44
Graft-Versus-Host Disease
• Table 44.1 lists risk factors for GVHD in HSCT recipients.
Table 44.1
Risk factors associated with the development of graft-versus-host disease (GVHD).
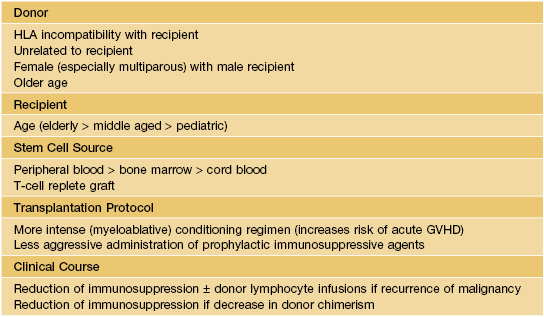
HLA, human leukocyte antigen.
• Divided into acute and chronic forms based on clinical features.
Acute GVHD
• Most often arises 4–6 weeks after HSCT with traditional regimens.
• Typically presents as a morbilliform exanthem, often with perifollicular accentuation.
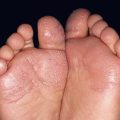
• Clinical staging is based on the proportion of the cutaneous surface involved: Stage 1, <25%; Stage 2, 25–50%; and Stage 3, >50%. Stage 4 represents erythroderma with bullae/epidermal detachment resembling toxic epidermal necrolysis (Fig. 44.1).
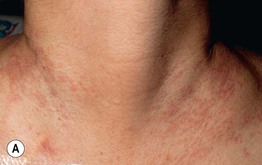
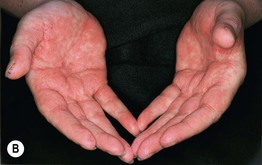
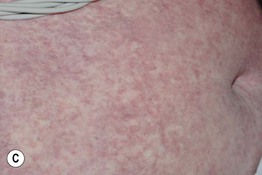
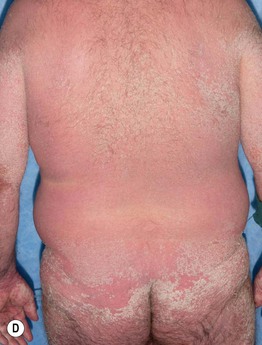
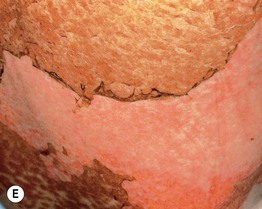
Fig. 44.1 Clinical spectrum of acute cutaneous graft-versus-host disease. A Stage 1 – discrete and coalescing small pink papules on the upper chest and neck of a woman 6 weeks following allogeneic bone marrow transplant. B, C Stage 2 – pink macules and papules of the palms that are becoming confluent 14 weeks post allogeneic bone marrow transplant and pink-violet macules and minimally elevated papules on the abdomen in a liver transplant recipient. D Stage 3 – diffuse erythema with desquamation, but without bullae formation. E Stage 4 – coalescence of bullae plus epidermal necrosis leading to large areas of denudation in a patient who had received an allogeneic bone marrow transplant; note the resemblance to toxic epidermal necrolysis. A, D, Courtesy, Edward Cowen, MD; B, Courtesy, Dennis Cooper, MD; C, Courtesy, Julie V. Schaffer, MD.
Chronic GVHD
• Skin and mucosal involvement are extremely common and highly variable in their presentations (Table 44.2; Fig. 44.2A), and almost any organ system can be affected (Fig. 44.2B).
Table 44.2
Mucocutaneous manifestations of chronic graft-versus-host disease (GVHD).
Diagnostic features are in bold; other signs and symptoms listed are not considered sufficient to establish a diagnosis of chronic GVHD without further testing (e.g. histologic assessment) or evidence of other organ system involvement.
