CHAPTER 15 Genetic Factors in Common Diseases
Medical genetics usually concentrates on the study of rare unifactorial chromosomal and single-gene disorders. Diseases such as diabetes, cancer, cardiovascular and coronary artery disease, mental health, and neurodegenerative disorders are responsible, however, for the majority of the morbidity and mortality in developed countries. These so-called common diseases are likely to be of even greater importance in the future, with the elderly accounting for an increasing proportion of the population.
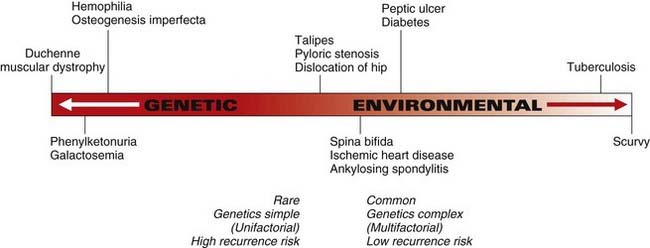
The common diseases do not usually show a simple pattern of inheritance. Instead, the contributing genetic factors are often multiple, interacting with each other and environmental factors in a complex manner. In fact, it is uncommon for either genetic or environmental factors to be entirely responsible for a particular common disorder or disease in a single individual. In most instances, both genetic and environmental factors are contributory, although sometimes one can appear more important than the other (Figure 15.1).
Genetic Susceptibility to Common Disease
For many of the common diseases, a small but significant proportion of cases have single-gene causes, but the major proportion of the genetic basis of common diseases can be considered to be the result of an inherited predisposition or genetic susceptibility. Common diseases result from a complex interaction of the effects of multiple different genes, or what is known as polygenic inheritance, with environmental factors and influences due to what is known as multifactorial inheritance (see Chapter 9).
Types and Mechanisms of Genetic Susceptibility
Genetic susceptibility for a particular disease can occur through single-gene inheritance of an abnormal gene product involved in a particular metabolic pathway, such as occurs in early coronary artery disease arising from familial hypercholesterolemia (FH) (p. 175). In an individual with a mutation in the FH gene, the genetic susceptibility is the main determinant of the development of coronary artery disease, but this can be modified by environmental alteration like reduction in dietary cholesterol and avoidance of other risk factors such as obesity, lack of exercise, and smoking.
Inheritance of single-gene susceptibility does not, however, necessarily lead to development of a disease. For some diseases, exposure to specific environmental factors will be the main determinant in the development of the disease (e.g., smoking or occupational dust exposure in the development of pulmonary emphysema in persons with α1-antitrypsin deficiency [p. 320, Table 23.1]).
In other instances, the mechanism of the genetic susceptibility is less clear-cut. This can involve inheritance of a single gene polymorphism (p. 67) that leads to differences in susceptibility to a disease (e.g., acetaldehyde dehydrogenase activity and alcoholism). In addition, inherited single-gene polymorphisms appear to determine the response to as yet undefined environmental factors—for example, the antigens of the major histocompatibility (HLA) complex and specific disease associations (p. 200) such as type 1 diabetes, rheumatoid arthritis, and coeliac disease. Lastly, genetic susceptibility can determine differences in responses to medical treatment; isoniazid inactivation status in the treatment of tuberculosis (p. 186) is a good example.
Approaches to Demonstrating Genetic Susceptibility to Common Diseases
In attempting to understand the genetics of a particular condition, the investigator can approach the problem in a number of ways (Box 15.1). These can include comparing the prevalence and incidence in various different population groups, the effects of migration, studying the incidence of the disease among relatives in family studies, comparing the incidence in identical and nonidentical twins, determining the effect of environmental changes by adoption studies, and studying the association of the disease with DNA polymorphisms. In addition, study can be made of the pathological components or biochemical factors of the disease in relatives (e.g., serum lipids among the relatives of patients with coronary artery disease). Study of diseases in animals that are homologous to diseases that occur in humans can also be helpful (p. 73). Before considering the use of these different approaches in a number of the common diseases in humans, specific aspects of some of these approaches will be discussed in more detail.
