CHAPTER 141 Glomus Tumors
The original oncotype is considered to be the “glomus cell,” a modified neuroblast of the neural crest; it differentiates into an epithelioid type that migrates during embryogenesis toward the autonomic ganglia1 and has the potential to store and secrete catecholamines in response to neuronal or chemical signals.2–4 The largest collection of these “neurocrine” cells is found in the adrenal medulla. Smaller colonies are physiologically present in the cardioaortic and laryngeal bodies near the carotid bifurcation, the adventitia of the jugular bulb, the promontory of the middle ear, and the vagal nerve, but their neoplastic representatives may be found in a wider range of locations.5–22
As a result of the various locations, a variety of eponyms have been coined to characterize this oncologic lineage. The term chemodectoma has mostly been used to refer to carotid body tumors because of their oxygen sensor,1,23,24 whereas the term paraganglioma or nonchromaffin paraganglioma and, in particular, glomus tumor or glomangioma have arbitrarily been used when dealing with other anatomic sites more familiar to neurosurgeons, otolaryngologists, and head and neck surgeons. Specifically, these sites include (1) the jugular bulb and foramen (“glomus jugulare”); (2) Jacobsons’s nerve, or the tympanic branch of the glossopharyngeal nerve (“glomus tympanicum”); and (3) Arnold’s nerve, or the auricular branch of the vagus nerve (“glomus vagale” or “juxtaovale”).23,25 Finally, the acronym “APUDOMA”26 or “intracranial pheochromocytoma”3 was created to define the limited fraction of secreting, non–chromaffin-containing paragangliomas that—similar to carcinoid tumors—maintain the potential for Amine Precursor Uptake and Decarboxylation, sometimes associated with carcinoid syndrome.27
Historical Background
The carotid body was first described by Van Haller in 1743,28 whereas the first surgical removal of the carotid body was performed by Riegner in 1880.29 The earliest reports on glomus tympanicum date back to Valentin (1840), who thought that the small cellular cluster near the tympanic nerve was a ganglion,30 and to Krause (1878), who was somehow impressed by the similarity between the glomus and the carotid body.31 In 1945, Rosenwasser, reporting on the surgical removal of a “carotid-body-like tumor” located in the middle ear, further stressed this consistent analogy.32 In 1941, Guild described the glomus jugulare in the adventitia of the jugular bulb.33
A few years later (1951), neoplastic lesions arising from paraganglionic cells in this area were called glomus jugulare tumour by Winship and Louzan.34 Finally, in 1953, Guild extensively analyzed a large series (88 specimens) of human temporal bone sections and substantially outlined the current glomus nosography by classifying the three major varieties as jugulare, tympanicum, and vagale.35
Surgical exploration of the jugular bulb was bravely initiated by Seiffert in 1934, Lundgren in 1949,36 and more skeptically by Weille and Lane in 1951.37
The suboccipital approach to this region was first reported in 1953 by Semmes38 and by Albernaz and Buzcy.39 During the following 2 decades, more aggressive surgical techniques were gradually introduced with the aim of removing even larger tumors usually considered inoperable, as well as to gain complete “anatomic control” of such a crucial area. The main achievements in this period, which were put into practice by Shapiro and Neues in 196440 and Gejrot in 1965,41 led to progressively more extended craniocervical incisions and wider skull base approaches, including either established techniques, such as labyrinthectomy (Gastpar, 196142) and radical mastoidectomy (Hilding and Greenberg, 197143), or sophisticated refinements such as mobilization and rerouting of the facial nerve for packing/resecting the sigmoid sinus or jugular bulb40–43 and eventually to temporal bilateral carotid artery occlusion with hypothermia.44 In 1969, House proposed his famous approach through the facial recess, which resulted in higher rates of cranial nerve preservation.45 This outstanding innovation was followed in the late seventies and eighties by the infratemporal fossa route proposed by Fisch and coworkers to overcome problems with exposure of the internal carotid artery46–48; the major variations of this technique were conceived to tackle a spectrum of anatomic extensions and different neoplasms of medium and large size.
In contrast, more limited approaches were devised for safe removal of smaller lesions: Mischke and Balkany’s “posterior-inferior” (1980) technique for sparing both the auditory and facial canal49 and Farrior’s (1984) “anterior hypotympanic” technique.50
In the mid-1980s, Brown published one of the largest surgical series,51 Donald and Chole described their transcervical-transmastoid approach,52 and above all, Gardner, Robertson, and colleagues reported their wide experience46,53; the latter repeatedly stressed the importance of comprehensive team management of these tumors and advocated cooperative effort by otologists, neurosurgeons, craniofacial surgeons, anesthesiologists, internal medicine specialists, and rehabilitation experts not only to optimize surgical performances but also to improve clinical and neurological outcomes. Their famous “lateral skull base approach” is a prime example of this multidisciplinary attitude.4,23,46,53
A wide spectrum of different grading systems were gradually introduced into the literature: Kinney54; Fisch (Table 141-1)47; Jackson, Glasscock, and coworkers (Table 141-2)55; Rockley and Hawke56 and Alford and Gilford57 (Table 141-3) (Figs. 141-1 and 141-2); and Spector and colleagues (Table 141-4).58
| CLASS | DESCRIPTION |
|---|---|
| A | Tumor limited to the middle ear cleft (glomus tympanicum) |
| B | Tumor limited to the tympanomastoid area with no involvement of the infralabyrinthine compartment |
| C | Tumor involving the infralabyrinthine compartment of the temporal bone and extending into the petrous apex |
| C1 | Tumor with limited involvement of the vertical portion of the carotid canal |
| C2 | Tumor invading the vertical portion of the carotid canal |
| C3 | Tumor invading the horizontal portion of the carotid canal |
| D | |
| D1 | Tumor with intracranial extension <2 cm in diameter |
| D2 | Tumor with intracranial extension >2 cm in diameter |
From Fisch U. The infratemporal fossa approach for extensive tumors of the temporal bone and base of the skull. In: Silverstein H, Norrell H, eds. Neurological Surgery of the Ear. Birmingham, AL: Aesculapius; 1977:34-53.
TABLE 141-2 Glasscock-Jackson Classification of Glomus Jugulare Tumors
| CLASS | DESCRIPTION |
|---|---|
| I | Small tumor involving the jugular bulb, middle ear, and mastoid |
| II | Tumor extending under the internal auditory canal; may extend into the intracranial canal |
| III | Tumor extending into the petrous apex; may extend into the intracranial canal |
| IV | Tumor extending beyond the petrous apex into the clivus or infratemporal fossa; may extend into the intracranial canal |
From Jackson CG, Glasscock ME 3rd, Nissen AJ, et al. Glomus tumor surgery: the approach, results, and problems. Otolaryngol Clin North Am. 1982;15:897-916.
TABLE 141-3 Synoptic Comparison of “Predictive” Gradings in the Rockley-Hawke56 (1990) and Alford-Gilford57 (1962) Categorization
| ROCKLEY-HAWKE | ALFORD-GILFORDPrognosis | |
|---|---|---|
| Type of Glomus Tumor | Arising From | |
| Glomus tympanicum | ||
Data from Rockley TJ, Hawke M. Glomus bodies in the temporal bone. J Otolaryngol. 1990;19:50-56; and Alford BR, Gilford FR. A comprebensive study of tumors of the glomus jugulare. Laryngoscope. 1962;72:765-805.
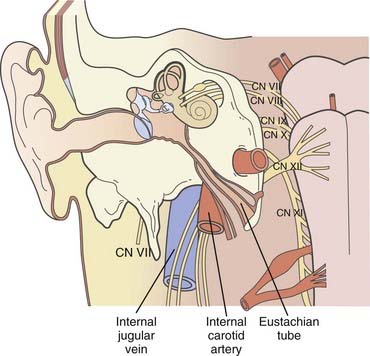
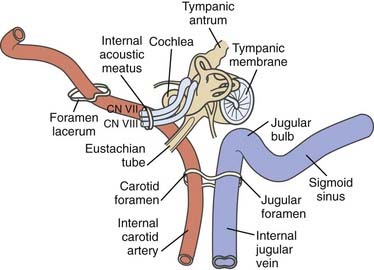
TABLE 141-4 Classification of Glomus Jugulare Tumor Spreading Patterns According to Spector and Colleagues
| CLASS | DESCRIPTION |
|---|---|
| I | Down the eustachian tube to the nasopharynx and foramina of the skull base |
| II | Along the carotid artery into the middle fossa |
| III | Along the jugular vein into the posterior fossa |
| IV | Through the tegmen tympani to the floor of the middle fossa |
| V | Through the round window of the labyrinth, invading the internal auditory canal, into the cerebellopontine angle |
From Spector GJ, Druck NS, Gado M. Neurologic manifestations of glomus tumors in the head and neck. Arch Neurol. 1976;33:270-274.
Despite their discrepancies in terms of specific coordinates, these grading systems have been extremely useful in planning surgical strategies and have been shown to reliably predict clinical outcomes. The use of more extensive, combined infratemporal and posterior fossa approaches to achieve adequate exposure, even with large lesions, as well as to avoid multiple-stage surgery, has repeatedly been emphasized in the last 2 decades. Al-Mefty and colleagues in 1987,59 Bordi and coworkers in 1989,60 Patel and associates in 1994,61 and Al-Mefty and Teixeira in 200262 have exhaustively shown the potential of integrated surgical routes (subtemporal/infratemporal/retrosigmoid–extreme lateral–transcondylar, and so on), particularly when dealing with the specifically defined “complex” variety of GTs,62 such as giant, multiple, secreting, or malignant lesions that have previously failed endovascular or radiation treatment.
