Graves’ Disease
Graves’ disease is now universally classified among the autoimmune organ-specific diseases because it fulfills all the criteria required for this definition (Table 9-1). The main pathogenic mechanism is stimulation of growth and function of the thyroid gland by circulating antibodies directed against the thyroid-stimulating hormone (TSH) receptor (TSHR), thereby mimicking the effects of TSH. Thus TSHR is the major autoantigen of Graves’ disease.
Table 9-1
Criteria for Organ-Specific Autoimmune Diseases and Their Presence in Graves’ Disease
| Criteria | Present in Graves’ Disease |
| Lymphocytic infiltration of the target organ | Yes |
| Identification of the specific antigen(s) | Yes |
| Production of humoral and/or cellular autoimmune responses (or both) in animals sensitized by autologous antigen | Yes |
| Presence of organ-specific lesions in autosensitized animals | Yes |
| Association with other autoimmune diseases | Yes |
Historical Notes
Graves’ disease is the eponym by which a syndrome characterized by diffuse goiter and hyperthyroidism is recognized in English-speaking countries. Robert James Graves (Fig. 9-1) was a brilliant and productive Irish physician who contributed in many ways to the development of medical science of his time.1 Credit for his prominent position is probably due to his description in 1835 of “… three cases of violent and long palpitations in females, in each of which the same peculiarity presented … enlargement of the thyroid gland”, which was the first report of toxic diffuse goiter.2 However, Caleb Hillier Parry, a less renowned physician of Bath, England, had described a similar syndrome earlier, in 1825: “There is one malady which I have in five cases seen coincident with what appears to be enlargement of the heart, and which, so far as I know has not been noticed in that connection by medical writers. This malady to which I allude is enlargement of the thyroid gland”.3 He also described protrusion of the eyes as a feature of the syndrome. Even earlier than that, in 1805, the Italian Giuseppe Flajani in Rome had reported two cases of diffuse swelling of the neck accompanied by palpitations.4 He failed to recognize the thyroidal origin of the swelling and named it “bronchocele.” In 1840, in Germany, Carl A. von Basedow described “Exophthalmos durch Hypertrophie del Zellgewebes in der Augenhohle,” or exophthalmos caused by hypertrophy of the cellular tissue of the orbit.5 This was in fact the first description of the complete syndrome that included the triad exophthalmos, goiter, and palpitations. Von Basedow was struck by the prominence of the eye changes and made exophthalmos the hallmark of the disease. His descriptions were widely disseminated at the time, so that in most non-English-speaking European countries, the disease is still called Basedow’s disease. In 1880, Ludwig Rehn performed the first thyroidectomy for toxic diffuse goiter, and in 1909, Kocher was awarded the Nobel Prize for his innovations in thyroid surgery.6 In 1886, Moebius proposed that exophthalmic goiter was due to an excessive function of the thyroid gland.6 In 1911, Marine proposed treatment of Graves’ disease with iodine in the form of Lugol’s solution.7 In the early 1940s, the antithyroid drugs thioureas were described,8 and Astwood introduced them into clinical use for the control of thyrotoxicosis.9 At the same time, physicists and physicians in Boston and in Berkeley started to treat thyrotoxic patients with radioiodine (131I).10 In the span of just a few years, the two mainstays of modern treatment of Graves’ disease were initiated. The following decade was marked by the discovery in 1956 of the long-acting thyroid stimulator (LATS) by Adams and Purves11 and by the subsequent identification of this stimulator as an antibody, thereby forming the basis for our current understanding of the pathogenic mechanisms of Graves’ disease. Cloning of TSHR12,13 is only the most recent and will certainly not be the last memorable event in the uncovering of a disease that has paralleled the development of modern medicine across 2 centuries.

Epidemiology
Graves’ disease is a relatively prevalent disorder, and it is the most frequent cause of thyrotoxicosis in iodine-sufficient countries.14
Several studies have attempted estimating the exact frequency of Graves’ disease in the general population. However, comparison of surveys is difficult because of the use of different criteria in population sampling, because of ethnic differences, and because diagnostic tools have changed over the years. In the United States, a large survey performed in the 1970s estimated the prevalence of Graves’ disease to be 0.4%.15 A similar prevalence (0.6%) was found in the Pescopagano study in Italy.16 The Whickham survey in the United Kingdom suggested a prevalence of 1.1% to 1.6% (i.e., about threefold to fourfold higher) for thyrotoxicosis of all causes, of which Graves’ disease was presumably the most frequent.17,18 A recent study performed in Sweden has shown an incidence of Graves’ disease of approximately 25 cases/100,000 per year, which is relatively high compared with other studies, a possible interpretation of which is a high iodine intake of the selected population (see later discussion).19 Overall, a meta-analysis of various studies has estimated the general prevalence of the disorder to be about 1%,20 which makes it one of the most frequent clinically relevant autoimmune disorders.
The dietary iodine supply appears to be a major factor in determining the frequency of Graves’ disease.21–28 For example, iodine supplementation of previously iodine-deficient Tasmania induced a threefold increase in the incidence of hyperthyroidism in 3 years.21 Although the increase was mainly due to iodine-induced thyrotoxicosis in patients with autonomously functioning nodules or goiters, it was also shown that LATSs or LATS protectors were present in a number of cases of thyrotoxicosis in the early supplementation period, thus suggesting that some of the iodine-induced cases of hyperthyroidism might have been due to Graves’ disease. Since the Tasmanian report, outbursts of iodine-induced thyrotoxicosis have been reported in many countries after the implementation of iodine supplementation programs.22–28 Thyrotoxicosis occurred primarily in older people with preexisting nodular goiter. However, in a study performed in Switzerland, a slight and transient increase in the incidence of Graves’ disease was noted after stepwise, full iodine supplementation (Fig. 9-2).23 Similar increases in the incidence of thyrotoxicosis have been reported in Sweden (16.6/100,000),24 New Zealand (15/100,000),25 Britain (23/100,000),26 and Denmark27, in the latter population especially in younger age groups.28 Population-based studies also show differences in the incidence of Graves’ disease in populations with different but relatively constant iodine intake. Nevertheless, this corresponds to a higher frequency of nonautoimmune thyrotoxicosis in iodine-deficient areas. Thus, by comparing two genetically similar populations that differed in terms of iodine intake (iodine-sufficient Iceland versus iodine-deficient East Jutland in Denmark), it was found that the incidence of Graves’ hyperthyroidism was slightly higher in the iodine-sufficient (20/100,000 inhabitants/year) than in the iodine-deficient population (15/100,000/year), but that the incidence of thyrotoxicosis of all causes was greater in the iodine-deficient (39/100,000/year) than in the iodine-sufficient (23/100,000/year) population.14 Clearly, these findings show that fear of an increased incidence of Graves’ disease should not prevent the implementation of iodine supplementation programs.
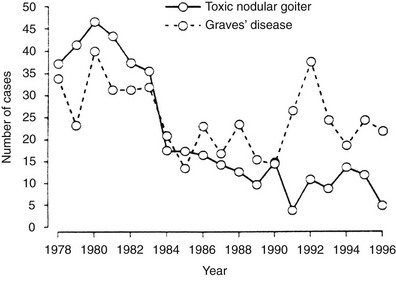
FIGURE 9-2 Increased incidence of hyperthyroidism from Graves’ disease and toxic nodular goiter in Switzerland after the introduction of iodine supplementation. (Modified from Burgi H, Kohler M, Morselli B: Thyrotoxicosis incidence in Switzerland and benefit of improved iodine supply. Lancet 352:1034, 1998.)
Although ethnic differences in susceptibility to Graves’ disease are likely to exist, they have not been consistently investigated in comparative studies. As with many other autoimmune disorders, Graves’ disease is about fivefold more prevalent in women than men. The reasons for this observation are understood only in part, but some hypotheses will be discussed later. The annual incidence is clearly and consistently related to age, with peaks in the fourth to sixth decades of life,28 although Graves’ disease can be observed in people of any age, including children.
Etiology
Most of the pathogenic mechanisms of Graves’ disease have been clarified since the first description of LATS.11 It is now well established that Graves’ disease is an organ-specific autoimmune disorder, with involvement of both T-cell and B-cell–mediated immunity against thyroid antigens. TSHR is the main antigen involved, and circulating autoantibodies against TSHR (TSH receptor antibodies [TRAbs]) that are capable of stimulating the receptor are responsible for the most distinctive features of the disease, namely hyperthyroidism and goiter. Nevertheless, in spite of advancements in understanding the pathogenic mechanisms of Graves’ disease, the ultimate cause of the disease remains elusive. The majority of investigators share the opinion that Graves’ disease is a multifactorial disease caused by a complex interplay of genetic, hormonal, and environmental influences that lead to the loss of immune tolerance to thyroid antigens and to the initiation of a sustained autoimmune reaction.
Genetics of Graves’ Disease
The strongest data in support of a genetic predisposition to Graves’ disease comes from twin studies.29,30 Dizygotic twins share on average 50% of their genome, whereas monozygotic twins share 100%. Moreover, twins are likely to share environmental factors more than any other kind of siblings. Several large twin studies have reported greater concordance rate of Graves’ disease in monozygotic than in dizygotic twins.29 Data obtained with modern diagnostic tools have shown a relatively low (~17% to 35%), but still significant, concordance in monozygotic twins.29,30 These findings clearly show a genetic influence, possibly characterized by low penetrance of the genes involved.