Family Studies
Genetic susceptibility to a disease can be suggested by the finding of a higher frequency of the disease in relatives than in the general population. The proportion of affected relatives of a specific relationship—first degree, second degree, and so forth—can provide information for empirical recurrence risks in genetic counseling (p. 346), as well as evidence supporting a genetic contribution (see Chapter 9). Familial aggregation does not, however, prove a genetic susceptibility, since families share a common environment. The frequency of the disease in spouses who share the same environment but who will usually have a different genetic background can be used as a control, particularly for possible environmental factors in adult life.
Twin Studies
Both members of a pair of twins are said to be concordant when either both are affected or neither is affected. The term discordant is used when only one member of a pair of twins is affected. Both types of twins will have a tendency to share the same environment but, whereas identical twins basically have identical genotypes (p. 106), nonidentical twins are no more similar genetically than brothers and sisters. If a disease is entirely genetically determined, then apart from rare events such as chromosome nondisjunction or a new mutation occurring in one of a twin pair, both members of a pair of identical twins will be similarly affected, but nonidentical twins are more likely to differ. If a disease is entirely caused by environmental factors, then identical and nonidentical twins will have similar concordance rates.
Polymorphism Association Studies
The widespread existence of inherited biochemical, protein, enzyme, and DNA variants (p. 147) allows the possibility of determining whether particular variants occur more commonly in individuals affected with a particular disease than in the population in general, or what is known as association. Although demonstration of a polymorphic association can suggest that the inherited variation is involved in the aetiology of the disorder, such as the demonstration of HLA associations in the immune response in the causation of the autoimmune disorders (p. 200), it may only reflect that a gene nearby in linkage disequilibrium (p. 138) is involved in causation of the disorder.
The human genome contains approximately 10 million single nucleotide polymorphisms (SNPs). Developments in high-throughput microarray SNP genotyping, together with information about SNP haplotypes (from the HapMap project [p. 148]) and the availability of large collections of DNA samples from patients with common diseases, have collectively enabled genome-wide association (GWA) studies (p. 149) to reliably identify numerous loci harbouring such variants.
Animal Models
Recognition of the same disease/disorder that occurs in humans and in another species such as the mouse allows the possibility of experimental studies that are often not possible in humans. In many instances, however, the disorder in the animal model will have a single-gene basis that has been identified or bred for. Nevertheless, spontaneously occurring or transgenic animal models (p. 73), experimentally induced for mutations in single genes involved in the metabolic processes or disease pathways of common diseases, will provide vital insights into the genetic contribution to these disorders.
Disease Models for Multifactorial Inheritance
The search for susceptibility loci and polygenes, sometimes also referred to as quantitative trait loci, in human multifactorial disorders has met with increasing success in recent years. This is largely due to the success of GWA studies (p. 149). Examples of recent research in some common conditions will be considered to illustrate the progress to date and the extent of the challenges that lie ahead.
Diabetes Mellitus
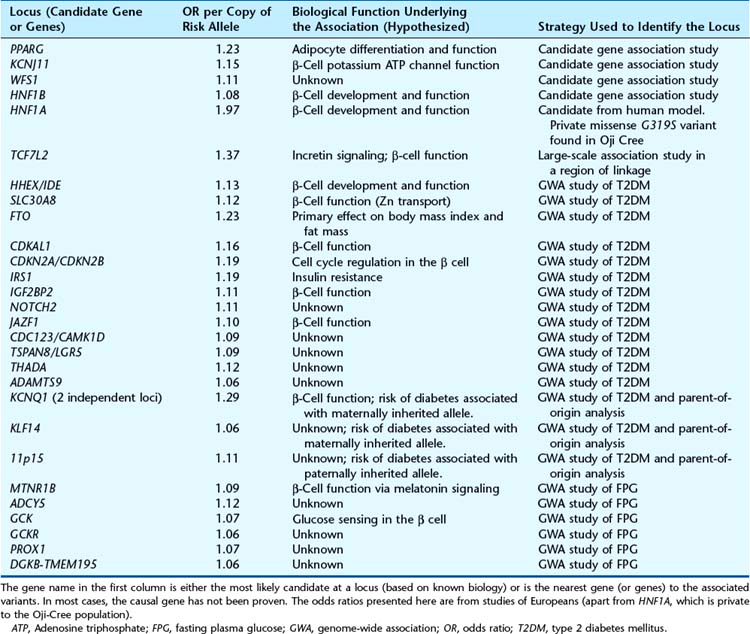
There are two main forms of diabetes mellitus (DM) that are clinically distinct. Type 1 (T1DM) is the rarer juvenile-onset, insulin-dependent form (previous abbreviation IDDM) which affects 0.4% of the population and shows a high incidence of potentially serious renal, retinal, and vascular complications. T1DM has a peak age of onset in adolescence and can only be controlled by regular injection of insulin. Type 2 is the more common later-onset, non–insulin-dependent form that affects up to 10% of the population. It usually affects older persons and may respond to simple dietary restriction of carbohydrate intake, although many persons with T2DM require oral hypoglycemic medication, and some require insulin. An additional 1% to 2% of persons with diabetes have monogenic (single gene) forms of diabetes (Table 15.1).

Diabetes can also occur secondary to a variety of other rare genetic syndromes and nongenetic disorders. Examples include Prader-Willi syndrome (p. 122), Bardet-Biedl syndrome, Wolfram syndrome, and Friedreich ataxia (Table 2.5, p. 24). Diabetes mellitus is therefore aetiologically heterogeneous.
Monogenic Forms of Diabetes
Rare forms of diabetes that show high penetrance within families are usually due to mutations in single genes. More than 20 monogenic forms of diabetes have been identified (see Table 15.1).
Maturity-Onset Diabetes of the Young
Mutations in five additional genes which encode transcription factors required for development of the β cell have been reported. The hepatocyte nuclear factor 1α (HNF1A) and hepatocyte nuclear factor 4α (HNF4A) genes were identified through positional cloning efforts and are associated with a more severe, progressive form of diabetes usually diagnosed during adolescence or early adulthood. These patients are sensitive to treatment with sulphonylurea tablets; this is an example of pharmacogenetics (see Chapter 12). Good glycemic control is important, as patients have a long duration of diabetes and may suffer from diabetic complications. Mutations in the HNF1A gene are the most common cause of MODY in most populations (65% of UK MODY), and HNF4A mutations are less frequent.
Neonatal Diabetes
Neonatal diabetes can be transient or permanent. More than 70% of cases of transient neonatal diabetes result from the overexpression of paternally expressed genes on chromosome 6q24. The inheritance and extent of this abnormality of imprinting (p. 121) are variable. However, in a small percentage of cases, patients have imprinting abnormalities besides 6q24 at multiple loci in the genome; these are associated with mutations in the zinc-finger transcription factor gene, ZFP57. Patients with a 6q24 abnormality are usually diagnosed in the first week of life and treated with insulin. Apparent remission occurs by 3 months, but there is a tendency for children to develop diabetes in later life.
Permanent neonatal diabetes does not remit, and until recently, patients were treated with insulin for life. The most common causes (>50%) are mutations in the KCNJ11 or ABCC8 genes which encode the Kir6.2 and SUR1 subunits of the adenosine triphosphate (ATP)-sensitive potassium (K-ATP) channel in the pancreatic β cell. Closure of these channels in response to ATP generated from glucose metabolism is the key signal for insulin release. The effect of activating mutations in these genes is to prevent channel closure by reducing the response to ATP, and hence insulin secretion. The most exciting aspect of this recent discovery is that most patients with this genetic aetiology can be treated with sulphonylurea drugs that bind to the channel and cause closure independently of ATP. Not only have patients been able to stop insulin injections and take sulphonylurea tablets instead, but they achieve better glycaemic control, which improves both their quality of life and later risk of diabetic complications.
Type 1 Diabetes
Initial research into the genetics of diabetes tended to focus on type 1 diabetes, where there is greater evidence for familial clustering (λs is 15 for T1DM versus 3.5 for T2DM [p. 146]). The concordance rates in monozygotic and dizygotic twins are around 50% and 12%, respectively. These observations point to a multifactorial aetiology with both environmental and genetic contributions. Known environmental factors include diet, viral exposure in early childhood, and certain drugs. The disease process involves irreversible destruction of insulin-producing islet β cells in the pancreas by the body’s own immune system, perhaps as a result of an interaction between infection and an abnormal genetically programmed immune response.
