[level-membership-for-neurosurgery-category]
CHAPTER 359 Genetics of Intracranial Aneurysms
The exact etiology and pathogenesis of intracranial aneurysms remain unclear. Several lines of evidence implicate acquired risk factors such as smoking or hypertension,1,2 whereas others support the role of genetic factors.3,4 The two main lines of evidence supporting the role of genetic factors are the association of intracranial aneurysms with heritable connective tissue disorders and the familial occurrence of intracranial aneurysms.
Heritable Connective Tissue Disorders
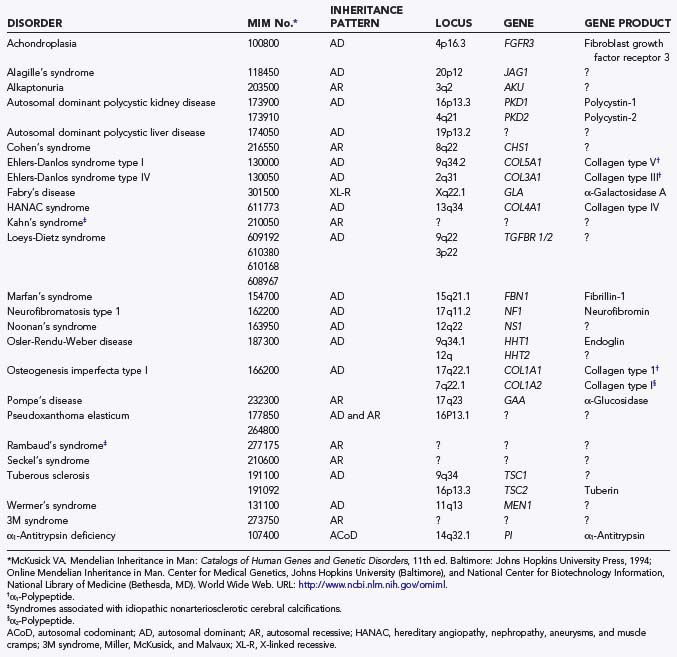
Numerous heritable connective tissue disorders have been associated with intracranial aneurysms, including polycystic kidney disease, Ehlers-Danlos syndrome type IV, Marfan’s syndrome, neurofibromatosis type 1, and bicuspid aortic valve (Table 359-1).3–5 To what extent these specific heritable disorders contribute to the entire population of patients with intracranial aneurysms is unknown. In one series of 100 consecutive hospitalized patients with intracranial aneurysms, 5 had an identifiable heritable connective tissue disorder.6 The true frequency of heritable connective tissue disorders in patients with aneurysms is probably higher because these disorders often remain undiagnosed as a result of the substantial variability in their phenotypic expression. The family history may also be negative because the disease can be caused by a new mutation. Nevertheless, identifiable heritable connective tissue disorders contribute to a relatively small percentage of intracranial aneurysms.
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) affects about 1 in 400 to 1000 persons and is the most common monogenetic disease in humans.7 It is inherited as an autosomal dominant trait with almost complete penetrance but with variable expression. The family history is negative in about 20% of patients, thus suggesting a fairly high spontaneous mutation rate.
ADPKD is a systemic disease, and cysts are present in the kidneys, liver, pancreas, spleen, ovaries, and seminal vesicles.7 Moreover, ADPKD should be included among the heritable connective tissue disorders.5 A wide variety of connective tissues may be involved,7 including the heart valves (mitral valve prolapse), vasculature (aneurysms and dissections), and meninges (arachnoid cysts). Patients with ADPKD are at increased risk for the development of gastrointestinal diverticula and inguinal hernias.7
Neurosurgical disorders that have been associated with ADPKD include intracranial aneurysms, cervicocephalic arterial dissections, intracranial dolichoectasia, intracranial arachnoid cysts, spinal meningeal diverticula/cerebrospinal fluid leaks, and chronic subdural hemorrhages.8–13 Intracranial aneurysms have long been known to be associated with ADPKD.14–21 Until the underlying connective tissue defect of the disease became well known, however, aneurysms were frequently attributed to the arterial hypertension that usually accompanies ADPKD. Intracranial aneurysms are detected in approximately a fourth of patients with ADPKD at autopsy; in most of these patients, aneurysmal rupture was the cause of death. Conversely, ADPKD accounts for 2% to 7% of all patients with intracranial aneurysms. Using magnetic resonance angiography or, less commonly, catheter angiography in patients with good renal function, several groups have screened adult ADPKD patients for asymptomatic intracranial aneurysms. The detection rate has ranged between 5% and 10%.15–19 Familial clustering of intracranial aneurysms occurs in ADPKD; the yield of screening increases to 10% to 25% in such families.15–19 The presence of polycystic liver disease in patients with ADPKD may also increase the development of intracranial aneurysms.16
Screening patients with ADPKD for asymptomatic aneurysms remains controversial but should certainly be considered for those with a family history of intracranial aneurysms. Most asymptomatic intracranial aneurysms detected with screening are less than 6 mm in diameter. In one study, none of these small aneurysms ruptured during 500 months of cumulative follow-up.17 It has been suggested that ADPKD patients are at an increased risk for the de novo development of aneurysms some time after their first intracranial aneurysm is discovered, but the exact significance of this risk remains to be determined.22 When compared with the general population, aneurysmal subarachnoid hemorrhage (SAH) in patients with ADPKD occurs at an earlier age, but the mortality rate is similar.8
ADPKD is a genetically heterogeneous disease. Several loci are involved, and mutations, which are responsible for at least 85% of cases, have been identified in a gene on chromosome 16 (PKD1), as well as in a gene on chromosome 4 (PDK2).23–26 In general, patients with mutations in the PKD1 gene are more severely affected than those with mutations in the PKD2 gene, but intracranial aneurysms are a manifestation of both types of ADPKD. Polycystin-1 and polycystin-2 are the proteins encoded by the PKD1 and PKD2 genes, respectively.23 Both proteins are integral membrane proteins with large extracellular domains, and they probably play a role in maintaining structural integrity of the connective tissue extracellular matrix.23
Autosomal dominant polycystic liver disease (ADPLD) is a familial form of isolated polycystic liver disease that is distinct from ADPKD.27 Patients with ADPLD may also be at high risk for the development of intracranial aneurysms.27
Ehlers-Danlos Syndrome Type IV
Ehlers-Danlos syndrome type IV is potentially one of the most deadly heritable connective tissue disorders that neurosurgeons may encounter. It is uncommon, with a prevalence of approximately 1 in 50,000 to 500,000 persons.28 It is inherited in an autosomal dominant fashion, but the family history is frequently noncontributory because of the high spontaneous mutation rate (approximately 50%).
Ehlers-Danlos syndrome type IV can be life-threatening because spontaneous rupture, dissection, or aneurysm formation on large and medium-sized arteries occurs in all areas of the body.5,28–30 These arterial complications cause death in most patients. Other well-described life-threatening complications of Ehlers-Danlos syndrome type IV are spontaneous rupture of the bowel or gravid uterus and spontaneous pneumothorax.28–30
An intracranial aneurysm may be the initial manifestation of Ehlers-Danlos syndrome type IV. Consequently, neurosurgeons may be the first physicians involved in these patients’ medical care. The syndrome is often difficult to recognize because its external features can be subtle.5,28–30 The more salient features of Ehlers-Danlos syndrome type IV are summarized in Table 359-2. The characteristic facial appearance was first described by Carl Graf, a neurosurgeon31; many striking examples have since been published.29,30 The facial features consist of (1) large expressive eyes with the sclera clearly visible around the iris, (2) a thin nose, (3) thin lips, and (4) lobeless ears. Many patients with Ehlers-Danlos syndrome type IV, however, do not exhibit this facial appearance. The characteristic cutaneous features include thin and fragile skin that is almost transparent and allows the subcutaneous veins to be clearly visible. Patients bruise easily, and multiple ecchymoses are common. Scars are often papyraceous and wide, or they may be complicated by keloid formation. The skin of some patients with Ehlers-Danlos syndrome type IV, however, appears normal. The joint hypermobility is often mild and limited to the fingers and toes. Identifying Ehlers-Danlos syndrome type IV in any patient with an intracranial aneurysm is important because vascular fragility can make any invasive procedure a hazardous undertaking.
TABLE 359-2 Characteristic Features of Ehlers-Danlos Syndrome Type IV
Intracranial aneurysms and spontaneous carotid cavernous fistulas are well-described vascular complications of Ehlers-Danlos syndrome type IV.5,28–33 In some patients, the carotid cavernous fistula is due to rupture of a cavernous carotid aneurysm, although the fistula may be caused by a simple tear in the artery in other patients. The importance of intracranial aneurysmal disease in this group of patients is well described. For example, in a cohort of 202 patients with Ehlers-Danlos syndrome type IV, 4 had ruptured intracranial aneurysms, 4 suffered an intracranial hemorrhage of undetermined cause, and 6 had carotid cavernous fistulas.28 The exact incidence of intracranial aneurysms in patients with Ehlers-Danlos syndrome type IV is unknown because screening for asymptomatic intracranial aneurysms is limited and systematic autopsy studies are unavailable. Screening for asymptomatic intracranial aneurysms in these patients is not recommended because safe treatment options are limited; arteriography, endovascular intervention, and surgical treatment are all associated with high complication rates.
