Chapter 62 Genetic Diseases
The Long QT Syndrome
Congenital long QT syndrome (LQTS) is a relatively uncommon but important clinical disorder. Since 1975, under the unifying name of the “long QT syndrome,” it has been considered to include two hereditary variants.1 Jervell and Lange-Nielsen syndrome (J-LN) is associated with deafness, and Romano-Ward syndrome (R-W) is not.2–5
Molecular Genetics of Long QT Syndrome
The list of LQTS genes continues to grow. At the latest count, 12 of them had been identified, and the number is certain to increase. For practical purposes, however, the first three genes identified continue to remain the most important. This chapter will review only essential concepts; for additional details, interested readers are referred to more extensive reviews.6
KCNQ1 (LQT1) and KCNE1 (LQT5)
Expression studies of mutated proteins have suggested multiple mechanisms of functional failure. Defective proteins may coassemble with wild-type proteins and exert a dominant negative effect. Other mutations lead to defective proteins that do not assemble with wild-type peptides, which results in a loss of function that reduces the IKs current by 50% or less (haplo-insufficiency). Finally, defective peptides may not even reach the membrane of the cardiac cell because the mutations interfere with intracellular protein trafficking.7
KCNH2 (LQT2) and KCNE2 (LQT6)
The KCNH2 and the KCNE2 gene encode the α (HERG)-subunit and the β (MIRP)-subunit, respectively, of the potassium channel conducting the IKr current. This is the second most common variant of LQTS accounting for 35% to 40% of mutations in patients with the LQTS genotype. Mutations in KCNH2 cause a reduction of IKr current. Defective proteins may cause a dominant negative effect on the wild-type subunits, or they may not interfere with the function of the normal subunits, thus causing haplo-insufficiency. Trafficking abnormalities are another consequence of KCNH2 mutations.7
SCN5A (LQT3)
The SCN5A gene encodes the protein of the cardiac sodium channel. In vitro expression studies have shown that LQTS-SCN5A mutations produce the LQTS phenotype by inducing a “gain of function” leading to increase in the Na+ inward current, which prolongs the action potential duration. The prevalence of LQT3 among LQTS patients is around 10%.8
CACNA1c (LQT8): Timothy Syndrome
LQT8 is a rare variant characterized by marked Q-T interval prolongation, often presenting with 2 : 1 functional atrioventricular (AV) block, macroscopic T-wave alternans, and syndactyly. LQT8 is highly malignant and 10 (59%) of 17 of the children reported by Splawski et al died at a mean age of 2.5 years. Additional abnormalities may be present in Timothy syndrome.9
This variant has been associated, so far, with one specific missense mutation (G406R) in the voltage-gated calcium channel gene (CACNA1c).9 G406R produces sustained inward Ca2+ currents by causing nearly complete loss of voltage-dependent inactivation.9 In the heart, prolonged Ca2+ current delays cardiomyocyte repolarization and increases the risk of arrhythmia.
Prevalence
In 1975, it was already suggested that LQTS “could be more unrecognized than rare.”1 However, until now the prevalence was assumed to be anywhere between 1 in 5000 and 1 in 20,000. None of these estimates was based on actual data.
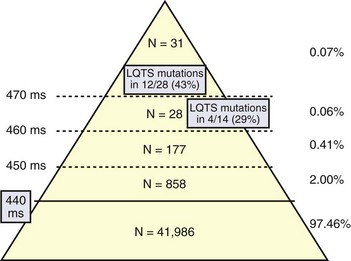
The first data-based assessment of the prevalence of LQTS comes from the largest prospective study of neonatal electrocardiography ever performed, which involved 44,596 infants of 3 to 4 weeks of age in 18 Italian maternity hospitals.10 An electrocardiogram (ECG) was performed in 44,596 infants 15 to 25 days old (43,080 whites). In infants with a QTc greater than 450 ms, the ECG was repeated within 1 to 2 weeks. Genetic analysis, by screening seven LQTS genes, was performed in 28 (90%) of 31 and in 14 (50%) of 28 infants with a QTc greater than 470 ms or between 461 and 470 ms, respectively, regarded as markedly prolonged by the European Task Force on Neonatal Electrocardiography.11 QTc readings of 451 to 460 ms, 461 to 470 ms, and greater than 470 ms were observed in 184 (0.41%), 28 (0.06%), and 31 (0.07%) infants, respectively (Figure 62-1). Among genotyped infants, disease-causing mutations were found in 12 (43%) of 28 with a QTc greater than 470 ms and in 4 (29%) of 14 with a QTc of 461 to 470 ms. One genotype-negative infant (QTc of 482 ms) was diagnosed to be affected by LQTS on clinical grounds. Among family members of genotype-positive infants, 51% were found to carry disease-causing mutations. In total, 17 of 43,080 white infants were affected by LQTS, which demonstrated a prevalence of at least 1 per 2534 apparently healthy live births (95% confidence interval [CI] 1:1 to 583 to 1:4 to 350). Among them, 1.4% had a QTc between 440 ms and 469 ms and 0.7 of 1000 had a QTc of 470 ms or greater. As 43% of the infants with QTc 470 ms or greater (0.7 per 1000) and 29% of the infants with a QTc between 460 ms and 469 ms have disease-causing mutations and as at least some of the infants with a QTc between 450 ms and 459 ms are also likely to be mutation carriers, it follows that the prevalence of LQTS must be close to 1 per 2000. This does not include the silent mutation carriers (QTc < 440 ms), a group ranging between 10% and 36% according to genotype.6 This is the first time that the prevalence of a cardiac disease of genetic origin has been quantified on the basis of actual data.
Clinical Presentation
The large increase in the number of patients diagnosed as affected has significantly modified the perception of the natural clinical history of the disease. Since the early 1970s, when the first consistent series of patients were reported, the typical clinical presentation of LQTS was considered to be the occurrence of syncope or CA, precipitated by emotional or physical stress, in a young individual with a prolonged Q-T interval on the surface ECG.1 If these symptomatic patients were left untreated, the syncopal episodes would recur and eventually prove fatal in the majority of cases. This concept was largely based on the fact that the patients initially diagnosed were those most severely affected. It has become progressively evident that this traditional picture represents an oversimplification, and it now appears that even though some patients have the severe manifestations described above, numerous patients have a very benign course as well. Unfortunately, the occasional occurrence of sudden death cannot yet be predicted. Evidence that modifier genes, some of them associated with the release of autonomic mediators, contribute to the highly different severity of the clinical manifestations of LQTS is growing.12–15
When family screening is performed, prolongation of the Q-T interval can be often detected, and a family history of fainting episodes or of sudden unexpected deaths in an early age is often present. Nonetheless, numerous sporadic cases (approximately 30%), that is, patients with syncope and a prolonged Q-T interval but without clinical evidence of familial involvement, do exist.6
The clinical history of repeated episodes of loss of consciousness under emotional or physical stress is typical and unique and is therefore unmistakable; however, the physician must be aware of the existence of LQTS to make the correct diagnosis. However, the clinical presentation is not always clear, so sometimes the diagnosis may be uncertain. The highly malignant J-LN syndrome will not be discussed here2; it must, however, be noted that its clinical course is very severe and that the patients become symptomatic at a very early age (Figure 62-2). Interested readers are referred to a comprehensive report on this variant.3
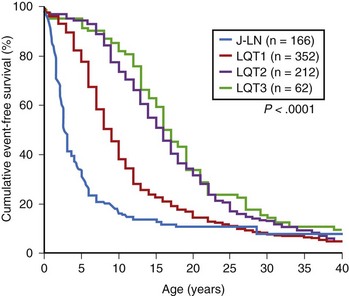
FIGURE 62-2 Kaplan-Meier curves of event-free survival. A, all patients with Jervell and Lange-Nielsen (J-LN) syndrome. B, All patients with J-LN, with a magnified view of the events occurring in the first 15 years of life. C, Patients with J-LN versus symptomatic patients with Romano-Ward syndrome of known genotype.10 D, Patients with J-LN versus symptomatic patients with LQT1 and an unselected LQT1 population.10,12
(From Schwartz PJ, Spazzolini C, Crotti L, et al: The Jervell and Lange-Nielsen syndrome. Natural history, molecular basis, and clinical outcome, Circulation 113:783–790, 2006.)
The two cardinal manifestations of LQTS are syncopal episodes and electrocardiographic abnormalities. The latter have been repeatedly described. Interested readers are referred to previous reviews.5 Here we will mention only T wave alternans and a specific echocardiographic pattern.
T-Wave Alternans
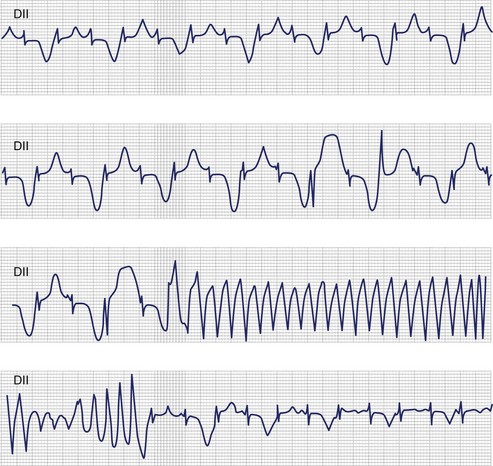
In 1975, Schwartz proposed that T-wave alternans represents a characteristic ECG feature of LQTS.16 Beat-to-beat alternation of the T wave, in polarity or amplitude, may be present at rest for brief moments but usually appears during emotional or physical stress and may precede torsades de pointes (TdP) (Figure 62-3). T-wave alternans is a marker of major electrical instability, and it identifies patients at particularly high risk. Its transient nature limits the possibility of its observation. This is a rather gross phenomenon that should not go unnoticed, when present. The observation of T-wave alternans in a patient with LQTS who is already on therapy strongly suggests the presence of a persisting high degree of cardiac electrical instability, which should prompt reassessment of therapy.
Echocardiographic Abnormalities
LQTS is still regarded by many as a purely electrical disease that does not involve any mechanical alterations. However, a case-control study demonstrated the frequent presence of highly unusual echocardiographic abnormalities among the patients.17 These abnormalities included an increased rate of thickening in the early phase of contraction and the presence of a slow movement in the late thickening phase with a plateau morphology sometimes accompanied by a second peak. Recently, these findings have been fully confirmed by a Norwegian study.18,19 LQTS is, therefore, not a purely electrical disease. These abnormalities were more frequent in symptomatic patients than in asymptomatic patients, which suggests that these abnormalities may reflect the presence of an arrhythmogenic mechanism. The evidence that the calcium-entry blocker verapamil completely normalizes the contraction pattern suggests that symptomatic LQTS patients may have an abnormal increase in the intracellular calcium concentration before relaxation is completed and that this may be related to an early after-depolarization (EAD): The contraction abnormality would be the mechanical equivalent of an EAD.20 The inward flux of calcium linked to the EAD may cause a small but rapid increase in intracellular calcium, which may be sufficient to trigger the calcium-induced release of calcium from the sarcoplasmic reticulum, causing a much greater increase in cytosolic calcium and the occurrence of a second contraction or the prolongation of the contraction itself.
Cardiac Events and Their Relationship to Genotype
The syncopal episodes are caused by TdP often degenerating into ventricular fibrillation. Long pauses facilitate the onset of TdP.21 Although most LQTS patients develop their symptoms under stress, sometimes these life-threatening cardiac events occur at rest. The reason(s) for these different patterns remained obscure until molecular biology was able to distinguish among different genotypes. In 1995, an unexpected observation was made that among a tiny group of genotyped individuals, LQT2 patients appeared to be at higher risk during emotional stress, whereas LQT3 patients had their events mostly at rest or during sleep; this fostered a large and targeted study that shed light on this issue of major clinical relevance.22,23
In 670 LQTS patients of known genotype and who all had symptoms (syncope, CA, or sudden death), Schwartz et al examined possible relationships between genotype and the conditions (“triggers”) associated with the events.23 As predicted by their impairment on the IKs current (essential for QT shortening during increases in heart rate), most of the events in LQT1 patients occurred during exercise or stress. Swimming is especially dangerous for them, and 99% of the events which occurred while swimming did involve LQT1 patients. Conversely, most of the events (including the lethal ones) in LQT2 patients occurred during emotional stress such as auditory stimuli (e.g., sudden noises and telephone ringing, especially occurring while at rest), and most of the events of LQT3 patients occurred while they were asleep or at rest (Figure 62-4).
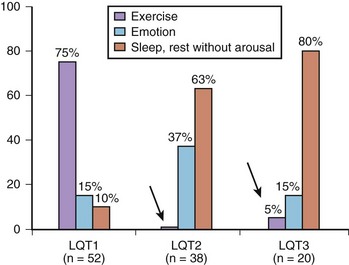
FIGURE 62-4 Lethal cardiac events according to triggers and genotype. Numbers in parentheses are triggers, not patients.
(Modified from Schwartz PJ, Priori SG, Spazzolini C, et al: Genotype-phenotype correlation in the long QT syndrome. Gene-specific triggers for life-threatening arrhythmias, Circulation 103:89–95, 2001.)
In women, even during the postpartum period, genotype is important because the risk is higher for patients with LQT2 than for those with LQT1.24,25 The higher risk for women with LQT2 is probably partly related to sleep disruption; accordingly, it is recommended that the spouses or other caregivers take on some of the night-time feeding of the infants, thus allowing a fair amount of uninterrupted sleep for the women with LQT2.
These significant genotype-phenotype correlations have an impact on management.
Molecular Diagnosis in Long QT Syndrome
Who Should Be Screened for Long QT Syndrome and Medico-legal Implications
Molecular diagnosis should always be attempted whenever LQTS has either been diagnosed or is suspected on sound clinical grounds. When successful (80% to 85% of cases in the author’s laboratory), it allows the rapid screening of all the family members and the identification of all the “silent mutation carriers” (individuals with a disease-causing mutation and a QTc less than 440-ms interval) whose prevalence increases from 10% for LQT3 to 37% for LQT1 patients.26
Molecular Genetics and Risk Stratification
Molecular genetics contribute importantly to risk stratification. In 2003, data on 647 patients of known genotype from 193 families indicated that the incidence of life-threatening events was lower among LQT1 patients, but this was partly because of the high prevalence of silent mutation carriers (QTc < 440 ms); the risk was higher among females with LQT2 compared with males with LQT2 and among males with LQT3 compared with females with LQT3. Independent of genotype, the risk of becoming symptomatic was strongly correlated with QTc and was markedly greater with QTc greater than 500 ms. Most of the patients with LQT1 go through life without ever having cardiac events; also, among patients with LQT2 and LQT3, almost half of them remain asymptomatic. The fact that this is often forgotten is shown by the growing preference for implantable cardioverter-defibrillator (ICD) implantations in asymptomatic individuals just because they have been diagnosed with LQTS.27
In 2002, Moss et al indicated that patients with LQT2 with mutations in the pore region were at higher risk compared with patients with LQT2 with mutations in different regions of the same gene.28 In 2007, Moss et al demonstrated in 600 patients with LQT1 that both the transmembrane location of the mutations and their dominant-negative effect are independent risk factors for cardiac events.29 Shortly thereafter, Crotti et al focused on the hot spot KCNQ1-A341V, a common mutation found responsible for a founder effect in 25 South African families, and demonstrated that the unusually high clinical severity already reported by Brink et al in the South African families is present also among patients with LQT1 from different ethnic backgrounds but carrying the same A341V mutation.30,31 Moreover, as KCNQ1-A341V has a mild dominant-negative effect (the current loss barely exceeds 50%), its striking clinically severe phenotype is explained neither by the location (transmembrane) nor by the functional consequence of the mutation (dominant-negative). This implies that the current biophysical assessments of the electrophysiological effects of LQTS-causing mutations do not provide all the information necessary to make a complete genotype-phenotype correlation. In this regard, the study by Crotti et al paves the way for a mutation-specific risk stratification.30
The risk stratification process is complicated by the presence of additional genetic variants that may modify clinical severity, as demonstrated in a family with LQT2 with C-terminal A1116V mutation, where the risk for life-threatening events was increased by the presence of the very common KCNH2-K897T polymorphism. Electrophysiological evidence did show that K897T produces an accentuation of the mutation-dependent IKr current loss resulting in the unmasking of a clinically latent C-terminal LQT2 mutation.13 This finding has very recently confirmed by Antzelevitch’s group in sudden infant deaths.32
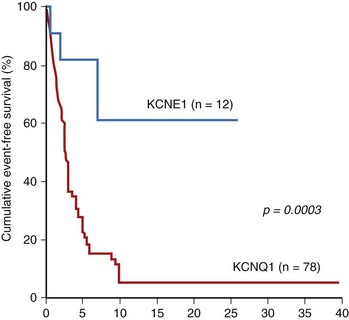
Genetic variations in NOS1AP, which encodes a nitric oxide synthase adaptor protein, contribute to Q-T interval duration in the general population.33 Accordingly, the author’s group tested in their enlarged South African founder population (500 subjects, 205 mutation carriers) the hypothesis that NOS1AP is a genetic modifier of LQTS. The main findings were that two NOS1AP variants (rs4657139 and rs16847548) were significantly associated with occurrence of symptoms, with clinical severity manifested by an almost double probability for CA and sudden death, and with a greater likelihood of having a Q-T interval in the top 40% of the values among all mutation carriers. This is the first evidence, demonstrated in subjects sharing the same mutation, that NOS1AP is a genetic modifier of LQTS and that some of its variants are associated with a greater risk for CA and sudden death.15 One of the best evidences for the value of molecular biology in risk stratification comes from J-LN syndrome, in which a striking difference in prognosis is seen, according to the gene involved, KCNQ1 or KCNE1 (Figure 62-5).3
Clinical Diagnosis
Given the characteristic features of LQTS, the typical cases present no diagnostic difficulty for the physicians who are aware of the disease. However, borderline cases are more complex and require the evaluation of multiple variables besides clinical history and ECG studies. Diagnostic criteria were proposed in 1985 and subsequently updated in 1993 and in 2006.4,34–36
The new diagnostic criteria are listed in Table 62-1. In the experience of the author of this chapter, the point score was arbitrarily divided into three probability categories: (1) ≤1 point = low probability of LQTS; (2) >1 to 3 points = intermediate probability of LQTS; and (3) ≥3.5 points = high probability of LQTS. Whenever a patient receives a score of 2 to 3 points, serial ECGs and especially several 24-hour Holter recordings should be obtained because the QTc value in patients with LQTS may vary from time to time and from day to night. In this group, with intermediate probability of LQTS, the presence of additional morphologic abnormalities may help in diagnostic decisions. Often, precordial leads are more informative.
Table 62-1 1993–2006 Long QT Syndrome (LQTS) Diagnostic Criteria
| POINTS | |
|---|---|
| ELECTROCARDIOGRAPHIC FINDINGS* | |
| >480 ms | 3 |
| A. QTc† | |
| 460–470 ms | 2 |
| 450–459 (male) ms | 1 |
| B. Torsade de pointes‡ | 2 |
| C. T-wave alternans | 1 |
| D. Notched T wave in three leads | 1 |
| E. Low heart rate for age§ | 0.5 |
| CLINICAL HISTORY | |
| With stress | 2 |
| A. Syncope‡ | |
| Without stress | 2 |
| B. Congenital deafness | 0.5 |
| FAMILY HISTORY¶ | |
| A. Family members with definite LQTS | 1 |
| B. Unexplained sudden cardiac death before age 30 years among immediate family members | 0.5 |
Score: ≤1 point = low probability of LQTS; >1 to 3 points = intermediate probability of LQTS; ≥3.5 points = high probability of LQTS.
* In the absence of medications or disorders known to affect these electrocardiographic features.
† QTc calculated by Bazett’s formula, where 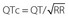 .
.
§ Resting heart rate below the second percentile for age.
Therapy
Antiadrenergic Interventions
The most significant information on therapy still comes from a 1985 study, which included 233 symptomatic patients with detailed clinical information on the time of first syncope and with an adequate follow-up.37 The mortality at 15 years after the first syncope was 9% in the group treated by antiadrenergic therapy (β-blockers, sympathectomy, or both) and more than 53% in the group not treated or treated by miscellaneous therapies not including β-blockers. These data conclusively demonstrated that pharmacologic antiadrenergic therapy, surgical antiadrenergic therapy, or both radically modify the prognosis for symptomatic patients with LQTS. In 2009, mortality among patients treated with β-blockers and left cardiac sympathetic denervation (LCSD) dropped to around 1%. The dramatic success of carefully executed therapies underscores the unacceptability of symptomatic patients with LQTS being either undiagnosed or incorrectly treated.
β-Blockers
In a large number of patients of unknown genotype, mortality on β-blocker therapy was 2%, and it was 1.6% when limited to patients with syncope (no cardiac arrest) and without events in the first year of life.38 Clear evidence that β-blockers are extremely effective in LQT1 patients does exist. Data from two large studies reported mortality around 0.5% and sudden death combined with CA up to 1%.23,39 The impairment in the IKs current makes these patients particularly sensitive to catecholamines and quite responsive to β-blockade. These patients seldom need more than antiadrenergic therapy.
Particularly, important information has come from a study published in 2009.40 Vincent et al performed a retrospective study of the details surrounding cardiac events in 216 patients with genotyped LQT1, who were treated with β-blockers and were followed up for 10 years. Before β-blocker therapy, cardiac events occurred in 157 patients (73%) at a median age of 9 years, with CA in 26 (12%). After β-blockers, 75% were asymptomatic, and the risk for life-threatening cardiac events was reduced by 97% (P < .001). Twelve patients (5.5%) had a CA or sudden death, but 11 (92%) of 12 were noncompliant (n = 8), were on a QT-prolonging drug (n = 2), or both (n = 1) at the time of the event. The risk for CA or sudden death in compliant patients not taking QT-prolonging drugs was dramatically less compared with noncompliant patients on QT-prolonging drugs (odds ratio, 0.03; 95% CI, 0.003 to 0.22; P < .001). None of the 26 patients with CA before β-blocker had CA or sudden death on β-blockers. The conclusion was that β-blockers are extremely effective in treating LQT1. Noncompliance with β-blocker therapy and use of QT-prolonging drugs are responsible for almost all life-threatening β-blocker “failures.”
Partly because of the small numbers available (which affect percentages), a concept unsupported by firm evidence has rapidly spread—that LQT3 patients are not protected at all by β-blockers or by antiadrenergic therapy. In fact, actual data point to a profound difference seen among these patients on the basis of the age of the first manifestation. On the one hand, if cardiac events occur during the first year of life, then β-blockers are insufficient and the disease has a highly malignant course. On the other hand, with a mean follow-up of 9 years, patients without cardiac events in the first year of life have done extremely well with β-blockers, LCSD, or both.41 Larger studies will have to be performed, but available evidence indicates that a molecular diagnosis of LQT3 in an asymptomatic patient should never lead to an automatic decision for ICD implantation.
Left Cardiac Sympathetic Denervation
A thorough description of LCSD has recently been published.42 Following a small incision in the left subclavicular region, LCSD is performed using an extrapleural approach, which makes thoracotomy unnecessary. The average time for surgery is 35 to 40 minutes. LCSD requires the removal of the first four thoracic ganglia. The cephalic portion of the left stellate ganglion is left intact to avoid Horner’s syndrome. In almost 30% of patients, a very modest (1 to 2 mm) ptosis, which can be noted only by close examination but fully escapes notice in normal social interactions, results.
The latest data published in 2004 include 147 patients with LQTS who underwent LCSD during the past 35 years.43 They represented a very high-risk group, as 99% were symptomatic, their mean QTc being very long (563 ± 65 ms), 48% had a CA, and especially 75% continued to have syncope despite full-dose β-blockers. The data most relevant to current clinical decisions are those regarding patients without cardiac arrest (who almost always should receive an ICD) who have syncope despite being treated with a full dose of β-blockers. During a mean follow-up of 8 years, a 91% reduction in cardiac events was observed. LCSD produced a mean QTc shortening of 39 ms, which indicates an action on the substrate as well as on the trigger. Mortality was 3% in this high-risk group. A postsurgery QTc less than 500 ms predicted a highly favorable outcome. Importantly, this series included five patients who underwent LCSD because they had experienced multiple ICD shocks and electrical storms: in this group, over a 4-year follow-up, a 95% decrease in the number of shocks (from an average of 29 shocks per year), with a dramatic improvement in the quality of life of the patients and of their families, was observed.
The major antifibrillatory efficacy of LCSD has been previously demonstrated in high-risk patients with post–myocardial infarction; and recently, it was demonstrated in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT), who were not protected by β-blockers and were receiving multiple appropriate shocks from the ICD.44,45 The Boston Children Hospital and the Mayo Clinic have recently published their highly successful experience with LCSD in both LQTS and CPVT.46–48
Implantable Cardioverter-Defibrillators
The U.S. and the European ICD-LQTS Registries provide the disquieting information that the majority of implanted patients had not had a CA and that many had not even failed β-blocker therapy.49,50 A recent multicenter study went so far as to indicate that the mere presence of an SCN5A mutation, even in a totally asymptomatic individual, is sufficient for immediate ICD implantation.27
It should not be forgotten that ICDs do not prevent occurrence of malignant arrhythmias and that TdP is frequently self-terminating in LQTS. The recurrence of electrical storms has led to suicide attempts by teenagers; the high incidence (>10%) of electrical storms in children has been considered “devastating” by a large group of experienced pediatric cardiologists.51 The massive release of catecholamines—triggered by pain and fear that follow an ICD discharge in a conscious patient, especially a young one—leads to further arrhythmias and to further discharges, all of which produce a dramatic vicious circle.
The European data on 233 patients with LQTS who had an ICD indicate that 9% of these patients were asymptomatic and that 41% received an ICD without having been first on LQTS therapy.50 Within 5 years 31% of the patients had adverse events. Appropriate ICD discharges (in 28% of patients) were predicted by age younger than 20 years at implantation, prior CA, and cardiac events despite therapy. Among patients without these factors, no one received appropriate shocks within 7 years, in contrast to 70% of those with all four factors.
Gene-Specific Therapy and Management
Patients with LQT1 are at higher risk during sympathetic activation, such as during exercise and emotions. They should not participate in competitive sports. Swimming is particularly dangerous, as 99% of the arrhythmic episodes in patients with LQT1 were associated with swimming.23
In patients with LQT2, some of whom have a tendency to lose potassium (K+), it is essential to preserve adequate K+ levels. Oral K+ supplements in combination with K+-sparing agents are a reasonable approach. As these patients are at higher risk, especially when aroused from sleep or rest by a sudden noise, it is recommended that telephones and alarm clocks be removed from their bedrooms; especially in the case of children, whenever they have to be wakened in the morning, it should be done gently and without yelling.23
The realization that SCN5A mutations producing LQT3 have a “gain-of-function” effect has lent support to the early suggestion by the author’s group to try sodium channel blockers, especially mexiletine, as possible adjuvants in the management of patients with LQT3.8,22,36 The author’s group tests the effectiveness of mexiletine in all patients with LQT3 by using the acute oral drug test technique (half the daily dose during continuous ECG monitoring). Within 90 minutes, the peak plasma concentration is reached; if the QTc is shortened by more than 40 ms, then mexiletine is added to β-blocker therapy. In fact, in most patients with LQT3, QTc is shortened by more than 70 to 80 ms with mexiletine (Figure 62-6). Even though no conclusive evidence for a beneficial effect exists and definite failures have occurred, evidence of significant benefit in a number of individual cases is growing. The highly malignant forms manifest in infancy because of mutations that cause extremely severe electrophysiological dysfunction, which is corrected by the combination of mexiletine and propranolol.52
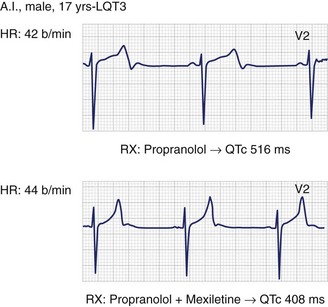
During heart rate increases, QTc shortens more in patients with LQT3 than among healthy controls; indeed, normal physical activity may not have to be restricted in patients with LQT3.22 They are at higher risk of death at rest, especially at night-time. When these patients sleep and are in a horizontal position, the onset of TdP produces a progressive but slow fall in blood pressure, which facilitates noisy gasping preceding death. Patients with LQT2 (also at risk while resting) as well as those with LQT3 have, on occasion, been saved by family members, for example, the spouse sleeping next to the patient. Therefore, it is recommended that patients with LQT2 and LQT3 keep an intercom system in their bedrooms and, if young, in the parents’ or other caregivers’ bedrooms.
Asymptomatic Patients with LQTS and Patients with a Normal QTc
As the first manifestation of LQTS in approximately 12% of cases is sudden death, β-blocker treatment should be initiated in all patients, including those still asymptomatic. Among these, reasonable exceptions appear to be males above age 20 to 25 years with LQT1—because very seldom does the first event occur later than this age—and adults with a QTc less than 500 ms. Women with LQT2 seem to remain at risk throughout life, and it is recommended that they receive treatment throughout their lives. Patients with a normal QTc (<440 ms) appear to be at very low risk for life-threatening arrhythmias. In the absence of any clear follow-up data, treatment is optional for them; the characterization of the mutation could influence this choice. Cardiac and noncardiac drugs that block the IKr current and thereby prolong the Q-T interval should always be avoided by patients with LQTS. A drug list is available at www.torsades.org. Such a list, updated every year, should be given to all patients with LQTS because their physicians may not be aware of these electrophysiological actions.
Key References
Crotti L, Lundquist AL, Insolia R, et al. KCNH2-K897T is a genetic modifier of latent congenital long QT syndrome. