[level-membership-for-neurology-category]
Chapter 8 Genetics of Restless Legs Syndrome
Clinical Genetics of Restless Legs Syndrome
Data from epidemiological and family studies clearly indicate that a genetic component plays an important role in the pathogenesis of restless legs syndrome (RLS).
Restless Legs Syndrome is a Common Neurological Disorder with Variable Prevalence in Different Populations
Although RLS was first systematically described by Ekbom,1 a Swedish neurologist, in 1945, accurate estimates of population incidence and prevalence only started to emerge in the late 1990s, following the first large-scale population survey by Montplaisir and associates.2 Numerous recent epidemiological studies further confirmed that RLS is a very prevalent disorder in the population (see Chapter 7), with a prevalence around 10% to 15% in Western populations,3–9 which is equivalent to the sum of prevalences for epilepsy (≈1%),10 Parkinson’s disease (≈1%),11,12 Alzheimer’s disease (≈1%),13 schizophrenia (≈1%),14,15 and other neuropsychatric disorders, and only comparable with a few common prevalent diseases, such as depression (≈10%),16 hypertension (≈20%),17 and obesity (≈16%).18 Further studies suggest that there are substantial prevalence variations of RLS in different populations and racial groups. The prevalence was estimated higher in certain geographically or socially isolated populations, such as the French Canadians in Quebec compared with those in other provinces of Canada.2 Conversely, RLS prevalence seems to be lower in Asian and African American populations compared with whites based on some preliminary results.19–24 However, one more recent survey of an adult community sample from east Baltimore suggests that there may be no appreciable difference of RLS prevalence between whites and African Americans.25 Further large-scale population studies with validated diagnostic instruments will be needed to clarify the racial difference in RLS. Although both genetic and environmental factors may account for the divergence of prevalence in different populations, significant variations in disease incidence and prevalence among different racial/ethnic groups are usually an indicator of a strong genetic effect at the population level, especially when different ethnic groups show variable disease frequencies within a similar environment. Certainly, the discrepancy of prevalence difference between studies may also have derived from the sensitivity and specificity of the different diagnostic instruments used in the different studies, as well as ascertainment bias and population substratification due to geographical or cultural reasons.
Familiality
RLS is not only prevalent in the general population but also aggregates in families. The familial character of RLS has long been recognized since its first clinical description,1 and it has been consistently reported by experienced clinicians.2,26–31 Family history has been recognized as a significant risk factor for RLS.32 The proportion of familial cases present in the overall RLS patient population has been estimated to be 60%,27–3033 but it could be as high as 80% to 90% among idiopathic RLS patients.27,34 In our extensive family studies of 244 probands with idiopathic RLS, only 57 probands do not have a positive family history, which translates into a familial rate of 76.6% in our sample (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). Family data from the Baltimore group also show that about 70% of their RLS patients have an interviewed first-degree relatives affected by RLS (W. A. Hening, preliminary data, 2006). A sample of RLS Foundation brain bank volunteers (N = 86; 82% women) had an even higher frequency of affected first-degree relatives: 79% (W. A. Hening, preliminary data, 2006).
Several large pedigrees were described in the literature where multiple members were affected with RLS over a span of three to five generations.28,31,35–41 According to our experience, this phenomenon is quite common in the general population, at least in French Canadians, further confirming its genetic nature.
Heritability
Adequate clinical data strongly indicate that RLS has an important genetic component. However, a crucial step before undertaking any molecular genetic studies in RLS is to determine the degree to which the phenotype is determined by the underlying genetic component. Data from population, family, and twin studies usually permit geneticists to estimate the magnitude of the genetic effect on the trait under study. The estimation of heritability is calculated by the expected correlation between family members for the phenotypic trait based on their degree of relationship. The prevalence of RLS in first-degree relatives of affected patients is estimated to be between 20% and 60%.26–2931 Montplaisir and colleagues28 examined 133 RLS patients who were studied using a standardized self-report questionnaire. In this sample, 63% had at least one first-degree relative affected with RLS, and 221 of 568 (38.9%) first-degree relatives were reported as affected.
Traditionally, the most powerful way to estimate the genetic and environmental components of phenotypic variance is to study monozygotic (MZ) and dizygotic (DZ) twins. However, due to its recent acceptance as a clinical entity and the development of accurate diagnostic criteria and instruments, there are few papers reporting limited data on twin studies about RLS that are up to date. One is from the Ondo group,42 which reported 12 pairs of MZ twins, among them 10 pairs concordant, 1 pair probably concordant, and 1 pair discordant on RLS symptoms. There were no concomitant data on DZ twins, making it impossible to differentiate the relative proportions and magnitude of genetic and environmental factors. The other large-scale population-based twin studies of RLS symptoms included 933 MZ and 1004 DZ twins who have completed two questions regarding their restless legs symptoms.43 This study showed that the concordance rate of possible RLS symptoms is 61% in MZ and 45% in DZ twins; the concordance rate for “involuntary leg jerks during the night,” a possible metaphor for periodic leg movements during sleep (PLMS), is 69% in MZ and 54% in DZ twins. Further genetic modeling with the same set of data by using the frequencies of disease-concordant and disease-discordant rates and applying to a multifactorial liability threshold model indicates that additive genetic factors and a unique environment best explained the variance in disease liability. However, the critical weakness of this study is that the RLS diagnosis was only based on a simplified two-question self-report survey, which did not include all four essential clinical features. Therefore, they did not study RLS, but rather possibly a component of the disease. Nevertheless, as demonstrated in numerous studies of other genetic disorders, twin studies are powerful genetic tools to quantify and define the genetic contribution, possible mode of inheritance, and potential gene–environment interaction. Our data from a population-based survey of 272 twin pairs from Canada show that the concordant rate of definite RLS is 53.7% and 19.0% in MZ and DZ twins, respectively.44 The estimated heritability is 69.4%, confirming the importance of the genetic factor in RLS etiology. However, in the World War II twin cohort (176 MZ and 135 DZ twin pairs), concordance was only slightly higher in MZ twins (23% versus 16%), leading to a much lower estimated heritability of 20%.8 This may reflect the much greater influence of environmental factors leading to secondary RLS in this aged, male-only cohort. The information obtained from additional appropriate twin studies will be critical for further molecular genetic studies aimed at identifying underlying causative or susceptibility genes for RLS.
It is well known in the field of genetic studies that the pattern of risk ratio (λR) in various degrees of relationship within a family may reflect the underlying genetic mechanism. The magnitude of λR can usually be used to predict the statistical power to detect linkage in gene mapping.45–47 For a single-locus model and an additive multilocus genetic model, λR decreases by a factor of 2 with each degree of relationship. However, for a multiplicative (epistasis) model, λR decreases more rapidly than by a factor of 2 with degree of relationship.45 For example, λR in first-, second-, and third-degree relatives of schizophrenia patients is 6% to 17%, 2% to 6%, and 2%, respectively,48 suggesting the presence of multiple interacting loci. Unfortunately, there are very limited data available regarding the prevalence of RLS in more distant relatives other than the first-degree relatives, due to the difficulty in disease ascertainment. In one study of 96 RLS families, Allen and associates30 reported that 19.9% of the first-degree relatives versus 4.1% of the second-degree relatives are affected compared with 3.5% versus 0.5% in 15 non-RLS control subjects.30 Compared with a prevalence of 3.5% in their control group, the relative risk ratio λR dropped quickly from 5.7 to 1.2 from the first-degree relatives to the second-degree relatives. In another study of 15 large multiplex families with RLS by the Baylor group, the λR for parent-offspring is 10.25 and the λR for siblings is 16.23, and the overall heritability in their sample was estimated at 60%.31 Undoubtedly, the λR estimated in this study will not reflect the relative risk in general population, because the sample is extremely biased toward only the heavily loaded RLS families. In the Johns Hopkins family study, the relative risk for first-degree relatives of RLS probands (N = 134) was 3.6 compared with both the general population figures9 and an age- and gender-matched control group (N = 58), with higher figures for younger-onset probands (W. A. Hening and colleagues, preliminary data).
