CHAPTER 393 Genetics of Cerebral Cavernous Malformations
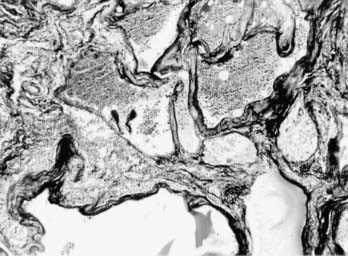
Cerebral cavernous malformations (CCMs) are some of the most common types of vascular malformations of the central nervous system (CNS), and they affect nearly 1 in 200 people. CCM lesions are characterized by grossly dilated vascular channels lined by a single layer of endothelium (Fig. 393-1).1 They typically affect the brain and spinal cord but may also involve the skin and retinal vasculature.2 CCMs lack normal vessel wall elements such as smooth muscle and are also devoid of intervening normal parenchyma.3
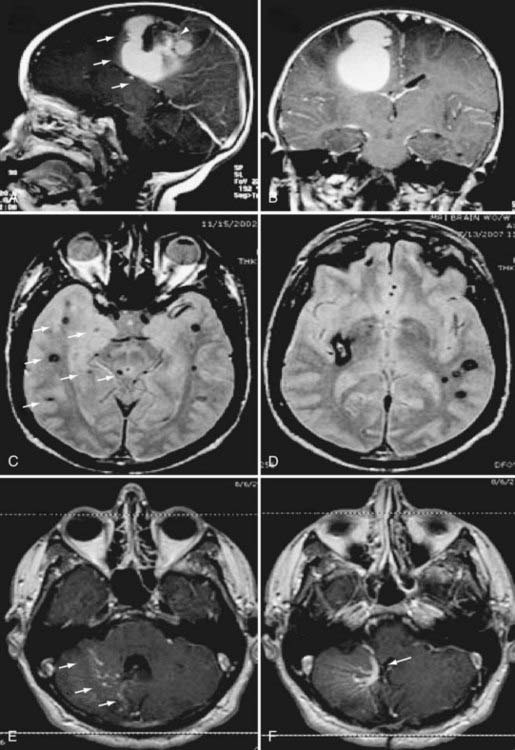
The CCM population prevalence of 0.5% is an estimate based on both magnetic resonance imaging (MRI) and autopsy studies. Although fairly common in the general population, only 20% to 30% of CCMs become symptomatic.4–7 Patients are typically seen initially in the third through fifth decades of life because of hemorrhagic stroke, headache, seizures, or focal neurological deficits.5 As a result of their rudimentary vessel wall and the low-pressure blood flow through the lesions, CCMs tend to “leak” and cause microhemorrhages as evidenced by a characteristic hemosiderin ring seen on MRI (Fig. 393-2).3 CCM lesions can also lead, albeit rarely, to gross macrohemorrhages causing clinical signs and symptoms reflecting the size and location of the bleeding. Catastrophic hemorrhages are much rarer, again being mainly dependent on the location. Treatment options for CCMs include observation for asymptomatic lesions, antiepileptic medication for seizures, and surgical excision of accessible lesions in patients who suffer from symptomatic hemorrhage or intractable seizures.8,9
Both sporadic and familial forms of CCM exist. The familial form is usually manifested as multiple lesions in the setting of a strong family history of neurological disease. Sporadic forms almost never involve more than two lesions, and a family history is absent. The heritable nature of CCMs has been well recognized since its original description10; however, the limited neuroradiologic data available to early investigators resulted in an incomplete appreciation for affected family members. The advent of MRI has led to the detection of asymptomatic CCMs, and in 1988, Rigamonti and coauthors reported a series of 13 Hispanic American families with CCM, complete with clinical and radiographic features,11 a study complemented in 1998 by the first large series of white, non-Hispanic patients with familial CCM.12 These and other studies have demonstrated that although only 10% to 20% of affected white individuals have a first-degree relative with the disorder, this figure is as high as 50% in Hispanic Americans, a population in which our group has demonstrated a strong founder mutation.10,13
It is now established that the genetic mode of inheritance of familial CCM is autosomal dominant with a high degree of penetrance, thus rendering it an ideal genetic model for human hemorrhagic stroke. Genetic linkage analyses have mapped three CCM loci to chromosomes 7q (CCM1), 7p (CCM2), and 3q (CCM3).14–19 All three causative genes have now been identified, which has allowed new insight into the pathophysiology of CCMs.
Molecular Genetic Approaches
In parametric linkage analysis, a single extended family pedigree, large enough to support linkage independently, is analyzed. A number of DNA markers, in the form of single nucleotide polymorphisms (SNPs) or short tandem repeats, of known location dispersed throughout the entire human genome are subsequently used to investigate the inheritance of polymorphic regions of the genome by statistically computing recombination frequencies. For each marker, evidence of linkage is sought by using cosegregation of the trait (and presumably the disease-causing gene) and a particular variant of the marker being analyzed. This is possible because the closer the disease and marker loci are located on a chromosome, the less likely it is that a recombination event between them will occur and the more likely that the specific marker variant will be present in all individuals who are affected with the disease. Thus, the genetic distance between the marker locus and the disease-causing gene can be estimated in the genetic unit of morgan (M), with one-one hundredth morgan (1 cM) corresponding to a recombination fraction of 1%.20
Statistically, the logarithm of the odds (LOD) score function is used to test for significance of the linkage21 and is a robust measure of its strength. The LOD score represents the logarithm of the odds that the DNA marker locus is linked to the disease trait and is calculated according to the following formula:

The availability of new molecular genetic technologies, including high-density SNP arrays, allowed the development of new methodology, such as CNV analysis. Recent studies using the CNV approach have allowed identification of segments of DNA in the genome ranging from kilobases to megabases in length that are lost (deleted) or gained (duplicated).22 Although CNVs are abundantly present and distributed throughout the human genome, thus suggesting that many benign variations exist,23 it is becoming evident that an increased burden of CNVs is observed in disease states.24–28 We and others have successfully applied this technology to identify rare disease-causing alleles in CCM.29,30
Genetics of Cerebral Cavernous Malformations
Familial CCM is an autosomal dominant disease with high, but incomplete penetrance. It should also be kept in mind, however, that mutations in one of the three identified CCM loci are evident in only 96% of familial CCM cases.12 Thus, the possibility of a fourth, to date unidentified CCM locus should not be excluded. The disease that ensues from mutations in the three loci is clinically and radiographically very similar.
The three CCM genes thus far identified are Krev1 interaction trapped protein-1 (KRIT1) located on chromosome 7q as the CCM1 gene, MGC4607 (or malcavernin) on chromosome 7p as the CCM2 gene, and programmed cell death-10 (PDCD10) on chromosome 3q as the CCM3 gene.31–38 The following sections focus on the molecular biology of these three genes.
CCM1
The CCM1 gene is located on chromosome 7q21 and contains 16 exons that encode a 736–amino acid protein, KRIT1. This is the first CCM gene that was identified and, consequently, has been the most studied in terms of its cellular function. Mutational analysis of this gene has revealed a founder mutation (C742T) that is responsible for more than 90% of familial cases in Hispanic American patients.13,32,33 Numerous studies have now identified in excess of 80 different mutations of this gene leading to clinically evident CCM. Nearly all are nonsense mutations in the form of stop codons or frameshift mutations that result in premature termination of translation.33,35,39–43 Thus, there is strong evidence to suggest that loss of function of the CCM1 protein leads to disease, which probably results from a two-hit mechanism.44 In the two-hit model, the first genetic “hit” is inherited, whereas the second is acquired in a subset of cells and rapidly leads to disease afterward because affected cells can no longer synthesize functionally intact proteins.