Another tool widely used to establish the existence of a genetic predisposition to any condition is family studies, in which the prevalence of the disease in relatives of index cases is compared with the prevalence in the general population. Early family studies showed a high prevalence of Graves’ disease and other thyroid abnormalities in first-degree relatives of patients with Graves’ disease and Hashimoto’s thyroiditis.31,32 The prevalence of circulating thyroid autoantibodies in siblings of patients was as high as 56% in some studies,31 which suggested a dominant mode of inheritance. These observations have been consistently confirmed in highly selected populations,33 but the results may not be applicable to the general population because of ascertainment bias. In an extensive segregation analysis with randomly ascertained probands, circulating antibodies were found in only 25% of the offspring of positive parents and in 14% of the offspring of negative parents, thus suggesting a multigenic model with less than 100% penetrance for the antibody trait.34 With the exception of very early studies, the prevalence of overt Graves’ disease has been found to be relatively low in siblings of patients.33,36 However, initial abnormalities in thyroid function compatible with subclinical hyperthyroidism or hypothyroidism have been reported.33,35 Villanueva et al. found that 36% of Graves’ patients with ophthalmopathy have a family history of either Graves’ disease or autoimmune thyroiditis, which in 23% of the cases affected first-degree relatives.37 Autoimmune thyroiditis is frequently observed in siblings of probands with Graves’ disease, as well as the contrary,33,37 suggesting that the two diseases may share some susceptibility genes that predispose to thyroid autoimmunity, but that the full expression of the phenotype depends on other genes and/or on environmental factors. Other organ-specific, non-thyroid-related autoimmune diseases may also be more prevalent in relatives of patients with Graves’ disease.33
These data and those obtained in twin studies are indicative of a complex multigenic pattern of inheritance of Graves’ disease. Some of the components of the phenotype, such as the presence of circulating antithyroglobulin and antithyroperoxidase antibodies, may be inherited in a dominant fashion with high penetrance.33 However, these genetic determinants do not appear to be sufficient for full expression of the disease. Clearly, other genes must be involved, which is in line with the complexity of the inheritance observed. Also, it appears from epidemiologic and experimental data that environmental factors (reviewed later in this chapter) play an important role by modulating the effect of an inherited predisposition. Based on the above evidence, a number of genes or loci have been investigated as candidates for predisposing factors (Table 9-2).33,38
Table 9-2
Genetic Determinants Associated With Graves’ Disease
| Gene | Possible Mechanism | Evidence in Favor of an Association |
| HLA-DR | Altered antigen presentation | Fair |
| CD40 | Altered antigen presentation | Fair |
| CTLA-4 | Altered antigen presentation | Good |
| PTPN22 | Altered T-cell activation | Fair |
| Thyroglobulin | Loss of tolerance | Good |
| TSH-R | Loss of tolerance | Poor |
Genes Predisposing to Graves’ Disease
In the last 20 years, the impressive advancement of biomedical research has allowed a remarkable expansion in genetics, which has led to the identification of several genes involved in the predisposition to Graves’ disease.33,38 Linkage, association, and candidate genes analyses, as well as whole-genome screening, have all been used to accomplish this goal. Based on the data available, genetic susceptibility to thyroid autoimmunity in general and to Graves’ disease in particular seems to be determined by a combination of a number of genes, some which most likely still remain to be identified.33,38,39
1:: The HLA Complex The HLA complex, which is located on the short arm of human chromosome 6, contains the sequence encoding about a hundred genes, most of which are involved in regulation of the immune response.40–42 The HLA genes are classically grouped into three major classes. Class I includes histocompatibility genes expressed on the surface of most cells (HLA-A, HLA-B, and HLA-C). Class II includes histocompatibility genes expressed exclusively on the surface of leukocytes and immune cells (HLA-DR). Class III includes a heterogeneous group of genes encoding molecules involved in the immune response, such as some complement factors, cytokines, and lymphocyte surface molecules. Other genes in this class are not clearly related with immunity. Most genes of the HLA complex are highly polymorphic, which makes them excellent candidates for disease susceptibility.
Experimental thyroiditis in the mouse was the first autoimmune disease to be associated with HLA.43,44 Early population studies in humans indicated an association of Graves’ disease with HLA-B8 and a relative risk of 3.9 in white patients.45 Subsequent studies also suggested an influence of that haplotype on the clinical course of the disease.33,44 However, HLA-DR3 (HLA-DRB1*03) was later shown to increase the risk to a greater extent and was considered to be the true determinant of the disease because it was in linkage disequilibrium with the B8 allele.33 Among Caucasians, HLA-DQA1*0501 was found to confer a relatively high risk within DR3 itself.33,44 This allele is in linkage disequilibrium with both B8 and DR3 and gives a relative risk of 3 to 4 for Graves’ disease in the white population. Sequencing of the DRβ-1 chain of HLA-DR3 allowed the identification of Arg74 as the critical amino acid conferring susceptibility to Graves’ disease.33,44
Different haplotypes seem to be involved in ethnic groups other than Caucasians: DQ3 in patients of African descent and Bw46 in those of Asian descent, although the data available are limited and have not always been reproducible.33,44
In general, HLA associations have been shown to confer a relatively low risk, even with alleles that have a high prevalence in the general population. Thus linkage analysis, a powerful tool for mapping essential predisposition genes, has been negative when the HLA region was examined using different polymorphisms at the same locus.33,44 Overall, it seems that the HLA locus explains a small fraction of the total genetic predisposition, but it is neither the major nor the only determinant, although it represents an established risk-increasing factor.
2:: CD40 CD40, a member of the tumor necrosis factor receptor family, is expressed in B cells and other antigen-presenting cells and is involved in B-cell activation and proliferation, antibody secretion, immunoglobulin class switching, affinity maturation and generation of memory cells.46 Linkage studies have shown an association of the CD40 gene with Graves’ disease and the subsequent sequencing of the gene led to the identification of a C/T polymorphism at the 5′ untranslated region of CD40 strongly associated with Graves’ disease.33 This polymorphism influences the translational efficiency of the gene, which may have functional consequences in the CD40 protein.
3:: CTLA-4 CTLA-4 is a T-lymphocyte surface protein with a major role in down-regulation of the immune response.47 Several studies have provided evidence that CTLA-4 is linked to Graves’ disease, autoimmune thyroiditis, and to the production of autoantibodies against thyroid antigens.33,44–48 Several variants of the CTLA-4 gene have been implicated in its possible causative role in Graves’ disease, among which a CTLA-4 polymorphism at position 60 was found to be the most suitable candidate in a large comprehensive analysis, the functional relevance of the polymorphism being possibly due to a reduced mRNA expression encoding the soluble form of the molecule.47 Nevertheless, a subsequent study did not confirm these data.48 Although CTLA-4 seems to be a genetic determinant of Graves’ disease, the causative variant remains to be identified with certainty, and it is possible that a haplotype consisting of more than one variant is responsible for the association.33
4:: The Protein Tyrosine Phosphatase 22 (PTPN22) Gene PTPN22 is a powerful inhibitor of T-cell activation.49 A single nucleotide polymorphism at codon 620 associated with other autoimmune diseases was found to be associated with both Graves’ disease and autoimmune thyroiditis, with significant ethnic differences in the association.49,50
5:: Thyroglobulin Thyroglobulin is the precursor of thyroid hormones and a major autoantigen in thyroid autoimmunity. Recent wide genome screens have provided evidence for a strong linkage between a locus on chromosome 8q24, where the thyroglobulin gene is located, and autoimmune thyroid diseases.33,51 Sequence analysis of the thyroglobulin gene has shown numerous single nucleotide missense polymorphism, suggesting that amino acid variants in the Tg proteins may contribute the pathogenesis of autoimmune thyroid diseases, including Graves’ disease.33,52
6:: TSH-R Despite the central role of TSH-R in the pathogenesis of Graves’ disease, the association of the disease with the gene encoding the receptor remains controversial.33 Although three common missense single nucleotide polymorphisms have been found to be associated with Graves’ disease, these associations were not always confirmed by other studies. The extent of the contribution of the TSH-R gene remains to be established.33
7:: Other Genes In the search for genetic determinants of Graves’ disease, several other genes have been studied over the years, namely genes involved in the immune response. The immunoglobulin genes were studied extensively, but conflicting results were observed in association studies.33 Other candidate immunoregulatory genes that have been studied include interleukin 1 (IL-1), IL-1 receptor antagonist, tumor necrosis factor receptor 2 (TNF-2), and interferon γ (INF-γ). None of these genes showed significant associations with Graves’ disease.33 Additional loci have been linked in families with Graves’ disease, including one on chromosome 14q31 (GD-1), one on chromosome 20 (GD-2), and one on chromosome Xq21-22 (GD-3).33
Environmental Factors and Graves’ Disease
The relative low penetrance in twins and first-degree relatives of patients with Graves’ disease suggests that environmental factors must play a major role in inducing the disease in genetically susceptible individuals.33,53,54 Several studies have shown that various nongenetic factors may in fact contribute to the development of Graves’ disease.
Infections
Over the years, both experimental and epidemiologic evidence has suggested that infections could play a role in the pathogenesis of Graves’ disease.54,55 Seasonal and geographic variations in the incidence of the disease have been reported,56,57 although seasonal variations have not been confirmed in other studies.58 Blood group nonsecretors, who are more prone than secretors to infections, are more frequently found among patients with Graves’ disease than in controls.59 This observation has been interpreted as indirect evidence that infectious pathogens may be involved in the etiology of Graves’ disease, although a direct genetic effect of the secretor status could also explain these results. Evidence of a recent viral infection has been reported in a high percentage of patients with Graves’ disease.54,55
Molecular mimicry has been invoked to explain the association between infections and Graves’ disease.60 Molecular mimicry is based on the hypothesis that cross-reactions of some microbial antigens with a self-antigen may cause an immune response to autoantigens. In Graves’ disease, the pathogen Yersinia enterocolitica has been thoroughly studied after reports of association of this microbe with the disease. A high prevalence of circulating antibodies against Y. enterocolitica has been observed in patients with Graves’ disease, and Yersinia antibodies were found to interact with thyroid structures.61–63 In a recent study from Denmark, it was shown that the occurrence of IgA and IgG antibodies against Yersinia not only is more frequent in Graves’ patients than in case controls but also in twins with Graves’ disease compared with their discordant twins.64 Saturable binding sites for TSH have been found in Yersinia and were also recognized by TRAbs from patients with Graves’ disease.65,66 In animals immunized with Yersinia proteins, antibodies developed against human thyroid epithelial cells and TSHR.67,68 Overall, the affinity of these cross-reactive antibodies to the thyroid was low, and immune responses were transient. Low-affinity binding sites for TSH have also been found in other bacteria, including some species of Leishmania and Mycoplasma.53,54 However, it must be noted that thyroid autoimmunity does not develop in most patients with Yersinia infections,69 so the evidence in favor of Yersinia infections as a precipitating cause of Graves’ disease awaits confirmation.