A major advance in surgical results is certainly related to the introduction of preoperative superselective embolization, with Hilal and Michelsen in 197563 and Brismar and Cronqvist in 197864 providing the original “basic” contributions and Simpson and coworkers publishing their experience with GT in 1979.65 Given the limited tumor volume coverage of current techniques and the significant rates of revascularization after endovascular occlusion, embolization per se represents a palliative option that is often effective in reducing tinnitus, vertigo, and dizziness.66,67 As confirmed by accurate analyses, this technique is essentially “ancillary” to surgery and provides a substantial reduction in both intraoperative blood loss and operating time while increasing the extent of surgical resection.64–6668 However, endovascular procedures do not seem to reduce clinical sequelae; in fact, they may entail additional neurological risk.25,68–70
Fractionated radiation therapy (FRT) was first proposed as an alternative to surgery for critical GTs in 1952: Capps treated four consecutive patients and achieved good results in all of them.70 Subsequently, “conventional” radiotherapy has been used for GTs either as a primary or salvage treatment modality.71 Mainly because of their documented dose-response correlation,72,73 GTs may be considered relatively radiosensitive oncotypes,74,75 although this concept remained controversial for decades after a skeptical analysis published by Spector and associates in 197376 and the ensuing debate on parameterized radiation dose/effect with these tumors.75,77
Stereotactic radiosurgery (SRS) has recently been proposed as a potential therapeutic option for GTs.77–82 Because of the particular conformity and selectivity of updated radiosurgical dose planning in these tumors, SRS may achieve local tumor control (LTC) rates and clinical responses equivalent to or even higher than those obtained with FRT, and the incidence of sequelae seems to be lower because of the smaller volume of normal tissue exposed to radiation.77,78,81,82
Epidemiology
GTs are rare, with an incidence of approximately 1.2 to 3 cases per million people77,84; a strict minority are genetically inherited.85–88 GTs account for 0.012% to 0.03% of all newly diagnosed neoplasms and 0.6% of all head and neck tumors.78,89–91 Clinical onset is typically in the sixth and seventh decades of life,51,77,78,80,81,92,93 with a marked predilection in women and a female-to-male ratio as high as 5 : 1 to 6 : 1.51,77,78,80,81,89,92–95 Furthermore, the incidence of GTs is higher in populations living at higher altitudes than in people living at sea level96 and in patients suffering from hypoxemic conditions such as chronic obstructive pulmonary disease.96–98
In the statistically predominant nonfamilial tumors, 10% of cases are multicentric.85–86 In the 20% to 40% familial forms, bilateral involvement is rather common,87,88 and multicentricity occurs in 25% to 78% of patients.89,99,100 The tumor development rate seems consistent with autosomal dominant inheritance.89,99–101
Clinical Signs and Symptoms
The vast majority of GTs are indolent, with slow, locally invasive and destructive growth. The estimated average interval between the first symptom and diagnosis is 4 to 6 years,75,77 and the median doubling time has been calculated to be 4.2 years.89 Neurological manifestations are primarily related to the specific “region” infiltrated, which may eventually involve neurovascular structures within the temporal bone, particularly the middle ear; distal structures such as the jugular foramen, hypoglossal canal, and clivus; and more cephalad, the intrapetrous carotid artery and cavernous sinus.92,102 GTs can quite rarely become an immediately life-threatening condition, but in the mid- to long-term range, they usually jeopardize quality of life.103
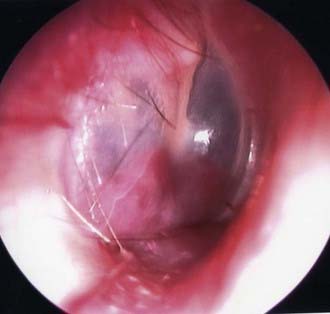
In the typical glomus tympanicum tumor the basic complaints at onset are mostly pulsating tinnitus, followed by conductive hearing loss and vertigo.23,104–106 On otomicroscopic examination, the lesion appears reddish to bluish beneath the eardrum (Fig. 141-3).
The tympanic membrane can be perforated by neoplastic invasion of the external auditory canal, and bloody otorrhea may occur. Involvement of the lower cranial nerves is usually the next step in the tympanic variety and a more precocious feature in the jugulare or vagale type. According to the literature, facial nerve dysfunction affects 8% to 33% of patients,107,108 paresis of the vagal nerve affects 6% to 30%,109 and dysfunction of the glossopharyngeal, spinal accessory, and hypoglossal nerves occurs less frequently.110,111
GTs usually have a nonsecreting phenotype.68,92,102,112,113 The approximately 1% to 3% catecholamine-secreting histotypes101,114 seem to be more common in males and younger patients.112 In such cases, the initial symptoms may include a serotonin hypersecretion–related carcinoid syndrome4,23,27 consisting of headache, facial flushing, mild hypertension, and diarrhea.23,27
A strict minority of these tumors may behave quite aggressively. Malignant variants account for 2% to 4.5% of all reported cases,57,93,115–117 with the preferred sites of colonization being the lymph nodes, liver, and lung.93,110,116–118
Pathology
However, several different anatomic-surgical classifications have been proposed. Because most of them are based either on controversial histogenetic issues or on the original extension and spreading pathways of the tumor (i.e., tumor growth topography), this particular nosography has frequently been used for predicting the natural history and surgical outcome. The preliminary study of Alford and Gilford (1962) was the first to differentiate the relatively more benign course of the tympanic form from the worse profile of the jugular variety.57 The concept was further stressed by Kinney,54 who devised one of the first staging systems for GTs based on intracranial extension, and by Rockley and Hawke in 1990, who based the prognosis of the three major varieties on the original site of the lesion.56
As mentioned earlier, Fisch47 (1977) and Jackson and colleagues55 (1982) exhaustively defined the main surgical-prognostic categories of GTs according to the size and location of the lesion, and their work received wide clinical and scientific consensus. This idea had been heralded by Spector and colleagues (1976)58 with their original classification of the main anatomic spreading pathways of these tumors (see Table 141-4). The degree of bony involvement was the crucial cutoff point for predictability.
Histology
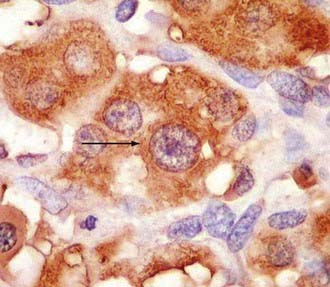
Glomus and carotid body tumors share common histologic patterns. The dominant oncotypes are large, polygonal epithelioid cells with scanty, eosinophilic, fine granular cytoplasm that may be positive for norepinephrine on immunohistochemistry. The rounded, hyperchromatic nuclei may sometimes have mitotic figures, as well as prominent nucleoli or other atypical features (Figs. 141-4 and 141-5). However, these cytologic hints of malignancy, easily trackable in most of these specimens, poorly correlate with the effective metastatic potential of the tumor, which is rarely displayed.4,23,89,117
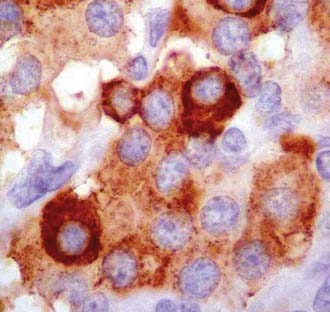
FIGURE 141-4 Typical oncotypes: large polygonal cells with scant eosinophilic, fine granular (chromogranin) cytoplasm.
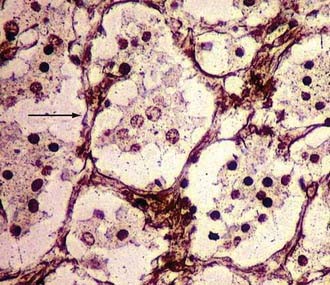
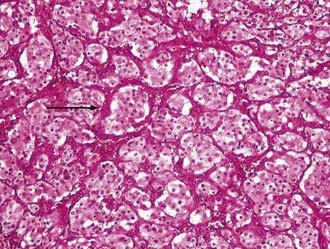
As regards histoarchitecture (Figs. 141-6 and 141-7), these cells usually form clusters or nests, hence the term Zellballen, interspersed among a reticulin network full of thin-walled capillaries. These capillaries provide an unusually rich blood supply that attracts vessels not only from the external carotid system but also from branches of the internal carotid artery.
Genetics and Molecular Biology
Familial GTs represent approximately 20% to 40% of reported cases, and the genetic defects are known just partially.24 In the largest subgroup (10% of all GTs), the cause has been documented to be sporadic germline mutations in two putative regions on chromosome 11: PGL1 at 11q23.1 and PGL2 at 11q13.1.119–122 Both regions encode for succinate dehydrogenase (SDH) subunits, the former for the putatively most relevant subunit “D,” which is localized in the mitochondrial membrane and participates in the Krebs cycle and the mitochondrial respiratory chain. Baysal and coauthors proposed that SDH is a critical component of a cellular oxygen-sensing system.123 Mutations in SDH may incapacitate this oxygen relay and lead to a hypoxic state that triggers cell proliferation. Support for this hypoxia-induced hyperplasia comes, as seen earlier, from epidemiologic studies showing an increased incidence of GTs in people living at higher altitude or suffering from chronic respiratory diseases.96–98124
An additional locus, PGL3, has been identified on chromosome 1q.125,126 Reliable genetic linkage explaining the observed association with neurofibromatosis has never been found.127 The pathogenesis of nonfamilial forms remains unexplained, but the most attractive theories involve angiogenesis mechanisms and apoptosis. Most angiogenesis-related molecules (fibroblast growth factor, platelet-derived growth factor, transforming growth factor-β1, and so on) are poorly represented in these tumors. In contrast, vascular endothelial growth factor (VEGF) and platelet-derived endothelial cell growth factor are extensively represented at the cellular (65%) and stromal (77%) levels, respectively.128 Similar observations have been reported for endothelin-1.129 Experimental protocols have shown that anti-VEGF treatment may reduce GT size in rats.130
Finally, with regard to apoptosis, high bcl-2 expression levels have been found in GTs.131 Because bcl-2 is an antiapoptotic homologue of bax and overexpression of it forestalls apoptosis and thus increases cell survival, this molecule, like murine double minute oncogene (MDM2), nuclear factor NF-κB,132 and other substances inhibiting apoptotic cascades, is currently being investigated to explore its role in the indolent course of these tumors.131
Diagnosis
In selected cases, a potential role of fine-needle aspiration biopsy is still recognized.133,134 However, to date, the basic diagnostic steps in these patients have essentially involved neuroradiology.