Mutations in the gene encoding the pro-α1-(III) chain of collagen type III (COL3A1) on chromosome 2 are the cause of Ehlers-Danlos syndrome type IV.29,30,33–36 This type of collagen is the major structural component of distensible tissues, including arteries, veins, hollow viscera, and the uterus. In addition, collagen type III may play an important role in the fibrillogenesis of collagen type I. Several studies have reported evidence of abnormal collagen type III metabolism in up to 50% of patients with intracranial aneurysms who do not have Ehlers-Danlos syndrome type IV.37–43 Mutations in the COL3A1 gene, however, are rare. For example, in a study of 40 patients with intracranial aneurysms, COL3A1 mutations were found in just 2 patients, and the functional consequences of these mutations were considered insignificant.44 The reasons for these conflicting data are unclear.
Marfan’s Syndrome
Marfan’s syndrome affects approximately 1 in 10,000 to 20,000 people and is characterized by abnormalities of the skeleton, cardiovascular system, eye, and spinal meninges.45,46 Aortic and mitral valve insufficiency is the most frequent cause of death in children with Marfan’s syndrome, and spontaneous aortic rupture and dissection are the most frequent causes of death in adults with the syndrome.45,46 Dissection of medium-sized arteries, however, is much less common.47 Although Marfan’s syndrome is easily recognized in patients who display the main features of the syndrome (particularly the skeletal manifestations of tall stature, dolichostenomelia, arachnodactyly, and anterior chest deformity), the variability in phenotypic expression is great and the diagnosis is seldom straightforward.45,46 For example, if the parents of a patient with Marfan’s syndrome are short, the affected person’s habitus may be comparatively normal.45 Ectopia lentis, the classic ocular manifestation of Marfan’s syndrome, is observed in only about half the cases.45 Dural ectasia, another major diagnostic criterion of the syndrome, is usually asymptomatic and requires computed tomography or magnetic resonance imaging for diagnosis.48,49 Other manifestations of Marfan’s syndrome include spontaneous pneumothorax, striae distensae, and retinal detachment.45,46
Intracranial aneurysms in patients with Marfan’s syndrome may be saccular or fusiform, and intracranial dissecting aneurysms have also been described.5,50–53 Similar to Ehlers-Danlos syndrome type IV, there is a propensity for proximal intracranial carotid artery involvement, although carotid cavernous fistulas seem to be rare.5 Connective tissue fragility is seldom a major problem in the neurosurgical treatment of patients with Marfan’s syndrome. The frequently observed ectasia and tortuosity of the extracranial carotid and vertebral arteries, however, may render endovascular treatment of intracranial aneurysms impossible. The association of Marfan’s syndrome and intracranial aneurysms has not been firmly established. In an autopsy series of 7 patients with Marfan’s syndrome collected during a 25-year period at the Mayo Clinic, intracranial aneurysms, one ruptured and one unruptured, were observed in 2 patients.51 Combining this autopsy study with one performed at Johns Hopkins University54 but excluding the one ruptured aneurysm, incidental aneurysms were found in 2 (6.5%) of 31 patients.55 This frequency is higher than would be expected in the general population, particularly in view of the young age of the patients.55 The results of screening for asymptomatic intracranial aneurysms in patients with Marfan’s syndrome have not been reported.
Mutations in the gene encoding fibrillin-1 (FBN1) cause Marfan’s syndrome.56 Fibrillin-1 is a fairly recently detected glycoprotein that is one of the major components of microfibrils.57 Microfibrils are important constituents of the extracellular matrix and are distributed throughout the body in elastic tissues such as the skin and aorta and in nonelastic tissues such as the ciliary zonules of the ocular lens. In elastic arteries such as the aorta, fibrillin-1 is found in all three layers of the arterial wall. It is thought that fibrillin-1 plays an important role in maintaining the structural integrity of connective tissues, in part by providing a scaffolding for the elastic fibers. Mutations in the FBN1 gene or abnormal fibrillin metabolism (“fibrillinopathy”) have also been detected in patients with isolated features of Marfan’s syndrome but without the classic syndrome.58–61
Neurofibromatosis Type 1
Neurofibromatosis type 1 is a progressive systemic disease that affects approximately 1 in 3000 to 5000 persons.62 The principal clinical features of neurofibromatosis type 1 are café au lait spots, neurofibromas, and Lisch’s nodules (hamartomas) of the iris.62 Although these features each occur in more than 90% of adults with neurofibromatosis type 1, the number of lesions is variable. Patients with neurofibromatosis type 1 are also at increased risk for the development of optic glioma, pheochromocytoma, dural ectasia, and skeletal abnormalities such as scoliosis and sphenoid wing dysplasia.62 Vascular complications of neurofibromatosis type 1 have been recognized since 1945 and are characterized by stenosis, rupture, and aneurysm or fistula formation in large and medium-sized arteries.63–65
Intracranial aneurysms in patients with neurofibromatosis type 1 may be saccular or fusiform, and some have the appearance of dissecting aneurysms.66–73 Surgical repair of these aneurysms may be complicated by excessive vascular fragility or distortion of anatomic landmarks caused by sphenoid wing dysplasia.70 The intracranial aneurysms associated with neurofibromatosis type 1 often coexist with intracranial arterial occlusive disease,74 thereby increasing the risk associated with the surgical and particularly endovascular treatment of these aneurysms. An increased probability of the development of intracranial aneurysms has not been clearly established for patients with neurofibromatosis type 1, but the number of reported cases continues to increase and some have advocated screening patients with neurofibromatosis for asymptomatic intracranial aneurysms.67 Among a group of 100 consecutive patients with intracranial aneurysms, 1 patient was revealed to have neurofibromatosis type 1.70 Conversely, intracranial aneurysms were detected in 2 of 22 patients with neurofibromatosis type 1 who underwent magnetic resonance imaging.72
Neurofibromatosis type 1 is caused by mutations in the gene (NF1) encoding neurofibromin, a protein with a centrally located domain homologous to guanosine triphosphatase–activating protein (GAP), similar to other tumor suppressor gene products.75,76 The GAP domain of neurofibromin colocalizes with cytoplasmic microtubules, and it has been postulated that neurofibromin may have a regulatory role in the development of various connective tissues, including vascular connective tissue, through an effect on microtubular function. In a mouse model of mutations in genes for GAP and neurofibromatosis type 1, Henkemeyer and colleagues demonstrated thinning and rupture of large and medium-sized arteries during embryonic development.77 The GAP domain of neurofibromin, however, encompasses only about 10% of the protein, and neurofibromin may have a variety of undiscovered functions.
Bicuspid Aortic Valve
A bicuspid aortic valve (BAV) is one of the most common forms of congenital heart disease in adults and is found in 1% to 2% of the population.78 Many patients with BAV remain asymptomatic throughout life, but aortic valve insufficiency or stenosis eventually develops in most patients. Familial clustering of BAV has been described, and screening for BAV is generally recommended for first-degree relatives. Mutations in NOTCH1 have been reported in some families with BAV, but BAV is probably genetically heterogeneous. Cystic medial necrosis of the aorta is found on microscopic examination in most patients with BAV, aortic root dilation is present in at least half the patients with BAV, and aortic dissection is the cause of death in about 5% of patients with BAV. In the past, these aortic abnormalities were believed to be due to postvalvular hemodynamic changes, but it has become well established that the aortic changes are primarily related to an underlying arteriopathy, with hemodynamic factors playing a secondary role. Therefore, BAV has been included among the heritable disorders of connective tissues, along with Marfan’s syndrome and Ehlers-Danlos syndrome. The arteriopathy of BAV does not appear to be confined to the aorta, and spontaneous dissection of the cervical and intracranial arteries has been reported in patients with BAV as well. In one recent study, intracranial aneurysms were detected in 6 of 61 patients with BAV (10%) and in 3 of 291 controls (1%).79
Familial Intracranial Aneurysms
With the exception of ADPKD and, rarely, Ehlers-Danlos syndrome type IV, Pompe’s disease, or syndromes characterized by idiopathic nonarteriosclerotic cerebral calcifications, familial intracranial aneurysms have not been associated with any of the known heritable connective tissue disorders. The familial aggregation of intracranial aneurysms was first described in 1954 by Chambers and associates.80 Since then, hundreds of families have been reported. During the past 2 decades, interest in familial intracranial aneurysms has been renewed. Several studies have been focused on their epidemiologic features, clinical characteristics, and presymptomatic detection with noninvasive screening methods.