Viruses could theoretically trigger autoimmunity through several mechanisms, including interactions with autoantigens, permanent expression of viral proteins on the surface of epithelial cells, aberrant induction of HLA antigens on epithelial cells (see later), and molecular mimicry.60,70 In 1989, the presence of retroviral (HIV-1 glycosaminoglycan protein) sequences in the thyroid and peripheral mononuclear cells of patients with Graves’ disease was reported,71 but viral sequences were not found in control thyroids. This finding, however, remained isolated and was not confirmed in subsequent studies.72,73 Human foamy virus antigens were shown by immunofluorescence to be present in the thyroid of patients with Graves’ disease.74 Again, further studies using more specific and sensitive techniques failed to identify foamy virus DNA and antiviral antibodies in the blood of affected subjects.75,76 Homology between another HIV-1 protein (Nef) and human TSHR has also been reported, although sera from patients with Graves’ disease did not react with the peptide bearing the highest degree of homology.77 Another retroviral protein, p15E, has been isolated from the thyroids of patients with Graves’ disease but not from control glands.78 In this regard, it is worthwhile emphasizing that retroviral-like proteins, including p15E, are encoded by the normal human genome. Although their function is unclear, they may be expressed in many epithelial tissues under certain conditions such as inflammation and may modulate but not initiate the immune response.79 The finding of retroviral sequences or proteins in the glands of patients with Graves’ disease may therefore represent a secondary rather than a causative phenomenon. Circulating antibodies against another retroviral particle, namely HIAP-1, have been found in as many as 87.5% of patients with Graves’ disease as compared with 10% to 15% of controls,80 but HIAP-1 particles were not detected when human T cells were co-cultured with Graves’ thyrocytes.81
A highly speculative hypothesis has been raised that involves superantigens.55,60 Superantigens are endogenous or exogenous proteins, such as microbial proteins, capable of stimulating a strong immune response through molecular interactions with nonvariant parts of the T-cell receptor and the HLA class II proteins. Through this mechanism, superantigens are in theory capable of stimulating the expansion of autoreactive T cells and therefore of driving an autoimmune response.55,60 Such a mechanism has been suggested in rheumatoid arthritis, and a similar mechanism was proposed for Graves’ disease. In vitro superantigen stimulation of glands with autoimmune thyroid disease induced expression of HLA class II molecules on thyrocytes, and this phenomenon was IFN-γ dependent.82 The interpretation of this observation was that superantigen-reactive T cells exist among the lymphocytes infiltrating the thyroid in autoimmune thyroid disease, and that these lymphocytes may have been activated after exposure to extrinsic superantigens.
The most recent hypothesis, the so-called “hygiene hypothesis of autoimmunity,” implies that infections may protect from, rather than precipitate, autoimmune diseases. Exposure of the immune system to infective agents may somehow allow better control of autoimmune responses. In this regard, improved living standards have been associated with decreased exposure to infections and an increased risk of autoimmune diseases. Kondrashova et al. reported a much reduced prevalence of thyroid autoantibodies in the lower-economic population, which may suggest the hygiene hypothesis may apply to thyroid autoimmune diseases.83 Further studies are needed to investigate whether this is in fact the case.
Stress
The suggestion that psychological stress may be a precipitating factor in Graves’ disease has been made as early as the first description of the disease.3 The occurrence of stressful events before the onset of Graves’ disease is a recurrent impression among clinicians, and by the end of the 19th century, Graves’ disease was considered to be a result of prolonged emotional disturbances. In cross-sectional questionnaire-based studies, some investigators have shown an increased prevalence of stressful life events in the months preceding the onset of Graves’ disease.84–87 However, some of the recorded events (such as arguments with spouses and in the workplace) could have been influenced by the behavior of patients with a yet undetected hyperthyroidism and therefore be a consequence rather than a cause of the disease. Other events, however, were largely independent of the patient’s behavior, such as unemployment and financial difficulties. In some studies, patients were asked to rate the stressfulness of life events, and patients with Graves’ disease ranked such events more stressful than did controls.84 Thus it is possible that the perception of life events is different in hyperthyroid patients. In cross-sectional studies, an increase in the prevalence of Graves’ disease was reported during World War II in Germany but not during the civil unrest in Ireland or during the German occupation of Belgium, which suggests that the stressful events of personal life may be more important than “social stress.”84 On the other hand, chronic stress due to panic disorder was not associated with the occurrence of Graves’ disease.88 Stress is associated with increased adrenocorticotropic hormone (ACTH) and cortisol secretion, which can in turn determine immune suppression, but additional non-ACTH-related immunosuppressive phenomena also occur.89,90 Recovery from such immune suppression can be associated with rebound immune hyperactivity, which could precipitate autoimmunity. The best example of such a phenomenon is perhaps the well-documented immune suppression of pregnancy, which can be followed by new or recurrent onset of autoimmune disorders, including Graves’ disease (see later discussion).91
Gender
Graves’ disease is typically but not exclusively a disease of women. In most series, the female-to-male ratio ranges from 5 to 10 at any age,16,17 although the difference may be smaller during childhood.92 The reason for the disproportionate prevalence of Graves’ disease in women is not known, but genetic and nongenetic factors must play a role. A number of studies indicate a stronger immune system in women.93,94 Autoimmune phenomena and diseases are in general more prevalent in women.93,94 A large body of evidence clearly indicates the existence of sexual dimorphism in normal and abnormal immune responses in spontaneous and experimental animal models, including models of autoimmune thyroiditis.93,94 In these models, male hormones appear to down-regulate immunity and therefore protect animals from autoimmunity, whereas the effect of estrogen is not always unequivocal. Despite the evidence obtained from animal studies, little evidence in the literature supports a role for sex hormones in the high prevalence of Graves’ disease in women.93,94 Women with normal baseline levels of estrogen, but with an increased sensitivity to the hormone as shown by the presence of melasma, had a higher prevalence of thyroid autoimmune disorders.95 However, a clear association between exogenous estrogen administration and Graves’ disease has never been reported. Moreover, thyroid autoimmunity is often found in patients with Turner’s syndrome, who typically have low estrogen levels.96 Conversely, male patients with primary hypogonadism such as in Klinefelter’s syndrome do not show a higher incidence of Graves’ disease or Hashimoto’s thyroiditis.97 In these human conditions, however, the chromosomal abnormality probably plays an important role that is largely independent of sex hormone levels.
Pregnancy is an important risk factor, and it is well recognized that in any woman the risk of development of Graves’ disease increases fourfold to eightfold in the postpartum year.91 The abrupt fall in the level of pregnancy-associated immunosuppressive factors immediately after delivery (rebound immunity) is likely to be the mechanism responsible for the precipitation of Graves’ disease.91 Nevertheless, in a recent retrospective study it was found that the relative frequencies of postpartum onset of Graves’ disease were similar in relation of increasing parity, which would not support a role of the postpartum period as a major risk factor for the first appearance of Graves’ disease.98 On the other hand, it was also shown by the same group that the postpartum period is indeed a risk factor for relapse of Graves’ thyrotoxicosis after withdrawal of antithyroid drugs.99 The factors involved in the immune alterations of the postpartum period may include but are not limited to estrogen and progestin.91,100
Besides sex hormones, factors on the X chromosome could explain the epidemiologic evidence of a female preponderance in Graves’ disease. A linkage analysis in families with Graves’ disease has located a putative Graves’ disease susceptibility locus on the long arm of the X chromosome.33 Although most X-linked disorders are expressed phenotypically only in men, it is possible that a gene with a dose-dependent effect may determine more relevant clinical effects in women. This finding could help explain the higher incidence of Graves’ disease observed in women and, possibly, in patients with Turner’s syndrome. Inactivation of the X chromosome, an epigenetic phenomenon, has also been suggested to be involved in the female predisposition to thyroid autoimmunity, and recently it was shown that this phenomenon is more frequently observed in patients with Graves’ disease or autoimmune thyroiditis than in healthy controls.101
Smoking
A number of studies have provided evidence for an association between smoking and thyroid diseases, including Graves’ disease.102,103 Retrospective analysis shows that in smokers there is an increased risk of Graves’ disease and ophthalmopathy, as well as of relapse of hyperthyroidism following anti-thyroid drug withdrawal, which is more pronounced in the female gender. The findings may be explained both by a direct action of smoking metabolites on the immune system or by damage induced by smoking metabolites on thyrocytes, which may determine exposure of thyroid antigens to the immune system.
Thyroid Damage
There are reports of Graves’ disease appearing after ethanol injections performed for treatment of autonomous thyroid nodules, which has been interpreted as due to the massive release of thyroid antigens, thereby triggering an autoimmune response to TSH-R in predisposed individuals.104 Furthermore, and possibly because of similar pathogenetic mechanisms (i.e., massive release of thyroid antigens), Graves’ disease or simply serum TRAb have been reported to appear following radioiodine treatment for toxic adenoma or toxic nodular goiter.105 We observed a patient with Graves’ disease that appeared after a neck injury (unpublished observation), which in theory may also reflect release of thyroid antigens.