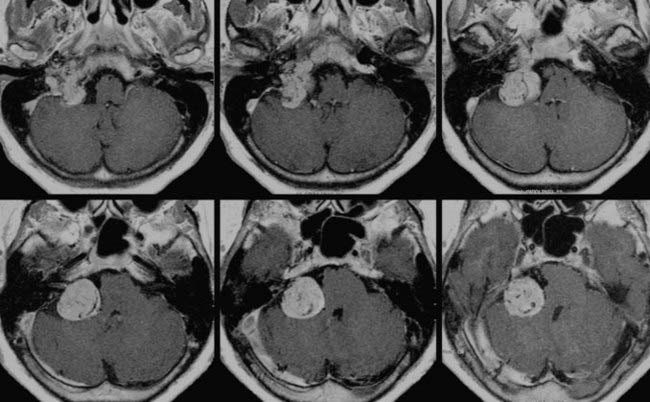
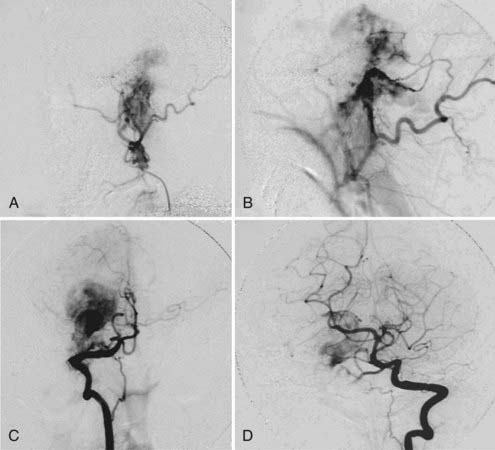
FIGURE 141-9 Same patient as in Figure 141-8, angiographic view. A, External carotid artery. B, Occipital artery. C, Right vertebral artery. D, Left vertebral artery.
Treatment Options
Endovascular Treatments: Potential and Problems
The option of superselective, preoperative endovascular embolization was initially added to the therapeutic armamentarium for GTs approximately 3 decades ago,65 and to date, combined treatment consisting of embolization and immediate surgery is still generally accepted to be the best approach for resectable GTs.4,23,25,65 In fact, more recently139–144 and particularly after the introduction of nonabsorbable embolic molecules, the potential for use of this technique has expanded, and the related limitations, pitfalls, and risks have been clarified (Table 141-5).
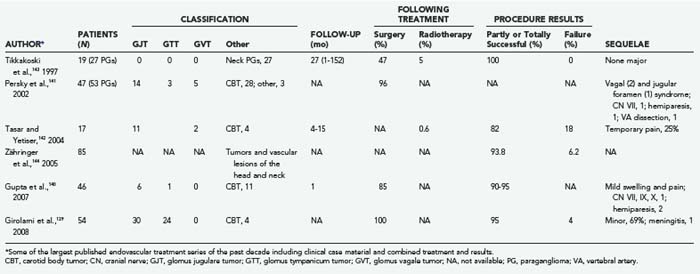
The major pros and cons of endovascular embolization can be summarized as follows:
Surgery
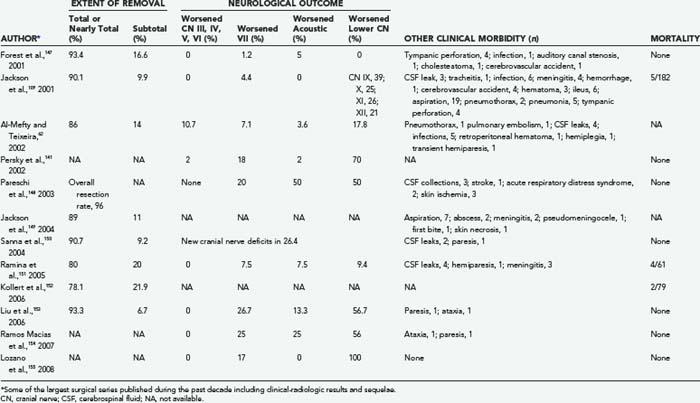
For more than a century surgery has been the therapeutic mainstay for patients harbouring GTs, particularly young healthy subjects, either as an unavoidable option, such as in presence of severe intracranial hypertension or brainstem compression, or as an elective procedure, frequently following endovascular treatments. Since the early 1990s, the introduction of combined approaches, as well as the availability of computerized intraoperative support, has gradually amplified the potential for this option in terms of radical resection and neuroprotection (Tables 141-6 and 141-7).60,109,141,147–155 Careful evaluation of eligibility criteria is still the preliminary step in this strategy.
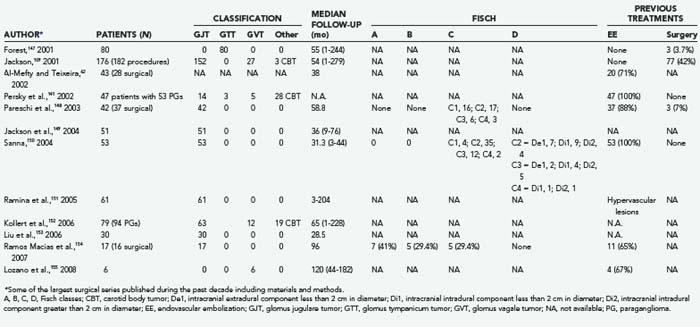
Consequently, a variety of preoperative classifications have been devised for the purpose of better defining the degree of resectability of these tumors and the expected risk for side effects. Most of the classifications are based on anatomic-surgical landmarks, such as the neurovascular encasement grading proposed in 1971 by Shamblin and associates for carotid body lesions.156 Other documented examples in this regard have been discussed earlier: the Glasscock-Jackson55 (see Table 141-2), Fisch47 (see Table 141-1), and Spector and colleagues’ (see Table 141-4). classifications.58
Additional factors may greatly influence the surgical outcome, as emphasized by Al-Mefty and Teixeira in their definition of “complex” GTs: giant size, multifocality, malignant oncotype, catecholamine secretion, and previously failed radiation treatments.60
As noted earlier, endovascular treatment performed immediately preoperatively has progressively become a quite rewarding preliminary phase, mainly because of the drastic reduction in hemorrhagic loss that can be achieved (see Table 141-6).60,141,148,150,154,155
Approaches
Lateral Skull Base Approach
The lateral skull base approach, popularized by Gardner, Robertson, and colleagues, is based on extensive, careful craniocervical dissection from the supra-auricular region to the anterior border of the sternocleidomastoid muscle.4,23,53,157 Its major components may be considered sectioning of the ear canal at the bone-cartilage junction, removal of the styloid processes and transverse process of C1, enlarged mastoidectomy, mobilization of the facial nerve, and finally ligation of the sigmoid sinus above the tumor mass and the jugular vein below (Fig. 141-10).
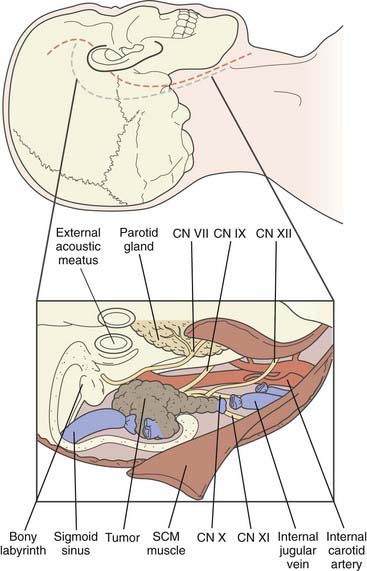
FIGURE 141-10 Top, Placement of a skin incision for the Gardner-Robertson lateral skull base approach (C-shaped incision, blue)4 and Fisch type A infratemporal fossa approach (S-shaped incision, red).47 Bottom, Exposure for removal of a glomus jugulare tumor. CN, cranial nerve; SCM, sternocleidomastoid.
Among several famous variants, it is worth mentioning, from the same authors,53,157 that the “modified lateral skull base approach” entails opening only the posterior wall of the ear canal, with sparing of the ossicles and tympanic membrane. The facial nerve is mobilized only in the mastoid portion not the tympanic portion. It seems preferable for smaller lesions in patients with preserved hearing.
Infratemporal Fossa Approach
The infratemporal fossa approach, advocated by Fisch,48 is considered mainly a wider lateral skull base approach extended rostrally to the ear canal to expose and isolate the carotid artery all the way to the cavernous sinus and up to the foramen ovale or, after sectioning the third trigeminal branch, to the foramen rotundum (see Fig. 141-10).
This particular technique, which has a wide spectrum of specific alternatives,158–161 probably offers the greatest chance for removing large GTs that grow predominantly toward the upper, rostral neighboring regions. Mainly because of their strict relationship with the petrous carotid loop, treatment of these lesions was for decades generally considered an extremely demanding or impossible task in view of the surgical options available, and patients were usually referred only for palliation. To date, extremely advanced infratemporal approaches, such as the combined transmastoid, retro-infralabyrinthine, transjugular-transcondylar-transtubercular, high-cervical approach by Sanna and Flanagan,162 can also provide a solid therapeutic option in such cases.
Otoneurosurgical Approach
The term otoneurosurgical approach had originally been used to define combined surgical techniques in which the most reliable experience of both specialties was integrated to remove GTs, as well as other neuro-otologic tumors, that had considerable to predominant intracranial extension with less infratemporal-cervical invasion.109,147,159,163 The rationale for combining the two fields was to increase the chance for radical tumor resection while reducing undue sequelae. Subsequently, this terminology has gradually included the vast majority of approaches routinely used when dealing with tumors extending from the temporal bone to the lateral skull base (e.g., retrosigmoid, presigmoid, retrolabyrinthine, translabyrinthine, transcochlear). Once again, the choice is basically related to the site and size of the lesion, as well as the impending cranial neuropathy.
