Chapter 43 Giant Cell Arteritis
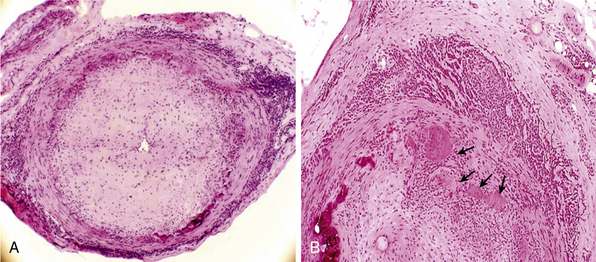
Giant-cell (temporal) arteritis (GCA) is a chronic inflammatory arteritis that preferentially involves large and medium-sized arteries and affects persons older than 50 years of age. Although autopsy studies have shown that the aorta and its major tributaries are almost invariably involved, most of the major clinical manifestations and complications of the disease arise from involvement of the carotid artery branches and include headaches, visual loss, and stroke.1–3 Aneurysms due to aortitis, as well as extremity arterial occlusions and claudication, can also be seen in GCA, especially in late disease.4,5 Histopathological diagnosis is usually determined from examination of a biopsy of the superficial temporal artery (Figs. 43-1 and 43-2).
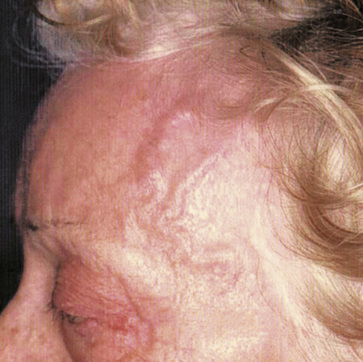
Some 50% of patients with GCA develop polymyalgia rheumatica (PMR), a clinically defined syndrome consisting of aching and stiffness in the neck, shoulders, or pelvic girdle. Polymyalgia rheumatica can also exist as a distinct entity with no evidence of vascular involvement.6
Epidemiology
Giant-cell arteritis characteristically occurs in people older than 50 years of age, and its frequency increases with age. Maximal incidence of GCA occurs in persons between 75 and 85, and it is twice as frequent in women as men. Although there is a markedly increased incidence of GCA among Caucasians in northern Europe and in populations with similar ethnic background,7 the disease can occur in all populations. Giant-cell arteritis is not an uncommon disease. Studies conducted in northern Europe and North America disclose an annual incidence of 19 to 32 cases per 100,000 people older than 508,9 and a rate of 49 per 100,000 for individuals in their 80s.7,9 In Mediterranean countries, the annual incidence is lower, about 6 to 10 cases per 100,000.10,11 Isolated PMR is even more frequent, with an average annual incidence of 52 per 100,000 among people older than age 50.6,7
Pathology
In the large and medium-sized arteries involved in GCA, the vessel wall is infiltrated by T lymphocytes and macrophages, frequently forming a granulomatous reaction with the presence of multinucleated giant cells12 (see Fig. 43-2). Scattered polymorphonuclear leukocytes can be occasionally identified. B lymphocytes are scarce and natural killer (NK) cells are virtually absent.13
Inflammatory infiltrates usually reach the artery through the adventitial layer and subsequently expand to involve the entire vessel thickness. Extent of inflammatory infiltrates is highly variable among patients and even varies in different sections of the same specimen. The internal elastic lamina is usually disrupted, and giant cells often accumulate in its vicinity but are not required for the diagnosis. In well-developed lesions, the lumen is occluded by intimal hyperplasia. In some specimens, inflammation of the vasa vasorum and small vessels surrounding the temporal artery is the only apparent abnormality.14 When inflammation of small branches is the only abnormality, other forms of systemic small- and medium-vessel vasculitis must be excluded.
Inflammatory lesions are typically segmental and preferentially distributed along the carotid and vertebral arteries. Intracranial vessels are usually spared. Small arteries and arterioles in the vicinity of the superficial temporal artery are almost invariably involved.14 Autopsy studies have demonstrated that inflammatory infiltrates are often detected in the aorta and its major tributaries.15
Histopathological findings in PMR include chronic synovitis and bursitis of proximal joints.16,17 Synovitis is usually mild, and inflammatory infiltrates contain CD4 T lymphocytes and macrophages, with scarce granulocytes. Muscle biopsies do not disclose specific abnormalities.
Pathogenesis
Genetic Predisposition
The predominance of GCA and PMR among Caucasians, especially those of northern European origin, strongly suggests a genetic component in the pathogenesis of GCA and PMR. A higher prevalence of HLA class II alleles DRB1*04 has been reported among patients with GCA and PMR compared to the general population.18 Certain polymorphisms of genes involved in immune and inflammatory responses, such as tumor necrosis factor (TNF)-α, vascular endothelial growth factor (VEGF),19,20 endothelial nitric oxide synthase (eNOS),21 intercellular adhesion molecule (ICAM)-1,22 interleukin (IL)-6, and mannose-binding lectin23 among others,24 appear to be more frequent in patients with GCA than in the general population. The influence of these polymorphisms in the development of GCA remains undefined but is under active investigation.
Possible Triggering Agents
Several observations suggest that GCA may be caused by an environmental triggering agent yet to be defined. Cyclic fluctuations in the incidence of GCA occurring every 6 or 7 years have been reported.9 Additionally, the characteristic granulomatous reaction with multinucleated giant cells has also prompted a search for an infectious agent that may cause a delayed-type hypersensitivity reaction. However, attempts to identify specific pathogens, including Chlamydia pneumoniae and parvovirus B19, have led to conflicting results,25,26 and the search for an infectious etiology of GCA continues.27
Immunopathogenic Mechanisms
It has been postulated that inflammatory lesions in GCA develop as a consequence of an antigen-specific immune response against antigens present in the vessel wall. Activated dendritic cells have been detected in inflammatory lesions of GCA,13,28 where they are thought to serve as antigen-presenting cells and provide co-stimulatory signals for CD4 T-cell activation. Innate immune system activation through Toll-like receptors (TLRs) seems to be relevant in the activation of dendritic cells.29 Immunopathological studies have demonstrated that inflammatory infiltrates in GCA mainly consist of activated macrophages and T lymphocytes, particularly of the CD4 subset.13 Expansion of CD4 T-lymphocyte clones derived from different areas of temporal artery biopsy specimens has shown that some of them share identical sequences at the third complementary determining region of the T-cell receptor,30 suggesting a specific immune recognition of a disease-relevant antigen. CD4 T lymphocytes undergo T helper 1 (TH1) differentiation, with vigorous production of interferon (IFN)-γ, a key cytokine in macrophage activation and granuloma formation.31 Interleukin-17 is also produced in GCA lesions, providing evidence for the participation of TH17-mediated responses in the pathophysiology of GCA.32
Mechanisms of Vascular Injury
Multiple processes have been implicated in the vascular injury seen in GCA. Activated macrophages produce oxygen radicals and proteolytic enzymes that participate in disruption of the vessel wall and tissue destruction.33,34 Increased levels of lipid peroxidation products, aldose reductase, inducible nitric oxide synthase (iNOS), and functionally active matrix metalloproteases MMP-2 and MMP-9 have been demonstrated in GCA.33,35
Systemic Inflammatory Response
Activated macrophages produce proinflammatory cytokines IL-1, TNF-α, and IL-6, which are major inducers of the systemic inflammatory response (fever, weight loss, anemia, and hepatic synthesis of acute-phase proteins) often prominent in patients with GCA. Expression of these cytokines in the arterial tissue of patients, as well as circulating levels of TNF-α and IL-6, correlate with the intensity of the systemic inflammatory reaction.36 As discussed later, these cytokines are able to activate proinflammatory cascades such as chemokine release, adhesion molecule expression, and angiogenesis. Although each of these cascades may amplify and maintain the inflammatory process,37 some of their effects on vessel wall components might be protective against vascular occlusion.
Vascular Response to Inflammation
Vessel wall components, particularly endothelial cells (ECs) and smooth muscle cells (SMCs), actively react to cytokines and growth factors released by infiltrating leukocytes. Vascular response to inflammation leads to amplification and perpetuation of the inflammatory process and vessel occlusion.37 Inflammation-induced angiogenesis is remarkable in GCA lesions and preferentially occurs at the adventitial layer and within inflammatory infiltrates, particularly in granulomatous areas at the intima media junction.38,39
Neovessels may have a protective role at distal sites where providing new blood supply may prevent organ ischemia.39 Additionally, neovessels provide a population of ECs that may exert a variety of proinflammatory activities.38 Vascular response to inflammation may eventually lead to vessel occlusion, with subsequent ischemia of the tissues supplied by involved vessels. In GCA, vessel occlusion results mostly from intimal hyperplasia driven by proliferation and matrix production by myointimal cells; platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β appear to be crucial cytokines in this process.37
Clinical Manifestations
Patients with GCA may present with a wide variety of clinical features, summarized in Table 43-1. Disease-related manifestations may appear rapidly or develop insidiously. A delay of weeks or even months between the onset of clinical symptoms and diagnosis is common, particularly in patients with predominantly systemic complaints in whom more prevalent diseases (e.g., infections, malignancies) are usually suspected. Ischemic complications tend to accumulate in some patients and are frequently early events during the course of the disease.40,41
Table 43-1 Clinical Findings in a Series of 250 Patients with Giant Cell Arteritis
| General Features | |
| Age (mean, range) | 75 (50-94) |
| Sex (female/male) | 178/72 |
| Weight loss | 61% |
| Fever | 47% |
| Cranial Symptoms | 86% |
| Headache | 77% |
| Temporal artery abnormality (swollen, tender, weak/absent pulse) | 74% |
| Jaw claudication | 44% |
| Scalp tenderness | 39% |
| Facial pain | 18% |
| Earache | 18% |
| Odynophagia | 12% |
| Ocular pain | 8% |
| Tongue pain | 5% |
| Carotodynia | 5% |
| Toothache | 5% |
| Trismus | 1% |
| Ophthalmic Events | 22% |
| Blindness (permanent) | 14% |
| Amaurosis fugax | 10% |
| Transient diplopia | 4% |
| Cerebrovascular Accident | 2% |
| Symptomatic Large Vessel Involvement (Claudication and/or Bruit) | 5% |
| Polymyalgia Rheumatica (PMR) | 48% |
Modified and reprinted from Cid MC , Hernandez-Rodriquez J, Grau JM: Vascular manifestations in giant-cell arteritis. In Asherson RA, Cervera R, editors: Vascular manifestations of systemic autoimmune diseases, London, 2001, CRC Press.