Pathology
It is now rare to observe the full pathologic changes that occur in the thyroid glands of untreated patients.106 On gross pathology the gland is significantly enlarged, with a smooth and hyperemic surface. A prominent pyramidal lobe is often visible, and the contour of the gland is irregular with multiple lobulations. Microscopically, both hypertrophy and hyperplasia are found. Follicles are small, with scanty colloid as a result of ongoing thyroid hormone secretion. The follicular epithelium presents a columnar aspect, with even a pseudopapillary appearance. Blood vessels are large and congested. Various degrees of lymphocytic infiltration can be found between the follicles. T cells predominate in the interstitium, whereas B cells and plasma cells predominate in the occasional lymphoid follicles. On electron microscopy, the cellular hyperactivity is demonstrated by an increase in the Golgi reticulum and the number of mitochondria and by the presence of prominent microvilli. With longstanding Graves’ disease, distinct nodularities with an adenomatous appearance may develop, and the lymphocytic infiltrate may become more prominent and resemble chronic thyroiditis.
This pathologic picture of active Graves’ disease is dramatically changed by antithyroid drugs and iodine treatment, which is now universally performed before surgery. Vascularity and vascular congestion are much less pronounced, and the follicles can be larger.106
Pathogenesis of Graves’ Disease
Although the ultimate cause of Graves’ disease is still unknown, over the years a large body of evidence has accumulated and provided important insight into the immune mechanisms that eventually lead to the clinical manifestations of the disease. Since their first description in 1956,11 much attention has been devoted to the study of TRAbs. It has also been recognized that although TRAbs are the ultimate cause of both goiter and hyperthyroidism, the nature of the immune dysfunction involves many aspects of the immune system, including changes in both B-cell and T-cell function. The follicular cell per se may also play an independent role.
Role of TSH Receptor Antibodies
The nomenclature of TRAbs is complex and largely dependent on the assay used to detect these antibodies in serum.107–111 Assays measuring displacement of radiolabeled TSH from its receptor by serum immunoglobulins detect TRAbs regardless of their functional activity. These antibodies have been termed TSH-binding inhibitory immunoglobulins (TBIIs). Assays for TSAbs use cellular systems carrying a functional TSHR and detect the release of cyclic adenosine monophosphate (cAMP) in the culture medium upon challenge with serum or purified immunoglobulins.107–111 These antibodies are essentially the cause of hyperthyroidism in Graves’ disease. In the same bioassay system, antibodies with blocking activity on the TSHR (TBAbs) can be detected.107–112 These antibodies characterize a subset of patients with atrophic autoimmune thyroiditis and hypothyroidism, but they can also occasionally be found in patients with Graves’ disease, in combination with TSAbs.112
Major Autoantigen in Graves’ Disease: Structure-Function Relationship of the TSHR
Definitive proof that TSHR is the target of TSAbs came from the cloning of this protein in the late 1980s.12,13,107–109 The receptor is a member of the G protein–coupled receptor superfamily. Its structure includes seven hydrophobic transmembrane domains, an extracellular N-terminal domain (ectodomain), and an intracellular C-terminal domain. Heavy glycosylation of the extracellular domain accounts for about 20% of its molecular weight of 84 kD. The primary structure of the protein consists of 744 residues. The gene is located on chromosome 14q31 and is formed by 10 exons that yield a single polypeptide. Shedding of the N-terminal extracellular 310 amino-acid residues (the so-called A subunit) may either initiate or amplify the immune response against TSHR, being immunoreactive epitopes partially sterically hindered on the holoreceptor on the plasma membrane.108,113,114 Binding sites for TSH and TRAbs are located in the extracellular domain and are mainly conformational.107,108 The major epitopes for TSAbs are located in the extreme N-terminal portion, whereas those for TBABs are mainly, but not exclusively, in the C-terminal portion of the extracellular domain, closer to the cell membrane.107,108,115–118 However, this should not be taken as a paradigm, especially since TBABs are more diverse and spread over the receptor extracellular domain.108 In fact, TRAbs are to some extent heterogeneous for epitope recognition, possibly due to epitope spreading during the immune response.108 Whether and to what extent glycosylation and dimerization of TSH receptor affects its recognition by antibodies is uncertain.108
Assays for TSH Receptor Antibody
The pioneering era of bioassays in vivo for TRAbs11,119 has been superseded by the present period, in which a number of in vitro assays are more readily performed by many laboratories and provide more reproducible and reliable measures of TRAb levels in the serum of patients. The radioreceptor assay uses TSHR from various sources: porcine or human recombinant TSHR from transfected cell lines.120–126 Although assay design varies in all these methods, they all rely on displacement of labeled TSH from solubilized TSHR from the serum of patients. Studies in hyperthyroid patients with Graves’ disease show positive TBII tests with these methods in 75% to 95% of patients. The recently designed second-generation radioreceptor assay attains even higher sensitivity while maintaining high specificity (99%)126 and is commonly used by the majority of laboratories for the routine detection of TRAb. An enzyme-linked assay is also available.127 Radioreceptor assays do not require permanent cell cultures; they are the most readily available (also commercially) and are therefore the most frequently used in clinical practice. The TBII test, however, does not give information on the functional properties of the antibody detected and can be positive in the presence of TBAb.
The functional stimulating properties of TRAbs can be studied by in vitro bioassays based on the measurement of cAMP production from cells with a functional TSHR.120 Human thyroid follicular cells,128 a rat thyroid cell strain (FRTL-5),129,130 and Chinese hamster ovary cells stably transfected with human TSHR (CHO-R)131–133 have all been used for this purpose. With these assays, TSAb can be detected in more than 90% of patients with untreated Graves’ disease (Fig. 9-3). The system using CHO-R cells has some advantages over the others: it is slightly more sensitive and relies on easier culture conditions, which also makes it more reproducible in different laboratories. As stated previously, the bioassays have the advantage of giving information on the functional properties of TRAbs and, in a modification of the assay, can also identify TBAbs.112,133 However, they require permanent cell culture equipment and pre-purification of the immunoglobulin fraction of serum, which makes these assays not readily available to routine endocrine laboratories. The latter problem has been overcome by very sensitive assays in which activation of a transfected firefly luciferase gene produces chemiluminescence in response to TSHR stimulation by whole serum.134,135 Even more recently, a coated-tube assay which discriminates TSAb from TBAb was developed.136 The assay is based on the use of a chimeric receptor, where a TSAb epitope 8-165 is replaced by comparable LH receptor residues. Binding of radiolabeled TSH to this chimera can be inhibited by sera containing TBAb up to 95%.
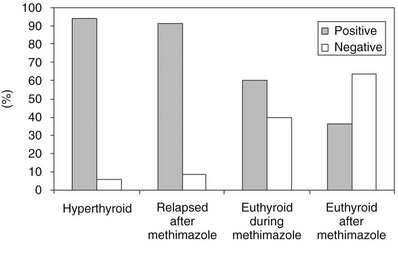
FIGURE 9-3 Prevalence of serum thyroid-stimulating antibody tests in Chinese hamster ovary cells transfected with recombinant human thyroid-stimulating hormone (TSH) receptor (CHO-R). Serum IgG was purified from untreated and treated patients with Graves’ disease. (Modified from Vitti P, Elisei R, Tonacchera M et al: Detection of thyroid-stimulating antibody using Chinese hamster ovary cells transfected with cloned human thyrotropin receptor. J Clin Endocrinol Metab 76:499–503, 1993.)
Thyroid-Stimulating Antibody in the Pathogenesis and Natural History of Graves’ Disease
Historically, the identification of TSAbs as the cause of hyperthyroidism and goiter in Graves’ disease came from the demonstration of a stimulating factor in the sera of hyperthyroid patients with a half-life much longer than that of TSH (LATS).11 Subsequently this factor was shown to be an autoantibody.137–139 TSAbs were shown to interact with TSHR in that they act as a potent agonist and thus cause hyperfunction of the thyroid gland.107–110 Definitive proof that TRAbs interact with TSHR eventually came from studies with the cloned protein.107–110 A clear-cut demonstration of the role of TSAb in the pathogenesis of hyperthyroidism is provided by the observation that the transplacental transfer of antibodies from TSAb-positive pregnant mothers to the fetus may cause a form of transient neonatal thyrotoxicosis that vanishes with the disappearance of TSAb from the serum of the newborn.140
TSAbs are oligo- or pauciclonal, and this observation has suggested a primary defect at the B-cell level.107–110 TSAbs appear to be produced mainly by thyroid-infiltrating lymphocytes and lymphocytes in the draining lymph nodes.141 Synthesis by peripheral blood lymphocytes has been documented as well.142–144 As mentioned earlier, TSAbs can be detected in more than 90% of patients with untreated Graves’ disease hyperthyroidism.120–126 The observation of a small proportion of patients with undetectable TBIIs or TSAbs has been attributed to the occurrence of these autoantibodies at a serum level too low to be detected by current methods. Alternatively, restricted intrathyroidal production of TRAbs has been hypothesized.145 A positive correlation between TSAb levels and serum triiodothyronine (T3) levels, serum thyroglobulin levels, and goiter size has been observed.107–110
TRAb levels usually fall during long-term treatment with antithyroid drugs.146,147 This phenomenon has been attributed to an immunosuppressive effect of antithyroid drugs,148 but it could also result from the correction of thyrotoxicosis or even reflect the natural history of the disease.
Other Antigens
In addition to TRAbs, autoantibodies against thyroglobulin and thyroperoxidase are commonly found in patients with Graves’ disease, although autoimmunity against these two antigens is generally believe to be a secondary phenomenon with no pathogenetic implications. A possible role in the pathogenesis of Graves’ disease, especially of its extrathyroidal manifestations (e.g., Graves’ ophthalmopathy), was recently attributed to the insulin-like growth factor 1 receptor (IGF1-R). The receptor is expressed in thyroid epithelial cells as well as in orbital fibroblasts, and autoantibodies against the receptor have been detected in patients with Graves’ disease.149 Whether autoimmunity against IGF1-R has any role in the pathogenesis of the disease is under investigation.