Mode of Inheritance
It is possible for geneticists to propose a genetic model for a given genetic trait, by deriving information from data obtained from population and family studies and by using specific statistical tools. Also, the simpler the underlying genetic structure, the more reliable their prediction becomes. However, the genetic study of RLS is at a very early stage compared with other hereditary forms of neurological and psychiatric disorders, such as epilepsy and schizophrenia, where many population and molecular genetic studies have been undertaken. In RLS, there are very limited reliable data on which to speculate. A few studies have attempted to model the genetic transmission of RLS. In most of the reported pedigrees, vertical transmission is predominant, making RLS mostly compatible with an autosomal dominant mode of inheritance with relatively high penetrance.31,35–37 The Winkelmann group from Germany performed complex segregation analyses on 238 families and predicted that a single autosomal allele acting dominantly can explain RLS in families with an early age at onset (AO) of symptoms, but it will not account for RLS with a later onset (>30 years).49 However, another recent segregation study by Mathias and coworkers50 indicates that a dominant model works for all families, not only younger-onset families. That study also found both a high frequency for the major gene and a high phenocopy rate (14%) fit better with the genetic model. That analysis also showed that there was genetic control of age at RLS onset, but without a major gene effect. Nevertheless, a careful examination of the detailed family histories in RLS pedigrees has always suggested a genetic model more complicated than a classic mendelian inheritance. In some reports, the percentage of RLS in first-degree relatives is greater than 50%34,37; we have also observed such segregation distortion in some of our collected RLS families (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). The observed ratio of more than one half of first-degree relatives being affected in some RLS families may be due to an ascertainment bias but warrants further study to either confirm or refute this unusual observation. Some dominant families also showed reduced AO in consecutive generations, indicating possible anticipation.36,37 Due to the high prevalence of RLS and possible assortative mating in the population, bilinear inheritance is not uncommon. Among 50 familial cases in the Ondo group’s study, three had both parents affected.42 In our family studies, we also frequently encounter affected spouses and observe bilinear inheritance (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). All pedigrees in the published linkage studies reported individuals carrying the predisposing haplotype but without the disease phenotype (nonpenetrants) and individuals without the haplotype but presenting with the disease symptoms (phenocopies).31,38,40 More surprisingly, our linkage study of one large French-Canadian family showed the most significant results under an autosomal recessive model with an unusually high common disease allele frequency (25%), although the pedigree appears to be dominant38; that is, a pseudodominant inheritance. In the systematic analysis of 15 autosomal dominant–looking multiplex families with 134 affected, 136 founders, and 317 nonfounders, Chen and coworkers31 demonstrated that the λR is higher in sib-pairs than in parent-offspring pairs (16.23 versus 10.25), which could also be explained by recessively or additively acting disease alleles. In summary, all available evidence suggests that genetic factors play an important role in the etiology of RLS; however, the mode of inheritance is probably more complex than it appears on cursory examination of available pedigrees. Results from complex segregation analysis need to be examined carefully and interpreted with caution because it is known to be problematic when dealing with complex traits.51,52
Variable Age at Onset and Bimodal Age at Onset Distribution
In general, RLS is an adult-onset neurological disorder with a prevalence that tends to increase linearly with age.3,7 In Ondo’s twin study, the earliest AO is 3 years of age, whereas the latest is 65 years of age. The AO varied by 40 years in two of the MZ twin pairs.42 The clinical manifestations of RLS also slowly progress with age in some patients with a severe form of RLS, which is similar to other late-onset neurodegenerative diseases. However, a certain percentage of patients and their family members may present with only a much milder form of RLS, never seeking medical attention and having a clinical course that is intermittent or waxing and waning. The limited number of neuropathological studies available also indicates that RLS is not caused by a traditional neurodegenerative processes, such as seen in τ- or α-synuclein brain pathologies.53,54
Several studies suggest a bimodal distribution of AO among RLS patients.28–30,49,55 The younger probands usually have a higher rate of positive family history and slower disease progression rate, suggesting a greater genetic contribution and possibly a distinctive clinical course compared with nongenetic forms of RLS cases.28–3055 Many complex and highly prevalent disorders exhibit a wide range of AO due to various genetic and environmental factors that contribute to the same phenotype. An earlier AO is often considered a sign of genetic predisposition in many diseases, such as Alzheimer’s disease,56,57 breast cancer,58 prostate cancer,59 and Parkinson’s disease.60,61 The use of AO information affects the power of gene mapping and identification. Several strategies can be applied to integrate AO information into molecular genetic studies. If a major gene effect is suspected that manifests itself at a specific range of age, the simplest way to use AO information is to stratify patients and families into early- and late-onset subgroups. Hopefully, in this way, the samples can be divided into more homogeneous subgroups to help circumvent the underlying genetic heterogeneity, or to purge the nongenetic cases. In parametric linkage analysis, adjustment of variable AO to different age-dependent liability classes within pedigrees is an important component of effective linkage analysis.62–64 Liability classes are used to define penetrance values for each of the possible genotypes of the trait locus and to classify each individual into different penetrance groups on the basis of their age at investigation. In complex traits, the effects of AO on penetrance model–free analyses are even more complicated and have been further investigated by Li and associates.65 These authors suggested that incorporating the AO information into affected sib-pairs (ASP) and transmission disequilibrium (TDT) tests, especially when focusing on sib-pairs both with early AO and TDT with all early AO trios, will greatly improve the power to detect the genetic signals. Alternatively, AO can be treated either as a covariate or quantitative trait by using variance component linkage analysis.66 This is usually applied to identify genetic variants that will influence age at disease onset. For example, this method has been successfully applied to map genes modifying disease onset for type 2 diabetes,67 Parkinson’s disease,68 and Alzheimer’s disease.69 Nevertheless, the preliminary data from complex segregation analyses by Mathias and colleagues50 suggest that there is complex genetic control of age at onset for RLS in their samples.
Variable Phenotypic Expressivity
One of the most striking features of RLS is the high degree of variable phenotypic expressivity,35,36 even within MZ twins.42 The cardinal clinical symptom of RLS is an imperative urge to move, which is very subjective without any reliable validation and measurement. Misdiagnosis and underdiagnosis are not uncommon in clinical practice. RLS is considered one of the most common and least diagnosed sleep disorders, as well as neurological disorders.70,71 Furthermore, as we mentioned earlier, frequently there are even milder forms among family members who never need medical attention and only get ascertained during family studies, quite often through telephone interviews only. We know that RLS can sometimes be very severe, causing intractable insomnia72; unfortunately, we do not know how mild symptoms should be interpreted, whether as mild RLS or as something completely unrelated. This is critically important in genetic studies. Until more reliable biological markers or laboratory tests become available, in current family linkage studies, different diagnostic criteria schemes can be applied to define the exact phenotype under study, from the most to the least stringent, to accommodate the wide range of phenotypic variations within the pedigree. Because the primary symptom of RLS is an imperative urge to move, other features, such as frequency and intensity of symptoms, can all be considered as measurements of severity. To fully address the phenotypic variations within and between families in gene mapping, the degree of severity, together with variable AO, can also be treated as covariate or quantitative traits by using a variance component method. Nonetheless, all these approaches require a significantly large number of participating families.
Phenocopies and Association with Other Common Medical Conditions
A phenotype that does not result, at least in part, from a specific gene or locus under study is called a phenocopy in genetic analysis. Phenocopies can be environmentally induced phenotypes that mimic the genetically determined phenotypes or they can be identical phenotypes that are not genetically controlled by the same gene under study. The literature reports that parametric linkage analysis is very sensitive to phenocopy rate.73,74 High phenocopy rate is considered as a significant obstacle to gene mapping.
RLS is a very common disorder; it is known that RLS symptoms can be caused or influenced by other nongenetic factors as well. RLS has been reported to be associated with several other common medical conditions, such as renal failure, anemia, and pregnancy. Patients suffering from arthritis, peripheral neuropathy, and spinal cord injury can present with exactly the same symptoms as idiopathic RLS patients. It remains unclear whether these medical conditions predispose to RLS through distinct pathological mechanisms or by interacting with common predisposing genetic variant(s). Therefore, idiopathic and nonidiopathic RLS should both be taken into consideration when searching for the common predisposing variant(s) and should be analyzed both as one entity and as separate disease groups. For example, RLS symptoms occur more frequently in pregnant women; their occurrence correlates with a stronger family history: 29% of women presenting with RLS during pregnancy reported a first-degree relative with RLS symptoms.75 Clinical observations also strongly suggest that iron metabolism might play an important role in RLS.53,76–78 Many studies indicate that there is relative iron deficiency in brains of RLS patients.53,76–78 It is unknown whether the RLS patients are more prone to anemia/iron insufficiency or they are just more sensitive to low iron levels in the body and/or in the brain.