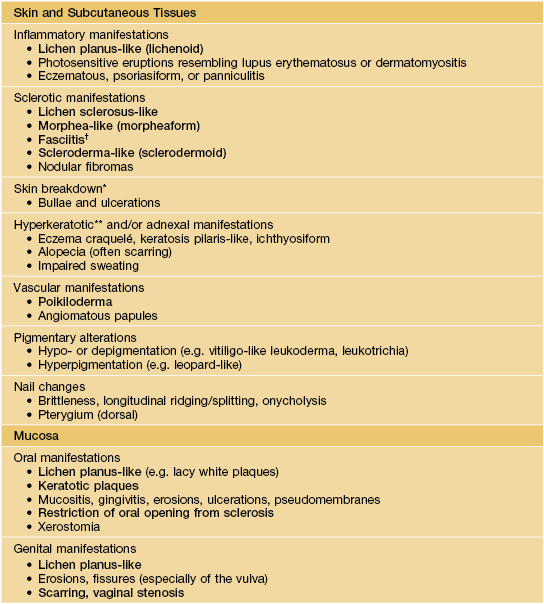
* Often overlying areas of cutaneous sclerosis.
† Magnetic resonance imaging can help to establish the diagnosis.
** May be related to destruction of eccrine glands.
Based on Filipovich AH, Weisdorf P, Pavletic S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. Diagnosis and Staging Working Group report. Biol. Blood Marrow Transplant. 2005;11:945–956. For manifestations in other organs (including the eye), see this reference as well as the text.
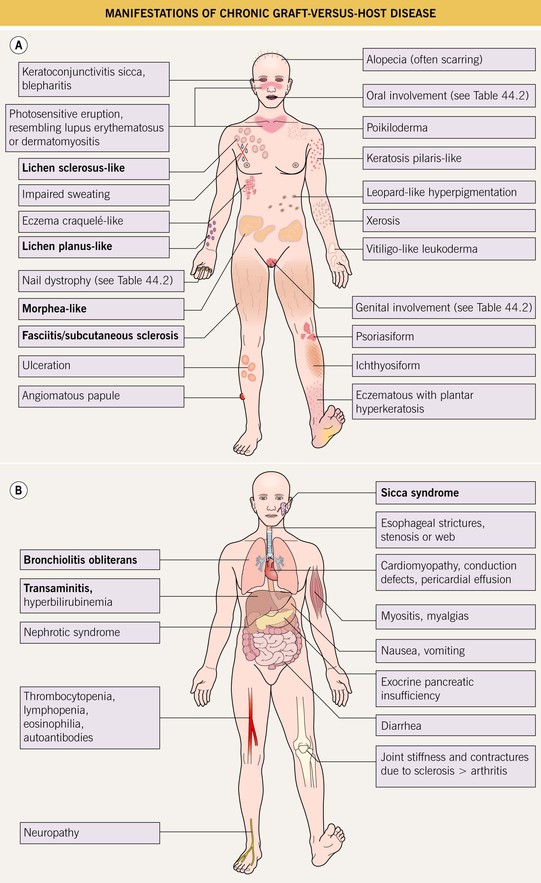
Fig. 44.2 Manifestations of chronic graft-versus-host disease. A Mucocutaneous findings. Lichen sclerosus-like lesions may be prominent on the back. Highly characteristic manifestations are in bold. B Internal involvement. The most common manifestations are in bold.
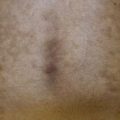
• Reticulate pink to violet papules with scale (lichen planus-like) favoring the dorsal hands and feet, forearms, and trunk represent one characteristic manifestation (Fig. 44.3A).