Role of Cellular Immunity
The primary requirement for a specific autoimmune (either humoral or cell-mediated) response is an antigen-specific T cell.150 Activation of T cells requires presentation of antigenic peptides in the context of HLA molecules. This task is accomplished by a specialized subset of immune cells called professional antigen-presenting cells. Once activated, helper (CD4+) T cells can be subdivided into two functional subtypes according to their cytokine production pattern150,151: the TH1 subset, mainly involved in delayed-type hypersensitivity reactions, and the TH2 subset, prominently involved in humoral immune responses. TH1 cells produce tumor necrosis factor-β, IFN-γ, and IL-2; TH2 cells secrete mainly IL-4, IL-5, IL-6, and IL-13. TH1 cells have been implicated in organ-specific autoimmune diseases, an action that seem to be exerted especially by the so-called TH17 subtype, which among TH1 cells produces uniquely IL-17.152 The subset of T lymphocytes primarily activated by specific antigens and the cytokine milieu produced by antigen-presenting cells determine the direction of the immune response toward a TH1 cell-mediated tissue-damaging reaction, a more prominent humoral reaction (TH2 mediated), or a balance of the two.152
Studies of patients with Graves’ disease showed activated T cells both in the peripheral circulation and in the thyroid gland.153–155 T cells infiltrating the thyroid gland in Graves’ disease have been studied by surface monoclonal antibody phenotyping. The percentage of CD8+ (suppressor/cytotoxic) T cells was found to be much lower in Graves’ disease than in Hashimoto’s thyroiditis.156–159 The phenotype of the CD4+ (helper/inducer) T-cell population was predominantly composed of memory cells.160,161 Further studies involved cloning of infiltrating T cells. Most of the resulting T-cell lines belonged to the memory (CD4+, CD29+) subtype and responded with significant growth and/or cytokine production to challenge with autologous thyroid follicular cells or thyroid antigens.162–166
As assessed by their cytokine profile, intrathyroidal T cells were found to be predominantly of the TH1 subtype,158,167 and this pattern was also true for TSHR-responsive clones.168 This finding is somewhat unexpected in a disease such as Graves’ disease, which is mainly characterized by the action of TSAbs producing thyroid hyperfunction and follicular cell growth. However, it is worthwhile noting that TH1 cells may also induce antibody production through secretion of IL-10, which in turn activates B cells.141 In keeping with this sequence of events, TRAbs of Graves’ disease are more often of the IgG1 subclass,151 which is selectively induced by TH1 cells. A significant proportion of TH0 (uncommitted) cells were also detected among TSHR autoreactive T cells.168
The basis for an organ-specific autoimmune process is the interaction of antigen-specific T cells with the target tissue itself in a way that leads to selection and clonal expansion of autoreactive cells. The specificity of this interaction is provided by the immense variability in mature T-cell antigen receptors caused by the somatic rearrangement of their variable (V) chain with the constant (C) and the junction (J) regions.169,170 When an immune response is initiated, T cells carrying the individual receptor specific for the antigen involved are stimulated and clonally expanded. In keeping with this concept, restricted use of T-cell receptor Vα and Vβ genes was observed in T lymphocytes obtained by fine-needle aspiration of Graves’ disease thyroids.171 When surgically obtained specimens of thyroids with Graves’ disease have been examined, selective use of T-cell receptor Vα and Vβ genes has been confirmed in some172–175 but not all studies.176,177 These observations indicate that a highly selective response to thyroid autoantigens is elicited in the thyroid glands of patients with Graves’ disease during the initial phase of the disease. Afterward, spreading of the autoimmune response occurs and leads to less restricted T-cell receptor gene usage.
The antigen specificity of autoreactive T cells has also been tested. Initial studies with thyroid follicular cells in culture or with their subcellular fractions suffered from contamination with thyroid autoantigens other than TSHR, so that the antigen specificity of T cells was questionable. Indeed, thyroid peroxidase–specific and thyroglobulin-specific T cells also exist within the thyroid gland of patients with Graves’ disease.168 Since the cloning of TSHR cDNA, however, a number of laboratories have searched for TSHR-specific T-cell clones and investigated their role in thyroid autoimmunity and Graves’ disease. Studies using TSHR peptides identified specific responses and showed positive stimulation indexes of peripheral blood mononuclear cells from patients with Graves’ disease, as well as from healthy controls.173 Considerable effort has also been expended in identifying immunodominant T-cell epitopes on the TSHR. Four distinct peptides were recognized by lymphocytes from most patients with Graves’ disease in one study.178 More recently, another set of immunodominant peptides has been described.179 It is possible that HLA haplotypes and other poorly understood factors play a role in determining which epitope is immunodominant in individual patients. Overall, these observations show that immunodominant T cell–dependent epitopes exist within the TSHR, and that these epitopes may be at least in part shared by different patients. Identification of T cell–dependent epitopes would be important in efforts to design immunologic approaches to the treatment of Graves’ disease, such as tolerizing vaccines or antigen-specific lymphocyte deletion.
Unlike in destructive autoimmune processes such as autoimmune thyroiditis, the network of factors and substances released by lymphocytes or macrophages should not play a major role in the pathogenesis of a disease such as Graves’ disease, in which autoantibodies have a more important role. Nevertheless, recent studies have underscored the possibility that chemokines may also be of some importance in Graves’ disease.180 Chemokines are a group of peptides that induce chemotaxis of different leukocyte subtypes. Their major function is the recruitment of leukocytes to inflammation sites. In the last few years, experimental evidence has accumulated supporting the concept that IFN-γ inducible chemokines (CXCL9, CXCL10, and CXCL11) and their receptor, CXCR3, play an important role in the initial stage of autoimmune disorders involving endocrine glands. After IFN-γ stimulation, endocrine cells secrete CXCL10, which recruits TH1 helper lymphocytes expressing CXCR3 and secreting IFN-γ, thus perpetuating the autoimmune inflammation. In Graves’ disease, serum levels of CXCL10 are higher in newly diagnosed hyperthyroid patients and decrease when euthyroidism is restored, which may be related to the active inflammatory phase of the disease.180
Role of Thyroid Follicular Cells
Whether primary defects in the thyroid gland contribute to the pathogenesis of thyroid autoimmunity has been pondered since the mid-1980s.181–183 The observation that thyroid cells from patients with Hashimoto’s thyroiditis and Graves’ disease express HLA class II antigen (DR), which is usually expressed by professional antigen-presenting cells, led to the hypothesis that aberrant expression of these molecules on thyroid cells could initiate thyroid autoimmunity via direct thyroid autoantigen presentation.184,185 It was later suggested that expression of HLA class II molecules on thyroid cells is a secondary rather than a primary phenomenon, determined by the cytokines released by the lymphocytic infiltrate.186,188 Thyroidal HLA-DR expression can be induced by cytokines such as IFN-γ, which is able to induce thyroiditis when administered to susceptible mice and to predisposed human subjects.186,187 In addition, overexpression of IFN-γ in transgenic mice results in hypothyroidism due to disruption of the thyroid structure.189,190 However, IFN-γ may not be the sole determinant; its genetic disruption reduced the severity of experimental thyroiditis but did not abrogate it.191 Moreover, in experimental models of thyroiditis, HLA class II expression appeared to be a late phenomenon that is present only after the infiltrate has appeared, although such observations may depend on the sensitivity of the detecting system used.192,193 Thus it would appear that HLA class II antigen expression is probably not a primary mechanism leading to Graves’ disease, but rather it is important for perpetuation and enhancement of the autoimmune reaction. In this regard, thyroid cells have been shown to be capable of stimulating T lymphocytes both in the presence and in the absence of professional antigen-presenting cells.194,195 Co-culture of peripheral blood mononuclear cells from Graves’ disease patients with homologous thyrocytes induced T-cell activation182 as well as IFN-γ production and HLA class II antigen expression on thyroid cells.196 There is evidence that genetic manipulation of HLA class II molecules may affect the susceptibility to the development of experimental autoimmune thyroiditis in mice,197 but such a phenomenon has not been tested in Graves’ disease. In a recent study, some of the HLA-DR natural ligands in Graves’-affected thyroid tissue were identified as thyroglobulin peptide, and it was suggested that binding between HLA-DR and thyroglobulin or thyroglobulin fragments may be involved in the maintenance of the autoimmune inflammatory process.198
Local production of chemokines by thyrocytes has been shown, which may be responsible for recruitment of immune cells.199 As mentioned previously, high levels of CXCL10 are present in the serum of Grave’s patients.180 Professional antigen-presenting cells, such as dendritic cells, macrophages, and even B cells, also exist within the thyroid lymphocytic infiltrate in close relationship with thyrocytes and are involved in thyroid autoantigen presentation.200,201
Professional antigen-presenting cells express on their surface a family of co-stimulatory proteins named B7 that interact with molecules (CD28 and CTLA-4) on the surface of helper (CD4+) T cells during antigen presentation.202 This co-stimulatory process is critical for determining the direction of the immune response, because its absence may result in anergy and/or deletion of antigen-specific T cells. The B7-1 and B7-2 molecules are not expressed on thyroid cells derived from patients with Graves’ disease,203 which suggests that these cells must rely on other co-stimulatory factors, possibly from professional antigen-presenting cells.202 Co-stimulatory molecules may also influence the resulting TH1 or TH2 T-cell phenotype.150 In this regard, studies on antigen presentation indicate that in the absence of co-stimulatory signals, such as those provided by B7 molecules, HLA class II expression by thyroid cells will lead to continued activation of T cells if the immune response has already been established by professional antigen-presenting cells, whereas it will induce peripheral tolerance of naive, not previously stimulated T cells.203
Animal Models of Graves’ Disease
No spontaneous animal diseases mimicking human Graves’ disease have been reported, and the absence of such an animal model has slowed the acquisition of knowledge on processes leading to this condition. Experimental immunization with soluble forms of TSHR has led only to antibodies deprived of stimulating activity that did not cause hyperthyroidism in the immunized animals. More recently, a different approach has been successful in inducing hyperthyroidism and goiter, thereby allowing researchers to gain some insights into the pathogenesis of Graves’ disease, especially concerning antigen presentation, the role of T cells and of humoral immunity, and the production of TSH-R monoclonal antibodies.204–207 In this model, mice are immunized by injecting living cells (professional or nonprofessional antigen-presenting cells) expressing TSHR or by DNA vaccination with TSHR cDNA in plasmid or adenovirus vectors.
