Chapter 54 Genetics and Cardiac Arrhythmia Syndromes
Cardiac arrhythmias are major causes of morbidity and mortality, including sudden cardiac death (SCD). SCD in the United States occurs with a reported incidence of more than 300,000 persons per year.1 Although coronary heart disease is a major cause of death, other etiologies contribute to this problem. In many of these non–ischemia-related cases, autopsies are unrevealing. Interest in identifying the underlying cause of the death in these instances has been focused on cases of unexpected arrhythmogenic death, which is estimated to represent 5% of all SCDs. In cases in which no structural heart disease can be identified, long QT syndrome (LQTS), ventricular pre-excitation (Wolff-Parkinson-White syndrome), and idiopathic ventricular fibrillation (IVF) or Brugada syndrome (characterized by ST-segment elevation in the right precordial leads with or without right bundle branch block [RBBB]) are most commonly considered as likely causes.1–3 Another important disease in which arrhythmias are believed to play a central role is sudden infant death syndrome (SIDS), a disorder with no structural abnormalities.4
Arrhythmogenic right ventricular dysplasia (ARVD) is also a significant cause of SCD and is considered a primary electrical disease despite being associated with fibrosis and fatty infiltration of the right ventricle.5 The arrhythmias associated with ARVD also occur in other disorders in which structurally normal myocardium is seen, such as catecholaminergic ventricular tachycardia (VT).6
This chapter describes the current understanding of the clinical and molecular genetic aspects of inherited diseases in which arrhythmias are prominent features. The discussion should serve as an introduction and overview to these conditions. Newer disorders are rapidly being added to this list and are discussed in more detail elsewhere in this textbook. A more detailed treatment of the current state of knowledge regarding the molecular basis and basic electrophysiological mechanisms of inherited arrhythmia syndromes is discussed in Chapters 6 and 7. Individual clinical syndromes are discussed in Chapters 62 to 65.
Long QT Syndrome
Clinical Description
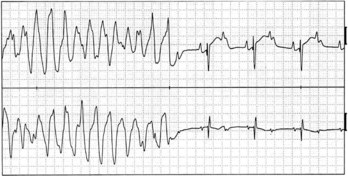
LQTS is an inherited or acquired disorder of repolarization identified by the electrocardiographic abnormalities of prolongation of the Q-T interval corrected for heart rate (QTc), usually above 460 to 480 ms; relative bradycardia; T-wave abnormalities (Figure 54-1); and episodic ventricular tachyarrhythmias, particularly torsades de pointes (Figure 54-2).7 The inherited form of LQTS is transmitted as an autosomal dominant or autosomal recessive trait. Acquired LQTS may be seen as a complication of various drug therapies or electrolyte abnormalities. Whether the abnormality is genetic or acquired, the clinical presentation is similar.1,7 The initial presentation of LQTS is heterogeneous and most commonly includes syncope, which, in many instances, is triggered by emotional stress, exercise, or auditory phenomena. Other presenting features include seizures or palpitations. SCD is the first symptom in some individuals, but some other cases are diagnosed by surface electrocardiogram (ECG) as a family screening evaluation necessitated by family history of LQTS or SCD.

Clinical Genetics
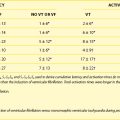
Two differently inherited forms of familial LQTS have been reported. Romano-Ward syndrome is the most common of the inherited forms of LQTS and appears to be transmitted as an autosomal dominant trait.8,9 In this disorder, the disease gene is transmitted to 50% of the offspring of an affected individual. However, low penetrance has been described, so gene carriers may, in fact, have no clinical features of disease.10 Individuals with Romano-Ward syndrome have the pure syndrome of prolonged Q-T interval on ECG, with the associated symptom complex of syncope, SCD, and, in some patients, seizures.11,12 Occasionally, other noncardiac abnormalities such as diabetes mellitus, asthma, or syndactyly may also be associated with QT prolongation.13–16 LQTS may also be involved in some cases of SIDS, which, in some cases, appear in several family members.5,17,18
Jervell and Lange-Nielsen syndrome (JLNS) is a relatively uncommon inherited form of LQTS. Classically, this disease has been described as having apparent autosomal recessive transmission.19–21 These patients have a clinical presentation identical to that in patients with Romano-Ward syndrome but also have associated sensorineural deafness. Clinically, patients with JLNS usually have longer Q-T intervals compared with individuals with Romano-Ward syndrome and also have a more malignant course. Priori and colleagues have reported autosomal recessive cases of Romano-Ward syndrome as well, thus changing one of the sine qua non of JLNS.22
Gene Identification in Romano-Ward Syndrome
KVLQT1 or KCNQ1: The LQT1 Gene
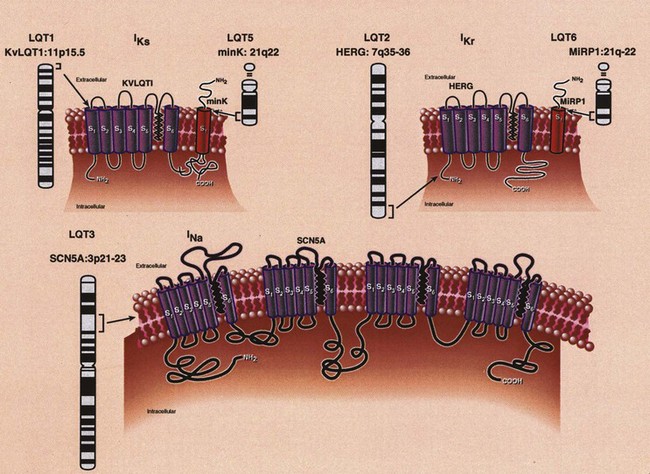
The first of the genes mapped for LQTS, termed LQT1, required 5 years from the time that mapping to chromosome 11p15.5 was first reported to gene cloning.23 This gene, originally named KVLQT1, but more recently called KCNQ1 (Table 54-1), is a novel potassium channel gene that consists of 16 exons, spans approximately 400 kb, and is widely expressed in human tissues, including the heart, inner ear, kidney, lung, placenta, and pancreas but not in the skeletal muscle, liver, or brain.24 Although most of the mutations are “private” (i.e., only seen in one family), at least one frequently mutated region (called a “hot spot”) of KVLQT1 exists.25–27 This gene is the most commonly mutated gene in LQTS.
Analysis of the predicted amino acid sequence of KVLQT1 suggests that it encodes a potassium channel α-subunit with a conserved potassium-selective pore-signature sequence flanked by six membrane-spanning segments similar to shaker-type channels (Figure 54-3).24,27–29 A putative voltage sensor is found in the fourth membrane-spanning domain (S4), and the selective pore loop is located between the fifth and sixth membrane-spanning domains (S5,S6). Biophysical characterization of the KVLQT1 protein confirmed that KVLQT1 is a voltage-gated potassium channel protein subunit, which requires coassembly with a β-subunit called minK to function properly.28,29 Expression of either KVLQT1 or minK alone results in either inefficient or no current development. When minK and KVLQT1 are co-expressed in either mammalian cell lines or Xenopus oocytes, however, the slowly activating potassium current (IKs) is developed in cardiac myocytes.28,29 The combination of normal and mutant KVLQT1 subunits forms abnormal IKs channels, and these mutations are believed to act through a dominant-negative mechanism (the mutant form of KVLQT1 interferes with the function of the normal wild-type form through a “poison pill” type mechanism) or a loss-of-function mechanism (only the mutant form loses activity).30
Because KVLQT1 and minK form a unit, mutations in minK could also be expected to cause LQTS. This fact was subsequently demonstrated (discussed below).31
HERG or KCNH2: The LQT2 Gene
The LQT2 gene was initially mapped to chromosome 7q35-36 by Jiang et al, and subsequently, candidate gene screening identified the disease-causing gene HERG (human ether-a-go-go-related gene), a cardiac potassium channel gene to be the LQT2 gene (see Table 54-1).27,32 HERG was originally cloned from a brain cDNA library and found to be expressed in neural crest–derived neurons, microglia, a wide variety of tumor cell lines, and the heart.33–37 LQTS-associated mutations were identified in HERG throughout the gene, including missense mutations, intragenic deletions, stop codons, and splicing mutations.27,37,38 Currently, this gene is thought to be the second most common gene mutated in LQTS (second to KVLQT1). As with KVLQT1, “private” mutations that are scattered throughout the entire gene without clustering preferentially are seen.
HERG consists of 16 exons and spans 55 kb of genomic sequence.37 The predicted topology of HERG (see Figure 54-3) is similar to that of KVLQT1. Unlike KVLQT1, HERG has extensive intracellular amino-and-carboxyl termini, with a region in the carboxyl-terminal domain having sequence similarity to nucleotide binding domains (NBDs).
Electrophysiological and biophysical characterization of expressed HERG in Xenopus oocytes established that HERG encodes the rapidly activating delayed rectifier potassium current IKr.39–41 Electrophysiological studies of LQTS-associated mutations showed that they act through either a loss of function or a dominant negative mechanism.41,42 In addition, protein trafficking abnormalities have been shown to occur.43,44 This channel has been shown to coassemble with β-subunits for normal function, similar to that seen in IKs. McDonald et al initially suggested that the complexing of HERG with minK is needed to regulate the IKr potassium current.45 Bianchi et al provided confirmatory evidence that minK is involved in the regulation of both IKs and IKr.46 Abbott et al identified MiRP1 as a β-subunit for HERG (discussed below).47
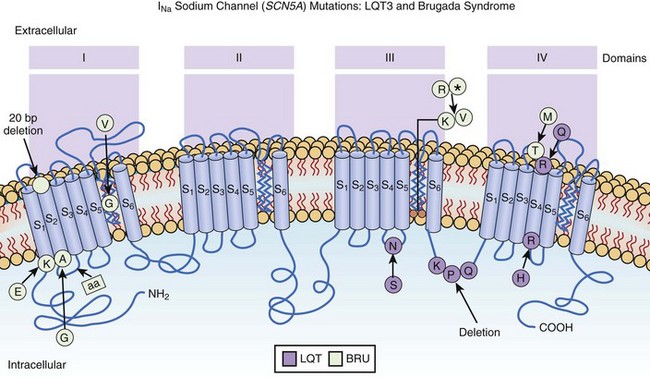
SCN5A: The LQT3 Gene
The positional candidate gene approach was also used to establish that the gene responsible for chromosome 3–linked LQTS (LQT3) is the cardiac sodium channel gene SCN5A (see Table 54-1).48,49 SCN5A is highly expressed in the human myocardium and brain but not in the skeletal muscle, liver, or uterus.50–52 It consists of 28 exons that span 80 kb and encodes a protein of 2016 amino acids with a putative structure that consists of four homologous domains (DI to DIV), each of which contains six membrane-spanning segments (S1 to S6) similar to the structure of the potassium channel α-subunits (see Figure 54-3).27,39 Linkage studies with LQT3 families and SCN5A initially demonstrated linkage to the LQT3 locus on chromosome 3p21-24, and multiple mutations were subsequently identified.50,51 Biophysical analysis of the initial three mutations were expressed in Xenopus oocytes, and it was found that all mutations generated a late phase of inactivation-resistant, mexiletine- and tetrodotoxin-sensitive whole-cell currents through multiple mechanisms.53,54 Two of the three mutations showed dispersed reopening after the initial transient current, but the other mutation showed both dispersed reopening and long-lasting bursts.54 These results suggested that SCN5A mutations act through a gain of function mechanism (the mutant channel functions normally, but with altered properties such as delayed inactivation) and that the mechanism of chromosome 3–linked LQTS is persistent non-inactivated sodium current in the plateau phase of the action potential. Later, An et al showed that not all mutations in SCN5A are associated with persistent current and demonstrated that SCN5A interacted with β-subunits.55
minK or KCNE1: The LQT5 Gene
minK (IsK or KCNE1) was initially localized to chromosome 21 (21q22.1) and found to consist of three exons that span approximately 40 kb (see Table 54-1). It encodes a short protein consisting of 130 amino acids and has only one transmembrane-spanning segment with small extracellular and intercellular regions (see Figure 54-3).30,31,56 When expressed in Xenopus oocytes, it produces potassium current that closely resembles the slowly activating delayed-rectifier potassium current IKs in cardiac cells.56,57 The fact that the minK clone was only expressed in Xenopus oocytes and not in mammalian cell lines raised the question whether minK is a human channel protein. With the cloning of KVLQT1 and the coexpression of KVLQT1 and minK in both mammalian cell lines and Xenopus oocytes, it became clear that KVLQT1 interacts with minK to form the cardiac slowly activating delayed rectifier IKs current.28,29 minK alone cannot form a functional channel but induces the IKs current by interacting with endogenous KVLQT1 protein in Xenopus oocytes and mammalian cells. Bianchi et al showed that mutant minK results in abnormalities of IKs, IKr as well as protein trafficking abnormalities.46 McDonald et al showed that minK also complexes with HERG to regulate the IKr potassium current.45 Splawski et al demonstrated that minK mutations cause LQT5 when they identified mutations in two families with LQTS.31 In both cases, missense mutations (S74L, D76N) were identified; they reduced IKs by shifting the voltage dependence of activation and accelerating channel deactivation. This was supported by the fact that a murine model of minK-defective LQTS was also created.58 The functional consequences of these mutations include delayed cardiac repolarization and, hence, an increased risk of arrhythmias.
MiRP1 or KCNE2: The LQT6 Gene
MiRP1, the minK-related peptide 1, or KCNE2 (see Table 54-1), is a novel potassium channel gene recently cloned and characterized by Abbott and colleagues.47 This small integral membrane subunit protein assembles with HERG (LQT2) to alter its function, enabling full development of the IKr current (see Figure 54-3). MiRP1 is a 123–amino acid channel protein with a single predicted transmembrane segment similar to that described for minK.56 Chromosomal localization studies mapped this KCNE2 gene to chromosome 21q22.1, within 79kb of KCNE1 (minK) and arrayed in opposite orientation.47 The open reading frames of these two genes share 34% identity, and both are contained in a single exon, suggesting that they are related through gene duplication and divergent evolution.
Three missense mutations associated with LQTS and ventricular fibrillation (VF) were identified in KCNE2 by Abbott et al, and biophysical analysis demonstrated that these mutants form channels that open slowly and close rapidly, thus diminishing potassium currents.47 In one case, the missense mutation, a C-to-G transversion at nucleotide 25, which produced a glutamine (Q) to glutamic acid (E) substitution at codon 9 (Q9E) in the putative extracellular domain of MiRP1, led to the development of torsades de pointes and VF after intravenous clarithromycin infusion (i.e., drug induced).
Genetics and Physiology of Autosomal Recessive Long QT Syndrome (Jervell and Lange-Nielsen Syndrome)
Neyroud et al reported the first molecular abnormality in patients with JLNS when they reported on two families in which three children were affected by JLNS and in whom a novel homozygous deletion-insertion mutation of KVLQT1 was found.59 A deletion of 7 bp and an insertion of 8 bp at the same location led to premature termination at the C-terminal end of the KVLQT1 channel. At the same time, Splawski et al identified a homozygous insertion of a single nucleotide that caused a frameshift in the coding sequence after the second putative transmembrane domain (S2) of KVLQT1.60 Together, these data strongly suggested that at least one form of JLNS is caused by homozygous mutations in KVLQT1 (see Table 54-1). This has been confirmed by others.27,30,61,62
As a general rule, heterozygous mutations in KVLQT1 cause Romano-Ward syndrome (LQTS only), whereas homozygous (or compound heterozygous) mutations in KVLQT1 cause JLNS (LQTS and deafness). The hypothetical explanation suggests that although heterozygous KVLQT1 mutations act by a dominant-negative mechanism, some functional KVLQT1 potassium channels still exist in the stria vascularis of the inner ear. Therefore congenital deafness is averted in patients with heterozygous KVLQT1 mutations. For patients with homozygous KVLQT1 mutations, no functional KVLQT1 potassium channels can be formed. It has been shown by in situ hybridization that KVLQT1 is expressed in the inner ear, suggesting that homozygous KVLQT1 mutations can cause the dysfunction of potassium secretion in the inner ear and lead to deafness.60 However, it should be noted that incomplete penetrance exists, and not all heterozygous or homozygous mutations follow this rule.11,22
As with Romano-Ward syndrome, if KVLQT1 mutations can cause the phenotype, it could be expected that minK mutations could also be causative of the phenotype (JLNS). Schulze-Bahr et al, in fact, showed that mutations in minK result in JLNS syndrome as well, and this was confirmed subsequently (see Table 54-1).60,63 Hence, abnormal IKs current, whether caused by homozygous or compound heterozygous mutations in KVLQT1 or mink, results in LQTS and deafness.
Genotype-Phenotype Correlations in Long QT Syndrome
Clinical Features
Kimbrough et al recently reported on the study of 211 probands with LQTS and classified the severity in the probands, affected parents, and siblings.64 Importantly, they showed that the severity of the disease in the proband did not correlate with the clinical severity seen in first-degree relatives, specifically their parents and siblings. In fact, variable intrafamily penetrance was noted, consistent with other genetic and environmental factors playing a role in modulating and modifying clinical manifestations in members of the same family. Several stratifiers were identified as important.
These findings complement the findings previously described by Zareba et al.65 In this study, the authors provided evidence of clinical outcome, age of onset, and frequency of events based on genotype. Patients with mutations in LQT1 had the earliest onset of events and the highest frequency of events followed by mutations in LQT2. The risk of SCD in these two groups was relatively low for any event. Mutations in LQT3 resulted in a paucity of syncopal events, but events commonly resulted in SCD. In addition, mutations in LQT3 resulted in the longest QTc duration. Mutations in LQT1 and LQT2 appeared to be associated with stress-induced symptoms, with LQT1 associated with exercise and swimming and LQT2 associated with auditory triggers.66–71 LQT3 appeared to be associated with sleep-associated symptoms and events.
Electrocardiographic and Biophysical Features
In 1995, Moss and colleagues provided the first evidence that mutations in different genes cause differing ECG features.72 Specifically, these authors focused on the different types of T waves seen in patients with LQT1 versus LQT2 versus LQT3. ECGs of patients with LQT1 were shown to display broad-based T waves, those with LQT2 had low-amplitude T waves, and those with LQT3 mutations had distinctive T waves with late onset. More recently, Zhang et al showed that there are actually four different ST-T wave patterns.73 Using these definitions, they were able to identify 88% of patients with LQT1 and LQT2 accurately by surface ECG and 65% of LQT3 carriers. Prospectively, these authors correctly predicted the genotype in 100% of patients.
Further insight into ECG findings and genotype were reported by Lupoglazoff et al using Holter monitor analysis.74 Analysis of 133 patients with LQT1 57 LQT2 carriers, and 100 control individuals, led the authors to conclude that T wave morphology was normal in most patients with LQT1 (92%) and in normal controls (96%), but the vast majority of patients with LQT2 had abnormal T waves (19% normal, 81% abnormal). In the largest percentage of patients with LQT2, T-wave notching was identified, with the T-wave protuberance seen above the horizontal, whereas another subset had a bulge at or below the horizontal. In the former case, young age, missense LQT2 mutations, and mutations in the core domain of HERG predicted morphology, whereas potential diagnostic clues gained by the latter morphology included amino-terminal or carboxy-terminal mutations or frameshifts in HERG.
Animal Models of Long QT Syndrome
By using an arterially perfused canine left ventricular wedge preparation developed pharmacologically, induced animal models of LQT1, LQT2, and LQT3 have been created.75,76 By using chromanol 293B, a specific IKs blocker, a model that mimics LQT1 was produced.75 In this model, IKs deficiency alone was not enough to induce torsades de pointes, but the addition of β-adrenergic influence (i.e., isoproterenol) predisposed the myocardium to torsade by increasing trans-mural dispersion of repolarization. The addition of a β-blocker or mexiletine reduced the ability to induce torsades de pointes, suggesting that these medications might improve patient outcomes.
Models for LQT2 and LQT3 were created by using D-sotalol (LQT2) or ATX-II (LQT3) in this wedge preparation.76 Both drugs preferentially prolong M cell action potential duration (APD), with ATX-II also causing a sharp rise in trans-mural dispersion. Mexiletine therapy abbreviated the Q-T interval prolongation in both models and reduced dispersion. Spontaneous torsades de pointes was suppressed, and the vulnerable window during which torsades de pointes induction occurs was also reduced in both models. These models support the current understanding of the different subtypes of LQTS and provide an explanation for potential therapies.
Therapeutic Options in Long QT Syndrome
Currently, the standard therapeutic approach in LQTS is the initiation of β-blockers at the time of diagnosis.7 Recently, Moss et al demonstrated significant reduction in cardiac events using β-blockers.77 However, syncope, aborted cardiac arrest, and SCD do continue to occur. When β-blockers cannot be used, such as in patients with asthma, other medications such as mexiletine have been tried.78 When medical therapy has failed, left sympathectomy or therapy with an implantable cardioverter-defibrillator (ICD) has been used.7
Genetics-based therapy has also been described. Schwartz et al showed that sodium channel blocking agents (i.e., mexiletine) shorten the QTc in patients with LQT3, whereas exogenous potassium supplementation or potassium channel openers have been shown to be potentially useful in patients with potassium channel defects.78–80 However, long-term potassium therapy with associated potassium-sparing agents has been unable to keep the serum potassium above 4 mmol/L because of renal potassium homeostasis. This suggests that long-term potassium therapy may not be useful. In addition, no definitive evidence that these approaches (i.e., sodium channel blockers, exogenous potassium, or potassium channel openers) improve survival has been published.
Andersen Syndrome (LQT7)
Clinical Aspects
Andersen and colleagues (1971)81 identified a complex phenotype, including ventricular arrhythmias, potassium-sensitive periodic paralysis, and dysmorphic features. The dysmorphisms included hypertelorism, broad nasal root, defects of the soft and hard palate, as well as short stature. More recently, skeletal abnormalities have broadened the phenotype (Andelfinger et al).82 The associated cardiac abnormalities include QTC prolongation, ventricular tachycardia (VT), ventricular fibrillation (VF), and atrial arrhythmias. Torsades de pointes and bi-directional VT have been seen. In addition, repolarization abnormalities affecting late repolarization and resembling giant U waves are common. SCD has not been reported as a major risk in this disorder.
Genetic Aspects
Andersen syndrome was originally mapped to chromosome 17q23-q24.2 by Plaster et al83 who used genome-wide linkage analysis. The critical region within this locus was narrowed, and candidate gene mutation screening identified mutations in KCNJ2, which encodes an inward rectifier potassium channel called Kir2.1 (Tristani-Firouzi et al).84 This channel is highly expressed in the heart and plays a role in phase 3 repolarization and in the resting membrane potential. Multiple gene mutations have been identified, to date, with relatively high penetrance noted. Functional studies have demonstrated reduction or suppression of Ik1, by a haplo insufficiency or dominant negative effect. This gene may play a role in developmental signaling pathways as well, which is believed to cause dysmorphisms.
Brugada Syndrome
Clinical Aspects of Brugada Syndrome
The first identification of the electrocardiographic pattern of RBBB with ST elevation in leads V1 to V3 was reported by Osher and Wolff.85 Shortly thereafter, Edeiken identified persistent ST elevation without RBBB in 10 asymptomatic males, and Levine et al described ST elevation in the right chest leads and conduction block in the right ventricle in patients with severe hyperkalemia.86,87 The first association of this ECG pattern with SCD was described by Martini et al and later by Aihara et al.88,89 This association was further confirmed in 1991 by Pedro and Josep Brugada, who described four patients with SCD and aborted SCD, with ECGs demonstrating RBBB and persistent ST elevation in leads V1 to V3 (Figure 54-4).90 In 1992 these authors characterized what they believed to be a distinct clinical and electrocardiographic syndrome.3
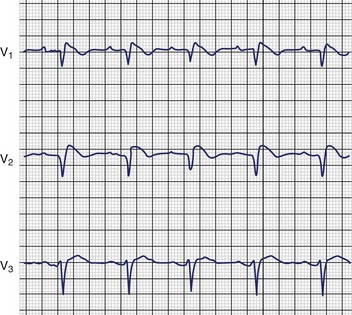
The finding of ST elevation in the right chest leads has been observed in various clinical and experimental settings and is not unique to or diagnostic of Brugada syndrome by itself.91 Situations in which these ECG findings occur include electrolyte or metabolic disorders, pulmonary or inflammatory diseases, and abnormalities of the central or peripheral nervous system. In the absence of these abnormalities, the term idiopathic ST elevation is often used and may identify patients with Brugada syndrome.
The ECG findings and associated sudden and unexpected death have been reported as common problems in Japan and Southeast Asia, where it most commonly affects men during sleep.92 This disorder, known as sudden and unexpected death syndrome (SUDS) or sudden unexpected nocturnal death syndrome (SUNDS), has many other names in Southeast Asia: bangungut (to rise and moan in sleep) in the Philippines; non-laitai (sleep-death) in Laos; lai-tai (died during sleep) in Thailand; and pokkuri (sudden and unexpectedly ceased phenomena) in Japan. General characteristics of SUNDS include young, healthy males in whom sudden death, preceded by a groan, occurs usually during sleep late at night. No precipitating factors are identified, and autopsy findings show no structural heart disease.93 Life-threatening ventricular tachyarrhythmias as a primary cause of SUNDS have been demonstrated, with VF occurring in most cases.94
The risk of SCD associated with Brugada syndrome and SUNDS in European and Southeast Asian individuals has been reported to be extremely high; approximately 75% of patients, as reported by Brugada et al, survived cardiac arrest.3,90,95 In addition, symptomatic and asymptomatic patients have been considered to be at equal risk. Priori et al have, however, disputed this claim.96 In a study of 60 patients with Brugada syndrome, asymptomatic patients had no episodes or events. The importance of this difference is its impact on therapeutic decision making, as currently all patients receive ICD therapy. Should the data of Priori et al hold up, selective use of ICDs would be appropriate.96 If selective use of ICDs were to be considered, other diagnostic tests for risk stratification would be necessary.
Kakishita et al studied a high-risk group of patients, 37% of whom had experienced spontaneous episodes of VF.97 As the majority of patients had ICDs, the authors were able to show that 65% of episodes were preceded by premature ventricular complexes (PVCs), which were essentially identical to the initiating PVCs of VF in morphology. In fact, the PVCs initiating all VF episodes arose from the terminal portions of the T wave, and pause-dependent arrhythmias were rare. This suggests that vigilant evaluation by Holter monitoring could identify at-risk patients. In addition, the authors suggested that the use of radiofrequency (RF) ablation targeting the initiating PVCs could be helpful in reducing risk and reducing the need for ICD placement.
Clinical Genetics of Brugada Syndrome
Most of the families thus far identified with Brugada syndrome have apparent autosomal dominant inheritance.98–100 In these families, approximately 50% of offspring of affected patients develop the disease. It is likely that the number of families reported has been small because of under-recognition as well as the occurrence of premature and unexpected deaths.91,101,102
Molecular Genetics of Brugada Syndrome
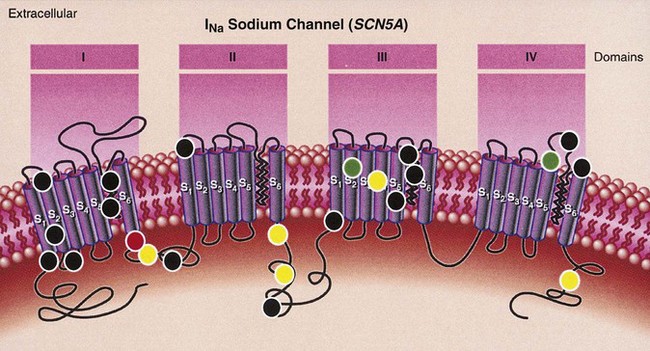
In 1998, our laboratory reported the findings on six families and several sporadic cases of Brugada syndrome.100 The families were initially studied by linkage analysis by using markers to the known ARVD loci and LQT loci, and linkage was excluded. Candidate gene screening using the mutation analysis approach of single-strand conformation polymorphism (SSCP) analysis and deoxyribonucleic acid (DNA) sequencing was performed, and SCN5A was chosen for study on the basis of the suggestions of Antzelevitch.91,99,101–103 In three families, mutations in SCN5A were identified (see Table 54-1): (1) a missense mutation (C-to-T base substitution) causing a substitution of a highly conserved threonine by methionine at codon 1620 (T1620M) in the extracellular loop between transmembrane segments S3 and S4 of domain IV (DIVS3-DIVS4), an area important for coupling of channel activation to fast inactivation; (2) a two-nucleotide insertion (AA), which disrupts the splice-donor sequence of intron 7 of SCN5A; and (3) a single nucleotide deletion (A) at codon 1397, which results in an in-frame stop codon that eliminates DIIIS6, DIVS1-DIVS6, and the carboxy-terminus of SCN5A (Figure 54-5).100 Since the initial report, multiple confirming mutations have been identified. SCN5A mutations have also been found in the case of SCD in children.104
Biophysical analysis of the mutants in Xenopus oocytes demonstrated a reduction in the number of functional sodium channels in both the splicing mutation and the one-nucleotide deletion mutation, which should promote development of re-entrant arrhythmias. In the missense mutation, sodium channels recover from inactivation more rapidly than normal. Subsequent experiments conducted in modified HEK cells revealed that at physiological temperatures (37° C), reactivation of the T1620M mutant channel was actually slower, whereas inactivation of the channel was significantly accelerated. These alterations leave the transient outward current unopposed and thus able to effect an all-or-none repolarization of the action potential at the end of phase 1.105 Failure of the sodium channel to express, as with the insertion and deletion mutations, results in similar electrophysiological changes. Reduction of the sodium channel INa current causes heterogeneous loss of the action potential dome in the right ventricular epicardium, leading to a marked dispersion of depolarization and refractoriness, an ideal substrate for development of re-entrant arrhythmias. Phase 2 re-entry produced by the same substrate is believed to provide the premature beat necessary for the initiation of the VT and VF responsible for symptoms in these patients. Interestingly, however, Kambouris et al identified a mutation in essentially the same region of SCN5A as the T1620M mutation (R1623H), but the clinical and biophysical features of this mutation were found to be consistent with LQT3 and not Brugada syndrome.106 More recently, mutations in SCN5A, in which both Brugada syndrome and LQT3 features were seen in the same patient, has been described.107 Hence, there clearly remains a gap in our understanding of these entities.
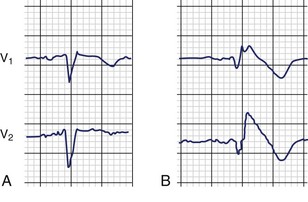
Risk Stratification in Brugada Syndrome
Most symptomatic or at-risk patients with Brugada syndrome manifest an ECG with a coved-type ST-segment elevation with or without provocation with sodium channel blocking agents such as ajmaline or flecainide (Figure 54-6).108 The other form of ST-segment elevation, the so-called saddle type, is not associated with definitive Brugada syndrome unless it transitions into a coved type by provocation or independently. Brugada et al have suggested, however, that the risk of SCD is not different between symptomatic patients and asymptomatic patients, including those with concealed forms of disease.90,95,108
Various other risk stratifiers have also been identified.109–112 Assessment of noninvasive markers by Ikeda et al demonstrated that late potentials noted using signal-averaged ECGs were present in 24 (73%) of 33 patients with a history of syncope or aborted SCD.110 Using multivariate logistic regression, the authors showed that the presence of late potentials were significantly correlated with the occurrence of life-threatening events in patients with Brugada syndrome. The evaluation of these same patients with microvolt T-wave alternans and corrected Q-T interval dispersion failed to correlate with outcome. These findings were supported by others as well.112
Finally, spontaneous episodes of VF in patients with Brugada syndrome were shown to be triggered by PVCs with specific morphologies. Kakishita et al suggested that the use of ICD therapy not only could be lifesaving but also could record the specific triggering events.97 They suggested that this knowledge could define risk and potentially lead to either ablative therapy or the ability to stratify the risk of SCD.
Cardiac Conduction Disease
Syncope and SCD may also be caused by bradycardia. The most common form of life-threatening bradycardias include disorders in which complete atrioventricular (AV) block occurs.113 These disorders require pacemaker implantation for continued well-being. Two major forms of conduction system disease in which no congenital heart disease is associated include isolated forms of conduction disease associated with dilated cardiomyopathy.113–115
Progressive cardiac conduction defect, also known as Lev-Lenègre disease, is one of the most common cardiac conduction disorders.113–116 This disorder is characterized by progressive alteration of conduction through the His-Purkinje system with development of RBBB or left bundle branch block (LBBB) with widening of the QRS complexes. Ultimately, complete AV block occurs, resulting in syncope and SCD. Lev-Lenègre disease represents the most common reason for pacemaker implantation worldwide, accounting for 0.15 implants per 1000 population yearly in developed countries. This disorder has been considered a primary degenerative disease, an exaggerated aging process with sclerosis of the conduction system, or an acquired disease. The first gene identified for Lev-Lenègre disease was reported in 1999 by Schott et al.116 They identified a missense mutation and deletion mutation, respectively, in SCN5A (see Table 54-1), the cardiac sodium channel gene, in two families with autosomal-dominant inheritance (Figure 54-7). Although the authors suggested that the biophysical abnormality was channel loss of function, no electrophysiological analysis was provided. As SCN5A also causes LQT3, Brugada syndrome, and SIDS (see Figure 54-7), all diseases in which ventricular tachyarrhythmias result in syncope and SCD, the association of conduction disturbance with SCN5A mutations was initially surprising.107,114,117,118 However, it is now known that conduction disturbance occurs in these disorders as well.
A similar disorder, known as isolated cardiac conduction disease, also results in complete AV block, syncope, and SCD. This disorder has been considered to be genetically inherited (autosomal dominant trait) and not acquired. Brink et al and de Meeus et al independently mapped a gene to chromosome 19q13.3 in families with isolated conduction disturbance in 1995, but the gene has remained elusive (see Table 54-1).119,120 Recently, however, Tan and colleagues identified a mutation in SCN5A in this disorder (see Figure 54-7) and also presented biophysical analysis (see Table 54-1).114 This mutation, a G-to-T transversion in exon 12 of SCN5A, resulted in a change from glycine to cysteine at position 514 (G514C) encoding an amino acid within the DI-DII intercellular linker of the cardiac sodium channel. Biophysical characterization of the mutant channel demonstrated abnormalities in voltage-dependent gating behavior. The sodium current (INa) was found to decay more rapidly than the wild-type channel. In the mutant, open-state inactivation was hastened, but closed state inactivation was reduced and destabilized. Computational analysis predicted that the gating defects selectively slowed myocardial conduction without provoking the rapid cardiac arrhythmias seen in LQTS and Brugada syndrome. When comparing Brugada syndrome, LQT3, and conduction disease biophysics, the following findings are notable. In Brugada syndrome, SCN5A mutations cause reduction in INa, hastening epicardial repolarization and causing the development of VT and VF. In contradistinction, LQT3 mutations in SCN5A result in excessive INa, delaying repolarization and torsades de pointes VT. Importantly, the G514C mutation evokes gating shifts reminiscent of both LQT3 and Brugada syndrome, including an activation gating shift responsible for reducing INa and destabilization of inactivation that causes an increase in INa. Tan et al showed that these voltage-dependent gating abnormalities may be partially corrected by dexamethasone, consistent with the known salutary effects of glucocorticoids on the clinical phenotype.114 It is also worth noting that some patients with LQT3 and Brugada syndrome have been reported to have conduction disturbances.
Finally, patients with conduction disease and dilated cardiomyopathy present a conundrum with regard to what comes first—conduction abnormalities leading to cardiomyopathy, or vice versa.115 Clinically, these patients tend to develop variable degrees of AV block in their teen years or 20s, with progression of this block over another 1 or 2 decades before developing the signs and symptoms of heart failure consistent with the cardiomyopathic phenotype. To date, only the gene lamin A/C located on chromosome 1q21 has been confirmed to cause this disease.121,122 Lamins A and C are members of the intermediate filament multigene family, which are encoded by a single gene. Lamins A and C polymerize to form part of the nuclear lamina, a structural filamentous network on the nucleoplasmic side of the inner nuclear membrane. The specific causes of conduction disease and myocardial dysfunction are not currently known but could be attributed to the progressive degeneration of cardiac tissue analogous to that described in Lev-Lenègre disease.
Sudden Infant Death Syndrome
SIDS is defined as the sudden death of an infant younger than 1 year of age that remains unexplained after performance of a complete autopsy, review of clinical and family histories, and examination of the death scene. Although the incidence of SIDS has been dramatically reduced from 1.6 per 1000 live births to 0.64 per 1000 live births as reported in 1998 in the United States, it is still one of the most common causes of death among babies between 1 and 6 months of age. Death usually occurs during sleep.123
The potential causes of sudden death in infants are many, including cardiac disorders, respiratory abnormalities, gastrointestinal diseases, metabolic disorders, traumatic injury, brain abnormality, or child abuse. One of the most referenced etiologic speculations was that described by Schwartz in 1976. He proposed that a developmental abnormality in cardiac sympathetic innervation predisposed some infants to lethal cardiac arrhythmias. Specifically, an imbalance in the sympathetic nervous system was speculated to result in prolongation of the Q-T interval on the ECG and in potentially lethal ventricular arrhythmias.17,18 In 1998, Schwartz et al published data collected from 1976 to 1994, in which ECGs were recorded on the third or fourth day of life in 34,442 Italian newborns.4 These babies were followed up for 1 year; during that period, 34 babies died. Evaluation of these 34 babies demonstrated that 24 died of SIDS. These 24 SIDS victims were found to have longer QTc measurements compared with controls or other infants dying of other causes. In 12 of these 24 cases, the QTc was clearly prolonged, and the authors suggested that QTc prolongation during the first week of life is associated with SIDS.125
Although this suggestion linking SIDS and LQTS was roundly criticized, the authors were subsequently able to identify a mutation in SCN5A (see Table 54-1) in one patient with aborted SIDS.118,126–132 In addition, Priori et al reported identification of an SCN5A mutation in an infant with Brugada syndrome.133 More recently, Ackerman et al reported a molecular epidemiology study of 95 cases of SIDS, in which the myocardium obtained at autopsy was screened for ion channel gene mutations.134 In 4 of 93 cases, mutations in SCN5A were identified in postmortem analysis, and the authors suggested that in 4.3% of this cohort SIDS was caused by mutations in this known arrhythmia-causing gene. Hence, it appears that ion channel mutations, particularly SCN5A, result in SIDS in some infants (see Figure 54-7). Biophysical analysis identified a sodium current characterized by slower delay, and a twofold to threefold increase in late sodium current similar to that seen in LQTS. The fact that these SIDS occurs during sleep is consistent with the features seen for this channel when mutations result in LQTS (LQT3). SCN5A mutations in SIDS have been further confirmed recently.134 It is likely that other ion channel gene abnormalities will be found in infants with SIDS and that there is wide etiologic heterogeneity.
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
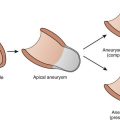
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is characterized by fatty infiltration of the right ventricle, fibrosis, and ultimately thinning of the wall with chamber dilatation (Figure 54-8).5 It is the most common cause of SCD in the young in Italy and is said to account for about 17% of SCD in the young in the Unites States.135,136 Rampazzo et al mapped this disease in two families, one to 1q42-q43 and the other on chromosome 14q23-q24.137,138 A third locus was mapped to 14q12.139 A large Greek family with ARVD and Naxos disease was recently mapped to 17q21.140 Two loci responsible for ARVD/C in North America were subsequently mapped at 3p23 and the other at 10p12.141,142
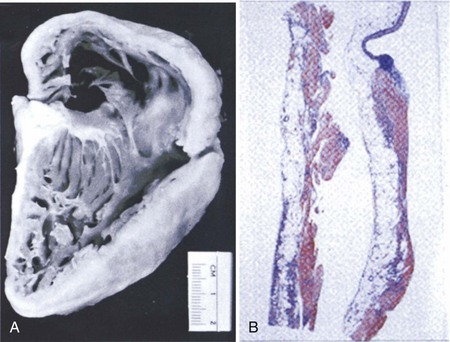
ARVD/C is a devastating disease because the first symptom is often SCD. Electrocardiographic abnormalities include inverted T waves in the right precordial leads, late potentials, and right ventricular arrhythmias with LBBB. In many cases, the ECG looks similar to that seen in Brugada syndrome, with ST elevation in V1 to V3.143 The issue of SCD is compounded by the great difficulty in making the diagnosis of ARVD/C even when occurring in a family with a history of the disease. Since the disease affects only the right ventricle, it is difficult to detect it by using most diagnostic modalities. No diagnostic definitive standard is available at present. The right ventricular biopsy may be definitive when positive but often gives a false-negative result, since the disease initiates in the epicardium and spreads to the endocardium of the right ventricular free wall, making it inaccessible to biopsy. A consensus diagnostic criteria that includes right ventricular biopsy, magnetic resonance imaging (MRI), echocardiography, and electrocardiography was developed.144
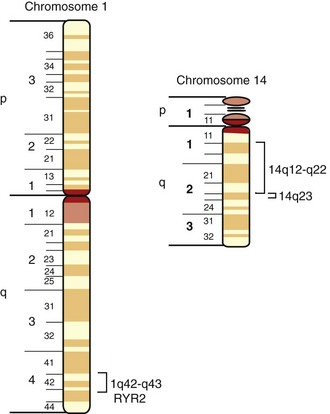
The genetic basis of ARVD/C has started to unravel recently. The first gene causing ARVD/C was identified by Tiso et al for the chromosome 1q42-1q43-linked ARVD2 locus in 2001 (Figure 54-9).145 This gene (see Table 54-1), the cardiac ryanodine receptor gene (RYR2), a 105 exon gene that encodes the 565 kd monomer of a tetrameric structure interacting with four FK-506 binding proteins called FKBP12.6, is fundamental for intracellular calcium homeostasis and for excitation–contraction coupling. This large protein physically links to the dihydropyridine (DHP) receptor of the t-tubule, where the DHP receptor protein, a voltage-dependent calcium channel, is activated by plasma membrane depolarization and induces a calcium influx.146,147 The RYR2 protein, activated by calcium, induces release of calcium from the sarcoplasmic reticulum into the cytosol. Hence, mutations in RYR2 would be expected to cause calcium homeostasis imbalance and result in abnormalities in rhythm as well as excitation-contraction coupling and myocardial dysfunction.147 This causative gene is therefore, in many ways, similar to the mutant genes responsible for the ventricular arrhythmias of LQTS, Brugada syndrome, and SIDS, in which ion channel mutations cause the clinical phenotype. In those instances, potassium channel dysfunction and sodium channel dysfunction occur, whereas in ARVD2, intracellular calcium channel dysfunction plays a central role.
Two other genes associated with arrhythmogenic cardiomyopathy have also been described. The first of these, plakoglobin, was shown to cause the chromosome 17q21-linked autosomal recessive disorder called Naxos disease (see Table 54-1).148 This disorder is characterized by ARVD/C in association with abnormalities of skin (palmoplantar keratoderma) and hair (woolly hair) and, therefore, is not exactly the same as isolated ARVD/C, being a more complex phenotype.
Plakoglobin is a cell adhesion protein thought to be important in providing functional integrity to the cell. This protein is found in many tissues, including the cytoplasmic plaque of cardiac junctions and the dermal-epidermal junctions of the epidermis, and it has a potential signaling role in the formation of desmosomal junctions. It is believed that plakoglobin serves as a linker molecule between the inner and outer portions of the desmosomal plaque by binding tightly to the cytoplasmic domains of cadherins. The mutations identified, a homozygous 2 bp deletion, resulted in a frameshift and premature termination of the protein.148 Support for this gene being causative of Naxos disease comes from a murine model with null mutations, which exhibit the heart and skin abnormalities seen in affected patients. The mutated protein is thought to cause disruption of myocyte integrity, leading to cell death and fibro-fatty replacement, with secondary arrhythmias being caused by the abnormal myocardial substrate.
The last gene identified, desmoplakin, is another desmosomal protein with similarities to plakoglobin (see Table 54-1).149 Homozygous mutations in this gene resulted in a Naxos-like disorder, although the cardiac features occurred in the left ventricle instead of the right ventricle. The affected protein is an important protein in cell adhesion and appears to function similarly to that described for plakoglobin. Although it is easy to speculate that mutation in this gene and in plakoglobin causes the myocardial abnormalities, it remains unclear why differences in ventricular chamber specificity occurs and how the ventricular tachyarrhythmias develop.
Brugada Syndrome and Arrhythmogenic Right Ventricular Dysplasia
Controversy exists concerning the possible association of Brugada syndrome and ARVD, with some investigators arguing that these are the same disorder or at least one is a forme fruste of the other.143,150–156 However, the classic echocardiographic, angiographic, and MRI findings of ARVD are not seen in patients with Brugada syndrome. In addition, patients with Brugada syndrome typically do not exhibit the histopathologic findings of ARVD. Further, the morphology of VT or VF differs.91,143 Finally, the genes identified so far differ.61,144,145,149,152
Polymorphic Ventricular Tachycardia
Familial polymorphic VT, an autosomal-dominant disorder characterized by episodes of bi-directional (Figure 54-10) and polymorphic VT, typically in relation to adrenergic stimulation or physical exercise, was first described by Coumel et al in 1978.155 This disorder most commonly occurs in childhood and adolescence, presenting with syncope and SCD.6,156 Mortality rates of 30% to 50% in patients aged 20 to 30 years have been reported, suggesting this is a highly malignant disorder. Autopsy data demonstrate this disorder to have no associated structural cardiac abnormalities.
Mutations in the cardiac ryanodine receptor (RYR2), the same gene responsible for ARVD2 (see Figure 54-9), were recently independently identified by Laitinen et al and Priori et al in multiple families linked to chromosome 1q42 (see Table 54-1).157–160 Interestingly, ARVD2 typically is considered to be the one form of ARVD/C in which catecholaminergic input is important in the development of symptoms. It is not clear at this time why patients with familial polymorphic VT have no associated structural cardiac abnormalities and patients with ARVD/C have classic fibro-fatty replacement in the right ventricle.
Mutations in another member of the ryanodine receptor gene family, RYR1, which is expressed in skeletal muscle, result in malignant hyperthermia and central core disease.161 The mutations in this gene appear to cluster in three regions of the gene, regions similar to the mutations found in RYR2 in the cases of VT reported, suggesting these to be functionally critical regions.
Wolff-Parkinson-White Syndrome
Wolff-Parkinson-White syndrome (WPW) is the second most common cause of paroxysmal supraventricular tachycardia (SVT), with a prevalence of 1.5 to 3.1 per 1000 individuals.2 In some parts of the world such as China, WPW is even more common, being responsible for up to 70% of cases of SVT.162 Tachycardia presents typically in a bi-modal fashion, with onset common in infancy as well as during the teen years. Symptoms most commonly include syncope, presyncope, shortness of breath, palpitations, and SCD.113
WPW has long been described to be caused by accessory pathways derived from muscle fibers providing direct continuity between the atrial and ventricular myocardia.2,163 These accessory pathways may be identified by the peculiar ECG findings seen in WPW, including short P-R interval, widened QRS complexes, and the classic δ wave in which an abnormal initial QRS vector is notable (Figure 54-11).2,163–165 In a significant percentage of patients, conduction abnormalities including high-grade sinoatrial or AV block occur, necessitating pacemaker implantation. In most patients with WPW and SVT, RF ablation of the accessory pathways is curative.166,167
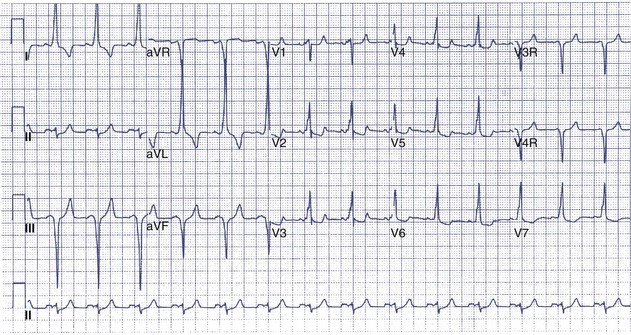
FIGURE 54-11 Electrocardiogram showing the short P-R interval and δ wave associated with Wolff-Parkinson-White syndrome.
Some cases of WPW are associated with other primary disorders such as hypertrophic cardiomyopathy or left ventricular noncompaction cardiomyopathy, or the congenital cardiac disorder Ebstein’s anomaly.166,168,169 Whether the underlying cause of WPW is similar in these cases compared with pure cases of WPW has been discussed for many years, but no definitive answers have been provided.
The first gene in patients with WPW was recently identified by Gollob et al and Blair et al independently in familial forms of WPW.170,171 In both reports, autosomal-dominant inheritance was reported. Interestingly, this gene, which maps to chromosome 7q34-7q36, was found to cause WPW and hypertrophic cardiomyopathy in a significant percentage of patients in both reports (see Table 54-1).168 The gene, the γ2 subunit of adenosine monophosphate–activated protein kinase, consists of 569 amino acids, is 63 kD in size, and functions as a metabolic sensor in cells, responding to cellular energy demands by regulating adenosine triphosphate (ATP) production and utilization.172 It is not clear whether this is a primary hypertrophic cardiomyopathy–causing gene or a WPW gene, particularly since the initial mapping of this locus was in patients with hypertrophic cardiomyopathy and associated WPW. Clearly, this is not the only gene responsible for WPW, and the functional and physiological abnormalities responsible for the resultant WPW are not yet obvious.
Summary
Ventricular arrhythmias appear to result from ion channel abnormalities. Whether this is necessarily a primary abnormality or a secondary one is becoming better understood. Therapeutic options including drug and device therapy are likely to be expanded once this knowledge has matured. These are detailed in Chapters 84, 94, and 95. Similarly, conduction system abnormalities have been shown to occur secondary to mutations in the ion channel gene and are widely but empirically treated with device therapy as discussed in Chapter 38. The individual disorders are now exhaustively discussed in Chapters 62 to 65.
Antzelevitch C. The Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:513-516.
Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden death. J Cardiovasc Electrophysiol. 1997;8:325-331.
Brugada P, Brugada J. Right bundle-branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396.
Corrado D, Nava A, Buja G, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443-448.
Gollob MH, Green MS, Tang AS-L, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823-1831.
Jervell A, Lange-Nielsen F. Congenital deaf-mutism, function heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
Keating MT, Atkinson D, Dunn C, et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704-706.
Moss AJ, Zareba W, Benhorin J, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long-QT syndrome. Circulation. 1995;92:2929-2934.
Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616-623.
Nademanee K, Veerakul G, Nimmannit S, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595-2600.
Priori SG, Barhanin J, Hauer RNW, et al. Genetic and molecular basis of cardiac arrhythmias: Impact on clinical management (Parts I and II). Circulation. 1999;99:518-528.
Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80-83.
Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20-21.
Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long QT syndrome. N Engl J Med. 2000;343:262-267.
Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709-1714.
Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129-133.
Towbin JA, Friedman RA. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1760-1761.
Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805-811.
Wilde AAM, Roden DM. Predicting the long-QT genotype from clinical data: From sense to science. Circulation. 2000;102:2796-2798.
1 Priori SG, Barhanin J, Hauer RNW, et al. Genetic and molecular basis of cardiac arrhythmias: Impact on clinical management (Parts I and II). Circulation. 1999;99:518-528.
2 Al-Khatib SM, Pritchett EL. Clinical features of Wolff-Parkinson-White syndrome. Am Heart J. 1999;138:403-413.
3 Brugada P, Brugada J. Right bundle-branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396.
4 Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709-1714.
5 Thiene G, Basso C, Danieli G, et al. Arrhythmogenic right ventricular cardiomyopathy. Trends Cardiovasc Med. 1997;7:84-90.
6 Fisher JD, Krikler D, Hallidie-Smith KA. Familial polymorphic ventricular arrhythmias: A quarter century of successful medical treatment based on serial exercise-pharmacologic testing. J Am Coll Cardiol. 1999;34:2015-2022.
7 Schwartz PJ, Locati EH, Napolitano C, Priori SG. The long QT syndrome. In: Zipes DP, Jallife J, editors. Cardiac electrophysiology: From cell to bedside. Philadelphia: WB Saunders, 1996.
8 Romano C, Gemme G, Pongiglione R. Antmie cardiache rare in eta pediatrica. Clin Pediatr. 1963;45:656-683.
9 Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103-106.
10 Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome. Clinical impact. Circulation. 1999;99:529-533.
11 Singer PA, Crampton RS, Bass NH. Familial Q-T prolongation syndrome: Convulsive seizures and paroxysmal ventricular fibrillation. Arch Neurol. 1974;31:64-66.
12 Ratshin RA, Hunt D, Russell ROJr., Rackley CE. QT-interval prolongation, paroxysmal ventricular arrhythmias, and convulsive syncope. Ann Intern Med. 1971;75:19-24.
13 Bellavere F, Ferri M, Guarini L, et al. Prolonged QT period in diabetic autonomic neuropathy: A possible role in sudden cardiac death. Br Heart J. 1988;59:379-383.
14 Ewing DJ, Boland O, Neilson JMM, et al. Autonomic neuropathy, QT interval lengthening, and unexpected deaths in male diabetic patients. Diabetologia. 1991;34:182-185.
15 Weintraub RG, Gow RM, Wilkinson JL. The congenital long QT syndromes in children. J Am Coll Cardiol. 1990;16:674-680.
16 Marks ML, Trippel DL, Keating MT. Long QT syndrome associated with syndactyly identified in females. Am J Cardiol. 1995;10:744-745.
17 Schwartz PJ, Segantini A. Cardiac innervation, neonatal electrocardiography and SIDS. A key for a novel preventive strategy? Ann N Y Acad Sci. 1988;533:210-220.
18 Schwartz PJ, Grotti L. Cardiac arrhythmias of genetic origin are important contributors to sudden infant death syndrome. Heart Rhythm. 2007;4:740-742.
19 Jervell A, Lange-Nielsen F. Congenital deaf-mutism, function heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
20 Jervell A. Surdocardiac and related syndromes in children. Adv Intern Med. 1971;17:425-438.
21 James TN. Congenital deafness and cardiac arrhythmias. Am J Cardiol. 1967;19:627-643.
22 Priori SG, Schwartz PJ, Napolitano C, et al. A recessive variant of the Romano-Ward long-QT syndrome. Circulation. 1998;97:2420-2425.
23 Keating MT, Atkinson D, Dunn C, et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704-706.
24 Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genet. 1996;12:17-23.
25 Li H, Chen Q, Moss AJ, et al. New mutations in the KVLQT1 potassium channel that cause long QT syndrome. Circulation. 1998;97:1264-1269.
26 Choube C, Neyroud N, Guicheney P, et al. Properties of KVLQTI K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J. 1997;16:5472-5479.
27 Chiang CE, Roden DM. The long QT syndromes: Genetic basis and clinical implications. J Am Coll Cardiol. 2000;36:1-12.
28 Barhanin J, Lesage F, Guillemare E, et al. KVLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78-80.
29 Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80-83.
30 Wollnick B, Schreeder BC, Kubish C, et al. Pathophysiological mechanisms of dominant and recessive KVLQTI K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6:1943-1949.
31 Splawski I, Tristani-Firouzi M, Lehmann MH, et al. Mutations in the minK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338-340.
32 Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosome 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994;8:141-147.
33 Warmke JE, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91:3438-3442.
34 Arcangeli A, Rosati B, Cherubini A, et al. HERG– and IRK-like inward rectifier currents are sequentially expressed during neuronal crest cells and their derivatives. Eur J Neurosci. 1997;9:2596-2604.
35 Pennefather PS, Zhou W, Decoursey TE. Idiosyncratic gating of HERG-like K+ channels in microglia. J Gen Physiol. 1998;111:795-805.
36 Bianchi L, Wible B, Arcangeli A, et al. HERG encodes a K+ current highly conserved in tumors of different histogenesis: A selective advantage for cancer cells? Cancer Res. 1998;58:815-822.
37 Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795-803.
38 Schulze-Bahr E, Haverkamp W, Funke H. The long-QT syndrome. N Engl J Med. 1995;333:1783-1784.
39 Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299-307.
40 Trudeau MC, Warmke J, Ganetzky B, Robertson G. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92-95.
41 Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum of HERG K+ channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci U S A. 1996;93:2208-2212.
42 Roden DM, Balser JR. A plethora of mechanisms in the HERG-related long QT syndrome genetics meets electrophysiology. Cardiovasc Res. 1999;44:242-246.
43 Furutani M, Trudeau MC, Hagiwara N, et al. Novel mechanism associated with an inherited cardiac arrhythmia. Defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation. 1999;99:2290-2294.
44 Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. J Biol Chem. 1998;263:21061-21066.
45 McDonald TV, Yu Z, Ming Z, et al. A minK-HERG complex regulates the cardiac potassium current IKr. Nature. 1997;388:289-292.
46 Bianchi L, Shen Z, Dennis AT, et al. Cellular dysfunction of LQT5-minK mutants: Abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum Mol Genet. 1999;8:1499-1507.
47 Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175-187.
48 Gellens M, George AL, Chen L, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:54-58.
49 George AL, Varkony TA, Drakin HA, et al. Assignment of the human heart tetrodotoxin-resistant voltage-gated Na channel-subunit gene (SCN5A) to band 3p21. Cytogenet Cell Genet. 1995;68:67-70.
50 Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805-811.
51 Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603-1607.
52 Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat Neurosci. 1999;2:593-595.
53 Bennett PB, Yazawa K, Makita N, George ALJr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683-685.
54 Dumaine R, Wang Q, Keating MT, et al. Multiple mechanisms of sodium channel-linked long QT syndrome. Circ Res. 1996;78:916-924.
55 An RH, Wang XL, Kerem B, et al. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta 1-subunits. Circ Res. 1998;83:141-146.
56 Honore E, Attali B, Heurteaux C, et al. Cloning, expression, pharmacology and regulation of a delayed rectifier K+ channel in mouse heart. EMBO J. 1991;10:2805-2811.
57 Arena JP, Kass RS. Block of heart potassium channels by clofilium and its tertiary analogs: Relationship between drug structure and type of channel blocked. Mol Pharmacol. 1988;34:60-66.
58 Vetter DE, Mann JR, Wangemann P, et al. Inner ear defects induced by null mutation of the IsK gene. Neuron. 1996;17:1251-1264.
59 Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186-189.
60 Splawski I, Timothy KW, Vincent GM, et al. Brief report: Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336:1562-1567.
61 Chen Q, Zhang D, Gingell RL, et al. Homozygous deletion in KVLQTI associated with Jervell and Lange-Nielsen syndrome. Circulation. 1999;99:1344-1347.
62 Tyson J, Tranebjaerg L, Bellman S, et al. IsK and KVLQTI: Mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Molec Genet. 1997;12:2179-2185.
63 Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267-268.
64 Kimbrough J, Moss AJ, Zareba W, et al. Clinical implications for affected parents and siblings of probands with long-QT syndrome. Circulation. 2001;104:557-562.
65 Zareba W, Moss AJ, Schwartz PJ, et al. Influence of the genotype on the clinical course of the long-QT syndrome. N Engl J Med. 1998;339:960-965.
66 Tanabe Y, Inagaki M, Kurita T, et al. Sympathetic stimulation produces a greater increase in both transmural and spatial dispersion of repolarization in LQT1 than LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol. 2001;37:911-919.
67 Ackermann MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74:1088-1094.
68 Moss AJ, Robinson JL, Gessman L, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999;84:876-933.
69 Wilde AAM, Jongbloed RJE, Doevendans PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1. J Am Coll Cardiol. 1999;33:327-332.
70 Wilde AAM, Roden DM. Predicting the long-QT genotype from clinical data: From sense to science. Circulation. 2000;102:2796-2798.
71 Ali RH, Zareba W, Moss AJ, et al. Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with the long QT syndrome. Am J Cardiol. 2000;85:457-461.
72 Moss AJ, Zareba W, Benhorin J, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long-QT syndrome. Circulation. 1995;92:2929-2934.
73 Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849-2855.
74 Lupoglazoff JM, Denjoy I, Berthet M, et al. Notched T waves on Holter recordings enhance detection of patients with LQT2 (HERG) mutations. Circulation. 2001;103:1095-1101.
75 Shimizu W, Antzelevitch C. Sodium channel block with Mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038-2047.
76 Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2, and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778-786.
77 Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616-623.
78 Schwartz PJ, Priori SG, Locati EH, et al. Long-QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate: Implications for gene-specific therapy. Circulation. 1995;92:3381-3386.
79 Compton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome: Correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-1022.
80 Shimizu W, Kurita T, Matsuo K, et al. Improvement of repolarization abnormalities by K channel opener in the long QT syndrome. Circulation. 1998;97:1581-1588.
81 Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. 1971;60:559-564.
82 Andelfinger G, Tapper AR, Welch RC, et al. KCN2 mutation results in Andersen syndrome with sex specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663-668.
83 Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511-519.
84 Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characteristics of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;222:381-388.
85 Osher HL, Wolff L. Electrocardiographic pattern simulating acute myocardial injury. Am J Med Sci. 1953;226:541-545.
86 Edeiken J. Elevation of RS-T segment, apparent or real in right precordial leads as probable normal variant. Am Heart J. 1954;48:331-339.
87 Levine HD, Wanzer SH, Merrill JP. Dialyzable currents of injury in potassium intoxication resembling acute myocardial infarction or pericarditis. Circulation. 1956;13:29-36.
88 Martini B, Nava A, Thiene G, et al. Ventricular fibrillation without apparent heart disease. Description of six cases. Am Heart J. 1989;118:1203-1209.
89 Aihara N, Ohe T, Kamakura S. Clinical and electrophysiologic characteristics of idiopathic ventricular fibrillation. Shinzo. 1990;22:80-86.
90 Brugada P, Brugada J. A distinct clinical and electrocardiographic syndrome: Right bundle-branch block, persistent ST segment elevation with normal QT interval and sudden cardiac death. Pacing Clin Electrophysiol. 1991;14:746.
91 Antzelevitch C, Brugada P, Brugada J. The Brugada syndrome. Armonk, NY: Futura Publishing; 1999.
92 Nademanee K, Veerakul G, Nimmannit S, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595-2600.
93 Gotoh K. A histopathological study on the conduction system of the so-called Pokkuri disease (sudden unexpected cardiac death of unknown origin in Japan). Jpn Circ J. 1976;40:753-768.
94 Hayashi M, Murata M, Satoh M, et al. Sudden nocturnal death in young males from ventricular flutter. Jpn Heart J. 1985;26:585-591.
95 Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden death. J Cardiovasc Electrophysiol. 1997;8:325-331.
96 Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102:2509-2515.
97 Kakishita M, Kurita T, Matsuo K, et al. Mode of onset of ventricular fibrillation in patients with Brugada syndrome detected by implantable cardioverter defibrillator therapy. J Am Coll Cardiol. 2000;36:1647-1653.
98 Kobayashi T, Shintani U, Yamamoto T, et al. Familial occurrence of electrocardiographic abnormalities of the Brugada-type. Intern Med. 1996;35:637-640.
99 Gussak I, Antzelevitch C, Bjerregaard P, et al. The Brugada syndrome: Clinical, electrophysiological, and genetic considerations. J Am Coll Cardiol. 1999;33:5-15.
100 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293-296.