Krit1 was initially identified in a yeast two-hybrid screen as an interactor of the small Ras family guanosine triphosphatase Krev1/Rap1a.45 Even though later studies challenged this finding,46,47 more recent studies have confirmed this interaction, thus suggesting that Rap1a signaling might be important in CCM pathophysiology.48,49
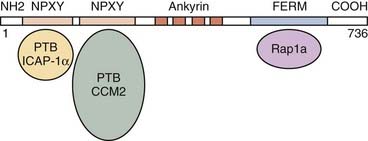
The KRIT1/CCM1 protein contains a number of domains implicated in protein-protein interactions, including a FERM domain at the C-terminal, which is typically critical for binding to the cell membrane.50 Recent studies have suggested this domain’s importance in CCM1’s binding cell-cell junctional proteins (see later).48,49 KRIT1 also contains a series of ankyrin repeats, but the proteins that interact with CCM1 through this domain remain unknown.45 Finally, CCM1 contains three NPXY domains that are known to interact with phosphotyrosine-binding (PTB) regions. Two of these domains create a binding site for integrin cytoplasmic domain–associated protein-1α (ICAP-1α)46,47 and CCM2 (see later) (Fig. 393-3).51
Our group was the first to demonstrate that CCM1 interacts with the plus end of microtubules.52 This finding was later confirmed by Beraud-Dufour and colleagues, who also demonstrated that CCM1 can adopt both an open and a closed conformation, depending on the interaction of the N-terminal NPXY motif with its C-terminal.48 In vitro, CCM1 interacts with microtubules in its closed conformation, which would in turn help deliver CCM1 to the plasma membrane, where it would bind to ICAP-1α in its open conformation. Even though both the small G-protein Rap1a and ICAP-1α can disrupt CCM1’s association with microtubules, thereby localizing it to endothelial cell-cell junctions, only ICAP-1α appears to be able to unfold CCM1.48
ICAP-1α is known to bind to β1 integrin, again through an NPXY/PTB domain interaction.53,54 Integrins, specifically β1 integrin, are important proteins that regulate endothelial cell adhesion and migration throughout angiogenesis.55,56 These observations imply a role for CCM1 in angiogenesis with its involvement in the Rap1a/Krev1 and integrin pathways. More recent studies suggested that after localizing to the cell membrane, CCM1 can also bind to β-catenin and play a role in stabilizing adherens cell junctions and regulating endothelial cell permeability.49
In addition, CCM1 binds to both CCM2 and CCM3.51,57 The exact nature of the interaction between the three CCM molecules is currently unknown, but among these three proteins, CCM2 appears to act as a scaffolding protein58 and is potentially recruited to cell-cell junctions through its interactions with CCM1.
Even though the molecules involved in the downstream signaling of CCM molecules are unknown, in vitro studies previously suggested that CCM1 translocates to the nucleus upon cellular stimulation,51 and recent work in Caenorhabditis elegans confirmed this finding by showing interaction of the CCM1 ortholog kri-1 with a transcription factor daf16.59 Mutations in kri-1 result in increased life span, mediated though its interactions with daf16. The importance of this aspect of CCM signaling in humans is currently unknown, but dissecting this molecular pathway will have important implications in understanding disease pathophysiology.
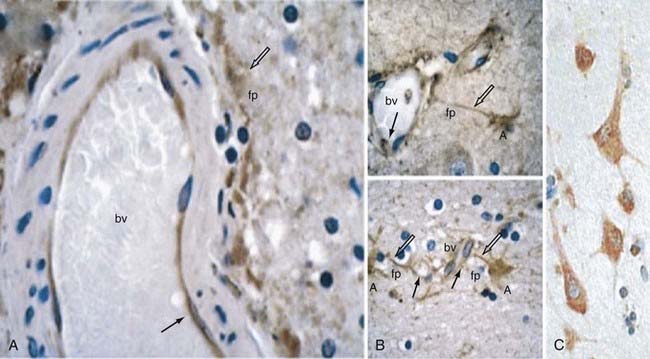
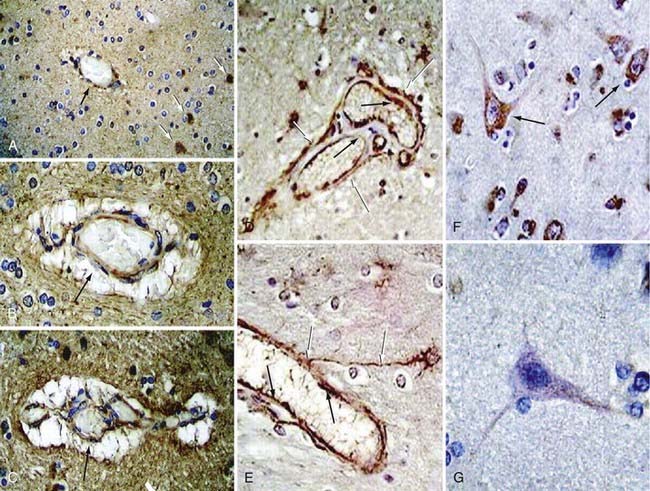
In an attempt to gain some insight into CCM pathophysiology, our group focused on the temporal and spatial expression patterns of the three CCM molecules. Our initial studies demonstrated CCM1 to be expressed in many human tissues, including the heart, brain, kidney, liver, pancreas, and spleen.60 Furthermore, CCM1 is expressed specifically in the endothelium of capillaries and arterioles, but not in the venous system.61 CCM1 (but also CCM2 and CCM3; see later) is abundantly expressed in neurons and astrocytic foot processes (Fig. 393-4), an observation potentially related to the finding that CCM lesions almost exclusively form within the CNS.61
In vivo studies based on mouse models have revealed an essential role for Ccm1 in vascular development: complete loss of Ccm1 results in embryonic lethality at midgestation.62 Ccm1−/− embryos demonstrate vascular defects incompatible with life, namely, dilated precursor brain vessels, an enlarged caudal dorsal aorta (probably caused by increased endothelial proliferation), and variable obstruction of branchial arch arteries and the proximal dorsal aorta.62 All these findings suggest a primary defect in arterial morphogenesis. Ccm1+/− animals are phenotypically normal with no apparent vascular phenotype and do not form CCM lesions. Interestingly, when these Ccm1+/− animals are crossed with p53−/− mice, vascular lesions resembling cavernous malformations in the brain develop in approximately 55% of the compound Ccm1+/−;p53−/− mutants.63 The mechanism underlying the enhanced phenotype in the absence of p53 is not known but is speculated to be an increased rate of somatic mutations leading to more second hits and thereby resulting in the formation of CCM lesions in the compound animals.