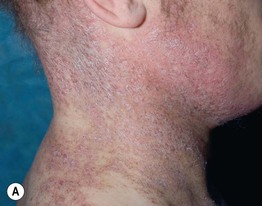
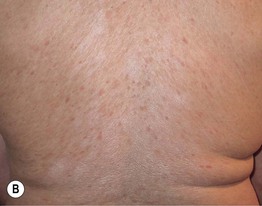
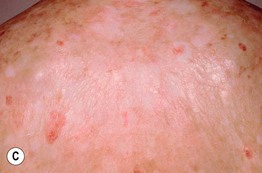
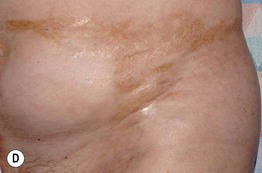
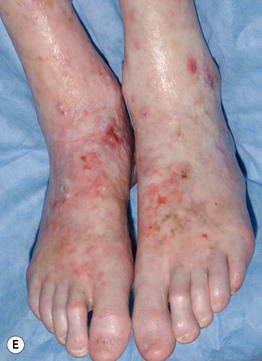
Fig. 44.3 Clinical spectrum of chronic cutaneous graft-versus-host disease. A Lichen planus-like – thin pink-violet papules and plaques with scale, admixed with post-inflammatory hyperpigmentation. B Lichen sclerosus-like (early) – multiple gray-white thin plaques with obvious wrinkling are present on the mid back. C Lichen sclerosus-like (late) – thicker shiny white plaques on the upper back admixed with erosions. D Morphea-like (morpheaform) – shiny, hyperpigmented, sclerotic plaque extending circumferentially around the beltline and into the inguinal area. E Scleroderma-like (sclerodermoid) – the skin is shiny and bound-down with dyspigmentation, hair loss, and multiple erosions; nail loss, small angiomatous nodules, and marked reduction in range of motion of the ankles are also present. F Eosinophilic fasciitis-like – a rippled appearance and irregular nodular texture to the skin is indicative of involvement of subcutaneous tissues; extension of the elbow is limited. Especially in the earlier, more edematous phase, the presence of hypereosinophilia is a clue to the diagnosis. A, D-F, Courtesy, Edward Cowen, MD; B, Courtesy, Dennis Cooper, MD; C, Courtesy, Joyce Rico, MD.
• The spectrum of sclerotic involvement includes.
– Lichen sclerosus-like shiny, wrinkled, gray-white plaques, ± follicular plugging, favoring the upper trunk (Fig. 44.3B,C).
– Localized or widespread morphea-like indurated, hyperpigmented to skin-colored plaques favoring the lower trunk (Fig. 44.3D).
– Eosinophilic fasciitis-like presentations with acute edema and pain evolving into areas of firm skin with subcutaneous rippling (‘pseudo-cellulite’; Fig. 44.3F) and linear depressions following a vein or between muscle groups (groove sign); favors the extremities (sparing the hands and feet) and may result in joint contractures.
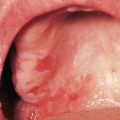
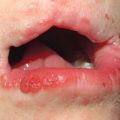
• Often affects the nails and the oral and genital mucosa (Fig. 44.4; see Table 44.2).
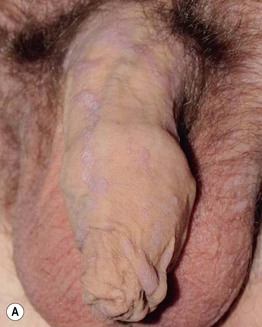
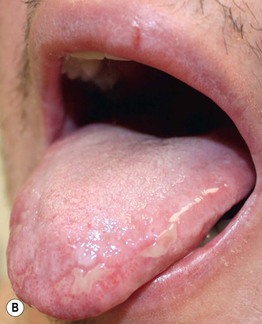
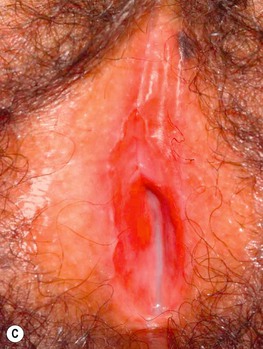
Fig. 44.4 Orogenital involvement in chronic cutaneous graft-versus-host disease. A, B Lichen planus-like – flat-topped violet papules on the penis and several ulcers on the tongue, also with a lacy white pattern on the upper vermilion lip and the distal dorsal tongue. C Severe erosive disease of the vulva, with nearly total resorption of the labia minora and agglutination of the lips of the clitoral hood. The vulvar introitus is also markedly narrowed. A, C, Courtesy, Edward Cowen, MD. B, Courtesy, Jean L. Bolognia, MD.
For further information see Ch. 52. From Dermatology, Third Edition.