Clinical Aspects of Graves’ Disease
The hallmarks of Graves’ disease are a diffuse goiter associated with the symptoms and signs of thyrotoxicosis and the typical orbitopathy.208 More rarely, pretibial myxedema and acropachy are present. The onset of symptoms is usually gradual over a period of weeks to months, but it can be abrupt in some cases. In other cases, mild symptoms can exist for years before a diagnosis is made.
Clinical Manifestations of Graves’ Hyperthyroidism
Thyrotoxic symptoms of Graves’ disease do not differ from those seen in thyrotoxicosis of other causes (Table 9-3).209 Most organs are sensitive to thyroid hormone action and are therefore altered by thyroid hormone excess. Symptoms of thyrotoxicosis consequently encompass a wide range of manifestations, each contributing to a clinical picture that can rarely be mistaken when all its components are present. However, the spectrum of manifestations of thyrotoxicosis may range widely from the classic picture to more subtle signs and symptoms, which depends on many variables, including age at onset, duration of thyrotoxicosis, severity of thyrotoxicosis, and possibly, poorly understood individual factors.

Thyroid
The thyroid gland is usually symmetrically enlarged (Fig. 9-4), although nodular glands can be seen, especially in geographic areas of iodine deficiency, where nodular goiter often preexisted. Sometimes true nodules are difficult to distinguish from the lobulations typical of a hyperplasic gland. Goiter size is widely variable, and some patients can even have a gland of normal size. Large goiters can be associated with engorgement of the jugular veins and a positive Pemberton sign (swelling of the jugular veins upon elevation of the arms). By palpation, the consistency of the gland is generally firm, although softer than in Hashimoto’s thyroiditis. Thrills and bruits resulting from increased blood flow may be present on the gland, especially in the early phases of the disease and with a large goiter.
Skin and Appendages
The skin of a thyrotoxic patient is warm, thin, and moist; palmar erythema is common. Dermatographism and pruritus are often reported, but their significance is unclear.210 Urticaria may also be associated. Vitiligo is frequent, being not a consequence of thyrotoxicosis but rather an associated independent skin autoimmune disease.211 The hair is friable, diffuse alopecia of mild degree is often observed, but alopecia areata is rare. Nails are soft and friable with longitudinal striations, and onycholysis (detachment of the nail from the ungual bed) is observed in long-lasting cases. Pretibial myxedema and thyroid acropachy, described elsewhere in this chapter, can also be seen.
Cardiovascular System
Heart-related symptoms are a frequent initial complaint in patients with Graves’ disease.212,213 The most common are tachycardia and palpitations. Signs and symptoms of heart failure may also develop, and edema of the lower extremities is often found in elderly patients. The heart and vascular system are major targets of thyroid hormones, which explains the high prevalence of cardiovascular symptoms in thyrotoxic patients with Graves’ disease. Cardiac complications may be a major concern in the care of these patients. Thyrotoxicosis causes an increase in both inotropism and chronotropism of the heart. Overall, vascular resistance is decreased because of peripheral vasodilatation. The net effect of these changes is increased cardiac output, which is the major pathophysiologic event. Increased cardiac workload causes increased oxygen consumption, which in turn can precipitate angina pectoris in the presence of preexisting coronary artery disease. Peripheral edema can be observed in the absence of overt heart failure. Dyspnea on exertion or at rest and chest pain may also be present and are prominent features of thyrotoxic heart disease.
Electrocardiographic findings are nonspecific and include sinus tachycardia with ST elevation, QT shortening, and PR prolongation. Atrial fibrillation or flutter can occur in up to 10% to 15% of patients, especially if elderly. Ischemic changes can be found when underlying coronary artery disease is present. The observation of reversible heart failure in younger patients with thyrotoxicosis has raised the question whether a distinct thyrotoxic cardiomyopathy exists in the absence of preexisting detectable heart disease.212,213 Technically, the high-output heart failure typically observed in these cases may not be due to “failure” of the heart pump, but instead only to the changes in the peripheral circulation induced by vasodilatation and sodium-water retention. However, long-lasting tachyarrhythmias have been shown to impair the contractility of cardiac myocytes, and this mechanism has been proposed for the thyrotoxicosis-induced heart failure observed in young patients.214 In most cases, however, cardiac complications occur in elderly patients in whom underlying heart disease is likely to exist. In this setting, heart failure occurs mainly in the presence of atrial fibrillation or ischemic heart disease.212,213
Gastrointestinal Tract
Increased appetite associated with weight loss is a very common complaint and is due to increased catabolism. Increased gastrointestinal motility of the bowel leads to frequent bowel movements and, less often, to diarrhea.215 These symptoms can be associated with some degree of malabsorption and steatorrhea, which can contribute to weight loss.216 Atrophic gastritis and/or celiac disease of autoimmune origin may be associated with Graves’ disease.211 Major toxic effects of thyroid hormone on the liver have not been reported. Nonetheless, mild elevations of liver enzymes are often detected in thyrotoxic patients.217 Thyrotoxicosis-induced liver function abnormalities can last for months and need to be taken into account when examining patients during treatment with antithyroid drugs, because they can be mistaken for adverse reactions to thionamides.
Nervous System
Psychic and nervous symptoms are a prominent and relevant part of the clinical picture.218 Insomnia and irritability are the most frequent complaints. Patients appear restless and agitated, and logorrhea is often present and becomes clearly evident during history taking. Concentration ability is also decreased. This picture may sometimes confound the physician, and manic disorders have initially been diagnosed in many patients with Graves’ disease. Fatigability and asthenia are often present and are important in differentiation from true manic or bipolar disorders. In some cases, nervous signs take the form of “apathetic thyrotoxicosis” with severe apathy, lethargy, and pseudodementia, a profile more commonly observed in elderly patients.219 In rare cases, true psychoses can be precipitated by thyrotoxicosis and improve with restoration of euthyroidism.218
The peripheral nervous system is also deeply affected.220,221 Fine distal tremor is an almost universal finding and can also be observed on protrusion of the tongue or at the eyelids. Deep tendon reflexes are brisk, with a shortened relaxation time. Clonus can be sometimes elicited. The characteristic stare of a thyrotoxic patient is due to autonomic hyperstimulation of the elevator muscle of the lid and can also be found in the absence of Graves’ ophthalmopathy. True thyrotoxic neuropathy has occasionally been reported and is characterized by areflexic flaccid quadriparesis.220,221
Muscles
Thyrotoxic patients frequently report muscle weakness and easy exhaustion. In more severe cases, atrophy of variable degree can occur in the setting of a more general wasting syndrome. Specific diseases of the muscle can be associated with Graves’ disease. Less than 1% of patients with Graves’ disease have classic myasthenia gravis, although ocular myasthenia gravis may be more frequent in these patients.222–225 Conversely, about 3% of patients with myasthenia gravis have Graves’ disease.223 The pathogenic significance of this association is not known, but it is interesting that the two prototypic diseases characterized by cell surface-receptor autoimmunity can occur together. Recognition of this association is clinically important because thyrotoxic myopathy can worsen the muscular symptoms of myasthenia. Moreover, it is important to correctly distinguish the ocular manifestations of the two disorders (they both cause diplopia), because the treatment is different. Therefore, when the degree of ocular muscle dysfunction in a patient with Graves’ disease is disproportionate to the degree of proptosis and inflammatory changes, clinical and serologic (anti–acetylcholine receptor antibodies) tests for myasthenia gravis are warranted.
In some patients, Graves’ disease thyrotoxicosis can precipitate crises of periodic hypokalemic paralysis.226 The syndrome is in all regards identical to familial periodic paralysis, but thyrotoxicosis of various causes is invariably present. The reason for the more frequent association with Graves’ disease may merely be that Graves’ disease is the most frequent cause of severe and long-lasting thyrotoxicosis in susceptible populations. Periodic hypokalemic paralysis is much more frequent in Asian subjects, in whom an association with certain HLA haplotypes has been observed,227 but it has also been reported in white and Native American patients.226 Mutations of ionic (potassium) channel genes have been shown to be responsible for some cases of hypokalemic paralysis in thyrotoxic patients.228 Effective treatments include potassium replacement, β-blocking agents, and rapid correction of thyrotoxicosis. Relapse of the hypokalemic crisis has occasionally been reported in patients after definitive treatment of the thyrotoxicosis, but it is rare.226
Skeletal System
Thyrotoxicosis is known to be associated with an increased rate of bone remodeling.229–231 The disproportionate increase in bone resorption over new bone formation leads to net bone loss, and consequently hyperthyroid patients have a reduced bone mass. Bone density improves after attainment of euthyroidism, but it often remains below the normal range. The degree of osteoporosis depends on the duration of hyperthyroidism and the coexistence of other risk factors for osteoporosis. Hence postmenopausal women with a history of hyperthyroidism have an increased risk of fractures, and hyperthyroid women are found more frequently among women with fractures.229–231 The consequences of this effect of thyrotoxicosis on public health have been demonstrated in a large epidemiologic survey showing that fracture-related mortality is significantly increased among women with a history of hyperthyroidism.232 Mild hypercalcemia and increased levels of bone turnover markers can be observed in thyrotoxic patients. Their levels are closely correlated with those of serum thyroid hormones and return to normal after correction of thyrotoxicosis.229–231 It has been recently found that the effects of thyrotoxicosis on bone may reflect the loss of a protective action that TSH exerts via the TNF-α pathway, rather than (or in addition to) a direct action of thyroid hormones on bone.233–236
Hematopoietic System
Mild leukopenia with relative lymphocytosis is a relatively common finding in patients with thyrotoxic Graves’ disease, which needs to be distinguished from antithyroid drug–induced leucopenia or agranulocytosis.237 Normocytic anemia is rare, but it can occur.238 Pernicious anemia occurs in a small minority of patients with Graves’ disease, but circulating autoantibodies to gastric parietal cells are found in a much higher percentage of cases and are a sign of associated gastric autoimmunity.211,239 Aplastic anemia has also been reported.240 Graves’ disease is occasionally associated with autoimmune thrombocytopenic purpura, but nonimmunologic alterations in hemostasis have also been reported.211 Increases in factor VIII levels and fibrinogen have been reported most consistently, but the clinical relevance of these findings is unknown.241
Reproductive System
Females: In severe thyrotoxicosis, the menstrual cycle is often deranged and characterized by oligomenorrhea or amenorrhea.242–244 As a consequence of impaired ovulation, fertility is decreased, but pregnancy can still occur. The mechanisms for these alterations are poorly understood, but they almost exclusively occur in women with severe weight loss. In women (and in men) with thyrotoxicosis, sex hormone–binding globulin (SHBG) is increased, but the physiologic consequences of this increase are unclear.245 Thyrotoxicosis in pregnancy is associated with an increased incidence of miscarriage, low-birth-weight infants, and preeclampsia (see later discussion).