When viewed as a whole, these studies are beginning to shed light on the cellular function of the CCM1 protein and its involvement in angiogenesis. CCM1 probably exerts the majority of its effects at the level of the cerebrovascular unit, where it mediates communication between its components, neural cells, and vascular endothelium of the CNS (Fig. 393-5).
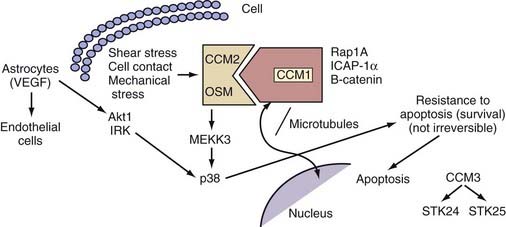
CCM2
In 2003, Liquori and colleagues identified MGC4607 (or malcavernin) as the causative gene in families linked to the CCM2 locus.30 MGC4607 is a 10-exon gene located on chromosome 7p13 that encodes the protein CCM2, or malcavernin.37 Similar to CCM1, almost all mutations identified to date in CCM2 are frameshift mutations or result in stop codons, again implying a loss-of-function mechanism, presumably via a two-hit model.
The CCM2 protein contains a putative PTB domain, also found in ICAP-1α, CCM1’s binding partner. Although binding of CCM1 to both ICAP-1α and CCM2 is mediated via PTB/NPXY domain interactions, the NPXY domains that interact with these two molecules have been shown to be distinct, thus potentially indicating concomitant effects of ICAP-1α and CCM2 on CCM1.51
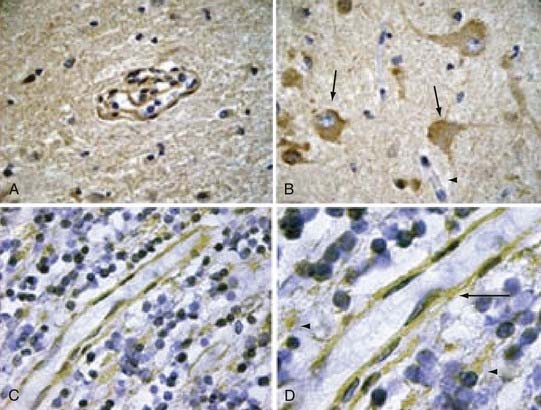
Interestingly, the temporal and spatial pattern of CCM2 expression parallels that of CCM1. CCM2, like CCM1, is expressed by arterial endothelium, neurons, and astrocytes (Fig. 393-6).64 This expression pattern supported the hypothesis that the two proteins may be involved in a common pathway. It should also be underscored that both proteins are restricted to the arterial endothelium, with exclusion from the venous circulation. Given that histologically CCMs are abnormally dilated microvessels, this finding is somewhat unexpected but has now been confirmed by in vivo studies as well.62,65
Considerable homology exists between CCM2 and the rodent protein OSM, which is involved in osmosensing and mechanosensing of the extracellular matrix.58 Like OSM, CCM2 has been shown to function as a scaffolding protein and signals through p38, a pathway that is involved in environmental stress signaling and plays a major role during angiogenesis.58,66 More recent in vitro assays have further elucidated this pathway and showed that CCM2, when overexpressed, has the potential to sequester CCM1 and form a complex with MAP kinase kinase kinase 3 (MEKK3).51 These results are the beginning of our understanding of CCM pathophysiology and imply that CCM2, as well as possibly CCM1 through interactions with CCM2, may assert its influence via the p38 pathway and could lead to disease by perturbing CNS angiogenesis.