Some data suggest that an initial presentation of GCA that includes signs and symptoms of systemic inflammation (elevated acute phase reactants, fever, anemia, weight loss) is associated with a specific pattern of clinical illness. For unclear reasons, patients with a strong systemic inflammatory response are at lower risk of ischemic events but are more refractory to therapy40,42 than patients with a seemingly weaker initial inflammatory response. Similarly, as with cranial ischemic complications, symptomatic stenosis of large vessels has been shown to be negatively associated with prominent inflammatory markers at the time of diagnosis.43
Systemic Manifestations
Patients with GCA or PMR frequently experience malaise, anorexia, weight loss, and depression.1,3,44 Nearly 50% of patients with GCA have fever, and in some patients, fever or constitutional symptoms are the most prominent findings. Approximately 10% of patients present with fever of unknown origin with subtle or no cranial manifestations.6
Clinical Manifestations of Cranial Arterial Involvement
Headache is one of the most common and classic symptoms and occurs in 60% to 98% of cases of GCA.6 Intensity and location of headache are highly variable, but it frequently predominates at the temporal areas. Scalp tenderness is also common. About 40% of patients experience jaw claudication when eating, a symptom highly specific for GCA but occasionally seen in other vascular diseases (e.g., necrotizing vasculitis, systemic amyloidosis). Other manifestations, such as facial swelling, tongue ischemia, or edema, are less frequently seen6,45 (see Table 43-1). Patients may also present with a variety of unusual pains in the craniofacial area, including ocular pain, earache, toothache, odynophagia, odontalgia, and carotidynia. When certain of these symptoms predominate, a substantial delay in diagnosis is common.
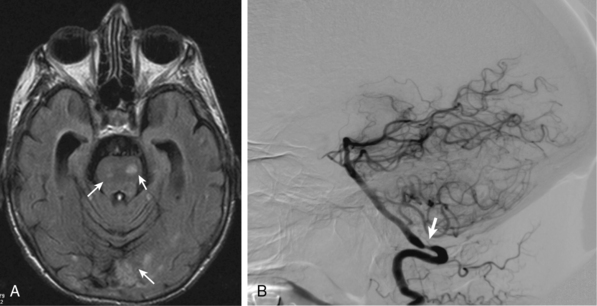
Less common ischemic complications include stroke due to involvement of carotid or vertebral tributaries, and scalp or tongue necrosis. Strokes, when they do occur, are more frequent in territories supplied by the vertebral arteries (Fig. 43-3). Additional ischemic manifestations include hearing loss and vestibular dysfunction.46
Cardiac, Aortic, and Peripheral Vascular Manifestations
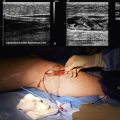
Aortitis and inflammatory involvement of the main branches of the aorta are more common in GCA than generally appreciated (Fig. 43-4). In a small autopsy series, aortic inflammation was observed in 12 out of 13 patients with GCA. Imaging techniques are now used to indirectly measure aortic inflammation in living individuals. Evidence of probable aortic involvement can be detected by positron emission tomography (PET) or computed tomographic angiography (CTA) in 50% to 65% of patients at the time of diagnosis47,48 (see Fig. 43-4). Aortic involvement is asymptomatic unless aortic dissection or dilation occurs (Fig. 43-5). Systematic screening of 54 patients revealed that aortic dilatation occurs in 22.5% of patients after a median follow-up of 5.6 years. Interestingly, unlike noninflammatory aortic disease, there is a markedly increased ratio of thoracic to abdominal aortic aneurysms in patients with GCA, and the risk of thoracic disease may be 17-fold higher among patients with a history of GCA.49
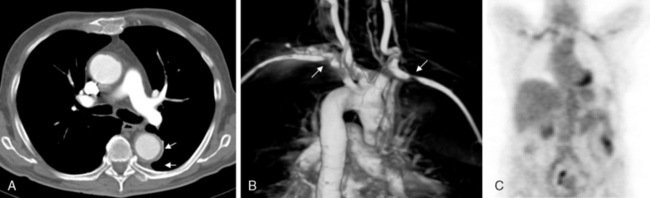
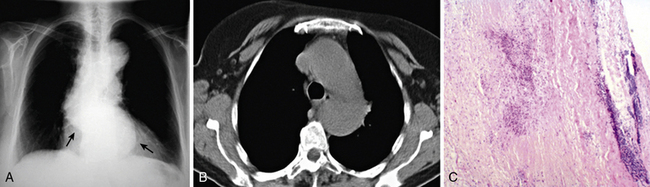
Figure 43-5 Aortic aneurysm in patient with giant cell arteritis (GCA) discovered 5 years after diagnosis.
Giant cell arteritis may also involve aortic branches (see Fig. 43-4). Positron emission tomography scan studies show that subclavian arteries may be involved in 70% of patients, axillary arteries in 40%, iliac arteries in 37%, and femoral arteries in 37%. Studies using color duplex ultrasonography (US) have disclosed abnormalities in these territories in about one third of patients.50 Distal lower-limb arteries (e.g., femoropopliteal, tibial, peroneal) may also be affected.5 Although in most instances, extremity involvement is asymptomatic, some patients develop claudication and even critical ischemia when significant vascular stenoses occur. Large-vessel occlusions are usually late manifestations of GCA, often occurring years after initial diagnosis. When they become clinically significant, they often are misdiagnosed as secondary to atherosclerotic disease. For this reason, the frequency of clinically relevant peripheral artery disease in GCA is not well known.
Myocardial or mesenteric infarction due to GCA is seen infrequently.51 Because GCA occurs in older people, some cases of inflammatory coronary or mesenteric arteritis may be misdiagnosed as due to atherosclerotic disease. Even among patients with GCA, however, atherosclerosis is a much more common cause of cardiac or mesenteric ischemia than vasculitis. With increased interest in the role of inflammation in coronary artery disease (CAD) and new tools to study the disease process, investigators are now better able to study the contributions, if any, of inflammatory arteritis to CAD among patients with GCA and other vasculitides.