Males: Gynecomastia may develop in men, and erectile dysfunction and reduced sperm count are not infrequent.246 Other features suggesting estrogenic excess include spider angiomas and reduced libido. Total testosterone is increased as a result of increased SHBG concentrations, but unbound and bioavailable testosterone levels remain in the normal range. The circulating estradiol level is increased, probably because of increased peripheral aromatization of testosterone. All these changes are fully reversible with treatment of thyrotoxicosis and require no other specific treatment. Treatment of reduced libido with testosterone may result in worsening of gynecomastia.
Metabolic Changes
Significant weight loss with normal or increased caloric intake is a hallmark of thyrotoxicosis.247 It is explained by an increased metabolic rate, with increased heat production as a net result. Mitochondrial oxygen consumption is increased by thyroid hormones in almost every tissue. Increased mitochondrial activity and numbers were also shown in several tissues in experimental thyrotoxicosis. The use of oxygen by mitochondria is inefficient in experimental thyrotoxicosis in that fewer molecules of high-energy substrates are produced per molecule of oxygen used. A widespread increase in the use of ATP by cation transporters in tissues has been proposed to explain the increased energy use, although this point is controversial.247,248 Whatever the mechanism, increased heat production and dispersion are manifested as a moderate rise in body temperature that is partially compensated for by increased sweating, heat intolerance, and weight loss.247
Peripheral utilization of carbohydrates is increased in thyrotoxicosis, in keeping with the enhanced energy consumption, and the primary mechanism seems to be an increased cellular transport of glucose.249 Thyrotoxicosis, however, also causes some degree of insulin resistance.249 Consequently, diabetes mellitus may be exacerbated by thyrotoxicosis. Type I diabetes mellitus can be associated with Graves’ disease within polyglandular autoimmune syndromes.211
Serum cholesterol and triglyceride levels are decreased in thyrotoxicosis, mainly because of a decrease in low-density lipoproteins (LDLs), in spite of an increase of hepatic lipogenesis.250,251 This reduction in lipids can result from the decrease in total body fat as a consequence of weight loss, but specific actions of thyroid hormones on lipid metabolism have also been described. Cholesterol conversion to bile acid in the liver is enhanced, and LDL receptor number on adipocytes is increased as well.250 These phenomena may account for the increased turnover of cholesterol and triglycerides. Thyroid function may alter adipokines, a number of biologically active substances produced by adipocyte with different physiologic functions, which include leptin, adiponectin, and resistin.251,252 Thyrotoxicosis was reported to be associated with elevated serum levels of adiponectin, but the finding was not confirmed.251–254 Serum leptin and resistin seem to be unaffected by thyrotoxicosis.251–254
Protein metabolism is altered during thyrotoxicosis, with both increased protein synthesis and degradation. In most cases, however, degradation predominates and causes negative nitrogen balance. This imbalance can be partly controlled by adequate calories and protein intake.247
Distinctive Manifestations of Graves’ Disease
Graves’ ophthalmopathy is the clinical manifestation of an inflammatory disorder of the orbit and is almost exclusively associated with Graves’ disease.255,256 The many aspects of this puzzling disorder are examined in detail elsewhere in this book. The prominence of the signs of Graves’ ophthalmopathy makes this feature the most striking physical finding in some patients (Fig. 9-5). Most of the manifestations of Graves’ ophthalmopathy are related to its central pathophysiologic event, an increase in the volume of retro-orbital tissue because of inflammation. As a consequence, the eye bulb is pushed forward and proptosis or exophthalmos results. Venous congestion causes swelling and edema of the periorbital tissue. Inflammatory changes also involve the extraocular muscles and may cause diplopia. The eyes are protruding, often asymmetrically. Lid retraction is seen and can be worsened by the concurrent thyrotoxicosis. Edema and swelling of the lids are also typical. The conjunctival mucosa is injected and edematous (chemosis). Lagophthalmos (incomplete palpebral closure) and lacrimal gland dysfunction may combine to cause drying of the mucosal and corneal surfaces and consequently irritation and (less often) corneal ulceration. Photophobia, burning sensation, retrobulbar pain, tearing, and a sandy sensation are common symptoms and may initially mislead clinicians into making a diagnosis of conjunctivitis. Optic neuropathy resulting from optic nerve compression by inflamed and swollen extraocular muscles at the orbital apex may occur in the most severe cases and cause reduction or loss of vision. When the proptosis is extremely severe, subluxation of the bulb outside the orbit may occur and pose an immediate threat to visual function.
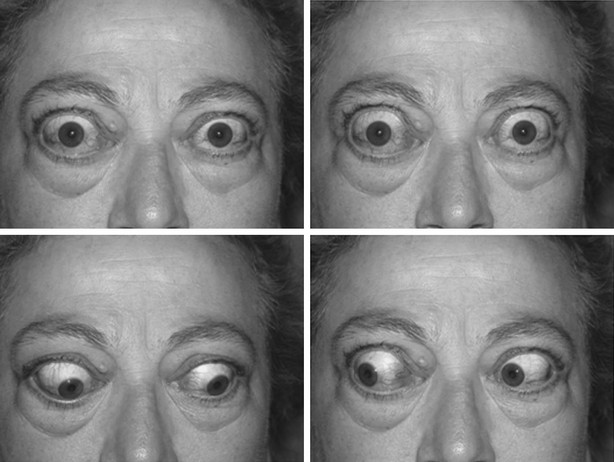
FIGURE 9-5 Typical ophthalmopathy in a patient with Graves’ disease. Proptosis, inflammatory signs (conjunctival injection, palpebral edema and redness, lacrimal gland edema), and eyelid retraction are quite evident.
Clinical signs or symptoms of ophthalmopathy are present in about 50% of Graves’ patients, with a wide variability of its degree of severity.255,256 When present, the onset of Graves’ ophthalmopathy coincides with the onset of thyrotoxicosis in about 40% of cases, follows it in another 40%, and precedes it in 20%.255,256 Even when the onset of the two disorders does not coincide, each occurs within 18 months from the onset of the first manifestation. As reported later in the chapter, the presence of ophthalmopathy may affect the choice of treatment of hyperthyroidism in patients with Graves’ disease.257
Pretibial (or Localized) Myxedema and Thyroid Acropachy
When von Basedow first described his cases of Graves’ disease,5 he also reported a puzzling manifestation of the skin characterized by a nonpitting swelling of the pretibial areas, brownish and reddish in color, well delimited, and containing little free fluid. This manifestation of unknown pathogenesis is relatively rare in patients with Graves’ disease and is almost invariably observed only when Graves’ ophthalmopathy is also present.258 Although most frequently localized to the pretibial regions, it has also been observed on the forearms and other areas.258 Different degrees of severity have been described (Fig. 9-6). Diffuse pretibial myxedema refers to the mildest form, with only superficial diffuse edema. Localized areas of more prominent infiltration that assume a papular aspect characterize the nodular form. In the most severe forms, elephantiasis occurs with extensive swelling and sometimes ulceration.
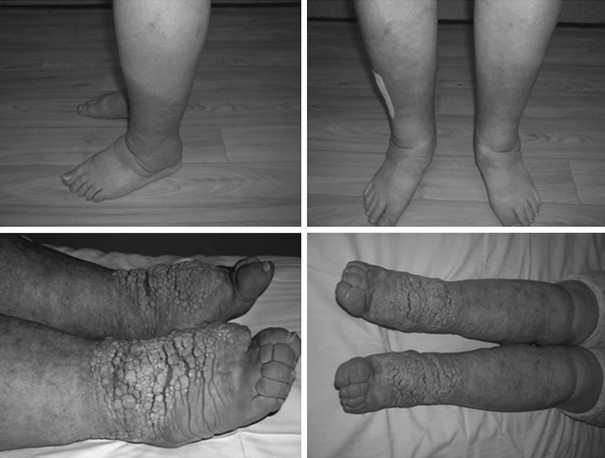
FIGURE 9-6 Two variants of pretibial myxedema patients with Graves’ disease. Upper panel: A classical form of pretibial myxedema: The skin of the pretibial area appears swollen, reddened, and typically wrinkled and takes the appearance of orange skin. Lower panel: Elephantiasic pretibial myxedema: The skin appears severely thickened with fibromatous-like lesions; inflammation is extended up to the knees.
