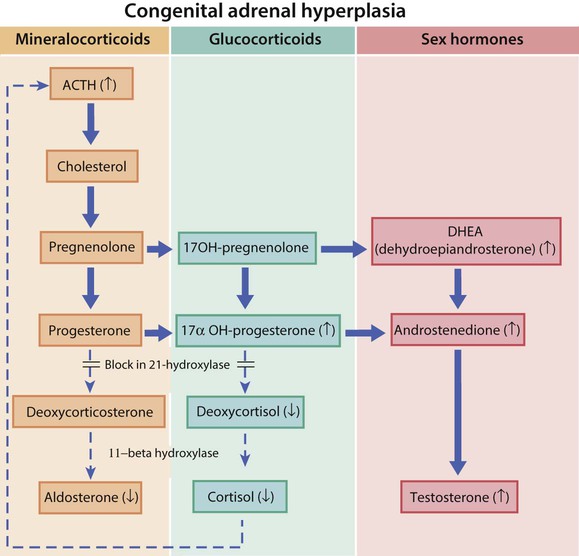
• Virilisation of the external genitalia in female infants, with clitoral hypertrophy and variable fusion of the labia (see Case History 11.4)
• In the infant male, the penis may be enlarged and the scrotum pigmented, but these changes are seldom identified
• A salt-losing adrenal crisis in the 80% of males who are salt losers; this occurs at 1–3 weeks of age, presenting with vomiting and weight loss, floppiness and circulatory collapse
• Tall stature in the 20% of male non-salt losers; both male and female non-salt losers also develop a muscular build, adult body odour, pubic hair and acne from excess androgen production, leading to precocious pubarche.
Management
The long-term management of both sexes is with:
• Lifelong glucocorticoids to suppress ACTH levels (and hence testosterone) to allow normal growth and maturation
• Mineralocorticoids (fludrocortisone) if there is salt loss; before weaning, infants may need added sodium chloride
• Monitoring of growth, skeletal maturity and plasma androgens and 17α-hydroxyprogesterone – insufficient hormone replacement results in increased ACTH secretion and androgen excess, which will cause rapid initial growth and skeletal maturation at the expense of final height; excessive hormonal replacement will result in skeletal delay and slow growth
• Additional hormone replacement to cover illness or surgery, as they are unable to mount a cortisol response.
Case History
11.4 Abnormal genitalia at birth
The appearance of this newborn infant’s genitalia is shown in Figure 11.19.
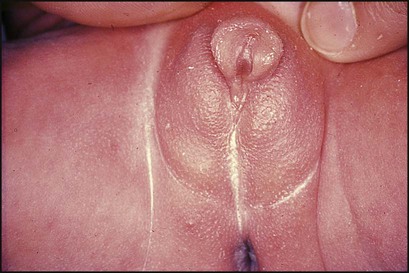
• A normal female karyotype, 46XX
• The presence of a uterus on ultrasound examination
• A markedly raised plasma 17α-hydroxy-progesterone concentration, confirming congenital adrenal hyperplasia.
• A low sodium and a high potassium level
• A low bicarbonate (metabolic acidosis) and high urea (dehydration).