Epidemiology
Familial intracranial aneurysms are much more common than has generally been appreciated. Four epidemiologic studies have examined the frequency of familial intracranial aneurysms and revealed that 7% to 20% of patients with aneurysmal SAH had first- or second-degree relatives with intracranial aneurysms (Table 359-3).81–84 However, this familial aggregation could have been fortuitous because at least 1% of adults harbor intracranial aneurysms and most of the reported families have included only two affected members. Whether relatives of patients with intracranial aneurysms have an increased risk for the development of SAH was therefore unknown.
TABLE 359-3 Frequency of Familial Intracranial Aneurysms in Patients with Aneurysmal Subarachnoid Hemorrhage
| STUDY | LOCATION | FREQUENCY OF FAMILIAL DISEASE (%)* |
|---|---|---|
| Norrgård et al., 1987† | Sweden | 6.6 (4.6-9.2) |
| Ronkainen et al., 1993† | East Finland | 9.8 (8.2-11.7) |
| Schievink et al., 1995‡ | Rochester, Minnesota | 20.0 (11.5-30.5) |
| de Braekeleer et al., 1996† | Quebec, Canada | 17.4 (14.2-20.6) |
† Includes one or more probands per family.
‡ Includes one proband per family only.
To address these issues, five independently conducted studies examined the risk for SAH in relatives of patients with SAH and reported comparable results despite widely differing analytic methods and patient populations (Table 359-4).81,84–87 In the population of King County, Washington, Wang and coworkers compared the frequency of familial SAH in patients with SAH with that in a control population.85 Patients with SAH were almost twice as likely to have an affected first-degree relative. However, this difference did not reach statistical significance, and a family history of SAH was never verified. Bromberg and colleagues compared the frequency of SAH in first-degree relatives with that in second-degree relatives of patients with SAH who were admitted to several hospitals in the Netherlands.86 Depending on how well certain diagnostic criteria for SAH were met, they observed a threefold to sevenfold increased risk. In a group of patients with aneurysmal SAH from Rochester, Minnesota, Schievink and associates compared the observed and expected number of first-degree relatives with aneurysmal SAH by using the well-established incidence rates of SAH in this community and observed about a fourfold increased risk.84 Despite this significantly increased risk, the overall absolute risk of first-degree relatives for the development of aneurysmal SAH did not reach 2% until the age of 70 years. In the inhabitants of the Saguenay Lac-Saint-Jean region of Quebec, Canada, de Braekeleer and coauthors compared the frequency of familial intracranial aneurysms in patients with ruptured and unruptured intracranial aneurysms with that of a control population and observed an approximately fivefold increased risk for first-degree relatives.81 Using data from two Danish national registries, Gaist and coworkers compared the incidence of SAH in first-degree relatives of patients with SAH with the incidence in the general population and found a threefold to fivefold increased risk.87
TABLE 359-4 Risk of Subarachnoid Hemorrhage in First-Degree Relatives of Patients with Subarachnoid Hemorrhage
| STUDY | LOCATION | RISK RATIO* |
|---|---|---|
| Wang et al., 1995† | King County, Washington | 1.8 (0.9-3.7) |
| Bromberg et al., 1995† | The Netherlands | 2.7 (1.4-5.5) 6.6 (2.0-21) |
| Schievink et al., 1995‡ | Rochester, Minnesota | 4.1 (2.1-7.4) |
| de Braekeleer et al., 1996‡ | Quebec, Canada | 4.7 (3.1-7.5) |
| Gaist et al., 2000‡ | Denmark | 2.9 (1.9-4.6) 4.5 (2.7-7.3) |
† Includes aneurysmal and nonaneurysmal subarachnoid hemorrhage.
Pattern of Inheritance
The inheritance pattern of familial intracranial aneurysms is unknown. The main difficulty in establishing the mode of transmission is that intracranial aneurysms are acquired lesions and often remain asymptomatic. At visual inspection, some pedigrees support autosomal dominant inheritance, whereas others support autosomal recessive or multifactorial transmission; in most, however, the inheritance pattern is unclear.81–84,87–89 In a segregation analysis of published pedigrees, no single mendelian model was the overall best fit. However, several possible patterns of inheritance were identified, and autosomal transmission was the most likely.88 This finding suggests that genetic heterogeneity is important in the genetics of intracranial aneurysms.88 Genetic heterogeneity had been suspected on the basis of the large number of heritable disorders that have been associated with intracranial aneurysms (see Table 359-1).
Although the Saguenay Lac-Saint-Jean region is well known for its large number of consanguineous marriages, de Braekeleer and associates observed that the coefficient of inbreeding was no higher in patients with intracranial aneurysms than in a control population.81 They also noted a decrease in the frequency of intracranial aneurysms in first-, second-, and third-degree relatives of affected patients. These observations suggest the presence of dominant instead of recessive genes in their reported kinships.
In families with two affected generations, children suffer SAH at a significantly younger age than the parents do.89,90 Although this age difference could be explained by ascertainment bias, onset of disease at earlier ages in later generations (genetic anticipation) is increasingly recognized as an expression of unstable DNA trinucleotide repeats expanding in subsequent generations. This genetic mechanism has been demonstrated in several dominantly inherited neurodegenerative diseases and may also underlie the inheritance of intracranial aneurysms in some families. A more recent study, however, could not demonstrate any evidence of genetic anticipation.91
Are Familial Aneurysms Different?
Numerous studies have compared the characteristics of familial intracranial aneurysms with those of nonfamilial (sporadic) aneurysms.84,89,92–96 These studies have consistently shown that familial aneurysms rupture, on average, about 5 years earlier than sporadic aneurysms do. In several populations of siblings, aneurysms more commonly rupture within the same decade of life. Aneurysms of the anterior communicating artery complex are underrepresented in patients with familial aneurysms. Although prospective studies are unavailable, studies suggest that familial aneurysms rupture at a smaller size than sporadic aneurysms do. The observed differences are small (1 to 2 mm) but may be important, particularly for the treatment of small asymptomatic aneurysms. De novo aneurysms may also develop in patients with familial aneurysms more often than in patients with sporadic aneurysms, although the number of observed cases is small. An increased proportion of multiple aneurysms is also found in patients with familial aneurysms. One hospital-based study suggested that the case fatality rate of SAH is higher in patients with familial aneurysms97; however, other studies have not supported this observation.98,99 At autopsy, patients with familial intracranial aneurysms have had changes in the media of the intracranial and extracranial arteries that were not present in patients with sporadic aneurysms.100 Together, these clinical and pathologic data suggest that familial intracranial aneurysms are different from nonfamilial intracranial aneurysms.
Screening
The benefits of screening for asymptomatic intracranial aneurysms have never been quantified. Several groups, however, have extensive experience in screening for familial intracranial aneurysms with magnetic resonance angiography (Table 359-5).101–106 In the absence of any clinical feature or biologic marker that can identify individuals in whom intracranial aneurysms are most likely to develop, screening for asymptomatic familial intracranial aneurysms may be recommended for first-degree relatives in families with two or more affected members. With this screening strategy, approximately 10% of individuals are found to have an intracranial aneurysm. In approximately a third of these patients, the aneurysms are larger than 5 mm in diameter. Some investigators have recommended screening individuals with only a single family member with an intracranial aneurysm. The absolute lifetime risk for first-degree relatives to suffer an aneurysmal SAH when there is only a single family member with an aneurysm is modest, however. Furthermore, the yield of such a screening strategy is fairly low (2% to 4%). Therefore, screening is seldom recommended for these patients. There are no guidelines for determining when aneurysm screening should be performed. Several investigators have applied sophisticated decision-making analytic models using similar data to determine the optimal age for screening. Widely differing age ranges have been suggested (e.g., Obuchowski and colleagues, <30 years107; Leblanc and colleagues, 20 to 50 years108; ter Berg and associates, 35 to 65 years).109 Ronkainen and coworkers limit their screening to individuals older than 30 years.110 To detect de novo aneurysms, repeated screening has been suggested at 6-month to 5-year intervals after the initial study.111
TABLE 359-5 Yield of Screening for Asymptomatic Intracranial Aneurysms
| POPULATION | RANGE OF YIELD (%) |
|---|---|
| General population | 0.5-2.0 |
| First-degree relatives in families with one affected member | 2-4 |
| Polycystic kidney disease | 5-10 |
| Polycystic kidney disease and a family history of intracranial aneurysms | 10-25 |
Evaluation of Patients with Familial Aneurysms
Patients with an intracranial aneurysm and a family history of intracranial aneurysms or SAH warrant further evaluation (Table 359-6). This evaluation primarily consists of obtaining a detailed medical and family history supplemented by a review of the available medical or autopsy records. A review of these records is important because a self-reported family history alone may not prove or refute a diagnosis of intracranial aneurysm or SAH. Consultation by a medical geneticist is often valuable in constructing a pedigree, obtaining records, and evaluating the patient for the presence of heritable disorders that have been associated with intracranial aneurysms. Apart from ADPKD90–97 and, occasionally, Ehlers-Danlos syndrome type IV,30,112 however, heritable connective tissue disorders are rarely identified in families with intracranial aneurysms. Familial intracranial aneurysms may be the first manifestation of ADPKD.113 Renal ultrasonography is a noninvasive and reliable technique and should therefore be considered to rule out ADPKD. When Ehlers-Danlos syndrome type IV is suspected, collagen type III analysis should be performed on cultured skin fibroblasts to confirm this diagnosis. Finally, adult first-degree relatives should be contacted and advised of the possibilities and uncertainties of invasive and noninvasive screening for asymptomatic intracranial aneurysms.