Histopathologic studies have shown that the swelling is caused by the inordinate accumulation of hyaluronic acid in the subcutaneous layers of the involved areas, strikingly similar to the diffuse myxedematous changes of hypothyroidism but also to some extent to the changes in orbital tissues in Graves’ ophthalmopathy. A lymphocytic infiltrate may be observed, but it is by no means a constant finding. The origin of the mucinous material (hyaluronic acid) appears to be the skin fibroblast.258
The cause of pretibial myxedema is unknown, as is its relationship to the pathogenetic events of Graves’ disease.258,259 Most experts in the field agree that pretibial myxedema is another autoimmune manifestation of Graves’ disease, because of their association and of the almost invariable presence of serum TRAbs in patients with myxedema.258,259 T-cell receptor V chain restriction has been observed in the lymphocytic infiltration of patients with early pretibial myxedema and suggests the presence of an ongoing antigen-specific immune response.260 As for Graves’ ophthalmopathy, TSHR is the obvious candidate antigen.258 Using patients’ serum as a tool, putative TSH-binding sites on pretibial fibroblasts were detected.261 In addition, expression of TSHR mRNA and TSHR immunoreactivity in human fibroblasts from the pretibial region were found, as well as immunoreactivity for TSHR in pretibial connective tissue from patients with myxedema, but not from normal subjects.262–264 In contrast, thyroglobulin, another candidate antigen which may contribute to the pathogenesis of Graves’ ophthalmopathy by reaching orbital tissues from the thyroid,265 was not found in pretibial myxedema (Marinò et al., unpublished). In summary, the pathogenesis of pretibial myxedema, similar to that of Graves’ ophthalmopathy, remains obscure, although most studies support the hypothesis of an autoimmune disorder because of the ectopic expression of thyroidal antigens or the presence of cross-reacting antigens localized to restricted regions of the human skin.
Thyroid acropachy is another rare manifestation of Graves’ disease that is observed most often in long-lasting and usually severe forms of Graves’ ophthalmopathy and pretibial myxedema; it is almost invariably associated with the presence of serum TRAbs.258,266 Thyroid acropachy is characterized by clubbing and soft tissue swelling of the last phalanx of the fingers and toes, all changes that are similar to those observed in chronic respiratory insufficiency. The overlying skin is often discolored and thickened. Microscopically, increased glycosaminoglycan deposition in the skin is observed. Subperiosteal new bone formation is also present. Again, the pathogenesis and the link with the immunologic changes of Graves’ disease are unknown, being the most “popular” hypothesis similar to those formulated for Graves’ ophthalmopathy and pretibial myxedema.258,266 The disease develops without symptoms over a period of years, and it often goes unnoticed by the patient. This manifestation of Graves’ disease is harmless and asymptomatic and generally requires no treatment.
Laboratory Diagnosis
Although laboratory assessment of thyroid function is treated in detail elsewhere in this book, it seems appropriate to review in this section the mainstays of the diagnosis of Graves’ hyperthyroidism. Suspicion of hyperthyroidism can be confirmed by thyroid hormone measurements. TSH is the single most useful test in confirming the presence of thyrotoxicosis.267 By sensitive assays, TSH should be undetectable or low in all patients with thyrotoxicosis of thyroidal origin. Low TSH levels can also be observed in a number of conditions such as nonthyroidal illnesses or endogenous or exogenous corticosteroid excess. Therefore, parallel measurement of thyroid hormone levels is recommended in all patients for a correct interpretation of a low TSH level. Total thyroxine (TT4) and triiodothyronine (TT3) measurements are relatively inexpensive and reliable, but they can yield high values in otherwise euthyroid subjects with conditions characterized by increased levels of serum thyroxine-binding globulin (TBG), most commonly pregnancy, oral contraceptive use, and chronic liver disease.267 Familial excess of TBG and familial dysalbuminemic hyperthyroxinemia are rare disorders that also cause elevated TT4 levels.268 Free thyroid hormone measurements, although not completely devoid of flaws, are therefore more satisfactory but more expensive.267 In most iodine-sufficient countries, a single free thyroxine (FT4) measurement is sufficient to confirm or reject the suspicion of thyrotoxicosis. After the TSH level, FT4 is the test most often used in North America for thyroid function screening and therefore the one that most clinicians are familiar with. However, in iodine-deficient countries, a significant proportion of hyperthyroid patients (up to 12%) may have normal FT4 levels with elevated free T3 (FT3) levels, a condition termed T3 toxicosis.267 Conversely, FT4 can be falsely elevated in conditions causing reduced peripheral conversion of T4 to T3, such as amiodarone administration or high-dose propranolol treatment. In our practice, we initially measure both FT4 and FT3 levels together with TSH, with little additional expense, to obtain a complete baseline assessment of thyroid function status. Circulating thyroglobulin concentrations are high in hyperthyroidism and low in factitious thyrotoxicosis. Measurement of thyroglobulin in serum is therefore useful in the differential diagnosis of thyrotoxicosis in patients with no goiter or ophthalmopathy.269
Circulating Autoantibodies
Although by no means necessary, tests for circulating thyroglobulin (TgAb) and thyroid peroxidase (TPOAb) antibodies may be useful in confirming the presence of thyroid autoimmunity. TPOAbs can be detected by commercial radioimmunoassays in up to 90% of patients with untreated Graves’ disease,270,271 whereas TgAbs are less frequently positive, in about 50% to 60% of cases, depending on the sensitivity of the method used.272,273 Both antibodies can be detected in a relatively high percentage (up to 25%) of normal subjects, especially elderly women or in patients with other nonautoimmune thyroid disorders such as nodular goiter or thyroid carcinoma.273,274 Therefore, tests for TPOAb and TgAb have limited diagnostic value and must be considered complementary to the diagnosis.
TRAb assay is very specific and sensitive for hyperthyroid Graves’ disease. Up to 98% of untreated patients are positive with second-generation radioreceptor assays, with very few false-positive results.126 The radioreceptor test for TRAb is widely available in commercial laboratories but quite expensive. It is needed in selected situations in which confirmation of the nature of thyrotoxicosis is required or when the clinical picture or thyroid function tests are not clear. These situations include the differential diagnosis of thyrotoxicosis in pregnancy, the nodular variants of Graves’ disease, which must be differentiated from toxic nodular goiter,275 and patients with exophthalmos without thyrotoxicosis (euthyroid Graves’ disease).224
Thyroid Radioiodine Uptake and Scanning
Before the introduction of accurate thyroid hormone and TSH measurements, a radioactive iodine uptake (RAIU) test was always required for the evaluation of thyrotoxicosis. Normal ranges for RAIU vary according to the status of iodine supply in the population.276,277 In iodine-replete countries, the upper limit of normal RAIU 24 hours after the administration of a tracer dose of radioiodine is around 25%, whereas it may reach 40% in areas with mild to moderate iodine deficiency. In patients with Graves’ disease, a high value is always found after 24 hours, and in some cases the value after 3 or 6 hours can be even higher as a consequence of the rapid iodine turnover typical of Graves’ disease glands. The RAIU test is not readily available as an office-based approach and is not always required. It can be very useful, however, to rule out silent or subacute thyroiditis, factitious thyrotoxicosis, and type II amiodarone-induced thyrotoxicosis278 (Table 9-4). RAIU results can also be used before radioiodine treatment of hyperthyroidism to calculate the dose needed (see relevant heading). RAIU, like any other radioisotopic in vivo procedure, is absolutely contraindicated during pregnancy.
Thyroid imaging with radioisotopes can be performed with radioiodine at the time that RAIU is performed or by using pertechnetate 99m (Fig. 9-7A). Thyroid scanning in Graves’ disease is useful when coexisting nodules are detected by palpation and their functional status needs to be evaluated.
FIGURE 9-7 Thyroid scanning (A), ultrasound (B), and color flow Doppler (C) findings in Graves’ disease. A, An enlarged thyroid with diffuse homogeneous and active uptake is observed at Pertechnetate 99m scanning. B, The thyroid is enlarged, and the tissue appears diffusely hypoechoic. C, Markedly increased signals show the diffuse increase in thyroid vascularity.
Thyroid Ultrasound
The thyroid gland is an excellent candidate for ultrasound studies, given its superficial anatomic location and the presence of a high number of liquid-solid interfaces (virtually one at every follicle/colloid-lining surface), which in normal conditions produce high reflectivity. Although the equipment is still relatively quite costly, it is now affordable at the office level and requires little maintenance. Thyroid ultrasound is an invaluable tool in the diagnosis of thyroid nodular disorders and has become more and more an extension of the physical examination for many endocrinologists. Its application to non-nodular disorders of the thyroid has come from early observations of typical changes of the gland’s echoic structure in thyroid autoimmune diseases.279 In hyperthyroid Graves’ disease, the echoic pattern undergoes diffuse changes. The tissue, possibly because of the reduction in colloid content, the increase in thyroid vascularity, and the lymphocytic infiltrate, becomes typically hypoechoic (Fig. 9-7B). This pattern is similar to the one observed in chronic thyroiditis and, when diffuse, is almost pathognomonic of thyroid autoimmunity.280 Therefore, thyroid ultrasound can be useful during the evaluation of thyrotoxicosis to confirm the suspicion of thyroid autoimmunity. Thyroid ultrasound also allows accurate measurement of thyroid size,281 information that can help in the decision-making process when definitive treatment is being planned (see later). Moreover, careful definition of coexisting nodules can be obtained when needed. Therefore, thyroid ultrasonography provides very useful information in the initial evaluation of patients with Graves’ disease, although it is seldom strictly required for purely diagnostic purposes.
Color flow Doppler (CFD) techniques have been applied to the study of glands with Graves’ disease. CFD allows semiquantitative measurement of blood flow to the thyroid gland.282 In untreated Graves’ disease, a distinct CFD pattern characterized by markedly increased signals with a patchy distribution is usually observed (Fig. 9-7C). This pattern, in conjunction with a hypoechoic pattern, allows distinction from Hashimoto’s thyroiditis. In this setting, CFD studies of the thyroid gland can be useful in distinguishing hyperthyroidism of Graves’ disease from thyrotoxicosis of other causes, such as amiodarone-induced thyrotoxicosis, subacute thyroiditis, painless thyroiditis and factitious thyrotoxicosis. For this reason, CFD evaluation may become a valid substitute for RAIU when these conditions are included in the differential diagnosis.