[level-membership-for-pediatrics-category]
Chapter 131 Graft Versus Host Disease (GVHD) and Rejection
Acute GVHD
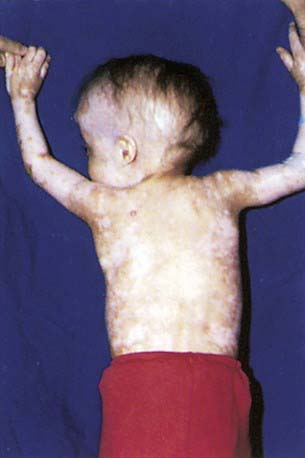
Acute GVHD usually develops from 2-5 wk post-transplantation. The primary manifestations are an erythematous maculopapular rash, persistent anorexia, vomiting and/or diarrhea, and liver disease with increased serum levels of bilirubin, alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase (Table 131-1). Diagnosis may benefit from skin, liver, or endoscopic biopsy for confirmation. Endothelial damage and lymphocytic infiltrates are seen in all affected organs. The epidermis and hair follicles of the skin are damaged, the hepatic small bile ducts show segmental disruption, and there is destruction of the crypts and mucosal ulceration of the gastrointestinal tract. Grade I acute GVHD (skin rash alone) has a favorable prognosis and often does not require treatment (Fig. 131-1). Grade II GVHD is a moderately severe multiorgan disease requiring therapy. Grade III GVHD is a severe multiorgan disease, and grade IV GVHD is a life-threatening, often fatal condition. The standard prophylaxis of GVHD relies mainly on post-transplant administration of immunosuppressive drugs such as cyclosporine or tacrolimus, often in combination with methotrexate or prednisone, anti–T-cell antibodies, mycophenolate mofetil, or other immunosuppressive agents. An alternative approach that has been widely used in clinical practice is the removal of T lymphocytes from the graft (T-cell depletion). Any form of GVHD prophylaxis in itself may impair post-transplant immunologic reconstitution, increasing the risk of infection-related deaths. T-cell depletion of the graft has also been associated with an increased risk of leukemia recurrence in patients transplanted from an HLA-identical sibling or an unrelated volunteer.

Chronic GVHD
Patients with chronic GVHD involving only the skin and liver have a favorable course (Fig. 131-2). Extensive multiorgan disease may be associated with a very poor quality of life, recurrent infections associated with prolonged immunosuppressive regimens to control GVHD, and a high mortality rate. Morbidity and mortality are highest in patients with a progressive onset of chronic GVHD that directly follows acute GVHD, intermediate in those with a quiescent onset after resolution of acute GVHD, and lowest in patients with de novo onset in the absence of acute GVHD. Single-agent prednisone is standard treatment at present, although other agents, including extracorporeal photopheresis, mycophenolate mofetil, anti-CD20 monoclonal antibody, and pentostatin, have been employed with variable success. Treatment with imatinib mesylate, which inhibits the synthesis of collagen, has been effective in patients with chronic GVHD and sclerotic features. As a consequence of prolonged immunosuppression, patients with chronic GVHD are particularly susceptible to infections and should receive appropriate antibiotic prophylaxis, including trimethoprim-sulfamethoxazole. Chronic GVHD resolves in most patients but may require 1-3 yr of immunosuppressive therapy before the drugs can be withdrawn without the disease recurring. Chronic GVHD also promotes the development of secondary neoplasms.
Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815-1822.
Ferrara JLM, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1560.
Fleischauer K, Locatelli F, Zecca M, et al. Graft rejection after unrelated donor hematopoietic stem cell transplantation for thalassemia is associated with non-permissive HLA-DPB1 disparity in host-versus-graft direction. Blood. 2006;107:2984-2992.
Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Developmental Committee of the European Group for Blood and Marrow Transplantation. Lancet. 2008;371:1579-1586.
Martin PJ. Choosing between GVHD and delayed engraftment. Nat Med. 2009;15:363-364.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 131 Graft Versus Host Disease (GVHD) and Rejection
Acute GVHD
Acute GVHD usually develops from 2-5 wk post-transplantation. The primary manifestations are an erythematous maculopapular rash, persistent anorexia, vomiting and/or diarrhea, and liver disease with increased serum levels of bilirubin, alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase (Table 131-1). Diagnosis may benefit from skin, liver, or endoscopic biopsy for confirmation. Endothelial damage and lymphocytic infiltrates are seen in all affected organs. The epidermis and hair follicles of the skin are damaged, the hepatic small bile ducts show segmental disruption, and there is destruction of the crypts and mucosal ulceration of the gastrointestinal tract. Grade I acute GVHD (skin rash alone) has a favorable prognosis and often does not require treatment (Fig. 131-1). Grade II GVHD is a moderately severe multiorgan disease requiring therapy. Grade III GVHD is a severe multiorgan disease, and grade IV GVHD is a life-threatening, often fatal condition. The standard prophylaxis of GVHD relies mainly on post-transplant administration of immunosuppressive drugs such as cyclosporine or tacrolimus, often in combination with methotrexate or prednisone, anti–T-cell antibodies, mycophenolate mofetil, or other immunosuppressive agents. An alternative approach that has been widely used in clinical practice is the removal of T lymphocytes from the graft (T-cell depletion). Any form of GVHD prophylaxis in itself may impair post-transplant immunologic reconstitution, increasing the risk of infection-related deaths. T-cell depletion of the graft has also been associated with an increased risk of leukemia recurrence in patients transplanted from an HLA-identical sibling or an unrelated volunteer.
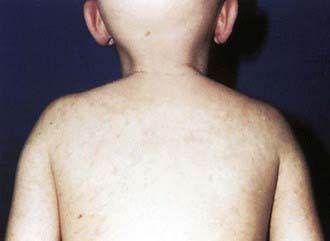
Figure 131-1 Acute graft versus host disease of the skin with ear, arm, shoulder, and trunk involvement.
(Courtesy of Evan Farmer, MD.)
[/not-level-membership-for-pediatrics-category]