101 Antzelevitch C. The Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:513-516.
102 Antzelevitch C. The Brugada syndrome: Diagnostic criteria and cellular mechanisms. Eur Heart J. 2001;22:356-363.
103 Antzelevitch C. Ion channels and ventricular arrhythmias: Cellular and ionic mechanisms underlying the Brugada syndrome. Curr Opin Cardiol. 1999;14:274-279.
104 Priori SG, Napolitano C, Giordano U, et al. Brugada syndrome and sudden cardiac death in children. Lancet. 2000;355:808-809.
105 Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803-809.
106 Kambouris NG, Nuss HB, Johns DC, et al. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation. 1998;97:640-644.
107 Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206-1213.
108 Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST segment elevation and right bundle branch block but structurally normal heart. Circulation. 2000;1:510-515.
109 RuDusky BM. Right bundle branch block, persistent ST-segment elevation, and sudden death. Am J Cardiol. 1998;82:407-408.
110 Ikeda T, Sakurada H, Sakabe K, et al. Assessment of noninvasive markers in identifying patients at risk in the Brugada syndrome: Insight into risk stratification. J Am Coll Cardiol. 2001;37:1628-1634.
111 Remme CA, Wever EFD, Wilde AAM, et al. Diagnosis and long-term follow-up of Brugada syndrome in patients with idiopathic ventricular fibrillation. Eur Heart J. 2001;22:400-409.
112 Gussak I, Bjerregaard P, Hammill SC. Clinical diagnosis and risk stratification in patients with Brugada syndrome. J Am Coll Cardiol. 2001;37:1635-1638.
113 Zipes DP, Wellens HJJ. Clinical cardiology: New frontiers. Sudden cardiac death. Circulation. 1998;98:2334-2351.
114 Tan HL, Bink-Boelkens MTE, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043-1047.
115 Kass S, MacRae C, Graber HL, et al. A gene defect that causes conduction system disease and dilated cardiomyopathy maps to chromosome 1p1-1q1. Nat Genet. 1994;7:546-551.
116 Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20-21.
117 Deschenes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55-65.
118 Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long QT syndrome. N Engl J Med. 2000;343:262-267.
119 Brink PA, Ferreira A, Moolman JC, et al. Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation. 1995;91:1633-1640.
120 de Meeus A, Stephan E, Debrus S, et al. An isolated cardiac conduction disease maps to chromosome 19q. Circ Res. 1995;77:735-740.
121 Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction system disease. N Engl J Med. 1999;341:1715-1724.
122 Brodsky GL, Muntoni F, Miocic S, et al. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473-476.
123 Towbin JA, Ackerman MJ. Editorial. Cardiac sodium channel gene mutations and SIDS. Circulation. 2001;104:1092-1093.
124 Schwartz PJ. Cardiac sympathetic innervation and the sudden infant death syndrome. A possible pathogenetic link. Am J Med. 1976;60:167-172.
125 Towbin JA, Friedman RA. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1760-1761.
126 Lucey JF. Comments on a sudden infant death article in another journal. Pediatrics. 1999;103:812.
127 Martin RJ, Miller MJ, Redline S. Screening for SIDS: A neonatal perspective. Pediatrics. 1999;103:812-813.
128 Guntheroth WG, Spiers PS. Prolongation of the QT interval and the sudden infant death syndrome. Pediatrics. 1999;103:813-814.
129 Hodgman JE. Prolonged QTc as a risk factor for SIDS. Pediatrics. 1999;103:814-815.
130 Hoffman JIE, Lister G. The implications of a relationship between prolonged QT interval and the sudden infant death syndrome. Pediatrics. 1999;103:815-817.
131 Shannon DC. Method of analyzing QT interval can’t support conclusions. Pediatrics. 1999;103:819.
132 Southall DP. Examine data in Schwartz article with extreme care. Pediatrics. 1999;103:819-820.
133 Ackerman MJ, Siu B, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001:2264-2269.
134 Wedekind H, Smits JPP, Schulze-Bahr E, et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 2001;104:1158-1164.
135 Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129-133.
136 Shen WK, Edwards WD, Hammill SC. Right ventricular dysplasia: A need for precise pathological definition for interpretation of sudden death. J Am Coll Cardiol. 1994;23:34.
137 Rampazzo A, Nava A, Erne P, et al. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet. 1995;4:2151-2154.
138 Rampazzo A, Nava A, Danieli GA, et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 1994;3:959-962.
139 Severini GM, Krajinovic M, Pinamonti B, et al. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics. 1996;31:193-200.
140 Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1996;97:2049-2058. 198
141 Ahmad F, Li D, Karibe A, et al. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation. 1998;98:2791-2795.
142 Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet. 2000;66:148-156.
143 Corrado D, Nava A, Buja G, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443-448.
144 McKenna WJ, Thiere G, Nava AA, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardio-myopathy. Br Heart J. 1994;71:215-218.
145 Tiso N, Stephan DA, Nava A, et al. Identification of mutations in cardiac ryanodine gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2. Hum Mol Genet. 2001;10:189-194.
146 Stokes DL, Wagenknecht T. Calcium transport across the sarcoplasmic reticulum—structure and function of Ca2+-ATPase and the ryanodine receptor. Eur J Biochem. 2000;267:5274-5279.
147 Missiaen L, Robberecht W, Van Den Bosch L, et al. Abnormal intracellular Ca2+ homeostasis and disease. Cell Calcium. 2000;28:1-21.
148 McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119-2124.
149 Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761-2766.
150 Naccarella F. Malignant ventricular arrhythmias in patients with a right bundle-branch block and persistent ST segment elevation in V1-V3: A probable arrhythmogenic cardiomyopathy of the right ventricle [editorial comment]. G Ital Cardiol. 1993;23:1219-1222.
151 Fontaine G. Familial cardiomyopathy associated with right bundle branch block, ST segment elevation and sudden death [letter]. J Am Coll Cardiol. 1996;28:540.
152 Scheinman MM. Is Brugada syndrome a distinct clinical entity? J Cardiovasc Electrophysiol. 1997;8:332-336.
153 Ohe T. Idiopathic ventricular fibrillation of the Brugada type—an atypical form of arrhythmogenic right ventricular cardio myopathy [editorial]. Intern Med. 1996;35:595.
154 Fontaine G, Piot O, Sohal P, et al. Right precordial leads and sudden death. Relation with arrhythmogenic right ventricular dysplasia. Arch Mal Coeur Vaiss. 1996;89:1323-1329.
155 Coumel P, Fidelle J, Lucet V, et al. Catecholaminergic-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: Report of four cases. Br Heart J. 1978;40(Suppl):28-37.
156 Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children: A 7-year follow-up of 21 patients. Circulation. 1995;91:1512-1519.
157 Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485-490.
158 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
159 Swan H, Piippo K, Viitasalo M, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035-2042.
160 Bauce B, Nava A, Rampazzo A, et al. Familial effort polymorphic ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy map to chromosome 1q42-43. Am J Cardiol. 2000;85:573-579.
161 McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15:410-417.
162 Wan Q, Wu N, Fan W, et al. Clinical manifestations and prevalence of different types of supraventricular tachycardia among Chinese. Chin Med J (Engl). 1992;105:284-288.
163 Gollob MH, Bharati S, Swerdlow CD. Accessory atrioventricular node with properties of a typical accessory pathway: Anatomic-electrophysiologic correlation. J Cardiovasc Electrophysiol. 2000;11:922-926.
164 Packard JM, Graettinger JS, Graybiel A. Analysis of the electrocardiograms obtained from 1000 young healthy aviators: Ten year follow-up. Circulation. 1954;10:384-400.
165 Hejtmancik MR, Hermann GR. The electrocardiographic syndrome of short P-R interval and broad QRS complexes: A clinical study of 80 cases. Am Heart J. 1957;54:708-721.
166 Khair GZ, Soni JS, Bamrah VS. Syncope in hypertrophic cardiomyopathy. II. Coexistence of atrioventricular block and Wolff-Parkinson-White syndrome. Am Heart J. 1985;110:1083-1086.
167 Jackman WM, Wang X, Friday KJ, et al. Catheter ablation of accessory atrioventricular pathways (Wolff-Parkinson-White syndrome) by radiofrequency current. N Engl J Med. 1991;324:1605-1611.
168 MacRae CA, Ghaisas N, Kass S, et al. Familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome maps to a locus on chromosome 7q3. J Clin Invest. 1995;96:1216-1220.
169 Ichida F, Hamamichi Y, Miyawaki T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34:233-240.
170 Gollob MH, Green MS, Tang AS-L, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823-1831.
171 Blair E, Redwood C, Ashratian H, et al. Mutations in the γ2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215-1220.
172 Lang T, Yu L, Tu Q, et al. Molecular cloning, genomic organization, and mapping of PRKAG2, a heart abundant γ2 subunit of 5’-AMP-activated protein kinase, to human chromosome 7q36. Genomics. 2000;70:258-263.
173 Hardie DG, Carling D. The AMP-activated protein kinase-fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259-273.
174 Kemp BE, Mitchelhill KI, Stapleton D, et al. Dealing with energy demand: The AMP-activated protein kinase. Trends Biochem Sci. 1999;24:22-25.
175 Winder WW, Holmes BF, Rubink DS, et al. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219-2226.
176 Cheung PC, Salt IP, Davies SP, et al. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346:659-669.