Results
Mid- to-long-term follow-up studies have shown increasing success with regard to radical surgical resection.60,62,80,113,164–167 Several recent studies (see Table 141-7) have reported gross total removal in 73% to 96% of cases,60,148,150,153 and 5-year actuarial LTC rates after resection vary from 44% to 90.7%.109,141,147,148,150–152,168
Neuroradiological follow-up seems to confirm these results,83,109,153,154,168 even in series with higher risk patients.60,150
Unfortunately, functional recovery (e.g., improvement in cranial neuropathy) is extremely rare or anecdotal,150 and the associated morbidity is still substantial60,62,71,75,78,92,101,169 (see Table 141-7). Specifically, (1) permanent postoperative deficits of the lower cranial nerves are reported in 9.4% to 100% of patients, with 19% needing repair of paralyzed vocal cords and 4% requiring gastrostomy60,109,141,147,148,150–155; (2) 1.2% to 26.7% of patients complain of facial nerve palsy, with tarsorrhaphy being required in 16% to 23% of cases;60,109,141,147,148,150–155; (3) deterioration or loss of hearing is recorded in 2% to 50% of patients60,109,141,147,148,150–155; (4) a minority may also have trigeminal or oculomotor damage60,141; (5) hemorrhagic sequelae (stroke, brainstem hematoma) may occur in 0.5% to 7% of cases and usually result in a motor deficit60,109,147,148; (6) cerebrospinal fluid leaks, with or without concomitant meningitis, are reported in 0.5% to 10% of patients60,109,148,150,151; and (7) unusual complications may include pulmonary embolism, tympanic perforation, skin necrosis, and others.60,109,147,148,152 Nevertheless, a proportion of such lesions should not be considered permanent because in a significant fraction of patients acceptable long-term recovery is eventually possible4,23 with medical therapy, reconstructive surgery, and rehabilitation.
Finally, several surgical series have reported non-negligible (1.2% to 6.4%) mortality rates.62,92,151,152,169
Fractionated Radiation Therapy
FRT has been used for either primary or salvage treatment of GTs (Table 141-8).170–173 In fact, since the early experience of Capps,70 routinely recognized indications were large, inoperable lesions, postoperative tumor recurrence, or unacceptable surgical risk.91,174,175 Mainly because of their documented dose-response correlation,71,72 GTs may be included in the relatively radiosensitive oncotypes,72,73,75 although the concept is still controversial.74,77 In fact, considerable tumor growth arrest has often been observed in treated patients,71,170–173 but substantial shrinkage of tumor volume or normalization of catecholamine secretion after FRT has never been reported.4,23,176 With routine treatment schedules (45- to 55-Gy total dose with 1.5- to 2.0-Gy daily fractions), the mechanism of action does not seem to be related to direct destruction of glomus cells; rather, it should involve radiation-induced fibrosis with obliteration of feeding vessels and ischemic necrosis of the tumor.73,169,177,178
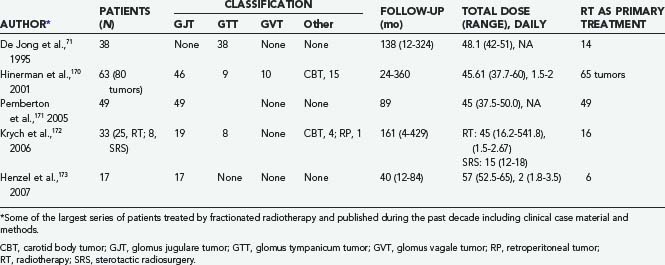
This mechanism might explain the acknowledged favorable results in long-term follow-up of the majority of these patients73,173,174,179–182 and, above all, the consistency in LTC rates (Table 141-9).73,75,177 These outstanding results have been confirmed by comparative studies of surgical and radiotherapeutic outcomes.73,91,174,175 Finally, adjuvant FRT seems to play a major role in the treatment of GTs with infiltrative bony involvement.73,175,179
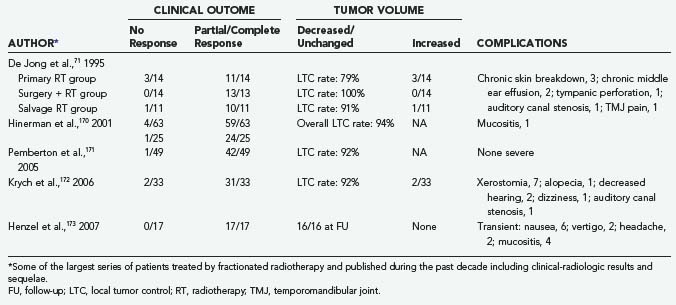
The incidence of complications (Table 141-9) is relatively low (0% to 10%), but the types of complications may range from focal parenchymal radionecrosis of the brain to osteonecrosis of the temporal bone, mastoiditis, mucositis, alopecia, and uncontrolled tumor regrowth.71,102,183 A radiation-induced case of malignant evolution has been reported.72 In addition, the increased risk for cerebrovascular accidents after wide-field radiation therapy (4% at 5 years) should never be minimized,74,78,184 as well as the higher surgical risk after FRT failure.4,23,60
Stereotactic Radiosurgery
Since the mid-1990s, SRS, particularly with Gamma Knife radiosurgery but also with a sophisticated micromultileaf linear accelerator (LINAC) and CyberKnife, has repeatedly been proposed as a reliable therapeutic option for GTs, either potentially as an alternative to surgery or as adjuvant therapy.71,74,77,78,81,82,94,174,184–186 This treatment strategy shares the same philosophy as conventional radiotherapy: long-term tumor control (i.e., tumor growth arrest or neoplastic regression) and preservation of neurological function (i.e., minimal morbidity and zero mortality). Such goals might be particularly relevant for these specific oncotypes in view of the fact that clinical onset typically occurs in the elderly decades and their slow cell kinetics makes it unlikely for patients to die of recurrence.80 Furthermore, because of the precise conformity and selectivity of current radiosurgical dose planning, SRS for these tumors has been shown to achieve, at least in short- to-mid-term follow-up studies, LTC rates and clinical responses equivalent to or even higher than those obtained with FRT, as well as an expectedly lower incidence of chronic complications.
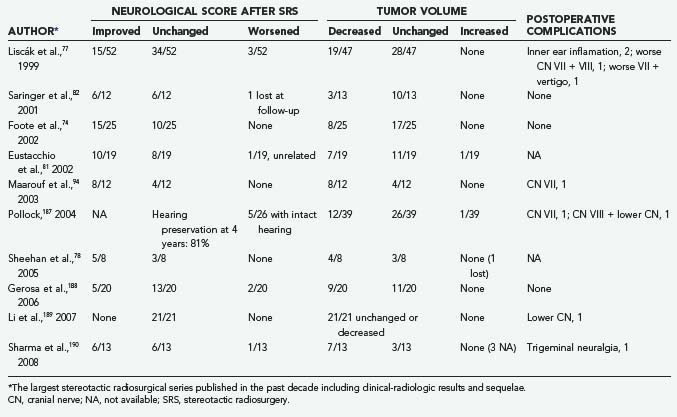
Since the original report by Foote and colleagues in 1997,79 these results have repeatedly been confirmed in an interesting series of retrospective Gamma Knife radiosurgical studies with an average follow-up of 24 to 86 months (Table 141-10).74,77,78,81,82,94,187–190 The relatively low number of cases (12 to 52) seems justified by the rarity of these tumors. In the multicenter European experience published by Liscák in 199977 of 52 patients with 95% of the lesions in Fisch groups B or C, LTC was achieved in 100% of patients, stable volume in 60%, and tumor shrinkage in 40%; 29% of patients improved clinically, 66% were unchanged, and 5% worsened.
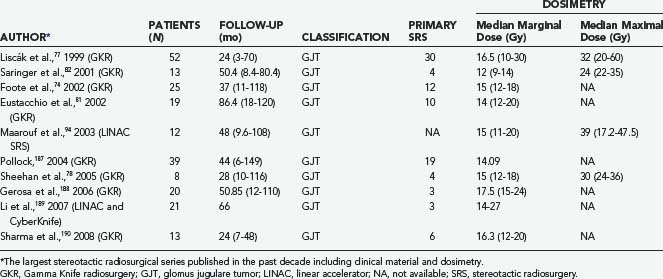
Similar results were reported by Pollock in 2004 in a group of 39 patients with consistently larger GTs (average tumor volume, 13.2 cc; mean follow-up period, 44 months): tumor shrinkage in 31%, unchanged size in 67%, and neoplastic progression in 1 patient (2%).187 The clinical outcome was satisfactory. However, 6 patients (15%) experienced new neurological deficits, mainly hearing loss.
Substantially equivalent findings have been documented in 3- to 7-year follow-up reports of Gamma Knife radiosurgery by Saringer,82 Eustacchio,81 Sheehan,78 Gerosa,188 and Sharma190 and their colleagues (Figs. 141-11 and 141-12), as well as in the LINAC SRS experience with smaller groups of patients by Poznanovic,186 Maarouf,94 and Li189 (CyberKnife) and their colleagues; in all cases, rates of complications were low (Table 141-11).74,77,78,81,82,94,187–190
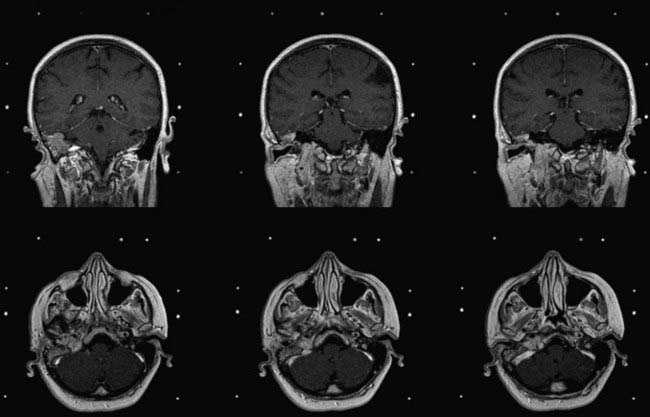
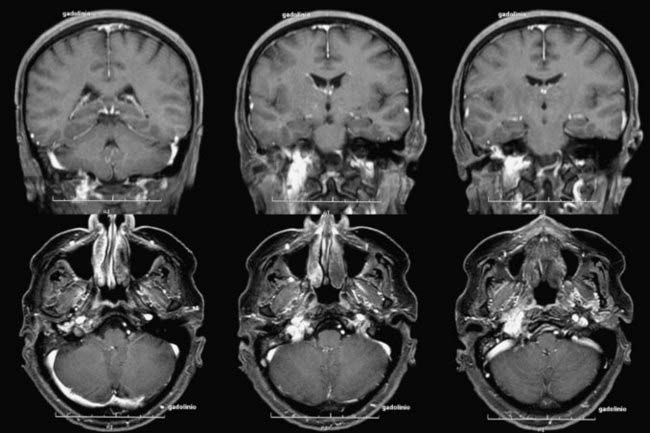
FIGURE 141-12 Same patient as in Figure 141-11, 6 months after Gamma Knife radiosurgery (16-Gy surface dose). Note the slight tumor shrinkage.