The most common medical condition associated with RLS is PLMS. Clinically, PLMS is the number one risk factor for RLS.32 It is reported that more than 80% to 90% of RLS patients have PLMS,28 and the presence of periodic limb movements has been used as a supportive clinical feature for RLS diagnosis.32,79 PLMS may nevertheless occur in healthy individuals, especially in the elderly (>65 years old).28 PLMS are also associated with several other sleep disorders, such as narcolepsy,80 obstructive sleep apnea,81 rapid eye movement (REM) sleep behavior disorder,82 and with Parkinson’s disease.83 RLS and PLMS are clinically distinguishable but significantly overlapping disorders. Due to their high prevalence in the general population, similar clinical features, and similar response to dopaminergic treatment, it has been proposed that they may share the same etiology and common pathways79; some clinicians have even proposed that PLMS is a milder form of RLS,40 or forme fruste. This creates a dilemma in RLS genetic studies: Are we mapping a gene causing/predisposing to RLS, or PLMS, or both? Not many studies have been conducted to characterize their co-occurrence in families, except for one of an Italian family mapped to chromosome 14q.40 Among 18 individuals in the studied family, 10 RLS patients had polysomnography (PSG) recordings done; all showed a PLMS index greater than 5. Four nonsymptomatic individuals had a normal PSG, and one individual showed a PLMS index greater than 5 but without RLS diagnosis. Therefore, RLS is highly but not completely correlated with PLMS in this family. In our study, significantly higher PLMS indices were observed for all RLS probands from families linked to the chromosome 12q locus.84 A study by the Baltimore group showed that PLMS are increased in first-degree relatives free of RLS symptoms or with mild RLS within studied RLS families, and PLMS increased with age in family members related to the probands with early AO.85 Their study further suggests that RLS and PLMS may share some common genetic component(s) or may represent the sensory and motor components of the disorder respectively. To consider both alternatives, we need to not only document RLS symptoms in individuals under study but also phenotype PLMS in all participants.
Molecular Genetics of Restless Legs Syndrome
Linkage Studies
To date, five loci have been reported to be linked to RLS. The first genetic locus for RLS was mapped to chromosome 12q by Desautels and colleagues38 in a large French Canadian family. However, the results were surprising, because the significant linkage was reached by using unusual parameters, that is, a recessive model and a very common disease allele frequency (25%), whereas inspection of the pedigree suggests an autosomal dominant–like segregation. Other groups failed to replicate the results.39,40 However, we have found further evidence supporting the linkage to this locus in additional RLS French Canadian families.84 Most recently, this locus has been confirmed in a large cohort of Icelandic samples reported by Hicks and associates.86 Their genome-wide linkage analysis of 1100 microsatellite markers in 382 affected individuals satisfying the four diagnostic criteria from 126 families yielded an LOD score of 2.4 by using an affected-only nonparametric method. Simultaneous re-analysis of the same genotype data with PLMS phenotype (dichotomous periodic leg movement index [PLMi] >10) generated an LOD score of 3.9 (p = 1.02 × 10−5, likelihood ratio z-score [Zlr] = 4.25). Although the results are not genome-wide significant after correction for multiple testing, it is supportive for the RLS1 locus on chromosome 12q. A further study by the German group suggests a low-penetrant allele was overtransmitted in 12 Bavarian RLS families with 70 patients with a confirmed diagnosis of RLS.87 Therefore, the RLS locus on chromosome 12q is the only linked locus currently replicated in additional samples and different populations, and thus it remains the major candidate region for RLS. However, the potential allelic heterogeneity, low disease penetrance, and high phenocopy rate have made follow-up studies difficult.
Two additional candidate loci for RLS have been reported: one on chromosome 14q (RLS2) in one Italian family40 and the other on chromosome 9p (RLS3) in two American families.31 The 14q (RLS2) locus was confirmed with suggestive linkage in one French Canadian family.88 The linkage to the RLS3 locus was also supported by TDT in one Bavarian family.89 In addition, we have identified a fourth locus for RLS on chromosome 20p13 (RLS4) in another large French Canadian family with a multipoint LOD score of 3.87 under an dominant model.90 Pichler and coworkers91 reported the fifth locus for RLS on chromosome 2q in a south Tyrolean population isolate. Table 8-1 provides a summary of these five loci.
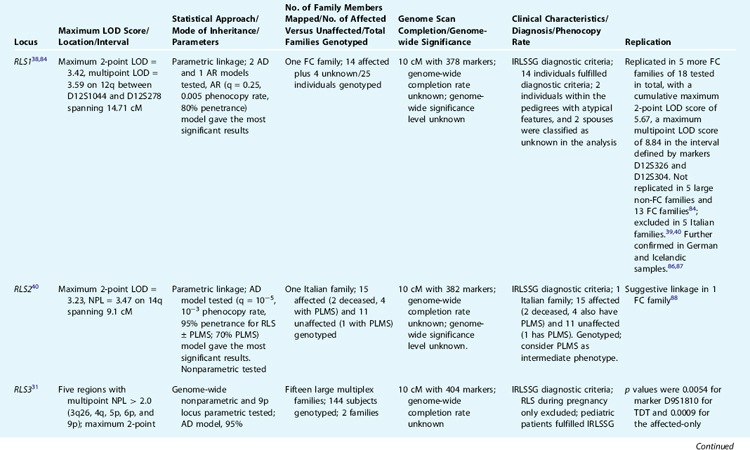

The chromosome 12q, 14q, and 20p loci were all identified using single families, whereas the third and fifth loci on 9p and 2q were discovered by a genome scan of multiple multiplex pedigrees. Overall, genome-wide significant level and completion rate of the genome scans were unreported. The study by the Baylor group is the second largest genome scan performed on RLS families to date. However, genome-wide nonparametric analyses did not detect any region with genome-wide significance. The follow-up of five regions with nonparametric linkage (NPL) greater than 2.0 led to the identification of two families (with eight and six affected individuals, respectively) with compatible linkage to the chromosome 9p region. However, these two linked families segregated two different haplotypes, indicating allelic genetic heterogeneity. More convincingly, 16 affected individuals from three different families showed haplotype sharing in the Tyrolean study for RLS5 on chromosome 2q.91 None of the linkage results reported to date can explain the high prevalence of RLS; on the contrary, they all point to, if the linkage results hold true, complex allelic and genetic heterogeneity.84
In the view of classic human genetics, diseases are divided into mendelian disorders and complex traits. Although the former are attributed to single gene mutations with a simple mode of inheritance, the latter are believed to result from environmental factors as well as multiple genes, each playing a small and interactive role in disease susceptibility in the general population. From the clinical perspective, a continuum of phenotypic manifestations may be observed. At one end of the spectrum are disorders caused by fully penetrant deleterious mutations; at the opposite end are diseases caused by pure environmental factors. Between these two extremes lie the incompletely penetrant and the polygenic disorders, creating a smooth transition from strictly genetic to multifactorial illnesses. Heterogeneity is the common denominator to some complex traits and can be considered the most important obstacle to overcome in studies aimed at finding predisposing genes. Identification of biomarkers or specific clinical features would allow stratification of cases into more homogeneous groups and therefore facilitate dissecting the complex heterogeneity. In this context, close scrutiny of RLS families linked to the four reported RLS loci did not reveal any specific clinical features or measurements that would allow sample stratification, except for the high PLMS measurements in the Italian RLS family linked to chromosome 14q31,38,40 and the French Canadian families linked to chromosome 12.84 Early AO has been implicated in genetic forms of RLS,28,49,55 but this clinical parameter has not yet been reported to be formally integrated in linkage analyses.