In vivo studies in zebrafish and mice have demonstrated further parallels between Ccm1 and Ccm2. In mice, similar to Ccm1, complete loss of Ccm2 is embryonically lethal as a result of vascular defects.65 Vascular lesions reminiscent of small cavernous malformations develop in approximately 10% of Ccm2+/− animals.65 Ccm2 inactivation in the endothelium causes cardiovascular pathology; however, both neural and smooth musclespecific conditional mutants appear normal.67–69 In zebrafish, recessive mutations of either the Ccm1 or Ccm2 orthologs santa and valentine result in a lethal cardiac phenotype with massive dilation of the cardiac chamber, apparently secondary to failure to generate appropriate concentric thickness of the myocardial wall.70
CCM3
The CCM3 gene was discovered in 2005 by Bergametti and colleagues to be PDCD10.34,36 Because it is the most recently discovered, this gene and its encoded protein are the least studied and understood. PDCD10 is a seven-exon gene located on 3q26 that encodes a 212–amino acid protein. Similar to CCM1 and CCM2, identified mutations of the CCM3 gene lead to stop codons and frameshifts and result in truncated protein products. In silico analyses have failed to demonstrate any known functional domains for CCM3.
Our group investigated the expression pattern of CCM3 in embryonic and postnatal mouse brain and in various human organs. Similar to CCM1 and CCM2, the CCM3 protein is expressed strongly in neurons, astrocytes, and arterial endothelium.71 CCM3 is either weakly present in the venous circulation or completely absent, thus underscoring the fact that CCM lesions are a result of arterial pathology (Fig. 393-7).
Little is known regarding the function of CCM3. It was first identified as being upregulated on deprivation of growth factor and induction of apoptosis in the TF-1 premyeloid cell line, as well as in fibroblast cell lines, which suggested a role for the protein in apoptotic pathways.72 In vitro, CCM3 appears to be both necessary and sufficient to induce apoptosis: overexpression of CCM3 (but not of mutated, biologically inactive forms of the protein identified in CCM patients) elicits activation of caspase 3 and cell death. Moreover, in the presence of proapoptotic stimuli, CCM3 levels are increased concomitantly with levels of activated caspase 3 and p38.71
An important discovery came from Voss and colleagues, who showed a potential link between the CCM3 and CCM2 proteins: in a yeast two-hybrid system, CCM3 binds to and is phosphorylated by serine/threonine kinase 25 (STK25), a molecule that also forms a complex with CCM2. Furthermore, CCM3 colocalizes and coprecipitates with CCM2. Interestingly, in the presence of CCM1, the amount of CCM3 immunoprecipitated with CCM2 increased.57 Hilder and coworkers hypothesized that the three CCM proteins are scaffold- or adaptor-type proteins that organize a large macromolecular complex and, with the use of a proteomics approach, demonstrated that the three proteins participate in a larger signaling complex.66 Taken together, these results pointed for the first time to an interaction among the three different CCM proteins by showing that CCM3 may be a part of the CCM1/CCM2 protein complex via its interactions with CCM2. This complex would have the potential to participate and influence integrin-mediated signaling and the p38 pathway and could significantly alter endothelial cell adhesion and migration during angiogenesis with resulting cavernous malformations. Global or endothelial deletion of Ccm3 causes embryonic lethality associated with defects in vascular development and VEGFR2 signaling,73 and in zebrafish, CCM3-mediated signaling through Ste20-like kinases is involved in cardiovascular development.74
Clinical Genetics
All three CCM genes have varying degrees of clinical penetrance. Studies estimate this penetrance to be approximately 70% for CCM1, 40% for CCM2, and 60% for CCM3.75 Thus, clinical disease will develop in different proportions of patients with mutations for each gene.
Among these patients, types of clinical manifestations might also vary. For example, hemorrhage rates might be lowest in the CCM1 group76 and highest in the CCM3 patients, particularly during childhood.75 Even though CCM1 patients may have a more benign course, there is evidence to support the fact that more lesions develop as they age.75 These early studies are beginning to demonstrate differences in the clinical behavior and radiographic features of the three CCM groups. To further complicate the issue, it is also known that the same mutation that segregates within a large family can lead to different disease manifestations, with some members of the family suffering from progressive bleeding and others remaining asymptomatic all their lives. These observations prove the complexity of the genetics of CCM and suggest that other modifier genes or yet unidentified environmental risk factors (or both) can modify progression of the disease. Further studies are currently aimed at understanding these genetic and nongenetic risk factors. Such studies may ultimately reveal certain subgroups of patients who harbor risk factors that render their lesions more prone to rupture and therefore may need to be treated more aggressively.
Conclusion and Future Directions
Because the families that harbor mutations in the three CCM genes are clinically and radiographically virtually indistinguishable, we had previously hypothesized that all three genes encode proteins that are involved in common or parallel pathways. In support of this hypothesis, our group has demonstrated that CCM1, CCM2, and CCM3 are very similar in their temporal and spatial expression pattern, specifically, in neurons, astrocytes, and arterial endothelium, with little, if any involvement of the venous circulation.61,64,71 Support of this theory also comes from in vivo studies in mice and zebrafish, which have shown that defects in Ccm1 and Ccm2 lead to similar lethal problems involving early angiogenesis.62,65,70
In addition, in vitro analyses have revealed CCM1 to bind to microtubules and travel between the cell membrane, where it interacts with molecules involved in cell adhesion48,52 and the nucleus. CCM1 also binds to CCM2, which acts as a scaffolding protein, and signals through the p38 mitogen-activated protein kinase signaling pathway, known to be important in diverse cellular processes, including the stress response, proliferation, apoptosis, and differentiation. More recent evidence adds CCM3 to the CCM1/CCM2 complex, thus lending further support to the hypothesis that all three CCM proteins are involved in the same pathway.57 Initial insight into CCM3 function suggests that its signaling is important within the neurovascular unit of the CNS and that it potentially affects cellular proliferation and apoptosis.51,66
Beraud-Dufour S, Gautier R, Albiges-Rizo C, et al. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 2007;274:5518-5532.
Bergametti F, Denier C, Labauge P, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42-51.
Clatterbuck RE, Eberhart CG, Crain BJ, et al. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. 2001;71:188-192.
Craig HD, Gunel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851-1858.
Denier C, Labauge P, Bergametti F, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550-556.
Dubovsky J, Zabramski JM, Kurth J, et al. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet. 1995;4:453-458.
Gault J, Sain S, Hu LJ, et al. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59:1278-1284.
Gunel M, Awad IA, Anson J, et al. Mapping a gene causing cerebral cavernous malformation to 7q11.2-q21. Proc Natl Acad Sci U S A. 1995;92:6620-6624.
Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med. 1996;334:946-951.
Gunel M, Laurans MS, Shin D, et al. Krit1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 2002;99:10677-10682.
Guzeloglu-Kayisli O, Amankulor NM, Voorhees J, et al. Krit1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. Neurosurgery. 2004;54:943-949.
Hayman LA, Evans RA, Ferrell RE, et al. Familial cavernous angiomas: natural history and genetic study over a 5-year period. Am J Med Genet. 1982;11:147-160.
Hilder TL, Malone MH, Bencharit S, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343-4355.
Labauge P, Laberge S, Brunereau L, et al. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 french families. Societe Française de Neurochirurgie. Lancet. 1998;352:1892-1897.
Liquori CL, Berg MJ, Siegel AM, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459-1464.
Plummer NW, Gallione CJ, Srinivasan S, et al. Loss of p53 sensitizes mice with a mutation in ccm1 (krit1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509-1518.
Plummer NW, Squire TL, Srinivasan S, et al. Neuronal expression of the ccm2 gene in a new mouse model of cerebral cavernous malformations. Mamm Genome. 2006;17:119-128.
Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319:343-347.
Seker A, Pricola KL, Guclu B, et al. CCM2 expression parallels that of CCM1. Stroke. 2006;37:518-523.
Tanriover G, Boylan AJ, Diluna ML, et al. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery. 2008;62:930-938.
Voss K, Stahl S, Schleider E, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249-256.
Whitehead KJ, Plummer NW, Adams JA, et al. CCM1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437-1448.
Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
Zawistowski JS, Stalheim L, Uhlik MT, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521-2531.
1 Russell DS, Rubenstein LJ. Pathology of Tumors of the Nervous System. Baltimore: Williams & Wilkins; 1989.
2 Gass JD. Cavernous hemangioma of the retina. A neuro-oculo-cutaneous syndrome. Am J Ophthalmol. 1971;71:799-814.
3 Clatterbuck RE, Eberhart CG, Crain BJ, et al. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. 2001;71:188-192.
4 Otten P, Pizzolato GP, Rilliet B, et al. [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies.]. Neurochirurgie. 1989;35:82-83. 128-131
5 Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709-714.
6 Sage MR, Brophy BP, Sweeney C, et al. Cavernous haemangiomas (angiomas) of the brain: clinically significant lesions. Australas Radiol. 1993;37:147-155.
7 Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
8 Maraire JN, Awad IA. Intracranial cavernous malformations: lesion behavior and management strategies. Neurosurgery. 1995;37:591-605.
9 Robinson JRJr, Awad IA, Magdinec M, et al. Factors predisposing to clinical disability in patients with cavernous malformations of the brain. Neurosurgery. 1993;32:730-735.
10 Hayman LA, Evans RA, Ferrell RE, et al. Familial cavernous angiomas: natural history and genetic study over a 5-year period. Am J Med Genet. 1982;11:147-160.
11 Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319:343-347.
12 Labauge P, Laberge S, Brunereau L, et al. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 french families. Societe Française de Neurochirurgie. Lancet. 1998;352:1892-1897.
13 Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med. 1996;334:946-951.
14 Dubovsky J, Zabramski JM, Kurth J, et al. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet. 1995;4:453-458.
15 Gunel M, Awad IA, Anson J, et al. Mapping a gene causing cerebral cavernous malformation to 7q11.2-q21. Proc Natl Acad Sci U S A. 1995;92:6620-6624.
16 Gil-Nagel A, Dubovsky J, Wilcox KJ, et al. Familial cerebral cavernous angioma: a gene localized to a 15-cM interval on chromosome 7q. Ann Neurol. 1996;39:807-810.
17 Notelet L, Chapon F, Khoury S, et al. Familial cavernous malformations in a large French kindred: mapping of the gene to the CCM1 locus on chromosome 7q. J Neurol Neurosurg Psychiatry. 1997;63:40-45.
18 Johnson EW, Iyer LM, Rich SS, et al. Refined localization of the cerebral cavernous malformation gene (CCM1) to a 4-cM interval of chromosome 7q contained in a well-defined yac contig. Genome Res. 1995;5:368-380.
19 Craig HD, Gunel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851-1858.
20 Vink JM, Boomsma DI. Gene finding strategies. Biol Psychol. 2002;61:53-71.
21 Morton NE. Sequential tests for the detection of linkage. Am J Hum Genet. 1955;7:277-318.
22 Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525-528.
23 Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444-454.
24 Consortium IS. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237-241.
25 Ferreira MA, O’Donovan MC, Meng YA, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1c in bipolar disorder. Nat Genet. 2008;40:1056-1058.
26 Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene–containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434-1440.
27 O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053-1055.
28 Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24-26.
29 Bilguvar K, Bydon M, Bayrakli F, et al. A novel syndrome of cerebral cavernous malformation and Greig cephalopolysyndactyly. Laboratory investigation. J Neurosurg. 2007;107:495-499.
30 Liquori CL, Berg MJ, Siegel AM, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459-1464.
31 Sahoo T, Goenaga-Diaz E, Serebriiskii IG, et al. Computational and experimental analyses reveal previously undetected coding exons of the KRIT1 (CCM1) gene. Genomics. 2001;71:123-126.
32 Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a Krev-1/Rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325-2333.
33 Laurans MS, DiLuna ML, Shin D, et al. Mutational analysis of 206 families with cavernous malformations. J Neurosurg. 2003;99:38-43.
34 Bergametti F, Denier C, Labauge P, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42-51.
35 Laberge-le Couteulx S, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189-193.
36 Guclu B, Ozturk AK, Pricola KL, et al. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery. 2005;57:1008-1013.
37 Denier C, Goutagny S, Labauge P, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326-337.
38 Liquori CL, Berg MJ, Siegel AM, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459-1464.
39 Verlaan DJ, Davenport WJ, Stefan H, et al. Cerebral cavernous malformations: mutations in KRIT1. Neurology. 2002;58:853-857.
40 Couteulx SL, Brezin AP, Fontaine B, et al. A novel KRIT1/CCM1 truncating mutation in a patient with cerebral and retinal cavernous angiomas. Arch Ophthalmol. 2002;120:217-218.
41 Chen DH, Lipe HP, Qin Z, et al. Cerebral cavernous malformation: novel mutation in a chinese family and evidence for heterogeneity. J Neurol Sci. 2002;196:91-96.
42 Davenport WJ, Siegel AM, Dichgans J, et al. CCM1 gene mutations in families segregating cerebral cavernous malformations. Neurology. 2001;56:540-543.
43 Zhang J, Clatterbuck RE, Rigamonti D, et al. Mutations in KRIT1 in familial cerebral cavernous malformations. Neurosurgery. 2000;46:1272-1277.
44 Gault J, Shenkar R, Recksiek P, et al. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005;36:872-874.
45 Serebriiskii I, Estojak J, Sonoda G, et al. Association of Krev-1/Rap1a with KRIT1, a novel ankyrin repeat–containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15:1043-1049.
46 Zhang J, Clatterbuck RE, Rigamonti D, et al. Interaction between KRIT1 and ICAP1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001;10:2953-2960.
47 Zawistowski JS, Serebriiskii IG, Lee MF, et al. KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet. 2002;11:389-396.
48 Beraud-Dufour S, Gautier R, Albiges-Rizo C, et al. KRIT 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 2007;274:5518-5532.
49 Glading A, Han J, Stockton RA, et al. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell-cell junctions. J Cell Biol. 2007;179:247-254.
50 Chishti AH, Kim AC, Marfatia SM, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281-282.
51 Zawistowski JS, Stalheim L, Uhlik MT, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521-2531.
52 Gunel M, Laurans MS, Shin D, et al. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 2002;99:10677-10682.
53 Zhang XA, Hemler ME. Interaction of the integrin beta1 cytoplasmic domain with ICAP-1 protein. J Biol Chem. 1999;274:11-19.
54 Chang DD, Wong C, Smith H, et al. ICAP-1, a novel beta1 integrin cytoplasmic domain–associated protein, binds to a conserved and functionally important NPXY sequence motif of beta1 integrin. J Cell Biol. 1997;138:1149-1157.
55 Brooks PC. Role of integrins in angiogenesis. Eur J Cancer. 1996;32A:2423-2429.
56 Bloch W, Forsberg E, Lentini S, et al. Beta 1 integrin is essential for teratoma growth and angiogenesis. J Cell Biol. 1997;139:265-278.
57 Voss K, Stahl S, Schleider E, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249-256.
58 Uhlik MT, Abell AN, Johnson NL, et al. RAC-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104-1110.
59 Berman JR, Kenyon C. Germ-cell loss extends C. elegans life span through regulation of DAF-16 by KRI-1 and lipophilic-hormone signaling. Cell. 2006;124:1055-1068.
60 Guzeloglu-Kayisli O, Kayisli UA, Amankulor NM, et al. Krev1 interaction trapped-1/cerebral cavernous malformation-1 protein expression during early angiogenesis. J Neurosurg. 2004;100:481-487.
61 Guzeloglu-Kayisli O, Amankulor NM, Voorhees J, et al. KRIT1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. Neurosurgery. 2004;54:943-949.
62 Whitehead KJ, Plummer NW, Adams JA, et al. CCM1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437-1448.
63 Plummer NW, Gallione CJ, Srinivasan S, et al. Loss of p53 sensitizes mice with a mutation in ccm1 (krit1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509-1518.
64 Seker A, Pricola KL, Guclu B, et al. CCM2 expression parallels that of CCM1. Stroke. 2006;37:518-523.
65 Plummer NW, Squire TL, Srinivasan S, et al. Neuronal expression of the ccm2 gene in a new mouse model of cerebral cavernous malformations. Mamm Genome. 2006;17:119-128.
66 Hilder TL, Malone MH, Bencharit S, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343-4355.
67 Whitehead KJ, Chan AC, Navankasattusas S, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177-184.
68 Boulday G, Blécon A, Petit N, et al. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech. 2009;2:168-177.
69 Kleaveland B, Zheng X, Liu JJ, et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15:169-176.
70 Mably JD, Chuang LP, Serluca FC, et al. Santa and Valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133:3139-3146.
71 Tanriover G, Boylan AJ, Diluna ML, et al. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery. 2008;62:930-938.
72 Wang YG, Liu HT, Zhang YM, et al. CDNA cloning and expression of an apoptosis-related gene, human TFAR-15 gene. Sci China C Life Sci. 1999;42:323-329.
73 He Y, Zhang H, Yu L, Gunel M, et al. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010 Apr;3:ra26.
74 Zheng X, Xu C, Di Lorenzo A, Kleaveland B, et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J Clin Invest. 2010;120:2795-2804. doi: 10.1172/JCI39679
75 Denier C, Labauge P, Bergametti F, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550-556.
76 Gault J, Sain S, Hu LJ, et al. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59:1278-1284.