TABLE 359-6 Evaluation of Patients with Familial Intracranial Aneurysms
| Medical history |
| Family history |
| Physical examination |
| Review of medical/autopsy records |
| Medical genetics consultation |
| Renal ultrasonography |
| Collagen analysis |
| Screening of first-degree relatives |
Gene-Environment Interactions
The etiology and pathogenesis of intracranial aneurysms are clearly multifactorial, and acquired (environmental) factors play a major role. Cigarette smoking is the most important modifiable environmental risk factor.1,2 Smokers have at a 3- to 10-fold increased risk for aneurysmal SAH.1,2 Those who continue to smoke may be at particularly high risk for the de novo development of aneurysms.1 Other risk factors include hypertension and excessive alcohol use. The gene-environmental interactions underlying the development of intracranial aneurysms are complex and poorly understood, but genetic components may predominate in younger patients and environmental components may predominate in the older population. It is possible, at least partially, that the familial aggregation of intracranial aneurysms can be explained by certain environmental factors that are shared by affected family members, such as cigarette smoking. For example, a Dutch study suggested that hypertension is a familial factor contributing to the increased risk for aneurysmal SAH in first-degree relatives, but it could not explain the entire excess risk.114
Intracranial Aneurysm Gene?
Another method for studying linkage is to analyze variations in the sharing of marker alleles among affected sibling pairs only. Although this method requires no knowledge of the mode of transmission and only affected family members are studied, very large sample sizes are often required. An alternative approach to locate intracranial aneurysm genes is candidate gene sequence analysis. This analysis involves evaluating the sequence of a gene for a protein plausibly involved in the development of intracranial aneurysms (e.g., PKD1 or COL3A1, see Table 359-1) and determining whether a mutant sequence variation occurs more frequently in affected patients than is predicted by chance. Several laboratories are currently directing their efforts at locating intracranial aneurysm genes with one or more of these approaches.
Polymorphisms of several genes have now been investigated in patients with intracranial aneurysms.115–117 Certain polymorphisms of the angiotensin I–converting enzyme116 and endoglin115 genes may be associated with an increased risk for aneurysm development, but these findings need to be confirmed in other groups of patients with aneurysms. Olson and colleagues performed a sibling-pair linkage analysis in Finnish patients with intracranial aneurysms and identified a susceptibility locus at 19q13.118,119 Onda and associates performed genome-wide linkage and haplotype association studies that demonstrated the 7q11 locus as a potential site of increased risk for intracranial aneurysms.120 Farnhan and coworkers demonstrated similar findings in a North American family.121 Candidate genes at this location include COL1A2 and the gene for elastin, which are important for blood vessel wall morphology. In a study of a large kindred with nonsyndromic intracranial aneurysms, Nahed and associates found a single locus, 1p34.3-p36.13, that was transmitted in autosomal fashion with high penetrance in a mendelian pattern of inheritance.122 More recent comprehensive studies have found susceptibility loci for the pathogenesis of intracranial aneurysms at 2q, 8q, and 9p and suggest that several loci may be involved (Table 359-7).123–125 Thus, many more studies need to be conducted to provide a molecular marker for identifying patients at increased risk for the development and rupture of intracranial aneurysms.
| 1p36 | 8q |
| 2q | 9p21 |
| 4q | 12p |
| 7p | 19q13 |
| 7q11 | Xp22 |
Bilguvar K, Yasuno K, Niemelä M, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 2008;40:1472-1477.
Bromberg JEC, Rinkel GJE, Algra A, et al. Subarachnoid haemorrhage in first and second degree relatives of patients with subarachnoid haemorrhage. BMJ. 1995;311:288-289.
Foroud T, Sauerbeck L, Brown R, et al. FIA Study Investigators: Genome screen in familial intracranial aneurysm. BMC Med Genet. 2009;10:3.
Gaist D, Vaeth M, Tsiropoulos I, et al. Risk of subarachnoid haemorrhage in first degree relatives of patients with subarachnoid haemorrhage: follow up study based on national registries in Denmark. BMJ. 2000;320:141-145.
Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217-224.
Huston JIII, Torres VE, Wiebers DO, et al. Follow-up of intracranial aneurysms in autosomal dominant polycystic kidney disease by magnetic resonance angiography. J Am Soc Nephrol. 1996;7:2135-2141.
Olson JM, Vongpunsawad S, Kuivaniemi H, et al. Search for intracranial aneurysm susceptibility gene(s) using Finnish families. BMC Med Genet. 2002;3:7.
Pepin M, Schwarze U, Superti-Furga A, et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673-680.
Pope FM, Kendall BE, Slapak GI, et al. Type III collagen mutations cause fragile cerebral arteries. Br J Neurosurg. 1991;5:551-574.
Ronkainen A, Hernesniemi J, Puranen M, et al. Familial intracranial aneurysms. Lancet. 1997;349:380-384.
Ronkainen A, Puranen MI, Hernesniemi JA, et al. Intracranial aneurysms: MR angiographic screening in 400 asymptomatic individuals with increased familial risk. Radiology. 1995;195:35-40.
Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol. 2005;4:179-189.
Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of heritable connective tissue disorders. A review. Stroke. 1994;25:889-903.
Schievink WI, Parisi JE, Piepgras DG, et al. Intracranial aneurysms in Marfan’s syndrome: an autopsy study. Neurosurgery. 1997;41:866-871.
Schievink WI, Riedinger M, Maya MM. Frequency of incidental intracranial aneurysms in neurofibromatosis type 1. Am J Med Genet A. 2005;134:45-48.
Schievink WI, Schaid DJ, Michels VV, et al. Familial aneurysmal subarachnoid hemorrhage: a community-based study. J Neurosurg. 1995;83:426-429.
Schievink WI, Schaid DJ, Rogers HM, et al. On the inheritance of intracranial aneurysms. Stroke. 1994;25:2028-2037.
The Magnetic Resonance Angiography in Relatives of Patients with Subarachnoid Hemorrhage Study Group. Risks and benefits of screening for intracranial aneurysms in first-degree relatives of patients with sporadic subarachnoid hemorrhage. N Engl J Med. 1999;341:1344-1350.
van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124:249-278.
1 Schievink WI. Intracranial aneurysms. N Engl J Med. 1997;336:28-40.
2 van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124:249-278.
3 Schievink WI. Genetics and aneurysm formation. Neurosurg Clin N Am. 1998;9:485-495.
4 Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol. 2005;4:179-189.
5 Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of heritable connective tissue disorders. A review. Stroke. 1994;25:889-903.
6 Schievink WI, Katzmann JA, Piepgras DG, et al. Alpha-1-antitrypsin phenotypes among patients with intracranial aneurysms. J Neurosurg. 1996;84:781-784.
7 Fick GM, Gabow PA. Natural history of autosomal dominant polycystic kidney disease. Annu Rev Med. 1994;45:23-29.
8 Schievink WI, Huston JIII, Torres VE, et al. Intracranial cysts in autosomal dominant polycystic kidney disease. J Neurosurg. 1995;83:1004-1007.
9 Schievink WI, Torres VE. Spinal meningeal diverticula in autosomal dominant polycystic kidney disease. Lancet. 1997;349:1223-1224.
10 Schievink WI, Torres VE, Piepgras DG, et al. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3:88-95.
11 Schievink WI, Torres VE, Wiebers DO, et al. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1997;8:1298-1303.
12 Wijdicks EFM, Torres VE, Schievink WI. Chronic subdural hematoma in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;35:40-43.
13 Schievink WI, Palestrant D, Maya MM, et al. Spontaneous spinal cerebrospinal fluid leak as a cause of coma after craniotomy for clipping of an unruptured intracranial aneurysm. J Neurosurg. 2009;110:521-524.
14 Suter W. Das kongenitale Aneurysma der basalen Hirnarterien und Cystennieren. Schweiz Med Wochenschr. 1949;79:471-476.
15 Chapman AB, Rubinstein D, Hughes R, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med. 1992;327:916-920.
16 Huston JIII, Torres VE, Sullivan PP, et al. Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1871-1877.
17 Huston JIII, Torres VE, Wiebers DO, et al. Follow-up of intracranial aneurysms in autosomal dominant polycystic kidney disease by magnetic resonance angiography. J Am Soc Nephrol. 1996;7:2135-2141.