It is worth stressing that in elderly patients, SRS has been proposed as an adjuvant-integrated treatment after carefully planned, limited surgical resection.191 This small cohort of five patients had a 100% LTC rate, without any worsening of cranial neuropathy.
Optimal management of GTs is still controversial, as brilliantly shown by the comparative analysis of SRS and microsurgery by Gottfried and associates.192 Eight SRS series (142 patients; mean follow-up, 39.4 months) were matched with seven surgical series (374 patients; mean follow-up, 49.2 months), and easily predictable conclusions were reached. In fact, surgery actually achieved an 88% radical tumor resection rate, although at a higher incidence of cranial nerve morbidity and 1.3% mortality. SRS was not associated with any mortality and had lower morbidity, but 61% of the tumors were unchanged in volume at follow-up. The authors consequently emphasize the higher risk for late recurrence in these cases.
However, long-term results of SRS-treated series are now becoming available and apparently support the role of this modality. A recent study from the Mayo Clinic172 analyzed 33 GT patients treated by either FRT or SRS with a median follow-up of 13 years and reported a 10-year LTC rate of 92%, with negligible side effects and no cases of radiation-induced malignancy.
Al-Mefty O, Fox JL, Rifai A, et al. A combined infratemporal and posterior fossa approach for the removal of giant glomus tumors and chondrosarcomas. Surg Neurol. 1987;28:423-431.
Al-Mefty O, Teixeira A. Complex tumors of the glomus jugulare: criteria, treatment, and outcome. J Neurosurg. 2002;97:1356-1366.
Balatsouras DG, Eliopoulos PN, Economou CN. Multiple glomus tumours. J Laryngol Otol. 1992;106:538-543.
Bordi LT, Cheesman AD, Symon L. The surgical management of glomus jugulare tumors, description of a single-staged posterolateral combined otoneurosurgical approach. Br J Neurosurg. 1989;3:21-30.
Brismar J, Cronqvist S. Therapeutic embolization in the external carotid artery region. Acta Radiol Diagn (Stockh). 1978;19:715-731.
Fisch U. The infratemporal fossa approach for extensive tumors of the temporal bone and base of the skull. In: Silverstein H, Norrell H, editors. Neurological Surgery of the Ear. Birmingham, AL: Aesculapius; 1977:34-53.
Gardner G, Robertson JT, Robertson JH, et al. Glomus jugular tumor: skull base surgery. In: Schmidek HH, Sweet WH, editors. Operative Neurosurgical Techniques. Orlando, FL: Grune & Stratton; 1987:739-752.
Gerosa M, Visca A, Rizzo P, et al. Glomus jugulare tumors: the option of Gamma Knife radiosurgery. Neurosurgery. 2006;59:561-569.
Heth J. The basic science of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E2.
Hilal SK, Michelsen JW. Therapeutic percutaneous embolization for extra-axial vascular lesions of the head, neck, and spine. J Neurosurg. 1975;43:275-287.
Jackson CG, Glasscock ME3rd, Nissen AJ, et al. Glomus tumor surgery: the approach, results, and problems. Otolaryngol Clin North Am. 1982;15:897-916.
Jansen JC, van den Berg R, Kuiper A, et al. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88(12):2811-2816.
Johnston F, Symon L. Malignant paraganglioma of the glomus jugulare: a case report. Br J Neurosurg. 1992;6:255-259.
Jyung RW, LeClair EE, Bernat RA, et al. Expression of angiogenic growth factors in paragangliomas. Laryngoscope. 2000;110:161-167.
Liscák R, Vladyka V, Wowra B, et al. Gamma Knife Radiosurgery of the glomus jugulare tumour: early multicentre experience. Acta Neurochir (Wien). 1999;141:1141-1146.
Michael LM2nd, Robertson MD. Glomus jugulare tumors: historical overview of the management of this disease. Neurosurg Focus. 2004;17(2):E1.
Netterville JL, Jackson CG, Miller FR, et al. Vagal paragangliom: a review of 46 patients treated during a 20-year period. Arch Otolaryngol Head Neck Surg. 1998;124:1133-1140.
Patel SJ, Sekhar LN, Cass SP, et al. Combined approaches for resection of extensive glomus jugulare tumors. A review of 12 cases. J Neurosurg. 1994;80:1026-1038.
Pávai Z, Orosz Z, Horváth E, et al. Immunohistochemical features of paragangliomas. J Cell Mol Med. 2001;5:311-316.
Petropoulos AE, Luetje CM, Camarata PJ, et al. Genetic analysis in the diagnosis of familial paragangliomas. Laryngoscope. 2000;110:1225-1229.
Pollock BE. Stereotactic radiosurgery in patients with glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E10.
Robertson JH, Brodkey JA, Wang PP. Glomus jugulare tumors. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: WB Saunders; 2004:1295-1310.
Robertson JT. Chemodectomas. In: Wilkins RH, Rengachary S, editors. Neurosurgery. New York: McGraw-Hill; 1985:785-790.
Simpson GT2nd, Konrad HR, Takahashi M, et al. Immediate postembolization excision of glomus jugulare tumors: advantages of new combined techniques. Arch Otolaryngol. 1979;105:639-643.
Springate SC, Haraf D, Weichselbaum RR. Temporal bone chemodectomas: comparing surgery and radiation therapy. Oncology (Willinston Park). 1991;5:131-137.
1 Lawson W. The neuroendocrine nature of the glomus cells: an experimental, ultrastructural, and histochemical tissue culture study. Laryngoscope. 1980;90:120-144.
2 Wasserman PG, Savrgaonkar P. Paragangliomas: classification, pathology, and differential diagnosis. Otolaryngol Clin North Am. 2001;34:845-862.
3 Parkinson D. Intracranial pheochromocytoma (active glomus jugulare): case report. J Neurosurg. 1969;31:94-100.
4 Robertson JH, Brodkey JA, Wang PP. Glomus jugulare tumors. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: WB Saunders; 2004:1295-1310.
5 Dalfior D, Parisi A, Cannizzaro C, et al. Pulmonary glomus tumor. Int J Surg Pathol. 2008;16:81-84.
6 Alempijevic T, Knezevic S, Knezevic D, et al. Gastric multicentric glomangioma: a case report of this rare cause of abdominal pain. Med Sci Monit. 2008;14:CS5-8.
7 Urbanczyk K, Stachura J, Papla B, et al. Gastric solid glomus tumor and multiple glomangiomyomas of the large bowel with intravascular spread, multifocal perivascular proliferations and liver involvement. Pol J Pathol. 2007;58:207-214.
8 Kapur U, Helenowski M, Zayaad A, et al. Pulmonary glomus tumor. Ann Diagn Pathol. 2007;11:457-459.
9 Koç O, Kivrak AS, Paksoy Y. Subungual glomus tumour: magnetic resonance imaging findings. Australas Radiol. 2007;51:B107-B109.
10 Akata S, Yoshimura M, Park J, et al. Glomus tumor of the left main bronchus. Lung Cancer. 2008;60:132-135.
11 Alam K, Maheshwari V, Sabir F, et al. Glomus tumor of lesser omentum, a case report. Indian J Pathol Microbiol. 2007;50:543-544.
12 Senol MG, Oguz M, Haholu A, et al. Glomus tumor of forearm: a rare cause of neuralgia. Acta Neurol Belg. 2007;107:58-59.
13 Hu XY, Hu CH, Fang XM, et al. Glomus tumor of the gastric body: helical CT findings. Chin Med J (Engl). 2007;120:1289-1291.
14 Takahashi N, Oizumi H, Yanagawa N, et al. A bronchial glomus tumor surgically treated with segmental resection. Interact Cardiovasc Thorac Surg. 2006;5:258-260.
15 Svajdler M, Bohus P, Závacký P, et al. Paraganglioma of the mesenterium: a case report. Cesk Pathol. 2007;43:153-156.
16 Yuan WQ, Wang WQ, Su TW, et al. A primary right atrium paraganglioma in a 15-year-old patient. Endocrine. 2007;32:245-248.
17 Kim HS, Chang WI, Kim YC, et al. Catecholamine cardiomyopathy associated with paraganglioma rescued by percutaneous cardiopulmonary support: inverted Takotsubo contractile pattern. Circ J. 2007;71:1993-1995.
18 Yau KK, Siu WT, Li MK. Unusual case of acute abdomen, ruptured retroperitoneal paraganglioma. Asian J Surg. 2008;31:32-35.
19 Haresh KP, Prabhakar R, Anand Rajan KD, et al. A rare case of paraganglioma of the sella with bone metastases. Pituitary. 2009;12:276-279.
20 Gonnella C, Messa FC, Confessore P, et al. Angiographic evidence of pigmented cardiac paraganglioma. J Cardiovasc Med (Hagerstown). 2008;9:319.
21 Garrel R, Raynaud P, Raingeard I, et al. An unusual succinate dehydrogenase gene mutation C in a case of laryngeal paraganglioma. J Laryngol Otol. 2008;11:1-4.
22 Kim KN, Lee KN, Roh MS, et al. Pulmonary paraganglioma manifesting as an endobronchial mass. Korean J Radiol. 2008;9:87-90.
23 Robertson JT. Chemodectomas. In: Wilkins RH, Rengachary S, editors. Neurosurgery. New York: McGraw-Hill; 1985:785-790.
24 Heth J. The basic science of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E2.
25 Michael LM2nd, Robertson MD. Glomus jugulare tumors: historical overview of the management of this disease. Neurosurg Focus. 2004;17(2):E1.
26 Pearse AGE. The APUD cell concept and its implications in pathology. Pathol Annu. 1974;9:27-41.
27 Farrior JB3rd, Hyams VJ, Benke RH, et al. Carcinoid apudoma arising in a glomus jugulare tumor: review of endocrine activity in glomus jugulare tumors. Laryngoscope. 1980;90:110-119.
28 Dickinson AM, Traver CA. Carotid body tumors: review of the literature with report of two cases. Am J Surg. 1945;69:9-11.
29 Lahey FH, Warren KW. A long term appraisal of carotid body tumors with remarks on their removal. Surg Gynecol Obstet. 1951;92:481-491.
30 Valentin G. Ueber eine gangliose Auschwellung in der Jacobsonchen Anastomose des Menschen. Arch Anat Physiol Lpz. 1840;16:287-290.
31 Krause W. Die Glandule tympanica des Menschen. Zentrabl Med Wissenesch. 1878;16:737-739.
32 Rosenwasser H. Carotid body tumor of the middle ear and mastoid. Arch Otolaryngol. 1945;41:64-67.
33 Guild SR. A hitherto unrecognized structure: the glomus jugularis in man. Anat Rec. 1941;79(suppl 2):28-29.
34 Winship T, Louzan J. Tumours of the glomus jugulare not associated with the jugular vein. Arch Otolaryngol. 1951;54:378-383.
35 Guild SR. Glomus jugulare in man. Ann Otol Rhinol Laryngol. 1953;62:1045-1071.
36 Lundgren N. Tympanic body tumors in the middle ear: tumors of carotid body type. Acta Otolayngol. 1949;37:366.
37 Weille FL, Lane LSJr. Surgical problems involved in the removal of glomus-jugulare tumors. Laryngoscope. 1951;61:448.
38 Semmes RE. Discussion of Alexander E Jr, Adams S. Tumor of the glomus jugulare: follow-up study two years after roentgen therapy. J Neurosurg. 1953;10:672-674.
39 Albernaz JG, Buzcy PC. Nonchromaffin paraganglioma of the jugular foramen. J Neurosurg. 1953;10:663-671.
40 Shapiro MJ, Neues DK. Technique for removal of glomus jugulare tumors. Arch Otolaryngol. 1964;79:219-224.
41 Gejrot T. Surgical treatment of glomus jugulare tumors: With special reference to the diagnostic value of retrograde jugularography. Acta Otolaryngol. 1965;60:150-168.
42 Gastpar H. Die Tumoren des Glomus caroticum, Glomus jugulare tympanicum, und Glomus vagale. Acta Otolaryngol (Stockh). 1961;167(suppl):1-45.
43 Hilding DA, Greenberg A. Surgery for large glomus jugulare tumor. Arch Otolaryngol. 1971;93:227-231.
44 Michelson RP, Connolly JE. Removal of glomus jugulare tumor utilizing complete occlusion of the cerebral circulation. Laryngoscope. 1962;72:788-805.
45 House W. Management of glomus tumors. Arch Otolaryngol. 1969;89:170-178.
46 Gardner G, Cocke EWJr, Robertson JT, et al. Combined approach surgery for removal of glomus jugulare tumors. Laryngoscope. 1977;87:665-688.
47 Fisch U. The infratemporal fossa approach for extensive tumors of the temporal bone and base of the skull. In: Silverstein H, Norrell H, editors. Neurological Surgery of the Ear. Birmingham: Aesculapius; 1977:34-53.
48 Fisch U, Fagan P, Valavanis A. The infratemporal fossa approach for the lateral skull base. Otolaryngol Clin North Am. 1984;17:513-552.
49 Mischke RE, Balkany TJ. Skull base approach to glomus jugulare. Laryngoscope. 1984;90:89-94.
50 Farrior JB. Anterior hypotympanic approach for glomus tumor of the infratemporal fossa. Laryngoscope. 1984;94:1016-1021.
51 Brown JS. Glomus jugulare tumors revisited: a ten-year statistical follow-up of 231 cases. Laryngoscope. 1985;95:284-288.
52 Donald PJ, Chole RA. Transcervical transmastoid approach to lesions of the jugular bulb. Arch Otolaryngol. 1984;110:309-314.
53 Gardner G, Cocke EWJr, Robertson JH, et al. Skull base surgery for glomus jugulare tumors. Am J Otol (Suppl). 1985;Nov:126-134.
54 Kinney SE. Glomus jugulare tumor surgery with intracranial extension. Otolaryngol Head Neck Surg. 1980;88:531-535.
55 Jackson CG, Glasscock ME3rd, Nissen AJ, et al. Glomus tumor surgery: the approach, results, and problems. Otolaryngol Clin North Am. 1982;15:897-916.
56 Rockley TJ, Hawke M. Glomus bodies in the temporal bone. J Otolaryngol. 1990;19:50-56.
57 Alford BR, Gilford FR. A comprehensive study of tumors of the glomus jugulare. Laryngoscope. 1962;72:765-805.
58 Spector GJ, Druck NS, Gado M. Neurologic manifestations of glomus tumors in the head and neck. Arch Neurol. 1976;33:270-274.
59 Al-Mefty O, Fox JL, Rifai A, et al. A combined infratemporal and posterior fossa approach for the removal of giant glomus tumors and chondrosarcomas. Surg Neurol. 1987;28:423-431.
60 Bordi LT, Cheesman AD, Symon L. The surgical management of glomus jugulare tumors, description of a single-staged posterolateral combined otoneurosurgical approach. Br J Neurosurg. 1989;3:21-30.
61 Patel SJ, Sekhar LN, Cass SP, et al. Combined approaches for resection of extensive glomus jugulare tumors. A review of 12 cases. J Neurosurg. 1994;80:1026-1038.
62 Al-Mefty O, Teixeira A. Complex tumors of the glomus jugulare: criteria, treatment, and outcome. J Neurosurg. 2002;97:1356-1366.
63 Hilal SK, Michelsen JW. Therapeutic percutaneous embolization for extra-axial vascular lesions of the head, neck, and spine. J Neurosurg. 1975;43:275-287.
64 Brismar J, Cronqvist S. Therapeutic embolization in the external carotid artery region. Acta Radiol Diagn (Stockh). 1978;19:715-731.
65 Simpson GT2nd, Konrad HR, Takahashi M, et al. Immediate postembolization excision of glomus jugulare tumors: advantages of new combined techniques. Arch Otolaryngol. 1979;105:639-643.
66 Turowski B, Zanella FE. Interventional neuroradiology of the head and neck. Neuroimaging Clin N Am. 2003;13:619-645.
67 Pecorari G, Roccia F, Nadalin J, et al. Combined endovascular and surgical treatment of carotid body tumor in a patient with thoracic situs insolitus. Head Neck. 2008;30:1523-1526.
68 Anand VK, Leonetti JP, Al-Mefty O. Neurovascular considerations in surgery of glomus tumors with intracranial extensions. Laryngoscope. 1993;103(suppl 60):722-728.
69 LaRouere MJ, Zappia JJ, Wilner HI, et al. Selective embolization of glomus jugulare tumors. Skull Base Surg. 1994;4:21-25.
70 Capps FC. Glomus jugulare tumors of the middle ear. J Laryngol Otol. 1952;66:302-314.
71 De Jong AL, Coker NJ, Jenkins HA, et al. Radiation therapy in the management of paragangliomas of the temporal bone. Am J Otolaryngol. 1995;16:283-289.
72 Lalwani AK, Jackler RK, Gutin PH. Lethal fibrosarcoma complicating radiation therapy for benign glomus jugulare tumors. Am J Otol. 1993;14:398-402.
73 Cole JM, Beiler D. Long-term results of treatment for glomus jugulare and glomus vagale tumors with radiotherapy. Laryngoscope. 1994;104:1461-1465.
74 Foote RL, Pollock BE, Gorman DA, et al. Glomus jugulare tumor: tumor control and complications after stereotactic radiosurgery. Head Neck. 2002;24:332-339.
75 Springate SC, Haraf D, Weichselbaum RR. Temporal bone chemodectomas: comparing surgery and radiation therapy. Oncology (Willinston Park). 1991;5:131-137.
76 Spector GJ, Maisel RH, Ogura JH. Glomus tumors in the middle ear. I. An analysis of 46 patients. Laryngoscope. 1973;83:1652-1672.
77 Liscák R, Vladyka V, Wowra B, et al. Gamma Knife Radiosurgery of the glomus jugulare tumour: early multicentre experience. Acta Neurochir (Wien). 1999;141:1141-1146.
78 Sheehan J, Kondziolka D, Flickinger J, et al. Gamma knife surgery for glomus jugulare tumors: an intermediate report on efficacy and safety. J Neurosurg. 2005;102(suppl):241-246.
79 Foote RL, Coffey RJ, Gorman DA, et al. Stereotactic radiosurgery for glomus jugulare tumors: a preliminary report. Int J Radiation Oncol Biol Phys. 1997;38:491-495.
80 van Der Mey AG, Frijns JH, Cornelisse CJ, et al. Does intervention improve the natural course of glomus tumours? A series of 108 patients seen in a 32-year period. Ann Otol Rhinol Laryngol. 1992;101:635-641.
81 Eustacchio S, Trummer M, Unger F, et al. The role of Gamma Knife Radiosurgery in the management of glomus jugulare tumors. Acta Neurochir (Suppl). 2002;84:91-97.
82 Saringer W, Khayal H, Ertl A, et al. Efficiency of gamma knife radiosurgery in the treatment of glomus jugulare tumors. Minim Invasive Neurosurg. 2001;44:141-146.
83 Thedinger BA, Glasscock ME3rd, Cueva RA, et al. Postoperative radiographic evaluation after acoustic neuroma and glomus jugulare tumor removal. Laryngoscope. 1992;102:261-266.
84 Marimann EC, van Beersum SE, Cremers CW, et al. Analysis of a second family with hereditary non-chromaffin paragangliomas locates the underlying gene at the proximal region of chromosome 11q. Hum Genet. 1995;95:56-62.
85 Blumenfeld J, Cohen N, Anwar M, et al. Hypertension and a tumor of the glomus jugulare region. Evidence for epinephrine biosynthesis. Am J Hypertens. 1993;6:382-387.
86 Spector GJ, Ciralsky R, Maisel RH, et al. IV. Multiple glomus tumors in the head and neck. Laryngoscope. 1975;85:1066-1075.
87 Balatsouras DG, Eliopoulos PN, Economou CN. Multiple glomus tumours. J Laryngol Otol. 1992;106:538-543.
88 Petropoulos AE, Luetje CM, Camarata PJ, et al. Genetic analysis in the diagnosis of familial paragangliomas. Laryngoscope. 2000;110:1225-1229.
89 Jansen JC, van den Berg R, Kuiper A, et al. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88:2811-2816.
90 Lack EE, Cubilla AL, Woodruff JM, et al. Paragangliomas of the head and neck region: a clinical study of 69 patients. Cancer. 1977;39:397-409.
91 Boyle JO, Shimm DS, Coulthard SW. Radiation therapy for paragangliomas of the temporal bone. Laryngoscope. 1990;100:896-901.
92 Green JD, Brackmann DE, Nguyen CD, et al. Surgical management of previously untreated glomus jugulare tumors. Laryngoscope. 1994;104:917-920.
93 Gulya AJ. The glomus tumor and its biology. Laryngoscope. 1993;103(suppl 60):7-15.
94 Maarouf M, Voges J, Landwehr P, et al. Stereotactic linear accelerator–based radiosurgery for the treatment of patients with glomus jugulare tumors. Cancer. 2003;97:1093-1098.
95 Zulch KJ. Brain Tumors. New York: Springer; 1986.
96 Saldana MJ, Salem LE, Travezan R. High altitude hypoxia and chemodectomas. Hum Pathol. 1973;4:251-263.
97 Lack EE. Hyperplasia of vagal and carotid body paraganglia in patients with chronic hypoxemia. Am J Pathol. 1978;91:497-516.
98 Lack EE, Perez-Atayde AR, Young JB. Carotid body hyperplasia in cystic fibrosis and cyanotic heart disease. A combined morphometric, ultrastructural, and biochemical study. Am J Pathol. 1985;119:301-314.
99 van der Mey AG, Masswinkel-Mooy PD, Cornelisse CJ, et al. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 1989;2:1291-1294.
100 DeMarino DP, Mueller DP, Yuh WT, et al. Multiple paragangliomas. Ann Otol Rhinol Laryngol. 1990;99:85-86.
101 Netterville JL, Jackson CG, Miller FR, et al. Vagal paraganglioma: a review of 46 patients treated during a 20-year period. Arch Otolaryngol Head Neck Surg. 1998;124:1133-1140.
102 Bojrab DI, Bhansali SA, Zarbo RJ. Management of metastatic glomus jugulare tumors. Skull Base Surg. 1992;2:1-5.
103 Havekes B, van der Klaauw AA, Hoftijzer HC, et al. Reduced quality of life in patients with head and neck paragangliomas. Eur J Endocrinol. 2008;158:247-253.
104 Gardner G, Cocke EWJr, Robertson JT, et al. Glomus jugulare tumours—combined treatment: Part I. J Laryngol Otol. 1981;95:437-454.
105 Gardner G, Cocke EWJr, Robertson JT, et al. Glomus jugulare tumours—combined treatment: Part II. J. Laryngol Otol. 1981;95:567-580.
106 Gardner G, Robertson JT, Cocke EWJr. Surgical therapy of tumors of the glomus jugulare. In: Schmidek HH, Sweet WH, editors. Current Techniques in Operative Neurosurgery. New York: Grune & Stratton; 1977:57-66.
107 Makek M, Franklin DJ, Zhao JC, et al. Neural infiltration of glomus jugulare tumors. Am J Otol. 1990;11:1-5.
108 Jackson CG, Harris PF, Glasscock ME3rd, et al. Diagnosis and management of paragangliomas of the skull base. Am J Surg. 1990;159:389-393.
109 Jackson CG, McGrew BM, Forest JA, et al. Lateral skull base surgery for glomus tumors: long-term control. Otol Neurotol. 2001;22:377-382.
110 Welsh LW, Welsh JJ, Huck GFJr. Glomus jugulare tumor. Disseminated form in the central nervous system. Arch Otolaryngol. 1976;102:507-510.
111 Beck DW, Kasselll NF, Drake CG. Glomus jugulare tumor presenting with increased intracranial pressure. Case report. J Neurosurg. 1979;50:823-825.
112 Farrior JB, Hyams VJ, Benke RH. Carcinoid apudomas arising in a glomus jugulare tumor: review of endocrine activity in glomus jugulare tumors. Laryngoscope. 1980;90:110-119.
113 Woods CI, Strasnik B, Jackson CG. Surgery for glomus tumors: The Otology Group experience. Layngoscope. 1993;103(suppl 60):65-70.
114 Pávai Z, Orosz Z, Horváth E, et al. Immunohistochemical features of paragangliomas. J Cell Mol Med. 2001;5:311-316.
115 Borsanyi S. Glomus jugulare tumors. Laryngoscope. 1962;72:1336-1345.
116 Gabriel EM, Sampson JH, Dodd LG, et al. Glomus jugulare tumor metastatic to the sacrum after high-dose radiation therapy: case report. Neurosurgery. 1995;37:1001-1005.
117 Johnston F, Symon L. Malignant paraganglioma of the glomus jugulare: a case report. Br J Neurosurg. 1992;6:255-259.
118 Brewis C, Bottrill ID, Wharton SB, et al. Metastases from glomus jugulare tumors. J Laryngol Otol. 2000;114:17-23.
119 Devilee P, van Schothorst EM, Bardoel AF, et al. Allelotype of head and neck paragangliomas: allelic imbalance is confined to the long arm of chromosome 11, the site of the predisposing locus PGL. Genes Chromosomes Cancer. 1994;11:71-78.
120 Heutink P, van der Mey AG, Sandkuijl LA, et al. A gene subject to genomic imprinting and responsible for hereditary paragangliomas maps to chromosome 11q23-qter. Hum Mol Genet. 1992;1:7-10.
121 Heutink P, van Schothorst EM, van der Mey AG, et al. Further localization of the gene for hereditary paragangliomas and evidence for linkage in unrelated families. Eur J Hum Genet. 1994;2:148-158.
122 Struycken PM, Cremers CW, Mariman EC, et al. Glomus tumours and genomic imprinting: influence of inheritance along the paternal or maternal line. Clin Otolaryngol Allied Sci. 1997;22:71-76.
123 Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848-851.
124 Rodriguez-Cuevas S, Lopez-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 metres above sea level. Head Neck. 1998;20:374-378.
125 Niemann S, Becker-Follmann J, et al. Assignment of PGL3 to chromosome 1 (q21-q23) in a family with autosomal dominant non-chromaffin paraganglioma. Am J Med Genet. 2001;98:32-36.
126 Niemann S, Steinberger D, Muller U. PGL3, a third, non maternally imprinted locus in autosomal dominant paraganglioma. Neurogenetics. 1999;2:167-170.
127 DeAngelis LM, Kelleher MB, Post KD, et al. Multiple paragangliomas in neurofibromatosis: a new neuroendocrine neoplasia. Neurology. 1987;37:129-133.
128 Jyung RW, LeClair EE, Bernat RA, et al. Expression of angiogenic growth factors in paragangliomas. Laryngoscope. 2000;110:161-167.
129 Watanabe K, Hiraki H, Hasegawa H, et al. Immunohistochemical localization of endothelin-1, endothelin-3 and endothelin receptors in human pheochromocytoma and paraganglioma. Pathol Int. 1997;47:540-546.
130 Zielke A, Middeke M, Hoffmann S, et al. VEGF-mediated angiogenesis of human pheochromocytomas is associated to malignancy and inhibited by anti-VEGF antibodies in experimental tumors. Surgery. 2002;132:1056-1063.
131 Wang DG, Johnston CF, Barros D’Sa AA, et al. Expression of apoptosis-suppressing gene bcl-2 in human carotid body tumours. J Pathol. 1997;183:218-221.
132 Erlandsson N, Baumann B, Rössler OG, et al. Lack of correlation between NF-kappa B activation and induction of programmed cell death in PC12 pheochromocytoma cells treated with 6-hydroxydopamine or the cannabinoid receptor 1-agonist CP55,940. Biochem Pharmacol. 2002;64:487-495.
133 Rosa M, Sahoo S. Bilateral carotid body tumor: The role of fine-needle aspiration biopsy in the preoperative diagnosis. Diagn Cytopathol. 2008;36:178-180.
134 Jayaram G, Hayati JN, Yip CH, et al. Cytologic, histologic and immunocytochemical features in fine needle aspiration of paraganglioma-like variant of medullary carcinoma. Acta Cytol. 2008;52:119-121.
135 Neves F, Huwart L, Jourdan G, et al. Head and neck paragangliomas: value of contrast-enhanced 3D MR angiography. AJNR Am J Neuroradiol. 2008;29:883-889.
136 Azzarelli B, Felten S, Muller J, et al. Dopamine in paragangliomas of the glomus jugulare. Laryngoscope. 1988;98:573-578.
137 Schwaber MK, Glasscock ME, Nissen AJ, et al. Diagnosis and management of catecholamine secreting glomus tumors. Laryngoscope. 1984;94:1008-1015.
138 Troughton RW, Fry D, Allison RS, et al. Depression, palpitations, and unilateral pulsatile tinnitus due to a dopamine-secreting glomus jugulare tumor. Am J Med. 1998;104:310-311.
139 Girolami G, Heuser L, Hildmann H, et al. Selective embolization and surgical resection for head and neck glomus tumours—clinical outcome analysis. Laryngorhinootologie. 2008;87:181-185.
140 Gupta AK, Purkayashta S, Bodhey NK, et al. Preoperative embolization of hypervascular head and neck tumours. Australas Radiol. 2007;51:446-452.
141 Persky MS, Setton A, Niimi Y, et al. Combined endovascular and surgical treatment of head and neck paragangliomas—a team approach. Head Neck. 2002;24:423-431.
142 Tasar M, Yetiser S. Glomus tumors: therapeutic role of selective embolization. J Craniofacial Surg. 2004;15:497-505.
143 Tikkakoski T, Luotonen J, Leinonen S, et al. Preoperative embolization in the management of neck paragangliomas. Laryngoscope. 1997;107:821-826.
144 Zähringer M, Guntinas-Lichius O, Gossmann A, et al. Percutaneous embolization for cervicofacial neoplasms and haemorrhages. ORL J Otorhinolaryngol Relat Spec. 2005;67:348-360.
145 Murphy TP, Brackmann DE. Effects of preoperative embolization on glomus jugulare tumors. Laryngoscope. 1989;99:1244-1247.
146 Brismar J, Cronqvist S. Therapeutic embolization in the external carotid artery region. Acta Radiol Diagan (Stockh). 1978;19(5):715-731.
147 Forest JA3rd, Jackson CG, McGrew BM. Long-term control of surgically treated glomus tympanicum tumors. Otol Neurotol. 2001;22:232-236.
148 Pareschi R, Righini S, Destito D, et al. Surgery of glomus jugulare tumors. Skull Base. 2003;13:149-157.
149 Jackson CG, Kaylie DM, Coppit G, et al. Glomus jugulare tumors with intracranial extension. Neurosurg Focus. 2004;17(2):E7.
150 Sanna M, Jain Y, De Donato G, et al. Management of jugular paragangliomas: the Gruppo Otologico experience. Otol Neurotol. 2004;25:797-804.
151 Ramina R, Maniglia JJ, Fernandes YB, et al. Tumors of the jugular foramen: diagnosis and management. Neurosurgery. 2005;57(1 suppl):59-68.
152 Kollert M, Minovi AA, Draf W, et al. Cervical paragangliomas—tumor control and long-term functional results after surgery. Skull Base. 2006;16:185-191.
153 Liu JK, Sameshima T, Gottfried ON, et al. The combined transmastoid retro- and infralabyrinthine transjugular transcondylar transtubercular high cervical approach for resection of glomus jugulare tumors. Neurosurgery. 2006;59(suppl 1):ONS115-125.
154 Ramos Macías A, Borkoski Barreiros S, Pérez Plasencia D, et al. Glomus tumours of temporal bone origin. Study of 17 cases. Acta Otorrinolaringol Esp. 2007;58:358-361.
155 Lozano FS, Gómez JL, Mondillo MC, et al. Surgery of vagal paragangliomas: six patients and review of literature. Surg Oncol. 2008;17:281-287.
156 Shamblin WR, ReMine WH, Sheps SG, et al. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am J Surg. 1971;122:732-739.
157 Gardner G, Robertson JT, Robertson JH, et al. Glomus jugular tumor: skull base surgery. In: Schmidek HH, Sweet WH, editors. Operative Neurosurgical Techniques. Orlando, FL: Grune & Stratton; 1987:739-752.
158 Kim CS, Suh MW. Skull base surgery for removal of temporal bone tumors. Acta Otolaryngol Suppl. 2007;558:4-14.
159 Kaylie DM, Wittkopf JE, Coppit G, et al. Revision lateral skull base surgery. Otol Neurotol. 2006;27:225-233.
160 Witiak DG, Pensak ML. Limitations to mobilizing the intrapetrous carotid artery. Ann Otol Rhinol Laryngol. 2002;111:343-348.
161 Sanna M, De Donato G, Taibah A, et al. Infratemporal fossa approaches to the lateral skull base. Keio J Med. 1999;48:189-200.
162 Sanna M, Flanagan S. The combined transmastoid retro- and infralabyrinthine transjugular transcondylar transtubercular high cervical approach for resection of glomus jugulare tumors. Neurosurgery. 2007;61:E1340.
163 Parhizkar N, Hiltzik DH, Selesnick SH. Facial nerve rerouting in skull base surgery. Otolaryngol Clin North Am. 2005;38:685-710.
164 Briner HR, Linder TE, Pauw B, et al. Long-term results of surgery for temporal bone paragangliomas. Laryngoscope. 1999;109:577-583.
165 Gjuric M, Seidinger L, Wigand ME. Long-term results of surgery for temporal bone paraganglioma. Skull Base Surg. 1996;6:147-152.
166 Gstoettner W, Matula C, Hamzavi J, et al. Long-term results of different treatment modalities in 37 patients with glomus jugulare tumors. Eur Arch Otorhinolaryngol. 1999;256:350-355.
167 Order SE, Hellman S, von Essen CF, et al. Improvement in quality of survival following whole-brain irradiation for brain metastasis. Radiology. 1968;91:149-153.
168 Erickson D, Kudva YC, Ebersold MJ, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210-5216.
169 Watkins LD, Mendoza N, Cheesman AD, et al. Glomus jugulare tumours: a review of 61 cases. Acta Neurochir (Wien). 1994;130(1-4):66-70.
170 Hinerman RW, Mendenhall WM, Amdur RJ, et al. Definitive radiotherapy in the management of chemodectomas arising in the temporal bone, carotid body, and glomus vagale. Head Neck. 2001;23:363-371.
171 Pemberton LS, Swindell R, Sykes AJ. Radical radiotherapy alone for glomus jugulare and tympanicum tumours. Oncol Rep. 2005;14:1631-1633.
172 Krych AJ, Foote RL, Brown PD, et al. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys. 2006;65:1063-1066.
173 Henzel M, Hamm K, Gross MW, et al. Fractionated stereotactic radiotherapy of glomus jugulare tumors. Local control, toxicity, symptomatology, and quality of life. Strahlenther Onkol. 2007;183:557-562.
174 Elshaikh MA, Mahmoud-Ahmed AS, Kinney SE, et al. Recurrent head and neck chemodectomas: a comparison of surgical and radiotherapeutic results. Int J Radiat Oncol Biol Phys. 2002;52:953-956.
175 Springate SC, Weichselbaum RR. Radiation or surgery for chemodectoma of the temporal bone: a review of local control and complications. Head Neck. 1990;12:303-307.
176 Schwaber MK, Gussack GS, Kirkpatrick W. The role of radiation therapy in the management of catecholamine secreting glomus tumors. Otolaryngol Head Neck Surg. 1998;98:150-154.
177 Schild SE, Foote RL, Buskirk SJ, et al. Results of radiotherapy for chemodectomas. Mayo Clin Proc. 1992;67:537-540.
178 Mukherji SK, Kasper ME, Tart RP, et al. Irradiated paragangliomas of the head and neck: CT and MR appearance. AJNR Am J Neuroradiol. 1994;15:357-363.
179 Larner JM, Hahn SS, Spaulding CA, et al. Glomus jugulare tumors. Long-term control by radiation therapy. Cancer. 1992;69:1813-1817.
180 Wang ML, Hussey DH, Doornbos JF, et al. Chemodectoma of the temporal bone: a comparison of surgical and radiotherapeutic results. Int J Radiat Oncol Biol Phys. 1988;14:643-648.
181 Cole JM. Panel discussion: glomus jugulare tumors of the temporal bone. Radiation of glomus tumors of the temporal bone. Laryngoscope. 1979;89:1623-1627.
182 Simko TG, Griffin TW, Gerdes AJ, et al. The role of radiation therapy in the treatment of glomus jugulare tumors. Cancer. 1978;42:104-106.
183 Carrasco V, Rosenman J. Radiation therapy of glomus jugulare tumors. Laryngoscope. 1993;103(suppl 60):23-27.
184 Jordan JA, Roland PS, McManus C, et al. Stereotactic radiosurgery for glomus jugulare tumors. Laryngoscope. 2000;110:35-38.
185 Linskey ME, Johnstone PA, O’Leary M, et al. Radiation exposure of the normal temporal bone structures during stereotactically guided gamma knife surgery for vestibular schwannomas. J Neurosurg. 2003;98:800-806.
186 Poznanovic SA, Cass SP, Kavanagh BD. Short-term tumor control and acute toxicity after sterotactic radiosurgery for glomus jugulare tumors. Otolaryngol Head Neck Surg. 2006;134:437-442.
187 Pollock BE. Stereotactic radiosurgery in patients with glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E10.
188 Gerosa M, Visca A, Rizzo P, et al. Glomus jugulare tumors: the option of Gamma Knife radiosurgery. Neurosurgery. 2006;59:561-569.
189 Li G, Chang S, Adler JRJr, et al. Irradiation of glomus jugulare tumors: a historical perspective. Neurosurg Focus. 2007;23(6):E13.
190 Sharma MS, Gupta A, Kale SS, et al. Gamma knife radiosurgery for glomus jugulare tumors: therapeutic advantages of minimalism in the skull base. Neurol India. 2008;56:57-61.
191 Willen SN, Einstein DB, Maciunas RJ, et al. Treatment of glomus jugulare tumors in patients with advanced age: planned limited surgical resection followed by staged gamma knife radiosurgery: a preliminary report. Otol Neurotol. 2005;26:1229-1234.
192 Gottfried ON, Liu JK, Couldwell WT. Comparison of radiosurgery and conventional surgery for the treatment of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E4.