Polymyalgia Rheumatica
Approximately 50% of patients with GCA present with symptoms of PMR, a syndrome clinically defined by the presence of aching and stiffness in the neck, shoulders, or pelvic girdle. Pain is exacerbated with movement. Morning stiffness is a prominent finding and may last for many hours. Proximal muscles are usually tender, but true weakness or myopathy is not seen. Ultrasonography, magnetic resonance imaging (MRI), and PET studies indicate that the underlying abnormalities are synovitis and bursitis of proximal joints.44,52
Polymyalgia rheumatica can appear simultaneously with cranial symptoms or may precede development of GCA symptoms by months or even years. Some patients without PMR develop PMR symptoms during GCA relapses, and PMR may sometimes be the only clinical manifestation of GCA.53
Patients with GCA may develop peripheral synovitis; knees, wrists, and metacarpophalangeal joints are most frequently involved. Peripheral manifestations usually occur in patients with PMR but may also occasionally appear in patients without proximal symptoms of PMR. Other associated manifestations include tenosynovitis, carpal tunnel syndrome, and distal swelling with pitting edema.44,54 Some patients develop a clinical picture indistinguishable from seronegative rheumatoid arthritis.44
Incidentally Discovered Giant Cell Arteritis
Occasionally, GCA may be unexpectedly diagnosed when vascular surgical specimens reveal arteritis with or without giant cells. In these patients, retrospective investigation may reveal prior signs or symptoms attributable to GCA. Several retrospective surgical or autopsy series of aortic specimens have demonstrated rates of nonsyphilitic aortitis ranging from 1% to 15%.55,56 In most instances, aortitis involved the thoracic aorta, and the majority of cases were in women. Fewer than 50% of these cases were associated with an identifiable clinical syndrome such as GCA. In a series of 1204 aortic surgical specimens from one institution, 52 (4.3%) demonstrated idiopathic aortitis, and only 12 of the 52 were found to have a non-GCA inflammatory disease.55 Because a subset of patients with idiopathic aortitis may go on to develop additional clinically important manifestations of GCA, it seems prudent to evaluate and follow such patients as one would cases of more clearly diagnosed GCA.57
Laboratory Findings
With few exceptions, both GCA and PMR are characterized by a strong acute-phase reaction. The erythrocyte sedimentation rate (ESR) is usually markedly elevated, frequently around 100 mm/h (Westergren method). Plasma concentrations of acute-phase proteins such as C-reactive protein (CRP), haptoglobin, and fibrinogen are also elevated. Protein electrophoresis shows an increase in α2 globulins. Thrombocytosis and chronic disease–type anemia are common, and some patients have abnormal liver function tests, particularly increased levels of alkaline phosphatase.6 Hyperbilirubinemia with visible jaundice is rare but may also occur. Symptomatic anemia may occasionally be the first clinical manifestation of GCA. Nonspecific immunological abnormalities, such as decreased numbers of circulating CD8 lymphocytes and elevated levels of soluble IL-2 receptors, are common in GCA and PMR.6
Several monocyte and EC activation products can be detected at increased concentrations in plasma from patients with GCA and PMR. These include cytokines such as IL-6 and TNF-α, soluble adhesion molecules such as ICAM-1, and von Willebrand factor (vWF) antigen.6,37,58,59 The role these or other cytokines or cellular markers may play in the diagnosis and management of GCA is an area of active investigation.
Diagnosis
Temporal Artery Biopsy
Histopathological examination of a temporal artery biopsy often provides the definitive diagnosis of GCA.12 The area to be excised is carefully selected, guided by symptoms and physical examination findings. At least a 2- to 3-cm fragment should be removed, and multiple sections examined histologically. When the initial biopsy is negative for evidence of GCA, excision of the contralateral artery is not routinely recommended but may increase diagnostic sensitivity in selected cases. Temporal artery biopsy is highly sensitive for the diagnosis of GCA.60 Occasionally, the temporal artery may be involved in the context of other systemic vasculitides or other disorders such as systemic amyloidosis.12,61,62 When systemic necrotizing vasculitis involves the temporal artery or its tributaries, it may present with cranial symptoms and complications similar to GCA.
Although the diagnostic yield of a temporal artery biopsy (if performed appropriately) is high, a normal temporal artery biopsy does not necessarily exclude GCA, owing to the segmental distribution of inflammatory infiltrates or involvement of other arteries. In only 10% of patients with negative temporal artery biopsy results obtained from biopsy performed and processed under optimal conditions is clinical suspicion strong enough to recommend long-term glucocorticoid therapy.60 Given the frequent existence of overlapping features among vasculitides, criteria sets have been established to classify patients with vasculitis into specific categories. The most commonly used classification criteria are those of the American College of Rheumatology63; the criteria for GCA are outlined in Box 43-1. Although not intended for use diagnostically, these criteria are useful when evaluating patients. Also, they are adopted for use as inclusion criteria for most research studies of GCA. However, to ensure that patients with nonvasculitic conditions are not mistakenly labeled as having GCA, caution must be exercised when applying these criteria. Classification criteria for this and other vasculitides are currently under reconsideration.64
 Box 43-1 American College of Rheumatology Criteria for Classification of Giant Cell (Temporal) Arteritis
Box 43-1 American College of Rheumatology Criteria for Classification of Giant Cell (Temporal) Arteritis
1. Age at disease onset ≥50 years
2. New onset of headache or new type of localized pain in the head
3. Temporal artery tenderness or decreased pulsation
5. Temporal artery biopsy showing vasculitis with a predominance of mononuclear cells or granulomatous inflammation, usually with multinucleated giant cells
ESR, erythrocyte sedimentation rate.
From Hunder GG, Bloch DA, Michel BA, et al: The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 33:1122–1128, 1990.
Diagnostic Imaging
A variety of imaging modalities are under investigation for use in the diagnosis and long-term management of GCA.65 These modalities include ultrasound, MRI, PET or PET-computed tomography (CT), and conventional contrast angiography (see Fig. 43-4). Lack of standardization for these modalities, wide variations in available equipment, absence of proper validation studies or long-term data, necessity of differentiating GCA findings from those of atherosclerotic disease, and the high cost of some studies are all limiting factors to more widespread adoption, but are all areas where progress is anticipated in the next decade. Furthermore, as these imaging techniques continue to be used extensively for evaluation of patients with fever of unknown origin, presumed cancer, or atherosclerotic disease, additional patients who actually have inflammatory vascular disease are likely to be encountered and diagnosed. Thus, vascular medicine specialists, vascular surgeons, and vascular radiologists will need to consider GCA more when reviewing such imaging studies.
Color duplex US and high-resolution MRI of the cranial arteries may be particularly useful in diagnosing GCA. The ultrasonographic finding of a dark hypoechoic halo surrounding the lumen, or MRI evidence of thickening and contrast enhancement of the artery wall, have remarkable specificity.66 Although temporal artery biopsy is the gold standard for diagnosis, these other techniques may be useful for biopsy site selection. They are increasingly considered as potential surrogates when biopsy is not feasible or when other arteries such as the occipital or axillary arteries are involved.50 Imaging techniques also may have a complementary role in evaluating other vascular territories; this is an area of active investigation.
MRI/MRA and CT/CTA are increasingly used imaging modalities for screening and evaluating large-vessel disease in GCA.65,67,68 Although MR or CT can detect luminal narrowing, arterial wall thickness, and wall enhancement, corresponding to inflammation, specificity of these findings as indicators of active vasculitis is unclear, and the tests are not sufficiently reliable to be the sole basis of treatment decisions. Nevertheless, MR is a relatively low risk method to screen and monitor patients for large-vessel disease in GCA and is increasingly part of the standard management for such patients.
Fluorodeoxyglucose-18 (18F-FDG) PET is another promising modality for evaluating patients with suspected GCA. Positron emission tomography scans may demonstrate FDG uptake in the aorta and its branches in patients with GCA, and also in some patients thought to have just PMR.69 As with MRI/MRA testing, the prognostic importance of detecting large arterial changes by 18F-FDG PET in asymptomatic patients with GCA is unclear. Furthermore, FDG uptake in the abdominal aorta and arteries of the lower limbs also can be seen in severe atherosclerosis, so specificity is lower in these locations.70 Its diagnostic sensitivity and specificity have to be tested in larger studies, but 18F-FDG PET may have a role in evaluation of vascular inflammation in patients with atypical symptoms, patients with fever of unknown origin, and when assessing vascular involvement in patients with apparently isolated PMR. The combination of PET with CT imaging may also have a role in evaluating these patients because it takes advantage of the properties of both techniques.
Diagnosis of Polymyalgia Rheumatica
Diagnosis of PMR relies at present on clinical criteria; imaging modalities may provide supportive evidence.71 Magnetic resonance imaging, PET, and ultrasound are able to detect subdeltoid or trochanteric bursitis, biceps tenosynovitis, or glenohumeral or hip synovitis, the sources of many polymyalgic symptoms.44,52,71,72 Recently an American College of Rheumatology/European League Against Rheumatic Disease classification algorithm71 has been proposed (Table 43-2). Polymyalgia rheumatica diagnosis requires evaluation to exclude other disorders, particularly rheumatoid arthritis, but also inflammatory myopathies, other vasculitides, and infections that occasionally present with similar symptoms.
Table 43-2 Polymyalgia Rheumatica Classification Criteria Scoring Algorithm*
| Criteria | Points without Ultrasound (0-6) | Points with Ultrasound (0-8)† |
|---|---|---|
| Morning stiffness >45 minutes | 2 | 2 |
| Hip pain or limited range of motion | 1 | 1 |
| Normal RF or ACPA | 2 | 2 |
| Absence of other joint movement | 1 | 1 |
| At least one shoulder with subdeltoid bursitis and/or biceps tenosynovitis and/or glenohumeral synovitis (either posterior or axillary) and At least one hip with synovitis and/or trochanteric bursitis |
NA | 1 |
| Both shoulders with subdeltoid bursitis, biceps tenosynovitis, or glenohumeral synovitis | NA | 1 |
ACPA, anticitrullinated protein antibody; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; NA, not applicable; RF, rheumatoid factor.
* Required criteria: age ≥50 years, bilateral shoulder aching, and abnormal CRP and/or ESR. A score of 4 or more is categorized as polymyalgia rheumatica (PMR) in the algorithm without ultrasound and a score of 5 or more is categorized as PMR in the algorithm with ultrasound.
† Optional ultrasound criteria.
From Dasgupta B, Cimmino MA, Maradit-Kremers H, et al: International Polymyalgia Rheumatica Classification Criteria Work Group. European League Against Rheumatism/American College of Rheumatology classification criteria for polymyalgia rheumatica. Ann Rheum Dis 71: 484–492, 2012.
Treatment and Management
Glucocorticoid therapy is the treatment of choice for GCA and in most cases induces a dramatic amelioration of disease manifestations within a few days. The most widely recommended initial dose is 40 to 60 mg/day of prednisone (or equivalent glucocorticoid). Presence of transient ocular manifestations (e.g., amaurosis fugax, diplopia, blurred vision) should be considered a medical emergency. Treatment should be started immediately, even before histological confirmation of GCA is obtained. Glucocorticoid treatment for several days, and even weeks, does not clear the inflammatory infiltrates and therefore should not hinder histopathological diagnosis.73–75 When visual loss is established, glucocorticoid pulses of 1 g/day of methylprednisolone (or equivalent glucocorticoid) for 3 days is frequently recommended, although it has not been clearly demonstrated that this dose regimen is more effective than the standard oral treatment. Treatment within the first 12 to 24 hours appears to be the major determinant of visual recovery, which can be expected in only 4% to 12% of cases.74–76 It is reasonable to recommend antiplatelet drugs, but their efficacy is not proven. Some patients with visual symptoms experience further visual loss during the first 1 to 2 weeks of glucocorticoid treatment. Visual loss beyond this point or during controlled relapses is rare.
The starting dose of prednisone (or equivalent) is maintained for 2 to 4 weeks. The daily dose is then tapered progressively by approximately 5 mg/wk. Although most patients do well with a daily maintenance dose of 7.5 to 10 mg, some patients require higher doses. Tapering is guided primarily by clinical evaluation. The ESR is a useful parameter for following patients with GCA, but therapeutic decisions must not rely solely on ESR values.6 The usual initial dose of prednisone for patients with isolated PMR is 10 to 20 mg/day. Guidelines for reduction are similar to those recommended for GCA. Some patients with PMR with mild symptoms may respond to nonsteroidal antiinflammatory drugs.54
Total duration of therapy may vary, but most patients require treatment for about 2 to 3 years. Reduction in dose below the maintenance doses must be made gradually to avoid relapses, which are common during the first 2 years after diagnosis. Approximately 40% to 60% of patients require low-dose glucocorticoid therapy for longer periods of time, some perhaps indefinitely.77
In the majority of patients, the ESR normalizes quickly after initiation of glucocorticoid therapy. Other inflammatory markers, however (e.g., IL-6, C-reactive protein, haptoglobin, vWF antigen) are elevated persistently in many patients who appear to be in clinical remission, possibly indicating persistent low-level inflammatory activity.37,78 Long-term consequences of persistent subclinical activity are unknown. Subclinical inflammatory activity does not appear to be associated with a higher incidence of delayed complications of GCA, such as aneurysm or vascular occlusion.4 Persistent modest elevation of inflammatory markers should not lead to an increase in glucocorticosteroid dose in the setting of clinical remission.
Other immunosuppressive drugs may be considered as “steroid sparing agents” to reduce the cumulative toxicity of glucocorticoid therapy. The most carefully investigated of these agents is methotrexate, which was evaluated in three randomized placebo-controlled double-blind studies.79–81 An individual patient–level meta-analysis of these trials demonstrated modest but significant benefits of methotrexate, including prevention of relapses and reduction of glucocorticoid use.82 Although small case series reported beneficial effects of TNF blockers for GCA, TNF-α blockade with infliximab was not superior to placebo in maintaining glucocorticoid-induced remission in an international randomized double-masked placebo-controlled trial.83 The efficacy in GCA of interfering with CD28-mediated T-cell activation with abatacept is currently being tested in a clinical trial. Although the potential for treatment of GCA through B-cell depletion (rituximab) or by neutralizing IL-6 (tocilizumab) are intriguing, case reports alone are not adequate evidence of efficacy, and randomized trials are needed to establish the roles, if any, of these or other agents.
1 Hunder G. Giant cell arteritis and polymyalgia rheumatica. Med Clin North Am. 1997;81:195–219.
2 Cid M.C., Coll-Vinent B., Grau J.M. Large vessel vasculitides. Curr Opin Rheumatol. 1998;10:18–28.
3 Salvarani C., Cantini F., Boiardi L. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med. 2002;347:261–271.
4 Garcia-Martinez A., Hernandez-Rodriguez J., Arguis P., et al. Development of aortic aneurysm/dilatation during the followup of patients with giant cell arteritis: a cross-sectional screening of fifty-four prospectively followed patients. Arthritis Rheum. 2008;59:422–430.
5 Assie C., Janvresse A., Plissonnier D. Long-term follow-up of upper and lower extremity vasculitis related to giant cell arteritis: a series of 36 patients. Medicine (Baltimore). 2011;90:40–51.
6 Salvarani C., Cantini F., Hunder G.G. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372:234–245.
7 Hunder G.G. Epidemiology of giant-cell arteritis. Cleve Clin J Med. 2002;69(Suppl 2):SII79–SII82.
8 Baldursson O., Steinsson K., Bjornsson J. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum. 1994;37:1007–1012.
9 Salvarani C., Gabriel S.E., O’Fallon W.M. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med. 1995;123:192–194.
10 Gonzalez-Gay M.A., Vazquez-Rodriguez T.R., Lopez-Diaz M.J., et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2009;61:1454–1461.
11 Sonnenblick M., Nesher G., Friedlander Y. Giant cell arteritis in Jerusalem: a 12-year epidemiological study. Br J Rheumatol. 1994;33:938–941.
12 Lie J. Histopathologic specificity of systemic vasculitis. Rheum Dis Clin North Am. 1995;21:883–909.
13 Cid M.C., Campo E., Ercilla G., et al. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum. 1989;32:884–893.
14 Esteban M.J., Font C., Hernandez-Rodriguez J., et al. Small-vessel vasculitis surrounding a spared temporal artery: clinical and pathological findings in a series of twenty-eight patients. Arthritis Rheum. 2001;44:1387–1395.
15 Ostberg G. Temporal arteritis in a large necropsy series. Ann Rheum Dis. 1971;30:224–235.
16 Meliconi R., Pulsatelli L., Uguccioni M., et al. Leukocyte infiltration in synovial tissue from the shoulder of patients with polymyalgia rheumatica. Quantitative analysis and influence of corticosteroid treatment. Arthritis Rheum. 1996;39:1199–1207.
17 Meliconi R., Pulsatelli L., Melchiorri C., et al. Synovial expression of cell adhesion molecules in polymyalgia rheumatica. Clin Exp Immunol. 1997;107:494–500.
18 Weyand C.M., Hicok K.C., Hunder G.G. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest. 1992;90:2355–2361.
19 Mattey D.L., Hajeer A.H., Dababneh A., et al. Association of giant cell arteritis and polymyalgia rheumatica with different tumor necrosis factor microsatellite polymorphisms. Arthritis Rheum. 2000;43:1749–1755.
20 Boiardi L., Casali B., Nicoli D., et al. Vascular endothelial growth factor gene polymorphisms in giant cell arteritis. J Rheumatol. 2003;30:2160–2164.
21 Salvarani C., Casali B., Nicoli D., et al. Endothelial nitric oxide synthase gene polymorphisms in giant cell arteritis. Arthritis Rheum. 2003;48:3219–3223.
22 Salvarani C.C., Boiardi B., Ranzi L., et al. Intercellular adhesion molecule 1 gene polymorphisms in polymyalgia rheumatica/giant cell arteritis: association with disease risk and severity. J Rheumatol. 2000;27:1215–1221.
23 Jacobsen S., Baslund B., Madsen H.O., et al. Mannose-binding lectin variant alleles and HLA-DR4 alleles are associated with giant cell arteritis. J Rheumatol. 2002;29:2148–2153.
24 Martinez-Taboada V.M., Alvarez L., RuizSoto M., et al. Giant cell arteritis and polymyalgia rheumatica: role of cytokines in the pathogenesis and implications for treatment. Cytokine. 2008;44:207–220.
25 Regan M.J., Wood B.J., Hsieh Y.H., et al. Temporal arteritis and Chlamydia pneumoniae: failure to detect the organism by polymerase chain reaction in ninety cases and ninety controls. Arthritis Rheum. 2002;46:1056–1060.
26 Salvarani C., Farnetti E., Casali B., et al. Detection of parvovirus B19 DNA by polymerase chain reaction in giant cell arteritis: a case-control study. Arthritis Rheum. 2002;46:3099–3101.
27 Weck K.E., Dal Canto A.J., Gould J.D., et al. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus- induced vascular disease. Nat Med. 1997;3:1346–1353.
28 Krupa W.M., Dewan M., Jeon M.S., et al. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am J Pathol. 2002;161:1815–1823.
29 Palomino-Morales R., Torres O., Vazquez-Rodriguez T.R., et al. Association between toll-like receptor 4 gene polymorphism and biopsy-proven giant cell arteritis. J Rheumatol. 2009;36:1501–1506.
30 Weyand C.M., Schonberger J., Oppitz U., et al. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. 1994;179:951–960.
31 Weyand C.M., Tetzlaff N., Bjornsson J., et al. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum. 1997;40:19–26.
32 Deng J., Younge B.R., Olshen R.A., et al. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121:906–915.
33 Rittner H.L., Kaiser M., Brack A., et al. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84:1050–1058.
34 Weyand C.M., Goronzy J.J. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349:160–169.
35 Segarra M., Garcia-Martinez A., Sanchez M., et al. Gelatinase expression and proteolytic activity in giant-cell arteritis. Ann Rheum Dis. 2007;66:1429–1435.
36 Hernandez-Rodriguez J., Segarra M., Vilardell C., et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNF-alpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology (Oxford). 2004;43:294–301.
37 Lozano E., Segarra M., Garcia-Martinez A., et al. New therapeutic targets in giant-cell arteritis. Considerations based on the current pathogenic model and the availability of new therapeutic agents. Clin Exp Rheumatol. 2008;26:S141–S150.
38 Cid M.C., Cebrian M., Font C., et al. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte-endothelial cell interactions. Arthritis Rheum. 2000;43:184–194.
39 Cid M.C., Hernandez-Rodriguez J., Esteban M.J., et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant-cell arteritis. Circulation. 2002;106:1664–1671.
40 Cid M.C., Ercilla G., Vilaseca J., et al. Polymyalgia rheumatica: a syndrome associated with HLA-DR4 antigen. Arthritis Rheum. 1988;31:678–682.
41 Font C.C., Cid M.C., Coll-Vinent B., et al. Clinical features in patients with permanent visual loss due to biopsy–Proven giant cell arteritis. Br J Rheumatol. 1997;36:251–254.
42 Hernandez-Rodriguez J., Segarra M., Vilardell C., et al. Elevated production of interleukin-6 is associated with a lower incidence of disease-related ischemic events in patients with giant-cell arteritis: angiogenic activity of interleukin-6 as a potential protective mechanism. Circulation. 2003;107:2428–2434.
43 Nuenninghoff D.M., Hunder G.G., Christianson T.J., et al. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48:3522–3531.
44 Salvarani C., Macchioni P., Boiardi L. Polymyalgia rheumatica. Lancet. 1997;350:43–47.
45 Huston K.A., Hunder G.G., Lie J.T., et al. Temporal arteritis: a 25-year epidemiologic, clinical, and pathologic study. Ann Intern Med. 1978;88:162–167.
46 Amor-Dorado J.C., Llorca J., Garcia-Porrua C., et al. Audiovestibular manifestations in giant cell arteritis: a prospective study. Medicine (Baltimore). 2003;82:13–26.
47 Blockmans D., de Ceuninck L., Vanderschueren S., et al. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. 2006;55:131–137.
48 Cid M.C., Prieto-Gonzalez S., Arguis P., et al. The spectrum of vascular involvement in giant-cell arteritis: clinical consequences of detrimental vascular remodelling at different sites. APMIS Suppl. 2009:10–20.
49 Evans J.M., O’Fallon W.M., Hunder G.G. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med. 1995;122:502–507.
50 Schmidt W.A., Seifert A., Gromnica-Ihle E., et al. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology (Oxford). 2008;47:96–101.
51 Scola C.J., Li C., Upchurch K.S. Mesenteric involvement in giant cell arteritis. An underrecognized complication? Analysis of a case series with clinicoanatomic correlation. Medicine (Baltimore). 2008;87:45–51.
52 Camellino D., Cimmino M.A. Imaging of polymyalgia rheumatica: indications on its pathogenesis, diagnosis and prognosis. Rheumatology (Oxford). 2011. May 12 [Epub ahead of print]
53 Hernandez-Rodriguez J., Font C., Garcia-Martinez A., et al. Development of ischemic complications in patients with giant cell arteritis presenting with apparently isolated polymyalgia rheumatica: study of a series of 100 patients. Medicine (Baltimore). 2007;86:233–241.
54 Chuang T.Y., Hunder G.G., Ilstrup D.M., et al. Polymyalgia rheumatica: a 10-year epidemiologic and clinical study. Ann Intern Med. 1982;97:672–680.
55 Rojo-Leyva F., Ratliff N.B., Cosgrove D.M.3rd, et al. Study of 52 patients with idiopathic aortitis from a cohort of 1,204 surgical cases. Arthritis Rheum. 2000;43:901–907.
56 Liang K.P., Chowdhary V.R., Michet C.J., et al. Noninfectious ascending aortitis: a case series of 64 patients. J Rheumatol. 2009;36:2290–2297.
57 Merkel P.A. Noninfectious ascending aortitis: staying ahead of the curve. J Rheumatol. 2009;36:2137–2140.
58 Roche N.E., Fulbright J.W., Wagner A.D., et al. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36:1286–1294.
59 Coll-Vinent B., Vilardell C., Font C., et al. Circulating soluble adhesion molecules in patients with giant cell arteritis. Correlation between soluble intercellular adhesion molecule-1 (sICAM-1) concentrations and disease activity. Ann Rheum Dis. 1999;58:189–192.
60 Vilaseca J., Gonzalez A., Cid M.C., et al. Clinical usefulness of temporal artery biopsy. Ann Rheum Dis. 1987;46:282–285.
61 Genereau T., Lortholary O., Pottier M.A., et al. Temporal artery biopsy: a diagnostic tool for systemic necrotizing vasculitis. French Vasculitis Study Group. Arthritis Rheum. 1999;42:2674–2681.
62 Duran E., Merkel P.A., Sweet S., et al. ANCA-associated small vessel vasculitis presenting with ischemic optic neuropathy. Neurology. 2004;62:152–153.
63 Hunder G.G., Bloch D.A., Michel B.A., et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–1128.
64 Basu N., Watts R., Bajema I., et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis. 2010;69:1744–1750.
65 Kissin E.Y., Merkel P.A. Diagnostic imaging in Takayasu arteritis. Curr Opin Rheumatol. 2004;16:31–37.
66 Arida A., Kyprianou M., Kanakis M., et al. The diagnostic value of ultrasonography-derived edema of the temporal artery wall in giant cell arteritis: a second meta-analysis. BMC Musculoskelet Disord. 2010;11:44.
67 Blockmans D. Utility of imaging studies in assessment of vascular inflammation. Cleve Clin J Med. 2002;69(Suppl 2):SII95–SII99.
68 Tso E., Flamm S.D., White R.D., et al. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum. 2002;46:1634–1642.
69 Blockmans D., Stroobants S., Maes A., et al. Positron emission tomography in giant cell arteritis and polymyalgia rheumatica: evidence for inflammation of the aortic arch. Am J Med. 2000;108:246–249.
70 Yun M., Jang S., Cucchiara A., et al. 18F FDG uptake in the large arteries: a correlation study with the atherogenic risk factors. Semin Nucl Med. 2002;32:70–76.
71 Dasgupta B., Cimmino M.A., Maradit-Kremers H., et al. European League Against Rheumatism/American College of Rheumatology classification criteria for polymyalgia rheumatica. Ann Rheum Dis. 2012;71:484–492.
72 Cantini F., Salvarani C., Olivieri I., et al. Shoulder ultrasonography in the diagnosis of polymyalgia rheumatica: a case-control study. J Rheumatol. 2001;28:1049–1055.
73 Achkar A.A., Lie J.T., Hunder G.G., et al. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med. 1994;120:987–992.
74 Hayreh SP P.A., Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998;125:521–526.
75 Foroozan R., Deramo V.A., Buono L.M., et al. Recovery of visual function in patients with biopsy-proven giant cell arteritis. Ophthalmology. 2003;110:539–542.
76 Gonzalez-Gay M.A., Blanco R., Rodriguez-Valverde V., et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum. 1998;41:1497–1504.
77 Proven A., Gabriel S.E., Orces C., et al. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49:703–708.
78 Weyand C.M., Fulbright J.W., Hunder G.G., et al. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43:1041–1048.
79 Jover J.A., Hernandez-Garcia C., Morado I.C., et al. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;134:106–114.
80 Spiera R.F., Mitnick H.J., Kupersmith M., et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol. 2001;19:495–501.
81 Hoffman G., Cid M., Hellmann D., et al. A multicenter placebo-controlled study of methotrexate (MTX) in giant cell arteritis (GCA) [abstract]. Arthritis Rheum. 43, 2002.
82 Mahr A.D., Jover J.A., Spiera R.F., et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789–2797.
83 Hoffman G.S., Cid M.C., Rendt-Zagar K.E., et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–630.