18 Iida H, Naito T, Hondo H, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease detected by MR angiography: screening and treatment. Nippon Jinzo Gakkai Shi. 1998;40:42-47.
19 Ruggieri PM, Poulos N, Masaryk TJ, et al. Occult intracranial aneurysms in polycystic kidney disease: screening with MR angiography. Radiology. 1994;191:33-39.
20 Ronkainen A, Hernesniemi J, Puranen M, et al. Familial intracranial aneurysms. Lancet. 1997;349:380-384.
21 Ring T, Spiegelhalter D. Risk of intracranial aneurysm bleeding in autosomal-dominant polycystic kidney disease. Kidney Int.. 2007;72:1400-1402.
22 Hughes R, Chapman A, Rubinstein D, et al. Recurrent intracranial aneurysms (ICA) in autosomal dominant polycystic kidney disease (ADPKD). Stroke. 1996;27:178.
23 Harris PC. Autosomal dominant polycystic kidney disease: clues to pathogenesis. Hum Mol Genet. 1999;8:1861-1866.
24 Kimberling WJ, Fain PR, Kenyon JB, et al. Linkage heterogeneity of autosomal dominant polycystic kidney disease. N Engl J Med. 1988;319:913-918.
25 Ong ACM, Harris PC. Molecular basis of renal cyst formation—one hit or two? Lancet. 1997;349:1039-1040.
26 van Dijk MA, Chang PC, Peters DJM, et al. Intracranial aneurysms in polycystic kidney disease linked to chromosome 4. J Am Soc Nephrol. 1995;6:1670-1673.
27 Schievink WI, Spetzler RF. Screening for intracranial aneurysms in patients with isolated polycystic liver disease. J Neurosurg. 1998;89:719-721.
28 Byers PH. Ehlers-Danlos syndrome type IV: a genetic disorder in many guises. J Invest Dermatol. 1995;105:311-313.
29 Pope FM, Kendall BE, Slapak GI, et al. Type III collagen mutations cause fragile cerebral arteries. Br J Neurosurg. 1991;5:551-574.
30 Pope FM, Narcisi P, Nicholls AC, et al. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996;135:163-181.
31 Graf CJ. Spontaneous carotid-cavernous fistula. Ehlers-Danlos syndrome and related conditions. Arch Neurol. 1965;13:662-672.
32 North KN, Whiteman DAH, Pepin MG, et al. Cerebrovascular complications of Ehlers-Danlos syndrome type IV. Ann Neurol. 1995;38:960-964.
33 Pepin M, Schwarze U, Superti-Furga A, et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673-680.
34 Pope FM, Martin GR, Lichtenstein JR, et al. Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci U S A. 1975;72:1314-1316.
35 Liu X, Wu H, Byrne M, et al. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852-1856.
36 Schwarze U, Schievink WI, Petty E, et al. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989-1001.
37 Brega KE, Seltzer WK, Munro LG, et al. Genotypic variations of type III collagen in patients with cerebral aneurysms. Surg Neurol. 1996;46:253-257.
38 Majamaa K, Savolainen E-R, Myllalä VV. Synthesis of structurally unstable type III procollagen in patients with cerebral artery aneurysm. Biochim Biophys Acta. 1992;138:191-196.
39 Neil-Dwyer G, Bartlett JR, Nicholls AC, et al. Collagen deficiency and ruptured cerebral aneurysms. A clinical and biochemical study. J Neurosurg. 1983;59:16-20.
40 Østergaard JR, Oxlund H. Collagen type III deficiency in patients with rupture of intracranial saccular aneurysms. J Neurosurg. 1987;67:690-696.
41 Pope FM, Limburg M, Schievink WI. Familial cerebral aneurysms and type III collagen deficiency. J Neurosurg. 1990;72:156-158.
42 Pope FM, Nicholls AC, Narcisi P, et al. Some patients with cerebral aneurysms are deficient in type III collagen. Lancet. 1981;1:973-975.
43 van den Berg JSP, Pals G, Arwert F, et al. Type III collagen deficiency in saccular intracranial aneurysms. Defect in gene regulation? Stroke. 1999;30:1628-1631.
44 Kuivaniemi H, Prockop DJ, Wu Y, et al. Exclusion of mutations in the gene for type III collagen (COL3A1) as a common cause of intracranial aneurysms or cervical artery dissections: results from sequence analysis of the coding sequences of type III collagen from 55 unrelated patients. Neurology. 1993;43:2652-2658.
45 Godfrey M. The Marfan syndrome. In: Beighton P, editor. McKusick’s Heritable Disorders of Connective Tissue. 5th ed. St. Louis: CV Mosby; 1993:51-135.
46 Pyeritz RE. The Marfan syndrome. In: Royce PM, Steinmann B, editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. New York: Wiley-Liss; 1993:437-468.
47 Schievink WI, Björnsson J, Piepgras DG. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke. 1994;25:2492-2496.
48 Pyeritz RE, Fishman EK, Bernhardt BA, et al. Dural ectasia is a common feature of the Marfan syndrome. Am J Hum Genet. 1988;43:726-732.
49 Fattori R, Nienaber CA, Descovich B, et al. Importance of dural ectasia in phenotypic assessment of Marfan’s syndrome. Lancet. 1999;354:910-913.
50 Rose BS, Pretorius DL. Dissecting basilar artery aneurysm in Marfan syndrome: case report. AJNR Am J Neuroradiol. 1991;12:503-504.
51 Schievink WI, Parisi JE, Piepgras DG, et al. Intracranial aneurysms in Marfan’s syndrome: an autopsy study. Neurosurgery. 1997;41:866-871.
52 Sekhar LN, Bucur SD, Bank WO, et al. Venous and arterial bypass grafts for difficult tumors, aneurysms, and occlusive vascular lesions: evolution of surgical treatment and improved graft results. Neurosurgery. 1999;44:1207-1224.
53 Kubo Y, Ogasawara K, Kurose A, et al. Ruptured cerebral fusiform aneurysm with mucopolysaccharide deposits in the tunica media in a patient with Marfan syndrome. J Neurosurg. 2009;110:518-520.
54 Conway JE, Hutchins GM, Tamargo RJ. Marfan syndrome is not associated with intracranial aneurysms. Stroke. 1999;30:1632-1636.
55 Schievink WI. Intracranial aneurysms and Marfan syndrome. Stroke. 1999;30:2767-2768.
56 Ramirez F, Gayraud B, Pereira L. Marfan syndrome: new clues to genotype-phenotype correlations. Ann Med. 1999;31:202-207.
57 Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499-2509.
58 Aoyama T, Francke U, Gasner C, et al. Fibrillin abnormalities and prognosis in Marfan syndrome and related disorders. Am J Med Genet. 1995;58:169-176.
59 Francke U, Berg MA, Tynan K, et al. A Gly1127Ser mutation in an EGF-like domain of the fibrillin-1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56:1287-1296.
60 Milewicz DM, Grossfield J, Cao S-N, et al. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest. 1995;95:2372-2378.
61 Schrijver I, Schievink WI, Godfrey M, et al. Spontaneous spinal cerebrospinal fluid leaks and minor skeletal features of Marfan syndrome: a microfibrillopathy. J Neurosurg. 2002;96:483-489.
62 Riccardi VM. Neurofibromatosis: Phenotype, Natural History and Pathogenesis, 2nd ed. Baltimore: Johns Hopkins University; 1992.
63 Greene JFJr, Fitzwater JE, Burgess J. Arterial lesions associated with neurofibromatosis. Am J Clin Pathol. 1974;62:481-487.
64 Reubi F. Neurofibromatosis et lésions vasculaires. Schweiz Med Wochenschr. 1945;75:463-465.
65 Schievink WI, Piepgras DG. Cervical vertebral artery aneurysms and arteriovenous fistulae in neurofibromatosis type 1: case reports. Neurosurgery. 1991;29:760-765.
66 Benatar MG. Intracranial fusiform aneurysms in von Recklinghausen’s disease: case report and literature review. J Neurol Neurosurg Psychiatry. 1994;57:1279-1280.
67 Poli P, Peillon C, Lahda E, et al. Anévrysmes intracraniens multiples en rapport avec une maladie de Recklinghausen: a propos d’un cas. J Mal Vasc. 1994;19:253-255.
68 Sasaki J, Miura S, Ohishi H, et al. Neurofibromatosis associated with multiple intracranial vascular lesions: stenosis of the internal carotid artery and peripheral aneurysm of the Huebner’s artery; report of a case. No Shinkei Geka. 1995;23:813-817.
69 Urashini R, Ochiai C, Okuno S, et al. Cerebral aneurysms associated with von Recklinghausen neurofibromatosis: report of two cases. No Shinkei Geka. 1995;23:237-242.