Association Studies
Because converging lines of evidence also suggest that dopaminergic pathways may be involved in the pathogenesis of RLS, with a potential connection to an iron abnormality, our group has conducted a systematic association study of 10 dopaminergic genes on unrelated RLS patients and matched control subjects. Association was not found in any of the genotyped functional variants from each candidate gene (dopamine receptors D1 through D5, dopamine transporter [DAT], tyrosine hydroxylase [TH], dopamine β-hydroxylase [DBH], and monoamine oxidase B [MAOB],92 with the exception of the monoamine oxidase A (MAOA) candidate gene. In 96 RLS patients and 200 control subjects, females with the high-activity MAOA allele had a greater risk (odds ratio, 2.0; 95% confidence interval, 1.06 to 3.77) of being affected with RLS than females carrying the low-activity alleles; this effect was not seen in male subjects.93 Due to the earlier reports of possible anticipation in some RLS families and clinical association of RLS and spinocerebellar ataxia type 3 (SCA3),94 our group has also genotyped the SCA3 trinucleotide repeat in 125 extensively characterized RLS patients, as well as in 188 matched healthy controls. No association was found.95 Nevertheless, all the above-mentioned association studies were conducted at an earlier stage and in a small number of case-control samples, mostly with single potentially functional variants. Therefore, negative results cannot exclude minor roles of these candidate genes in RLS. Case-control tests for association are an important tool for identifying complex trait susceptibility genes. However, population structure can invalidate this approach, leading to apparent spurious associations at markers that are unlinked to disease loci.96 Family-based tests of association can circumvent this problem.97,98 A more comprehensive whole gene-based association study has been advocated, which considers all variations within a gene and its regulatory region identified through comprehensive screening of a substantial number of affected individuals; this is followed by genotyping additional subjects only in a select subset of the variants and performing a follow-up family-based association study.99,100 This strategy can also be combined with a bioinformatic approach, which can be used to predict potential functional variants in the regulatory regions, such as binding sites for transcriptional factors, splicing sites, and so forth. Applying these more comprehensive methods, we have investigated two candidate genes on chromosome 12q—the neurotensin gene (NTS), a dopamine modulator, and the divalent metal transporter 1 gene (DMT1), an iron transporter—based on their potential high functional relevance in RLS. Using the single nucleotide polymorphism (SNPs) identified in the gene sequencing study and also from the SNP database (dbSNP), we conducted family-based and case-control association studies. No significant association was found in either gene.101,102 Most recently, statistically compelling associations have been identified by genome-wide association studies (GWAS) across a variety of complex genetic traits, including Crohn’s disease, obesity, type 1 and type 2 diabetes, coronary heart disease, and prostate and breast cancers. These findings were facilitated by the International HapMap project, the advanced high through-put SNP genotyping technology and analytic tools. The German group103 and the deCODE group104 have applied the same strategy and identified genes implicated in RLS by GWAS. Winkelmann and colleagues,103 using the Affymetrix 500K Array to genotype 401 clinically diagnosed familial RLS cases and 1644 population controls, identified three genomic regions associated with RLS: intron 8 of the MEIS1 gene, intron 5 of the BTBD9 gene, and the overlapping regions between the genes MAP2K5 and LBXCOR1 on chromosome 2p, 6p, and 15q, respectively. Surprisingly, none of these genes lies within a previously reported linkage region, nor are any of these genes directly implicated in iron and/or dopamine pathways as previously proposed (see Table 8-2 for detailed biological function and relevance to RLS phenotype). The associations of these three candidate regions with RLS phenotype have been further replicated in two independent samples, including 255 French Canadian patients and 287 control subjects from Quebec.103 The deCODE group, using a different SNP genotype panel (Human Hap300; Illumina), independently identified and replicated a genome-wide significant association with the same variant within the BTBD9 gene on chromosome 6p21.2.104 They further demonstrated that the risk allele contributes specifically to PLMS instead of RLS phenotype and is associated with decreased serum ferritin (a decrease of 13% per allele). The population attributable risk of RLS/PLMS is approximately 50% with the BTBD9 locus in the Icelandic population and a small representative sample from the United States.104 The German group estimated that each genetic variant from the three candidate gene regions was associated with a greater than 50% increase in risk for RLS in individual carrier, and the combined allelic variants conferred more than one half of the risk in the studied populations.103


Speculation and Future Directions
Is RLS a Qualitative or Quantitative Trait?
Considering that the major clinical symptom of RLS is an imperative urge to move, all other clinical features, such as sleep disturbances, relief of the urge to move by activity, or worsening during the night, are all related to this primary symptom. Then RLS can be considered as a relatively homogeneous phenotype. Phenotypic variations include different AO, the intensity and frequency of the symptom, the response to treatment, and progression of the clinical course; all of these could be considered as measurements of severity of the cardinal symptom, that is, the urge to move with or without leg discomfort. It would be worthwhile to first determine whether RLS is a distinctive dichotomous qualitative or continuous quantitative trait. This would be different from the arbitrary dichotomous clinical diagnosis, which mainly aims to stratify patients into different categories based on symptom severity for the purpose of treatment. To determine the qualitative or quantitative nature of RLS symptoms, detailed phenotypic data need to be collected from all family members, ideally in large pedigrees. The phenotypic data can be a direct measurement of disease severity or other to-be-identified biometric measurements, such as the PLMS. For example, type 2 diabetes is generally considered as a dichotomous trait defined on the basis of fasting blood glucose level. Insulin resistance is strongly associated with the development of type II diabetes, and elevated insulin concentration is a significant risk factor for the disease.105 Therefore, the related quantitative measurements of blood insulin level were used to map the susceptibility genes in type 2 diabetes.106
Possible Genetic Mechanism for RLS
Although RLS is a major cause for sleep disorders and has a significant adverse impact on quality of life, it is a chronic disorder usually only recognized much later in life; most patients came to medical attention around the age of 50 years. The RLS phenotype has little influence on an individual’s survival and ability to reproduce. Therefore, we speculate the following possible genetic mechanism. First, it is a highly prevalent disease without any distinctive syndromic character in specific families or groups, suggesting a common genetic predisposition (common disease/common variant hypothesis).107 This has been well demonstrated in the recent two GWAS of RLS.103,104 Second, with no obvious direct negative selection on the affected individual’s survival and reproduction, the predisposing variants should be relatively frequent in the population. Third, given the racial differences in disease prevalence observed for RLS, the predisposing variants may interact with specific environments, possibly through a positive selection in the white populations. Nevertheless, some rare variants with high penetrance, possibly with complex allelic and genetic heterogeneity, may present in some large multiplex families.
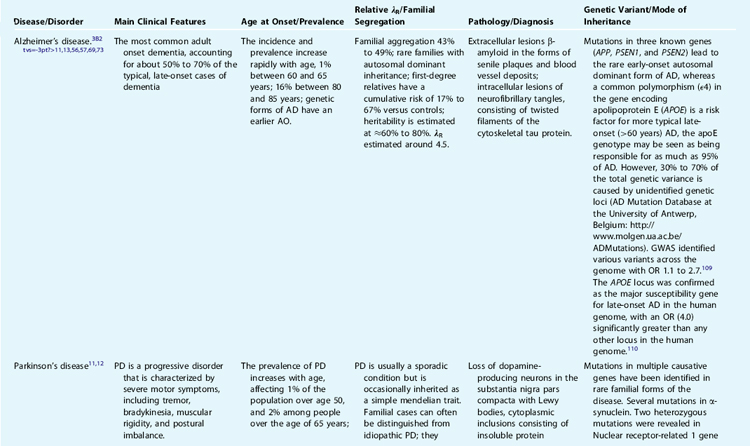
Experience From Genetic Studies on Alzheimer’s Disease and Parkinson’s Disease, Two Late-Onset Neurodegenerative Diseases
We can learn valuable lessons from previous studies of other genetic diseases with similar clinical features, especially when those studies are at a more advanced stage. Two good examples are Alzheimer’s disease (AD) and Parkinson’s disease (PD) (see Table 8-3 for detailed comparisons of AD, PD, and RLS). In AD and PD, as in other complex disorders, the success of gene mapping based on genome-wide scans depends on several factors. First, a precise definition of the phenotype is essential for reliability of results. In AD and PD, there is a characteristic pathology in most patients, which has greatly facilitated the gene mapping approach. Nevertheless, even with an excellent clinical evaluation and phenotype delineation, the presence of clinical variability, phenocopies, and age-dependent penetrance will complicate the selection of a homogeneous sample set. Consequently, without proper sample stratification, the effect of the potential loci will be “diluted.” In genetic studies of both AD and PD, stratifying the patients and families into early and late AO proved to be a successful strategy. The appropriate selection of the choice of samples to collect (extended pedigrees, affected sib-pairs, affected relative pairs) and the method of analysis are also crucial factors in successful gene identification. When a high frequency of a disease-causing allele is suspected, or a wide range of phenotypic variations are observed in large extended pedigrees, studying nuclear families with affected sib-pairs may prove to be an effective alternative strategy.

Sequence-Based Forward Genetics Approach: Direct Candidate Gene Investigation Based on Known or Hypothetical Pathways and Genome-wide Association Studies
The vast amounts of data accumulated from genomic research of humans and other species result from functional genomic studies; high throughput technologies have also prompted us to reconsider our strategies of gene hunting for complex traits like RLS. Promising alternatives include a switch from the genetic map–based gene discovery (based on pure neutral genetic information) to the sequence-based gene discovery (based on combined genetic and functional biological information) and a switch from analysis of single gene (or locus) to the study of multiple genes in the same pathway or system simultaneously. For example, studies suggest that the alteration of iron and/or dopamine metabolisms in the central nervous system is implicated in the etiology of RLS.53,78 Therefore, the direct investigation of all genes genome-wide involved in iron and dopamine metabolism and regulation in the central nervous system could be an experimental shortcut or alternative to identifying underlying genetic factors for RLS. GWAS have greatly enhanced our understanding of the genetic basis of common and complex diseases. The two recent GWAS of RLS have set an important milestone for genetic research of RLS. Facilitated by fast advancing technology, the allelic spectrum or genetic architecture of RLS will soon unfold, leading to a new level of research into the genetics of RLS.
Pharmacogenetic Studies
Pharmacogenetics is defined as the study of genetic variations that cause a variable drug response, including the genetic polymorphism of drug transporters, drug-metabolizing enzymes, and drug receptors.108 Pharmacogenetics refers to the study of inherited differences (variations) in drug metabolism and response to drugs, compared with the more commonly used term pharmacogenomics, which refers to the general study of the many different genes that determine drug behavior.108 There is a large body of phenotype data that has been collected regarding the different responses to various medications in RLS patients, including dopaminergic agents, opiates, anticonvulsants and iron supplements. This valuable collection of data has not yet been correlated with the underlying corresponding genotypes or investigated in families. Further exploration of the relation between different responses to different types of medications and underlying genetic variants could be considered as a parallel approach to dissecting the genetics of RLS. Identification of genetic variants associated with specific drug responses (the more specific situation in RLS being response to and augmentation in dopaminergic agents) will help either to further stratify patients into more homogeneous groups or to indicate potential pathways and candidate genes involved in the pathogenesis of RLS.
1. Ekbom KA. Restless legs: A clinical study. Acta Med Scand. 1945;158:1-123.
2. Lavigne GJ, Montplaisir JY. Restless legs syndrome and sleep bruxism: Prevalence and association among Canadians. Sleep. 1994;17:739-743.
3. Phillips B, Young T, Finn L, et al. Epidemiology of restless legs symptoms in adults. Arch Intern Med. 2000;160:2137-2141.
4. Rothdach AJ, Trenkwalder C, Haberstock J, et al. Prevalence and risk factors of RLS in an elderly population: The MEMO Study. Memory and Morbidity in Augsburg Elderly. Neurology. 2000;54:1064-1068.
5. Ohayon MM, Roth T. Prevalence of restless legs syndrome and periodic limb movement disorder in the general population. J Psychosom Res. 2002;53:547-554.
6. Nichols DA, Allen RP, Grauke JH, et al. Restless legs syndrome symptoms in primary care: A prevalence study. Arch Intern Med. 2003;163:2323-2329.
7. Rijsman R, Neven A, Graffelman W, et al. Epidemiology of restless legs in the Netherlands. Eur J Neurol. 2004;11:607-611.
8. Hening WA, Plassman B, Allen RP, Earley CJ. Prevalence of restless legs symptoms in an elderly American veteran cohort (abstract). Sleep. 2003;26:A338.
9. Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST General Population Study. Arch Intern Med. 2005;165:1286-1292.
10. Sander JW, Shorvon SD. Epidemiology of the epilepsies. J Neurol Neurosurg Psychiatry. 1996;61:433-443.
11. Treves TA, Chandra V, Korczyn AD. Parkinson’s and Alzheimer’s diseases: Epidemiological comparison. 1. Descriptive aspects. Neuroepidemiology. 1993;12:336-344.
12. Tanner CM, Aston DA. Epidemiology of Parkinson’s disease and akinetic syndromes. Curr Opin Neurol. 2000;13:427-430.
13. Hendrie HC. Epidemiology of dementia and Alzheimer’s disease. Am J Geriatr Psychiatry. 1998;6:3-18.
14. Jablensky A, Sartorius N, Ernberg G, et al. Schizophrenia: Manifestations, incidence and course in different cultures. A World Health Organization Ten-Country Study. Psychol Med Monogr Suppl. 1992;20:1-97.
15. Goldner EM, Hsu L, Waraich P, et al. Prevalence and incidence studies of schizophrenic disorders: A systematic review of the literature. Can J Psychiatry. 2002;47:833-843.
16. Bloom BS. Prevalence and economic effects of depression. Manag Care. 2004;13:9-16.
17. Welch E. Hypertension. Nurs Stand. 2003;18:45-53.
18. Jebb SA, Rennie KL, Cole TJ. Prevalence of overweight and obesity among young people in Great Britain. Public Health Nutr. 2004;7:461-465.
19. Tan EK, Seah A, See SJ, et al. Restless legs syndrome in an Asian population: A study in Singapore. Mov Disord. 2001;16:577-579.
20. Kutner NG, Bliwise DL. Restless legs complaint in African-American and Caucasian hemodialysis patients. Sleep Med. 2002;3:497-500.
21. Allen RP. Race, iron status and restless legs syndrome. Sleep Med. 2002;3:467-468.
22. Sevim S, Dogu O, Camdeviren H, et al. Unexpectedly low prevalence and unusual characteristics of RLS in Mersin, Turkey. Neurology. 2003;61:1562-1569.
23. Bhowmik D, Bhatia M, Gupta S, et al. Restless legs syndrome in hemodialysis patients in India: A case controlled study. Sleep Med. 2003;4:143-146.
24. Bhowmik D, Bhatia M, Tiwari S, et al. Low prevalence of restless legs syndrome in patients with advanced chronic renal failure in the Indian population: A case controlled study. Ren Fail. 2004;26:69-72.
25. Lee H, Hening W, Allen R, et al. Race and restless legs syndrome symptoms in adult community sample in east Baltimore. Sleep Med. 2006;7:642-645.
26. Walters AS, Hickey K, Maltzman J, et al. A questionnaire study of 138 patients with restless legs syndrome: The ‘Night-Walkers’ survey. Neurology. 1996;46:92-95.
27. Ondo W, Jankovic J. Restless legs syndrome: Clinicoetiologic correlates. Neurology. 1996;47:1435-1441.
28. Montplaisir J, Boucher S, Poirier G, et al. Clinical, polysomnographic, and genetic characteristics of restless legs syndrome: A study of 133 patients diagnosed with new standard criteria. Mov Disord. 1997;12:61-65.
29. Winkelmann J, Wetter TC, Collado-Seidel V, et al. Clinical characteristics and frequency of the hereditary restless legs syndrome in a population of 300 patients. Sleep. 2000;23:597-602.
30. Allen RP, La Buda MC, Becker P, et al. Family history study of the restless legs syndrome. Sleep Med. 2002;3:S3-S7.
31. Chen S, Ondo WG, Rao S, et al. Genomewide linkage scan identifies a novel susceptibility locus for restless legs syndrome on chromosome 9p. Am J Hum Genet. 2004;74:876-885.
32. Allen RP, Picchietti D, Hening WA, et al. Restless legs syndrome: Diagnostic criteria, special considerations, and epidemiology: A report from the Restless Legs Syndrome Diagnosis and Epidemiology Workshop at the National Institutes of Health. Sleep Med. 2003;4:101-119.
33. Ondo W. Epidemiology of restless legs syndrome. Sleep Med. 2002;3:S13-S15.
34. Hening WA, Washburn T, Somel D. Restless legs patients with a younger age of onset have an increased frequency of affected relatives. Neurology. 2003;60:A11.
35. Walters AS, Picchietti D, Hening W, et al. Variable expressivity in familial restless legs syndrome. Arch Neurol. 1990;47:1219-1220.
36. Trenkwalder C, Seidel VC, Gasser T, et al. Clinical symptoms and possible anticipation in a large kindred of familial restless legs syndrome. Mov Disord. 1996;11:389-394.
37. Lazzarini A, Walters AS, Hickey K, et al. Studies of penetrance and anticipation in five autosomal-dominant restless legs syndrome pedigrees. Mov Disord. 1999;14:111-116.
38. Desautels A, Turecki G, Montplaisir J, et al. Identification of a major susceptibility locus for restless legs syndrome on chromosome 12q. Am J Hum Genet. 2001;69:1266-1270.
39. Kock N, Culjkovic B, Maniak S, et al. Mode of inheritance and susceptibility locus for restless legs syndrome, on chromosome 12q. Am J Hum Genet. 2002;71:205-208.
40. Bonati MT, Ferini-Strambi L, Aridon P, et al. Autosomal dominant restless legs syndrome maps on chromosome 14q. Brain. 2003;126:1485-1492.
41. Ferini-Strambi L, Bonati MT, Oldani A, et al. Genetics in restless legs syndrome. Sleep Med. 2004;5:301-304.
42. Ondo WG, Vuong KD, Wang Q. Restless legs syndrome in monozygotic twins: Clinical correlates. Neurology. 2000;55:1404-1406.
43. Desai AV, Cherkas LF, Spector TD, Williams AJ. Genetic influences in self-reported symptoms of obstructive sleep apnea and restless legs: A twin study. Twin Res. 2004;7:589-595.
44. Xiong L, Jang K, Montplaisir J, et al. Canadian Restless Legs Syndrome Twin Study. Neurology. 2007;68:1631-1633.
45. Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet. 1990;46:222-228.
46. Risch N. Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet. 1990;46:229-241.
47. Schliekelman P, Slatkin M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet. 2002;71:1369-1385.
48. McGue M, Gottesman I. The genetic epidemiology of schizophrenia and the design of linkage studies. Eur Arch Psychiatry Clin Neurosci. 1991;240:174-181.
49. Winkelmann J, Muller-Myhsok B, Wittchen HU, et al. Complex segregation analysis of restless legs syndrome provides evidence for an autosomal dominant mode of inheritance in early age at onset families. Ann Neurol. 2002;52:297-302.
50. Mathias RA, et al. Segregation analysis of restless legs syndrome: Possible evidence for a major gene in a family study using blinded diagnoses. Hum Hered. 2006;10:157-164.
51. Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037-2048.
52. Sham PC. Statistical methods in psychiatric genetics. Stat Methods Med Res. 1998;7:279-300.
53. Connor JR, Boyer PJ, Menzies SL, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology. 2003;61:304-309.
54. Pittock SJ, Parrett T, Adler CH, et al. Neuropathology of primary restless leg syndrome: Absence of specific tau- and alpha-synuclein pathology. Mov Disord. 2004;19:695-699.
55. Allen RP, Earley CJ. Defining the phenotype of the restless legs syndrome (RLS) using age-of-symptom-onset. Sleep Med. 2000;1:11-19.
56. Pericak-Vance MA, Bebout JL, Gaskell PC Jr, et al. Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034-1050.
57. Mullan M, Houlden H, Crawford F, et al. Age of onset in familial early onset Alzheimer’s disease correlates with genetic aetiology. Am J Med Genet. 1993;48:129-130.
58. Claus EB, Risch NJ, Thompson WD. Using age of onset to distinguish between subforms of breast cancer. Ann Hum Genet. 1990;54:169-177.
59. Carter BS, Beaty TH, Steinberg GD, et al. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992;89:3367-3371.
60. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-608.
61. Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-259.
62. Morton LA, Kidd KK. The effects of variable age-of-onset and diagnostic criteria on the estimates of linkage: An example using manic-depressive illness and color blindness. Soc Biol. 1980;27:1-10.
63. Haynes C, Pericak-Vance M, Dawson D. Analysis of Huntington disease linkage and age-of-onset distributions. Genet Epidemiol Suppl. 1986;1:235-239.
64. Terwilliger J, Ott J. Handbook for Human Genetic Linkage. Baltimore: Johns Hopkins University Press, 1994;52-74.
65. Li H, Hsu L. Effects of age at onset on the power of the affected sib pair and transmission/disequilibrium tests. Ann Hum Genet. 2000;64:239-254.
66. Merette C, King MC, Ott J. Heterogeneity analysis of breast cancer families by using age at onset as a covariate. Am J Hum Genet. 1992;50:515-519.
67. Duggirala R, Blangero J, Almasy L, et al. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127-1140.
68. DeStefano AL, Lew MF, Golbe LI, et al. PARK3 influences age at onset in Parkinson disease: A genome scan in the GenePD study. Am J Hum Genet. 2002;70:1089-1095.
69. Ertekin-Taner N, Ronald J, Asahara H, et al. Fine mapping of the alpha-T catenin gene to a quantitative trait locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Hum Mol Genet. 2003;12:3133-3143.
70. Chokroverty S. Editor’s corner: Restless legs syndrome, a common disease uncommonly diagnosed. Sleep Med. 2003;4:91-93.
71. Hening WA. Restless legs syndrome: The most common and least diagnosed sleep disorder. Sleep Med. 2004;5:429-430.
72. Arnulf I, Konofal E, Gauthier C, et al. Severe restless legs syndrome presenting as intractable insomnia. Neurology. 2004;62:E19.
73. Xu JF, Taylor EW, Lung FW, et al. The impact of some parameters on linkage analysis of Alzheimer’s disease. Genet Epidemiol. 1993;10:407-412.
74. Durner M, Greenberg DA, Hodge SE. Phenocopies versus genetic heterogeneity: Can we use phenocopy frequencies in linkage analysis to compensate for heterogeneity? Hum Hered. 1996;46:265-273.
75. Manconi M, Govoni V, De Vito A, et al. Restless legs syndrome and pregnancy. Neurology. 2004;63:1065-1069.
76. Allen RP, Barker PB, Wehrl F, et al. MRI measurement of brain iron in patients with restless legs syndrome. Neurology. 2001;56:263-265.
77. Earley CJ, Connor JR, Beard JL, et al. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology. 2000;54:1698-1700.
78. Earley CJ, Allen RP, Beard JL, et al. Insight into the pathophysiology of restless legs syndrome. J Neurosci Res. 2000;62:623-628.
79. Hening WA. Subjective and objective criteria in the diagnosis of the restless legs syndrome. Sleep Med. 2004;5:285-292.
80. Schenck CH, Mahowald MW. Motor dyscontrol in narcolepsy: Rapid-eye-movement (REM) sleep without atonia and REM sleep behavior disorder. Ann Neurol. 1992;32:3-10.
81. Guilleminault C, Philip P. Tiredness and somnolence despite initial treatment of obstructive sleep apnea syndrome (what to do when an OSAS patient stays hypersomnolent despite treatment). Sleep. 1996;19:117-122.
82. Lapierre O, Montplaisir J. Polysomnographic features of REM sleep behavior disorder: Development of a scoring method. Neurology. 1992;42:1371-1374.
83. Krishnan PR, Bhatia M, Behari M. Restless legs syndrome in Parkinson’s disease: A case-controlled study. Mov Disord. 2003;18:181-185.
84. Desautels A, Turecki G, Montplaisir J, et al. Restless legs syndrome: Confirmation of linkage to chromosome 12, genetic heterogeneity and evidence of complexity. Arch Neurol. 2005;62:591-596.
85. Birinyi PV, Allen RP, Hening W, et al. Undiagnosed individuals with first-degree relatives with restless legs syndrome have increased periodic limb movements. Sleep Med. 2006;7:480-485.
86. Hicks AA, Rye DB, Kristijansson K, et al. Population-based confirmation of the 12q RLS locus in Iceland (abstract). Mov Disord. 2005;20:S34.
87. Winkelmann J, Lichtner P, Putz B, et al. Evidence for further genetic locus heterogeneity and confirmation of RLS-1 in restless legs syndrome. Mov Disord. 2006;21:28-33.
88. Levchenko A, Montplaisir JY, Dube MP, et al. The 14q restless legs syndrome locus in the French Canadian population. Ann Neurol. 2004;55:887-891.
89. Liebetanz KM, Winkelmann J, Trenkwalder C, et al. RLS3: Fine-mapping of an autosomal dominant locus in a family with intrafamilial heterogeneity. Neurology. 2006;67:320-321.
90. Levchenko A, Provost S, Montplaisir JY, et al. A novel autosomal dominant restless legs syndrome locus maps to chromosome 20p13. Neurology. 2006;67:900-901.
91. Pichler I, Marroni F, Volpato CB, et al. Linkage analysis identifies a novel locus for restless legs syndrome on chromosome 2q in a South Tyrolean population isolate. Am J Hum Genet. 2006;79:716-723.
92. Desautels A, Turecki G, Montplaisir J, et al. Dopaminergic neurotransmission and restless legs syndrome: A genetic association analysis. Neurology. 2001;57:1304-1306.
93. Desautels A, Turecki G, Montplaisir J, et al. Evidence for a genetic association between monoamine oxidase A and restless legs syndrome. Neurology. 2002;59:215-219.
94. Abele M, Burk K, Laccone F, et al. Restless legs syndrome in spinocerebellar ataxia types 1, 2, and 3. J Neurol. 2001;248:311-314.
95. Desautels A, Turecki G, Montplaisir J, et al. Analysis of CAG repeat expansions in restless legs syndrome. Sleep. 2003;26:1055-1057.
96. Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet. 1999;65:220-228.
97. Pritchard JK, Donnelly P. Case-control studies of association in structured or admixed populations. Theor Popul Biol. 2001;60:227-237.
98. Horvath S, Xu X, Laird NM. The family based association test method: Strategies for studying general genotype–phenotype associations. Eur J Hum Genet. 2001;9:301-306.
99. Daly MJ, Rioux JD, Schaffner SF, et al. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229-232.
100. Neale BM, Sham PC. The future of association studies: Gene-based analysis and replication. Am J Hum Genet. 2004;75:353-362.
101. Xiong L, Dion P, Montplaisir J, et al. Genetic studies of DMT1 on 12q in French-Canadian restless legs syndrome patients and families. Am J Med Genet B Neuropsychiatry Genet. 2007;144:911-917.
102. Xiong L, Levchenko A, Montplaisir J, etal. Association studies of neurotensin and restless legs syndrome in French Canadians. Sleep Med 2007;July. Epub ahead of print.
103. Winkelmann J, Schormair B, Lichtner P, et al. Genome-wide association study in restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39:1000-1006.
104. Stefansson H, Rye DB, Hicks A, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med. 2007;357:703-705.
105. Lillioja S, Mott DM, Spraul M, et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med. 1993;329:1988-1992.
106. Watanabe RM, Ghosh S, Langefeld CD, et al. The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. II. An autosomal genome scan for diabetes-related quantitative-trait loci. Am J Hum Genet. 2000;67:1186-1200.
107. Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502-510.
108. Nebert DW. Pharmacogenetics and pharmacogenomics: Why is this relevant to the clinical geneticist? Clin Genet. 1999;56:247-258.
109. Grupe A, Abraham R, Li Y, et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007;16:865-873.
110. Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68:613-618.
111. Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158-1160.
112. Paisán-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8 linked Parkinson disease. Neuron. 2004;44:595-600.
113. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-607.
114. Fung HC, Scholz S, Matarin M, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2006;5:911-916.
115. Elbaz A, Nelson LM, Payami H, et al. Lack of replication of thirteen single-nucleotide polymorphisms implicated in Parkinson’s disease: A large-scale international study. Lancet Neurol. 2006;5:917-923.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 8 Genetics of Restless Legs Syndrome
Clinical Genetics of Restless Legs Syndrome
Data from epidemiological and family studies clearly indicate that a genetic component plays an important role in the pathogenesis of restless legs syndrome (RLS).
Restless Legs Syndrome is a Common Neurological Disorder with Variable Prevalence in Different Populations
Although RLS was first systematically described by Ekbom,1 a Swedish neurologist, in 1945, accurate estimates of population incidence and prevalence only started to emerge in the late 1990s, following the first large-scale population survey by Montplaisir and associates.2 Numerous recent epidemiological studies further confirmed that RLS is a very prevalent disorder in the population (see Chapter 7), with a prevalence around 10% to 15% in Western populations,3–9 which is equivalent to the sum of prevalences for epilepsy (≈1%),10 Parkinson’s disease (≈1%),11,12 Alzheimer’s disease (≈1%),13 schizophrenia (≈1%),14,15 and other neuropsychatric disorders, and only comparable with a few common prevalent diseases, such as depression (≈10%),16 hypertension (≈20%),17 and obesity (≈16%).18 Further studies suggest that there are substantial prevalence variations of RLS in different populations and racial groups. The prevalence was estimated higher in certain geographically or socially isolated populations, such as the French Canadians in Quebec compared with those in other provinces of Canada.2 Conversely, RLS prevalence seems to be lower in Asian and African American populations compared with whites based on some preliminary results.19–24 However, one more recent survey of an adult community sample from east Baltimore suggests that there may be no appreciable difference of RLS prevalence between whites and African Americans.25 Further large-scale population studies with validated diagnostic instruments will be needed to clarify the racial difference in RLS. Although both genetic and environmental factors may account for the divergence of prevalence in different populations, significant variations in disease incidence and prevalence among different racial/ethnic groups are usually an indicator of a strong genetic effect at the population level, especially when different ethnic groups show variable disease frequencies within a similar environment. Certainly, the discrepancy of prevalence difference between studies may also have derived from the sensitivity and specificity of the different diagnostic instruments used in the different studies, as well as ascertainment bias and population substratification due to geographical or cultural reasons.
Familiality
RLS is not only prevalent in the general population but also aggregates in families. The familial character of RLS has long been recognized since its first clinical description,1 and it has been consistently reported by experienced clinicians.2,26–31 Family history has been recognized as a significant risk factor for RLS.32 The proportion of familial cases present in the overall RLS patient population has been estimated to be 60%,27–3033 but it could be as high as 80% to 90% among idiopathic RLS patients.27,34 In our extensive family studies of 244 probands with idiopathic RLS, only 57 probands do not have a positive family history, which translates into a familial rate of 76.6% in our sample (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). Family data from the Baltimore group also show that about 70% of their RLS patients have an interviewed first-degree relatives affected by RLS (W. A. Hening, preliminary data, 2006). A sample of RLS Foundation brain bank volunteers (N = 86; 82% women) had an even higher frequency of affected first-degree relatives: 79% (W. A. Hening, preliminary data, 2006).
Several large pedigrees were described in the literature where multiple members were affected with RLS over a span of three to five generations.28,31,35–41 According to our experience, this phenomenon is quite common in the general population, at least in French Canadians, further confirming its genetic nature.
Heritability
Adequate clinical data strongly indicate that RLS has an important genetic component. However, a crucial step before undertaking any molecular genetic studies in RLS is to determine the degree to which the phenotype is determined by the underlying genetic component. Data from population, family, and twin studies usually permit geneticists to estimate the magnitude of the genetic effect on the trait under study. The estimation of heritability is calculated by the expected correlation between family members for the phenotypic trait based on their degree of relationship. The prevalence of RLS in first-degree relatives of affected patients is estimated to be between 20% and 60%.26–2931 Montplaisir and colleagues28 examined 133 RLS patients who were studied using a standardized self-report questionnaire. In this sample, 63% had at least one first-degree relative affected with RLS, and 221 of 568 (38.9%) first-degree relatives were reported as affected.
Traditionally, the most powerful way to estimate the genetic and environmental components of phenotypic variance is to study monozygotic (MZ) and dizygotic (DZ) twins. However, due to its recent acceptance as a clinical entity and the development of accurate diagnostic criteria and instruments, there are few papers reporting limited data on twin studies about RLS that are up to date. One is from the Ondo group,42 which reported 12 pairs of MZ twins, among them 10 pairs concordant, 1 pair probably concordant, and 1 pair discordant on RLS symptoms. There were no concomitant data on DZ twins, making it impossible to differentiate the relative proportions and magnitude of genetic and environmental factors. The other large-scale population-based twin studies of RLS symptoms included 933 MZ and 1004 DZ twins who have completed two questions regarding their restless legs symptoms.43 This study showed that the concordance rate of possible RLS symptoms is 61% in MZ and 45% in DZ twins; the concordance rate for “involuntary leg jerks during the night,” a possible metaphor for periodic leg movements during sleep (PLMS), is 69% in MZ and 54% in DZ twins. Further genetic modeling with the same set of data by using the frequencies of disease-concordant and disease-discordant rates and applying to a multifactorial liability threshold model indicates that additive genetic factors and a unique environment best explained the variance in disease liability. However, the critical weakness of this study is that the RLS diagnosis was only based on a simplified two-question self-report survey, which did not include all four essential clinical features. Therefore, they did not study RLS, but rather possibly a component of the disease. Nevertheless, as demonstrated in numerous studies of other genetic disorders, twin studies are powerful genetic tools to quantify and define the genetic contribution, possible mode of inheritance, and potential gene–environment interaction. Our data from a population-based survey of 272 twin pairs from Canada show that the concordant rate of definite RLS is 53.7% and 19.0% in MZ and DZ twins, respectively.44 The estimated heritability is 69.4%, confirming the importance of the genetic factor in RLS etiology. However, in the World War II twin cohort (176 MZ and 135 DZ twin pairs), concordance was only slightly higher in MZ twins (23% versus 16%), leading to a much lower estimated heritability of 20%.8 This may reflect the much greater influence of environmental factors leading to secondary RLS in this aged, male-only cohort. The information obtained from additional appropriate twin studies will be critical for further molecular genetic studies aimed at identifying underlying causative or susceptibility genes for RLS.
It is well known in the field of genetic studies that the pattern of risk ratio (λR) in various degrees of relationship within a family may reflect the underlying genetic mechanism. The magnitude of λR can usually be used to predict the statistical power to detect linkage in gene mapping.45–47 For a single-locus model and an additive multilocus genetic model, λR decreases by a factor of 2 with each degree of relationship. However, for a multiplicative (epistasis) model, λR decreases more rapidly than by a factor of 2 with degree of relationship.45 For example, λR in first-, second-, and third-degree relatives of schizophrenia patients is 6% to 17%, 2% to 6%, and 2%, respectively,48 suggesting the presence of multiple interacting loci. Unfortunately, there are very limited data available regarding the prevalence of RLS in more distant relatives other than the first-degree relatives, due to the difficulty in disease ascertainment. In one study of 96 RLS families, Allen and associates30 reported that 19.9% of the first-degree relatives versus 4.1% of the second-degree relatives are affected compared with 3.5% versus 0.5% in 15 non-RLS control subjects.30 Compared with a prevalence of 3.5% in their control group, the relative risk ratio λR dropped quickly from 5.7 to 1.2 from the first-degree relatives to the second-degree relatives. In another study of 15 large multiplex families with RLS by the Baylor group, the λR for parent-offspring is 10.25 and the λR for siblings is 16.23, and the overall heritability in their sample was estimated at 60%.31 Undoubtedly, the λR estimated in this study will not reflect the relative risk in general population, because the sample is extremely biased toward only the heavily loaded RLS families. In the Johns Hopkins family study, the relative risk for first-degree relatives of RLS probands (N = 134) was 3.6 compared with both the general population figures9 and an age- and gender-matched control group (N = 58), with higher figures for younger-onset probands (W. A. Hening and colleagues, preliminary data).
Mode of Inheritance
It is possible for geneticists to propose a genetic model for a given genetic trait, by deriving information from data obtained from population and family studies and by using specific statistical tools. Also, the simpler the underlying genetic structure, the more reliable their prediction becomes. However, the genetic study of RLS is at a very early stage compared with other hereditary forms of neurological and psychiatric disorders, such as epilepsy and schizophrenia, where many population and molecular genetic studies have been undertaken. In RLS, there are very limited reliable data on which to speculate. A few studies have attempted to model the genetic transmission of RLS. In most of the reported pedigrees, vertical transmission is predominant, making RLS mostly compatible with an autosomal dominant mode of inheritance with relatively high penetrance.31,35–37 The Winkelmann group from Germany performed complex segregation analyses on 238 families and predicted that a single autosomal allele acting dominantly can explain RLS in families with an early age at onset (AO) of symptoms, but it will not account for RLS with a later onset (>30 years).49 However, another recent segregation study by Mathias and coworkers50 indicates that a dominant model works for all families, not only younger-onset families. That study also found both a high frequency for the major gene and a high phenocopy rate (14%) fit better with the genetic model. That analysis also showed that there was genetic control of age at RLS onset, but without a major gene effect. Nevertheless, a careful examination of the detailed family histories in RLS pedigrees has always suggested a genetic model more complicated than a classic mendelian inheritance. In some reports, the percentage of RLS in first-degree relatives is greater than 50%34,37; we have also observed such segregation distortion in some of our collected RLS families (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). The observed ratio of more than one half of first-degree relatives being affected in some RLS families may be due to an ascertainment bias but warrants further study to either confirm or refute this unusual observation. Some dominant families also showed reduced AO in consecutive generations, indicating possible anticipation.36,37 Due to the high prevalence of RLS and possible assortative mating in the population, bilinear inheritance is not uncommon. Among 50 familial cases in the Ondo group’s study, three had both parents affected.42 In our family studies, we also frequently encounter affected spouses and observe bilinear inheritance (L. Xiong, G. A. Rouleau, and J. Montplaisir, unpublished data). All pedigrees in the published linkage studies reported individuals carrying the predisposing haplotype but without the disease phenotype (nonpenetrants) and individuals without the haplotype but presenting with the disease symptoms (phenocopies).31,38,40 More surprisingly, our linkage study of one large French-Canadian family showed the most significant results under an autosomal recessive model with an unusually high common disease allele frequency (25%), although the pedigree appears to be dominant38; that is, a pseudodominant inheritance. In the systematic analysis of 15 autosomal dominant–looking multiplex families with 134 affected, 136 founders, and 317 nonfounders, Chen and coworkers31 demonstrated that the λR is higher in sib-pairs than in parent-offspring pairs (16.23 versus 10.25), which could also be explained by recessively or additively acting disease alleles. In summary, all available evidence suggests that genetic factors play an important role in the etiology of RLS; however, the mode of inheritance is probably more complex than it appears on cursory examination of available pedigrees. Results from complex segregation analysis need to be examined carefully and interpreted with caution because it is known to be problematic when dealing with complex traits.51,52
Variable Age at Onset and Bimodal Age at Onset Distribution
In general, RLS is an adult-onset neurological disorder with a prevalence that tends to increase linearly with age.3,7 In Ondo’s twin study, the earliest AO is 3 years of age, whereas the latest is 65 years of age. The AO varied by 40 years in two of the MZ twin pairs.42 The clinical manifestations of RLS also slowly progress with age in some patients with a severe form of RLS, which is similar to other late-onset neurodegenerative diseases. However, a certain percentage of patients and their family members may present with only a much milder form of RLS, never seeking medical attention and having a clinical course that is intermittent or waxing and waning. The limited number of neuropathological studies available also indicates that RLS is not caused by a traditional neurodegenerative processes, such as seen in τ- or α-synuclein brain pathologies.53,54
Several studies suggest a bimodal distribution of AO among RLS patients.28–30,49,55 The younger probands usually have a higher rate of positive family history and slower disease progression rate, suggesting a greater genetic contribution and possibly a distinctive clinical course compared with nongenetic forms of RLS cases.28–3055 Many complex and highly prevalent disorders exhibit a wide range of AO due to various genetic and environmental factors that contribute to the same phenotype. An earlier AO is often considered a sign of genetic predisposition in many diseases, such as Alzheimer’s disease,56,57 breast cancer,58 prostate cancer,59 and Parkinson’s disease.60,61 The use of AO information affects the power of gene mapping and identification. Several strategies can be applied to integrate AO information into molecular genetic studies. If a major gene effect is suspected that manifests itself at a specific range of age, the simplest way to use AO information is to stratify patients and families into early- and late-onset subgroups. Hopefully, in this way, the samples can be divided into more homogeneous subgroups to help circumvent the underlying genetic heterogeneity, or to purge the nongenetic cases. In parametric linkage analysis, adjustment of variable AO to different age-dependent liability classes within pedigrees is an important component of effective linkage analysis.62–64 Liability classes are used to define penetrance values for each of the possible genotypes of the trait locus and to classify each individual into different penetrance groups on the basis of their age at investigation. In complex traits, the effects of AO on penetrance model–free analyses are even more complicated and have been further investigated by Li and associates.65 These authors suggested that incorporating the AO information into affected sib-pairs (ASP) and transmission disequilibrium (TDT) tests, especially when focusing on sib-pairs both with early AO and TDT with all early AO trios, will greatly improve the power to detect the genetic signals. Alternatively, AO can be treated either as a covariate or quantitative trait by using variance component linkage analysis.66 This is usually applied to identify genetic variants that will influence age at disease onset. For example, this method has been successfully applied to map genes modifying disease onset for type 2 diabetes,67 Parkinson’s disease,68 and Alzheimer’s disease.69 Nevertheless, the preliminary data from complex segregation analyses by Mathias and colleagues50 suggest that there is complex genetic control of age at onset for RLS in their samples.
Variable Phenotypic Expressivity
One of the most striking features of RLS is the high degree of variable phenotypic expressivity,35,36 even within MZ twins.42 The cardinal clinical symptom of RLS is an imperative urge to move, which is very subjective without any reliable validation and measurement. Misdiagnosis and underdiagnosis are not uncommon in clinical practice. RLS is considered one of the most common and least diagnosed sleep disorders, as well as neurological disorders.70,71 Furthermore, as we mentioned earlier, frequently there are even milder forms among family members who never need medical attention and only get ascertained during family studies, quite often through telephone interviews only. We know that RLS can sometimes be very severe, causing intractable insomnia72; unfortunately, we do not know how mild symptoms should be interpreted, whether as mild RLS or as something completely unrelated. This is critically important in genetic studies. Until more reliable biological markers or laboratory tests become available, in current family linkage studies, different diagnostic criteria schemes can be applied to define the exact phenotype under study, from the most to the least stringent, to accommodate the wide range of phenotypic variations within the pedigree. Because the primary symptom of RLS is an imperative urge to move, other features, such as frequency and intensity of symptoms, can all be considered as measurements of severity. To fully address the phenotypic variations within and between families in gene mapping, the degree of severity, together with variable AO, can also be treated as covariate or quantitative traits by using a variance component method. Nonetheless, all these approaches require a significantly large number of participating families.
Phenocopies and Association with Other Common Medical Conditions
A phenotype that does not result, at least in part, from a specific gene or locus under study is called a phenocopy in genetic analysis. Phenocopies can be environmentally induced phenotypes that mimic the genetically determined phenotypes or they can be identical phenotypes that are not genetically controlled by the same gene under study. The literature reports that parametric linkage analysis is very sensitive to phenocopy rate.73,74 High phenocopy rate is considered as a significant obstacle to gene mapping.
RLS is a very common disorder; it is known that RLS symptoms can be caused or influenced by other nongenetic factors as well. RLS has been reported to be associated with several other common medical conditions, such as renal failure, anemia, and pregnancy. Patients suffering from arthritis, peripheral neuropathy, and spinal cord injury can present with exactly the same symptoms as idiopathic RLS patients. It remains unclear whether these medical conditions predispose to RLS through distinct pathological mechanisms or by interacting with common predisposing genetic variant(s). Therefore, idiopathic and nonidiopathic RLS should both be taken into consideration when searching for the common predisposing variant(s) and should be analyzed both as one entity and as separate disease groups. For example, RLS symptoms occur more frequently in pregnant women; their occurrence correlates with a stronger family history: 29% of women presenting with RLS during pregnancy reported a first-degree relative with RLS symptoms.75 Clinical observations also strongly suggest that iron metabolism might play an important role in RLS.53,76–
[/not-level-membership-for-neurology-category]