The first major breakthrough came with the recognition of strong associations with the HLA region on chromosome 6p21. The original associations were with the HLA B8 and B15 antigens that are in linkage disequilibrium with the DR3 and DR4 alleles (pp. 147, 200). It is with these that the T1DM association is strongest, with 95% of affected individuals having DR3 and/or DR4 compared with 50% of the general population. Following the development of PCR analysis for the HLA region, it was shown that the HLA contribution to T1DM susceptibility is determined by the 57th amino acid residue at the DQ locus, where aspartic acid conveys protection, in contrast to other alleles that increase susceptibility.
These two loci contribute λsvalues of approximately 3 and 1.3, respectively. However, the total risk ratio for T1DM is around 15. Confirmation that other loci are involved came first of all from linkage analysis using breeding experiments with the nonobese diabetic (NOD) strain of mice. These mice show a very high incidence of T1DM, with immunopathological features similar to those seen in humans. These linkage data pointed to the existence of 9 or 10 different susceptibility loci in mice. Following this, the results of numerous genome-wide linkage scans in humans provided evidence for the existence of between 15 and 20 susceptibility loci. However, apart from HLA, many of the other regions were not consistently replicated in independent studies. The largest and most recent linkage study in 2009 (2496 families and 2658 affected sibling pairs, p. 147) provided evidence of linkage to only one previously identified region of linkage besides HLA and INS: a region near to CTLA4. This locus was one of the only three (others encompassing the PTPN22 and IL2RA [CD25] genes) identified and confirmed in candidate gene association studies conducted between 1996 and 2007.
Since 2006, GWA studies p. 149) of increasing size have led to an explosion in the number of T1DM susceptibility loci supported by robust statistical evidence, bringing the total to over 40 distinct genomic locations. It is likely that many more remain to be identified through future, even larger, efforts. Most of the identified loci confer a modest increase in the risk of T1DM, with odds ratios (p. 148) ranging from 1.1 to 1.3 for each inherited allele, in contrast with the much larger role of the HLA locus. In most cases, the causal genes and variants underlying the associations have yet to be identified. However, the regions of association often encompass strong biological candidates—for example, the interleukin genes, IL10, IL19, IL20, and IL27. In two notable cases, follow-up studies have already enabled the causal gene to be confirmed, deepening our understanding of the biological pathways behind the associations.
The first example was a study of the IL2RA (CD25) locus by Dendrou et al. (2009). It used the UK-based Cambridge BioResource, a collection of approximately 5000 volunteers who can be recalled to participate in research on the basis of their genotype. Using fewer than 200 of these individuals, and by means of flow cytometry to assay the levels of CD25 protein expressed on the surface of T-regulatory cells, the study showed that people with the T1DM-protective haplotype expressed higher CD25 levels. This confirmed that IL2RA is indeed the causal gene and that the genotype-phenotype association is mediated via differences in expression of the gene product.
Type 2 Diabetes
Table 15.2 lists the known susceptibility loci for T2DM. There is no overlap with the T1DM loci, illustrating that these two diseases have very different aetiologies. Unlike the HLA and INS VNTR loci in T1DM, there are no major predisposing loci associated with T2DM. Most odds ratios are modest (between 1.05 and 1.3 per allele). As a result, large candidate gene and GWA studies (p. 146) have enabled progress in identifying the loci, whereas linkage studies (p. 146) have been underpowered and therefore unsuccessful.
Crohn Disease
In 2001, two groups working independently and using different approaches identified disease-predisposing variants in the CARD15 gene (previously known as NOD2). One of the groups, Ogura et al., had previously identified a Toll-like receptor (p. 193), NOD2, which activates nuclear factor Kappa-B (NFκB) (p. 201), making it responsive to bacterial lipopolysaccharides. The CARD15 gene is located within the 16p12 region and was therefore a good positional and functional candidate. Sequence analysis revealed three variants (R702W, G908R and 3020insC) that were shown by case-control and transmission disequilibrium tests to be associated with Crohn disease. The second group, Hugot et al., fine-mapped the 16p12 region by genotyping SNPs within the 20Mb interval and also arrived at the same variants within the CARD15 gene. These variants are found in up to 15% of patients with Crohn disease but only 5% of controls. The relative risk conferred by heterozygous and homozygous genotypes was approximately 2.5 and 40, respectively. For therapy, drugs which target the NFκB complex (p. 201) are already the most effective drugs currently available.
Since 2006, GWA studies have identified over 30 susceptibility loci for Crohn disease, all of which confer more modest risks of disease than the CARD15 variants (odds ratios per allele between 1.1 and 2.5). Discoveries of loci containing the IRGM and ATG16L1 genes were particularly exciting findings, as these genes are essential for autophagy, a biological pathway whose relevance to the disease was previously unsuspected. Further studies of the IRGM locus by McCarroll and colleagues (2008) identified that the causal variant is a 20-kb deletion immediately upstream of IRGM, which is in linkage disequilibrium (p. 138) with the associated SNPs. The deletion results in altered patterns of gene expression, which in turn were shown to modulate the autophagy of bacteria inside cells. Efforts are already underway to translate this finding into therapeutic applications.
Hypertension
Hypertension (chronically elevated blood pressure) leads to increased morbidity and mortality through a greater risk of stroke and coronary artery and renal disease. Various studies have shown that between 10% and 25% of the population is hypertensive, but the prevalence is age dependent, with up to 40% of 75- to 79-year-olds being hypertensive. Elevated blood pressure may contribute up to 50% of the global cardiovascular disease epidemic. There is substantial evidence that treatment of hypertension prevents development of these complications.
Genetic Factors in Hypertension
Family and twin studies have shown that hypertension is familial (Table 15.3) and that blood pressure correlates with the degree of relationship (Table 15.4). These findings suggest the importance of genetic factors in the aetiology of hypertension. In addition, there are differences in the prevalence of hypertension between populations, hypertension being more common in persons of Afro-Caribbean origin and less common in Eskimos, Australian Aborigines, and Central and South American Indians.
| Group | % |
|---|---|
| Population | 5 |
| 2 Normotensive parents | 4 |
| 1 Hypertensive parent | 8–28 |
| 2 Hypertensive parents | 25–45 |
From Burke W, Motulsky AG 1992 Hypertension. Chapter 10 in King RA, Rotter JI, Motulsky AG eds The genetic basis of common diseases. New York: Oxford University Press
Table 15.4 Coefficient of Correlation for Blood Pressure in Various Relatives
| Group | Correlation Coefficient |
|---|---|
| Siblings | 0.12–0.34 |
| Parent/child | 0.12–0.37 |
| Dizygotic twins | 0.25–0.27 |
| Monozygotic twins | 0.55–0.72 |
From Burke W, Motulsky AG 1992 Hypertension. Chapter 10 in King RA, Rotter JI, Motulsky AG eds The genetic basis of common diseases. New York: Oxford University Press
Susceptibility Genes
Common genetic variants also influence normal blood pressure variation. In 2009, very large meta-analyses and replication of GWA studies (p. 149) were published by the Global Blood Pressure Genetics (Global BPgen) consortium (N > 100,000 Europeans and > 12,000 Indian Asians) and the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) consortium (N > 29,136 Europeans). Fourteen genetic loci were robustly associated with either systolic or diastolic blood pressure, and all showed evidence of association with hypertension risk. Although the causal genes have not yet been confirmed, the loci highlighted likely candidates, including CYP17A1, rare mutations of which cause a form of adrenal hyperplasia (p. 174) characterized by hypertension.
Coronary Artery Disease
Coronary artery disease is the most common cause of death in industrialized countries and is rapidly increasing in prevalence in developing countries. It results from atherosclerosis, a process taking place over many years which involves the deposition of fibrous plaques in the subendothelial space (intima) of arteries, with a consequent narrowing of their lumina. Narrowing of the coronary arteries compromises the metabolic needs of the heart muscle, leading to myocardial ischemia, which if severe, results in myocardial infarction.
Family and Twin Studies
The risk to a first-degree relative of a person with premature coronary artery disease, defined as occurring before age 55 in males and age 65 in females, varies between 2 and 7 times that for the general population (Table 15.5). Twin studies of concordance for coronary artery disease vary from 15% to 25% for dizygotic twins and from 39% to 48% for monozygotic twins. Although these figures support the involvement of genetic factors, the low concordance rate for monozygotic twins clearly supports the importance of environmental factors.
Table 15.5 Recurrence Risks for Premature Coronary Artery Disease
| Proband | Relative Risk |
|---|---|
| Male (<55 Years) | |
| Brother | 5 |
| Sister | 2.5 |
| Female (<65 Years) | |
| Siblings | 7 |
Data from Slack J, Evans KA 1966 The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J Med Genet 3:239–257
Familial Hypercholesterolemia
The best-known disorder of lipid metabolism is familial hypercholesterolemia (FH) (p. 175). FH is associated with a significantly increased risk of early coronary artery disease and is inherited as an autosomal dominant disorder. It has been estimated that about 1 person in 500 in the general population, and about 1 in 20 persons presenting with early coronary artery disease, are heterozygous for a mutation in the LDLR (low-density lipoprotein receptor) gene. Molecular studies in FH have revealed that it is due to a variety of defects in the number, function, or processing of the LDL receptors on the cell surface (p. 176).
Susceptibility Genes
Since 2007, numerous large-scale GWA and follow-up replication studies have identified 12 susceptibility loci for coronary artery disease and myocardial infarction. The strongest association identified is on chromosome 9p21 (odds ratio per allele ≈ 1.3). The nearest genes, CDKN2A and CDKN2B, are over 100 kb away. Interestingly, the SNPs most strongly associated with coronary artery disease are only 10 kb away from those associated with type 2 diabetes (see Table 15.2). However, the two disease associations are independent and not in linkage disequilibrium with one another. Much work is already being done to investigate the role of ANRIL, a large, noncoding RNA which overlaps with the coronary artery disease–associated haplotype. It is expressed in tissues associated with atherosclerosis, and initial studies have shown correlations between expression of ANRIL transcripts and severity of atherosclerosis. However, additional evidence from large-scale association studies has shown that the same haplotype on 9p21 is associated with abdominal aortic aneurysm and intracranial aneurysm, suggesting that its role is not limited to atherosclerotic disease. Together with the other 11 loci, the locus at 9p21 only explains a small fraction of the heritability of coronary artery disease, and it is likely that many more loci will be identified.
Progress in uncovering susceptibility loci has also come from large GWA studies of lipid levels. Common variants at at least 30 loci are now robustly associated with circulating levels of lipids, with over one-third of these associated with LDL levels. Kathiresan and colleagues (2008) showed that individuals who inherit a higher number of LDL-raising alleles at these loci are more likely to have clinically high LDL cholesterol (>160 mg/dL) than those who inherit few alleles. The frequency of these LDL-raising alleles is higher in patients with coronary artery disease than in controls, indicating that they predispose to the disease via their primary effect on LDL levels. In many cases, the genes implicated by the loci are already associated with single-gene disorders. For example, PCSK9 harbours a full spectrum of LDL-altering alleles, from rare mutations which cause large differences in LDL (>100 mg/dL), through low-frequency variants with more modest effects (e.g., PCSK9 R46L has a 1% minor allele frequency and a 16 mg/dL effect size), to common variants at 20% minor allele frequency which change LDL levels by less than 5 mg/dL. The resequencing of further loci is likely to uncover rarer variants and mutations at lipid trait loci, which may further explain genetic susceptibility to coronary artery disease.
Schizophrenia
Family and Twin Studies
The results of several studies of the prevalence of schizophrenia and schizoid disorder among the relatives of schizophrenics are summarized in Table 15.6. If only schizophrenia is considered, the concordance rate for identical twins is only 46%, suggesting the importance of environmental factors. If, however, schizophrenia and schizoid personality disorder are considered together, then almost 90% of identical co-twins are concordant.
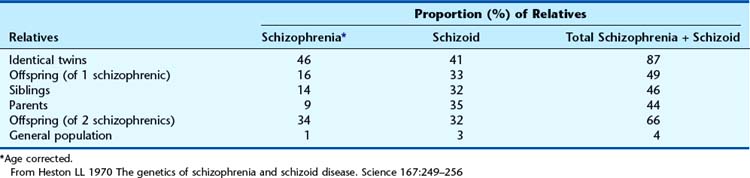
Susceptibility Genes
Genome-wide association studies of copy number variations (CNVs) have identified large (>500 kb) deletions associated with the condition—for example on chromosomes 1q21.1, 15q13.3, and 22q11.2 (p. 282). These deletions are rare but penetrant: the odds ratio for the 15q13.3 deletion has been estimated at between 16 and 18 in two independent studies. A key observation is that these deletions are not only associated with schizophrenia. The 1q21.1 deletion (pp. 284, 287) has also been associated with autism, learning disability, and epilepsy. Thus, current clinically defined disease boundaries are not mirrored by the underlying genetics. While these deletions explain some of the genetic susceptibility to schizophrenia, they also explain susceptibility to other conditions. It is likely that a better understanding of the genetics will lead to better definition of clinical phenotypes.
Common genetic variants are also implicated in the aetiology of schizophrenia. Recent meta-analyses of GWA studies have identified associations with the HLA region on chromosome 6p21.3-6p22.1, suggesting an immune system component to the risk of disease. Robust associations have also been observed with variants near the NRGN gene and in the TCF4 gene, which implicate biological pathways involved in brain development, cognition, and memory. Analyses of existing GWA data conducted by the International Schizophrenia Consortium in 2009 suggested that there are likely to be thousands more common variants of small effect that collectively explain much of the heritability of schizophrenia.
Alzheimer Disease
Dementia is characterized by an irreversible and progressive global impairment of intellect, memory, social skills, and control of emotional reactions in the presence of normal consciousness. Dementia is aetiologically heterogeneous, occurring secondarily to both a variety of nongenetic causes such as vascular disease and infections such as AIDS, as well as genetic causes. Alzheimer disease (AD) is the most common cause of dementia in persons with either early-onset dementia (less than the age of 60 years, or presenile) or late onset (greater than age 60 years, or senile). The classic neuropathological finding in persons with AD is the presence at postmortem examination of amyloid deposits in neurofibrillary tangles and neuronal or senile plaques. In addition, individuals with Down syndrome have an increased risk of developing dementia (p. 273), which at postmortem has identical CNS findings to those seen in persons with typical AD.
Epidemiology
Limited numbers of studies of the incidence and prevalence of AD are available, owing to problems of ascertainment. However, the risk of developing AD clearly increases dramatically with age (Table 15.7).
Table 15.7 Estimates of Age-Specific Cumulative Prevalence of Dementia
| Age Interval (Years) | Prevalence (%) |
|---|---|
| <70 | 1.3 |
| 70–74 | 2.3 |
| 75–79 | 6.4 |
| 80–84 | 15.3 |
| 85–89 | 23.7 |
| 90–94 | 42.9 |
| >95 | 50.9 |
From Heston LL 1992 Alzheimer’s disease. Chapter 39 in King RA, Rotter JI, Motulsky AG eds The genetic basis of common diseases. New York: Oxford University Press
Single-Gene Disorders
The identification of APP in the amyloid deposits of the neuronal plaques, its mapping in or near to the critical region of the distal part of chromosome 21q associated with the phenotypic features of Down syndrome (p. 273), and the increased risk of AD in persons with Down syndrome led to the suggestion that duplication of the APP gene could be a cause of AD. Evidence of linkage to the APP locus was found in studies of families with early-onset AD, and it is now known that mutations in the APP gene account for a small proportion of cases.
Evidence of linkage to early-onset AD was found for another locus on chromosome 14q. Mutations were identified in a proportion of affected individuals in one of a novel class of genes known as presenilin-1 (PSEN1), now known to be a component of the notch signaling pathway (p. 86). A large number of mutations in PSEN1 have now been identified and account for up to 70% of familial early-onset AD. A second gene, presenilin-2 (PSEN2), with homology to PSEN1, was mapped to chromosome 1q and has been shown to have mutations in a limited number of families with AD. PSEN1 and PSEN2 are integral membrane proteins containing multiple transmembrane domains that localize to the endoplasmic reticulum and the Golgi complex. All of the presenile dementias following autosomal dominant inheritance demonstrate high penetrance.
Susceptibility Genes
Polymorphisms in the apolipoprotein E (APOE) gene are the most important genetic risk factor identified for late-onset AD. The locus was initially identified in the early 1990s through linkage studies. The APOE gene has three major protein isoforms, ε2, ε3, and ε4. Numerous studies in various populations and ethnic groups have shown an increased frequency of the ε4 allele in persons with both sporadic and late-onset familial AD. In addition, the ε2 allele is associated with a decreased risk of the disease. The finding of apolipoprotein E in senile plaques and neurofibrillary tangles, along with its role in lipid transport, possibly in relation to the nerve injury and regeneration seen in AD, provides further evidence for a possible role in the acceleration of the neurodegenerative process in AD.
Hemochromatosis
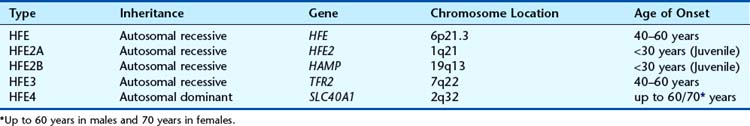
Genetic Heterogeneity
Hemochromatosis is a genetically heterogeneous disorder (Table 15.8), with mutations also reported in the transferrin receptor 2 (TFR2) gene and the SLC40A1 gene which encodes ferroportin. In addition to the common recessive adult-onset form, there is a rare juvenile form with iron overload and organ failure before the age of 30 years, which is lethal if untreated. Neonatal hemochromatosis is a severe form of unknown aetiology.
Venous Thrombosis
Venous thrombosis represents a major health problem worldwide, with increasing incidence from 1 in 100,000 during childhood to 1 in 100 in old age. Venous thromboembolism, including deep vein thrombosis and pulmonary embolism, is a complex disease that results from multiple interactions between inherited and acquired risk factors (Box 15.2). Inherited thrombophilias also increase the risk of fetal loss, both stillbirths and early miscarriages.
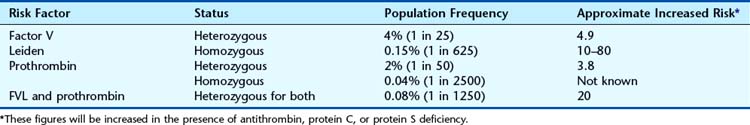
Identification of Genes
The identification of two low-frequency variants prevalent in Europeans has increased our understanding of venous thrombosis. The factor V Leiden variant (R506Q) renders the factor V protein resistant to cleavage by activated protein C, thus increasing generation of thrombin. The prothrombin variant G20210A is located in the 3’ UTR and is associated with increased prothrombin levels. These variants confer a four- to fivefold increased risk of thrombosis in the heterozygous state (Table 15.9), but individuals homozygous for one or heterozygous for both are at significantly elevated risk (up to 80-fold).
Age-Related Macular Degeneration
Associations have subsequently been confirmed with variants in the locus containing the complement component 2 (C2) and complement factor B (CFB) genes and in the complement component 3 (C3) gene. In addition, there is strong and well-replicated evidence for association at ARMS2 on chromosome 10q26. The known variants collectively account for a substantial proportion of the heritability of AMD, explaining at least half of the risk to siblings. Their cumulative effects on risk are additive, with no evidence of epistasis. Molecules involved in complement activation and its regulation are now prime targets for therapeutic intervention in AMD.
Adams PC. Hemochromatosis: clinical implications. Medscape Gastroenterology eJournal. 4, 2002.
A summary of clinical aspects of hemochromatosis and the role of genetic testing.
Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955-962.
Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055-1059.
Heston LL. Psychiatric disorders in foster home reared children of schizophrenic mothers. Br J Psychiatry. 1966;112:819-825.
A classic paper demonstrating genetic factors in the etiology of schizophrenia.
Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn disease. Nature. 2001;411:599-603.
Kathiresan S, Willer CJ, Peloso G, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2008;41:56-65.
UK Type 2 Diabetes Genetics ConsortiumLango H, Palmer CN, Morris AD, Zeggini E, et al. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57:2911-2914.
Recent publication describing the effects of multiple variants that predispose to type 2 diabetes.
Nejentsev S, Walker N, Riches D, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387-389.
Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with Crohn disease. Nature. 2001;411:603-606.
Prokopenko I, McCarthy MI, Lindgren C. Type 2 diabetes: new genes, new understanding. Trends Genet. 2008;24:613-621.
Seligsohn MD, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344:1222-1231.
A comprehensive review of hereditary thrombophilia.
Wicker LS, Clark J, Fraser HI, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25(Suppl):29-33.
Elements