70 Schievink WI. Genetics of intracranial aneurysms. Neurosurgery. 1997;40:651-663.
71 Zhao JZ, Han XD. Cerebral aneurysm associated with von Recklinghausen’s neurofibromatosis: a case report. Surg Neurol. 1998;50:592-596.
72 Schievink WI, Riedinger M, Maya MM. Frequency of incidental intracranial aneurysms neurofibromatosis type 1. Am J Med Genet A. 2005;134:45-48.
73 Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64:553-555.
74 Sobata E, Ohkuma H, Suzuki S. Cerebrovascular disorders associated with von Recklinghausen’s neurofibromatosis: a case report. Neurosurgery. 1988;22:544-549.
75 Gutmann DH, Collins FS. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron. 1993;10:335-343.
76 Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet. 1996;33:2-17.
77 Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
78 Tadros TM, Klein MD, Shapira OM. Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation. 2009;119:880-890.
79 Schievink WI, Raissi S, Maya MM. Screening for intracranial aneurysms in bicuspid aortic valve. Stroke. 2008;39:647.
80 Chambers WR, Harper BFJr, Simpson JR. Familial incidence of congenital aneurysms of cerebral arteries. JAMA. 1954;155:358-359.
81 De Braekeleer M, Pérusse L, Cantin L, et al. A study of inbreeding and kinship in intracranial aneurysms in the Saguenay Lac-Saint-Jean region (Quebec, Canada). Ann Hum Genet. 1996;60:99-104.
82 Norrgård Ö, Ångquist K-A, Fodstad H, et al. Intracranial aneurysms and heredity. Neurosurgery. 1987;20:236-239.
83 Ronkainen A, Hernesniemi J, Ryynänen M. Familial subarachnoid hemorrhage in east Finland, 1977-1990. Neurosurgery. 1993;33:787-797.
84 Schievink WI, Schaid DJ, Michels VV, et al. Familial aneurysmal subarachnoid hemorrhage: a community-based study. J Neurosurg. 1995;83:426-429.
85 Wang PS, Longstreth WTJr, Koepsell TD. Subarachnoid hemorrhage and family history. A population-based case-control study. Arch Neurol. 1995;52:202-204.
86 Bromberg JEC, Rinkel GJE, Algra A, et al. Subarachnoid haemorrhage in first and second degree relatives of patients with subarachnoid haemorrhage. BMJ. 1995;311:288-289.
87 Gaist D, Vaeth M, Tsiropoulos I, et al. Risk of subarachnoid haemorrhage in first degree relatives of patients with subarachnoid haemorrhage: follow up study based on national registries in Denmark. BMJ. 2000;320:141-145.
88 Schievink WI, Schaid DJ, Rogers HM, et al. On the inheritance of intracranial aneurysms. Stroke. 1994;25:2028-2037.
89 Bromberg JEC, Rinkel GJE, Algra A, et al. Familial subarachnoid hemorrhage: distinctive features and patterns of inheritance. Ann Neurol. 1995;38:929-934.
90 Bailey JC. Familial subarachnoid haemorrhage. Ulster Med J. 1993;62:119-126.
91 Woo D, Hornung R, Sauerbeck L, et al. FIA Investigators: Age at intracranial aneurysm rupture among generations: Familial Intracranial Aneurysm Study. Neurology. 2009;72:695-698.
92 Leblanc R, Melanson D, Tampieri D, et al. Familial cerebral aneurysms: a study of 13 families. Neurosurgery. 1995;37:633-639.
93 Lozano AM, Leblanc R. Familial intracranial aneurysms. J Neurosurg. 1987;66:522-528.
94 Ronkainen, Hernesniemi J, Tromp G. Special features of familial intracranial aneurysms: report of 215 familial aneurysms. Neurosurgery. 1995;37:43-47.
95 Motuo Fotso MJ, Brunon J, Outhel R, et al. Anéurysmes familiaux, anéurysmes multiples et anéurysmes ‘de novo’: a propos de deux observations. Neurochirurgie. 1993;39:225-230.
96 Leblanc R. De novo formation of familial cerebral aneurysms: case report. Neurosurgery. 1999;44:871-877.
97 Bromberg JEC, Rinkel GJE, Algra A, et al. Outcome in familial subarachnoid hemorrhage. Stroke. 1995;26:961-963.
98 Schievink WI, Schaid DJ. The prognosis of familial versus nonfamilial aneurysmal subarachnoid hemorrhage. Stroke. 1996;27:340-341.
99 Ronkainen A, Niskanen M, Piironen R, et al. Familial subarachnoid hemorrhage. Outcome study. Stroke. 1999;30:1099-1102.
100 Schievink WI, Parisi JE, Piepgras DG. Familial intracranial aneurysms: an autopsy study. Neurosurgery. 1997;41:1247-1252.
101 Brown BM, Soldevilla F. MR angiography and surgery for unruptured familial intracranial aneurysms in persons with a family history of cerebral aneurysms. AJR Am J Roentgenol. 1999;173:133-138.
102 Ronkainen A, Puranen MI, Hernesniemi JA, et al. Intracranial aneurysms: MR angiographic screening in 400 asymptomatic individuals with increased familial risk. Radiology. 1995;195:35-40.
103 Raaymakers TWM, Rinkel GJE, Ramos LMP. Initial and follow-up screening for aneurysms in families with familial subarachnoid hemorrhage. Neurology. 1998;51:1125-1130.
104 Ronkainen A, Miettinen H, Karkola K, et al. Risk of harboring an unruptured intracranial aneurysm. Stroke. 1998;29:359-362.
105 Raaymakers TW. Aneurysms in relatives of patients with subarachnoid hemorrhage: frequency and risk factors. MARS Study Group. Magnetic resonance angiography in relatives of patients with subarachnoid hemorrhage. Neurology. 1999;53:982-988.
106 The Magnetic Resonance Angiography in Relatives of Patients with Subarachnoid Hemorrhage Study Group. Risks and benefits of screening for intracranial aneurysms in first-degree relatives of patients with sporadic subarachnoid hemorrhage. N Engl J Med. 1999;341:1344-1350.
107 Obuchowski NA, Modic MT, Magdinec M. Current implications for the efficacy of noninvasive screening for occult intracranial aneurysms in patients with a family history of aneurysms. J Neurosurg. 1995;83:42-49.
108 Leblanc R, Worsley KJ, Melanson D, et al. Angiographic screening and elective surgery of familial cerebral aneurysms: A decision analysis. Neurosurgery. 1994;35:9-19.
109 ter Berg HWM, Dippel DWJ, Limburg M, et al. Familial intracranial aneurysms. A review. Stroke. 1992;23:1024-1030.
110 Ronkainen A, Hernesniemi J, Kuivaniemi H, et al. Current implications for the efficacy of noninvasive screening for occult intracranial aneurysms in patients with a family history of aneurysms. J Neurosurg. 1996;84:534-536.
111 Schievink WI, Limburg M, Dreissen JJR, et al. Screening for unruptured familial intracranial aneurysms: subarachnoid hemorrhage 2 years after angiography negative for aneurysms. Neurosurgery. 1991;29:434-438.
112 Pollack JS, Custer PL, Hart WM, et al. Ocular complications in Ehlers-Danlos syndrome type IV. Arch Ophthalmol. 1997;115:416-419.
113 McConnell RS, Hughes AE, Rubinsztein DEC, et al. Gene-environment interactions in familial clustering of cerebral aneurysm formation. J Neurol Neurosurg Psychiatry. 1997;63:128.
114 Bromberg JEC, Rinkel GJE, Algra A, et al. Hypertension, stroke, and coronary heart disease in relatives of patients with subarachnoid hemorrhage. Stroke. 1996;27:7-9.
115 Takenaka K, Sakai H, Yamakawa H, et al. Polymorphism of the endoglin gene in patients with intracranial saccular aneurysms. J Neurosurg. 1999;90:935-938.
116 Takenaka K, Yamakawa H, Sakai N, et al. Angiotensin I–converting enzyme gene polymorphism in intracranial saccular aneurysm individuals. Neurol Res. 1998;20:607-611.
117 Yoon S, Tromp G, Vongpunsawad S, et al. Genetic analysis of MMP3, MMP9, and PAI-1 in Finnish patients with abdominal aortic or intracranial aneurysms. Biochem Biophys Res Commun. 1999;265:563-568.
118 Olson J, Vongpunsawad S, Kuivaniemi H, et al. Genome scan for intracranial aneurysm susceptibility loci using Finnish families. Am J Hum Genet. 1998;63:A17.
119 Olson JM, Vongpunsawad S, Kuivaniemi H, et al. Search for intracranial aneurysm susceptibility gene(s) using Finnish families. BMC Med Genet. 2002;3:7.
120 Onda H, Kasuya H, Yoneyama T, et al. Genomewide-linkage and haplotype-association studies map intracranial aneurysm to chromosome 7q11. Am J Hum Genet. 2001;69:804-819.
121 Farnham JM, Camp NJ, Neuhausen SL, et al. Confirmation of chromosome 7q11 locus for predisposition to intracranial aneurysm. Hum Genet. 2003;114:250-255.
122 Nahed BV, Seker A, Guclu B, et al. Mapping a mendelian form of intracranial aneurysm to 1p34.3-p36.13. Am J Hum Genet. 2004;76:172-179.
123 Bilguvar K, Yasuno K, Niemelä M, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 2008;40:1472-1477.
124 Foroud T, Sauerbeck L, Brown R, et al. FIA Study Investigators: Genome screen in familial intracranial aneurysm. BMC Med Genet. 2009;10:3.
125 Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217-224.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 359 Genetics of Intracranial Aneurysms
The exact etiology and pathogenesis of intracranial aneurysms remain unclear. Several lines of evidence implicate acquired risk factors such as smoking or hypertension,1,2 whereas others support the role of genetic factors.3,4 The two main lines of evidence supporting the role of genetic factors are the association of intracranial aneurysms with heritable connective tissue disorders and the familial occurrence of intracranial aneurysms.
Heritable Connective Tissue Disorders
Numerous heritable connective tissue disorders have been associated with intracranial aneurysms, including polycystic kidney disease, Ehlers-Danlos syndrome type IV, Marfan’s syndrome, neurofibromatosis type 1, and bicuspid aortic valve (Table 359-1).3–5 To what extent these specific heritable disorders contribute to the entire population of patients with intracranial aneurysms is unknown. In one series of 100 consecutive hospitalized patients with intracranial aneurysms, 5 had an identifiable heritable connective tissue disorder.6 The true frequency of heritable connective tissue disorders in patients with aneurysms is probably higher because these disorders often remain undiagnosed as a result of the substantial variability in their phenotypic expression. The family history may also be negative because the disease can be caused by a new mutation. Nevertheless, identifiable heritable connective tissue disorders contribute to a relatively small percentage of intracranial aneurysms.
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) affects about 1 in 400 to 1000 persons and is the most common monogenetic disease in humans.7 It is inherited as an autosomal dominant trait with almost complete penetrance but with variable expression. The family history is negative in about 20% of patients, thus suggesting a fairly high spontaneous mutation rate.
ADPKD is a systemic disease, and cysts are present in the kidneys, liver, pancreas, spleen, ovaries, and seminal vesicles.7 Moreover, ADPKD should be included among the heritable connective tissue disorders.5 A wide variety of connective tissues may be involved,7 including the heart valves (mitral valve prolapse), vasculature (aneurysms and dissections), and meninges (arachnoid cysts). Patients with ADPKD are at increased risk for the development of gastrointestinal diverticula and inguinal hernias.7
Neurosurgical disorders that have been associated with ADPKD include intracranial aneurysms, cervicocephalic arterial dissections, intracranial dolichoectasia, intracranial arachnoid cysts, spinal meningeal diverticula/cerebrospinal fluid leaks, and chronic subdural hemorrhages.8–13 Intracranial aneurysms have long been known to be associated with ADPKD.14–21 Until the underlying connective tissue defect of the disease became well known, however, aneurysms were frequently attributed to the arterial hypertension that usually accompanies ADPKD. Intracranial aneurysms are detected in approximately a fourth of patients with ADPKD at autopsy; in most of these patients, aneurysmal rupture was the cause of death. Conversely, ADPKD accounts for 2% to 7% of all patients with intracranial aneurysms. Using magnetic resonance angiography or, less commonly, catheter angiography in patients with good renal function, several groups have screened adult ADPKD patients for asymptomatic intracranial aneurysms. The detection rate has ranged between 5% and 10%.15–19 Familial clustering of intracranial aneurysms occurs in ADPKD; the yield of screening increases to 10% to 25% in such families.15–19 The presence of polycystic liver disease in patients with ADPKD may also increase the development of intracranial aneurysms.16
Screening patients with ADPKD for asymptomatic aneurysms remains controversial but should certainly be considered for those with a family history of intracranial aneurysms. Most asymptomatic intracranial aneurysms detected with screening are less than 6 mm in diameter. In one study, none of these small aneurysms ruptured during 500 months of cumulative follow-up.17 It has been suggested that ADPKD patients are at an increased risk for the de novo development of aneurysms some time after their first intracranial aneurysm is discovered, but the exact significance of this risk remains to be determined.22 When compared with the general population, aneurysmal subarachnoid hemorrhage (SAH) in patients with ADPKD occurs at an earlier age, but the mortality rate is similar.8
ADPKD is a genetically heterogeneous disease. Several loci are involved, and mutations, which are responsible for at least 85% of cases, have been identified in a gene on chromosome 16 (PKD1), as well as in a gene on chromosome 4 (PDK2).23–26 In general, patients with mutations in the PKD1 gene are more severely affected than those with mutations in the PKD2 gene, but intracranial aneurysms are a manifestation of both types of ADPKD. Polycystin-1 and polycystin-2 are the proteins encoded by the PKD1 and PKD2 genes, respectively.23 Both proteins are integral membrane proteins with large extracellular domains, and they probably play a role in maintaining structural integrity of the connective tissue extracellular matrix.23
Autosomal dominant polycystic liver disease (ADPLD) is a familial form of isolated polycystic liver disease that is distinct from ADPKD.27 Patients with ADPLD may also be at high risk for the development of intracranial aneurysms.27
Ehlers-Danlos Syndrome Type IV
Ehlers-Danlos syndrome type IV is potentially one of the most deadly heritable connective tissue disorders that neurosurgeons may encounter. It is uncommon, with a prevalence of approximately 1 in 50,000 to 500,000 persons.28 It is inherited in an autosomal dominant fashion, but the family history is frequently noncontributory because of the high spontaneous mutation rate (approximately 50%).
Ehlers-Danlos syndrome type IV can be life-threatening because spontaneous rupture, dissection, or aneurysm formation on large and medium-sized arteries occurs in all areas of the body.5,28–30 These arterial complications cause death in most patients. Other well-described life-threatening complications of Ehlers-Danlos syndrome type IV are spontaneous rupture of the bowel or gravid uterus and spontaneous pneumothorax.28–30
An intracranial aneurysm may be the initial manifestation of Ehlers-Danlos syndrome type IV. Consequently, neurosurgeons may be the first physicians involved in these patients’ medical care. The syndrome is often difficult to recognize because its external features can be subtle.5,28–30 The more salient features of Ehlers-Danlos syndrome type IV are summarized in Table 359-2. The characteristic facial appearance was first described by Carl Graf, a neurosurgeon31; many striking examples have since been published.29,30 The facial features consist of (1) large expressive eyes with the sclera clearly visible around the iris, (2) a thin nose, (3) thin lips, and (4) lobeless ears. Many patients with Ehlers-Danlos syndrome type IV, however, do not exhibit this facial appearance. The characteristic cutaneous features include thin and fragile skin that is almost transparent and allows the subcutaneous veins to be clearly visible. Patients bruise easily, and multiple ecchymoses are common. Scars are often papyraceous and wide, or they may be complicated by keloid formation. The skin of some patients with Ehlers-Danlos syndrome type IV, however, appears normal. The joint hypermobility is often mild and limited to the fingers and toes. Identifying Ehlers-Danlos syndrome type IV in any patient with an intracranial aneurysm is important because vascular fragility can make any invasive procedure a hazardous undertaking.
TABLE 359-2 Characteristic Features of Ehlers-Danlos Syndrome Type IV
Intracranial aneurysms and spontaneous carotid cavernous fistulas are well-described vascular complications of Ehlers-Danlos syndrome type IV.5,28–33 In some patients, the carotid cavernous fistula is due to rupture of a cavernous carotid aneurysm, although the fistula may be caused by a simple tear in the artery in other patients. The importance of intracranial aneurysmal disease in this group of patients is well described. For example, in a cohort of 202 patients with Ehlers-Danlos syndrome type IV, 4 had ruptured intracranial aneurysms, 4 suffered an intracranial hemorrhage of undetermined cause, and 6 had carotid cavernous fistulas.28 The exact incidence of intracranial aneurysms in patients with Ehlers-Danlos syndrome type IV is unknown because screening for asymptomatic intracranial aneurysms is limited and systematic autopsy studies are unavailable. Screening for asymptomatic intracranial aneurysms in these patients is not recommended because safe treatment options are limited; arteriography, endovascular intervention, and surgical treatment are all associated with high complication rates.
Mutations in the gene encoding the pro-α1-(III) chain of collagen type III (COL3A1) on chromosome 2 are the cause of Ehlers-Danlos syndrome type IV.29,30,33–36 This type of collagen is the major structural component of distensible tissues, including arteries, veins, hollow viscera, and the uterus. In addition, collagen type III may play an important role in the fibrillogenesis of collagen type I. Several studies have reported evidence of abnormal collagen type III metabolism in up to 50% of patients with intracranial aneurysms who do not have Ehlers-Danlos syndrome type IV.37–43 Mutations in the COL3A1 gene, however, are rare. For example, in a study of 40 patients with intracranial aneurysms, COL3A1 mutations were found in just 2 patients, and the functional consequences of these mutations were considered insignificant.44 The reasons for these conflicting data are unclear.
Marfan’s Syndrome
Marfan’s syndrome affects approximately 1 in 10,000 to 20,000 people and is characterized by abnormalities of the skeleton, cardiovascular system, eye, and spinal meninges.45,46 Aortic and mitral valve insufficiency is the most frequent cause of death in children with Marfan’s syndrome, and spontaneous aortic rupture and dissection are the most frequent causes of death in adults with the syndrome.45,46 Dissection of medium-sized arteries, however, is much less common.47 Although Marfan’s syndrome is easily recognized in patients who display the main features of the syndrome (particularly the skeletal manifestations of tall stature, dolichostenomelia, arachnodactyly, and anterior chest deformity), the variability in phenotypic expression is great and the diagnosis is seldom straightforward.45,46 For example, if the parents of a patient with Marfan’s syndrome are short, the affected person’s habitus may be comparatively normal.45 Ectopia lentis, the classic ocular manifestation of Marfan’s syndrome, is observed in only about half the cases.45 Dural ectasia, another major diagnostic criterion of the syndrome, is usually asymptomatic and requires computed tomography or magnetic resonance imaging for diagnosis.48,49 Other manifestations of Marfan’s syndrome include spontaneous pneumothorax, striae distensae, and retinal detachment.45,46
Intracranial aneurysms in patients with Marfan’s syndrome may be saccular or fusiform, and intracranial dissecting aneurysms have also been described.5,50–53 Similar to Ehlers-Danlos syndrome type IV, there is a propensity for proximal intracranial carotid artery involvement, although carotid cavernous fistulas seem to be rare.5 Connective tissue fragility is seldom a major problem in the neurosurgical treatment of patients with Marfan’s syndrome. The frequently observed ectasia and tortuosity of the extracranial carotid and vertebral arteries, however, may render endovascular treatment of intracranial aneurysms impossible. The association of Marfan’s syndrome and intracranial aneurysms has not been firmly established. In an autopsy series of 7 patients with Marfan’s syndrome collected during a 25-year period at the Mayo Clinic, intracranial aneurysms, one ruptured and one unruptured, were observed in 2 patients.51 Combining this autopsy study with one performed at Johns Hopkins University54 but excluding the one ruptured aneurysm, incidental aneurysms were found in 2 (6.5%) of 31 patients.55 This frequency is higher than would be expected in the general population, particularly in view of the young age of the patients.55 The results of screening for asymptomatic intracranial aneurysms in patients with Marfan’s syndrome have not been reported.
Mutations in the gene encoding fibrillin-1 (FBN1) cause Marfan’s syndrome.56 Fibrillin-1 is a fairly recently detected glycoprotein that is one of the major components of microfibrils.57 Microfibrils are important constituents of the extracellular matrix and are distributed throughout the body in elastic tissues such as the skin and aorta and in nonelastic tissues such as the ciliary zonules of the ocular lens. In elastic arteries such as the aorta, fibrillin-1 is found in all three layers of the arterial wall. It is thought that fibrillin-1 plays an important role in maintaining the structural integrity of connective tissues, in part by providing a scaffolding for the elastic fibers. Mutations in the FBN1 gene or abnormal fibrillin metabolism (“fibrillinopathy”) have also been detected in patients with isolated features of Marfan’s syndrome but without the classic syndrome.58–61
Neurofibromatosis Type 1
Neurofibromatosis type 1 is a progressive systemic disease that affects approximately 1 in 3000 to 5000 persons.62 The principal clinical features of neurofibromatosis type 1 are café au lait spots, neurofibromas, and Lisch’s nodules (hamartomas) of the iris.62 Although these features each occur in more than 90% of adults with neurofibromatosis type 1, the number of lesions is variable. Patients with neurofibromatosis type 1 are also at increased risk for the development of optic glioma, pheochromocytoma, dural ectasia, and skeletal abnormalities such as scoliosis and sphenoid wing dysplasia.62 Vascular complications of neurofibromatosis type 1 have been recognized since 1945 and are characterized by stenosis, rupture, and aneurysm or fistula formation in large and medium-sized arteries.63–65
Intracranial aneurysms in patients with neurofibromatosis type 1 may be saccular or fusiform, and some have the appearance of dissecting aneurysms.66–73 Surgical repair of these aneurysms may be complicated by excessive vascular fragility or distortion of anatomic landmarks caused by sphenoid wing dysplasia.70 The intracranial aneurysms associated with neurofibromatosis type 1 often coexist with intracranial arterial occlusive disease,74 thereby increasing the risk associated with the surgical and particularly endovascular treatment of these aneurysms. An increased probability of the development of intracranial aneurysms has not been clearly established for patients with neurofibromatosis type 1, but the number of reported cases continues to increase and some have advocated screening patients with neurofibromatosis for asymptomatic intracranial aneurysms.67 Among a group of 100 consecutive patients with intracranial aneurysms, 1 patient was revealed to have neurofibromatosis type 1.70 Conversely, intracranial aneurysms were detected in 2 of 22 patients with neurofibromatosis type 1 who underwent magnetic resonance imaging.72
Neurofibromatosis type 1 is caused by mutations in the gene (NF1) encoding neurofibromin, a protein with a centrally located domain homologous to guanosine triphosphatase–activating protein (GAP), similar to other tumor suppressor gene products.75,76 The GAP domain of neurofibromin colocalizes with cytoplasmic microtubules, and it has been postulated that neurofibromin may have a regulatory role in the development of various connective tissues, including vascular connective tissue, through an effect on microtubular function. In a mouse model of mutations in genes for GAP and neurofibromatosis type 1, Henkemeyer and colleagues demonstrated thinning and rupture of large and medium-sized arteries during embryonic development.77 The GAP domain of neurofibromin, however, encompasses only about 10% of the protein, and neurofibromin may have a variety of undiscovered functions.
Bicuspid Aortic Valve
A bicuspid aortic valve (BAV) is one of the most common forms of congenital heart disease in adults and is found in 1% to 2% of the population.78 Many patients with BAV remain asymptomatic throughout life, but aortic valve insufficiency or stenosis eventually develops in most patients. Familial clustering of BAV has been described, and screening for BAV is generally recommended for first-degree relatives. Mutations in NOTCH1 have been reported in some families with BAV, but BAV is probably genetically heterogeneous. Cystic medial necrosis of the aorta is found on microscopic examination in most patients with BAV, aortic root dilation is present in at least half the patients with BAV, and aortic dissection is the cause of death in about 5% of patients with BAV. In the past, these aortic abnormalities were believed to be due to postvalvular hemodynamic changes, but it has become well established that the aortic changes are primarily related to an underlying arteriopathy, with hemodynamic factors playing a secondary role. Therefore, BAV has been included among the heritable disorders of connective tissues, along with Marfan’s syndrome and Ehlers-Danlos syndrome. The arteriopathy of BAV does not appear to be confined to the aorta, and spontaneous dissection of the cervical and intracranial arteries has been reported in patients with BAV as well. In one recent study, intracranial aneurysms were detected in 6 of 61 patients with BAV (10%) and in 3 of 291 controls (1%).79
Familial Intracranial Aneurysms
With the exception of ADPKD and, rarely, Ehlers-Danlos syndrome type IV, Pompe’s disease, or syndromes characterized by idiopathic nonarteriosclerotic cerebral calcifications, familial intracranial aneurysms have not been associated with any of the known heritable connective tissue disorders. The familial aggregation of intracranial aneurysms was first described in 1954 by Chambers and associates.80 Since then, hundreds of families have been reported. During the past 2 decades, interest in familial intracranial aneurysms has been renewed. Several studies have been focused on their epidemiologic features, clinical characteristics, and presymptomatic detection with noninvasive screening methods.
Epidemiology
Familial intracranial aneurysms are much more common than has generally been appreciated. Four epidemiologic studies have examined the frequency of familial intracranial aneurysms and revealed that 7% to 20% of patients with aneurysmal SAH had first- or second-degree relatives with intracranial aneurysms (Table 359-3).81–84 However, this familial aggregation could have been fortuitous because at least 1% of adults harbor intracranial aneurysms and most of the reported families have included only two affected members. Whether relatives of patients with intracranial aneurysms have an increased risk for the development of SAH was therefore unknown.
TABLE 359-3 Frequency of Familial Intracranial Aneurysms in Patients with Aneurysmal Subarachnoid Hemorrhage
[/not-level-membership-for-neurosurgery-category]
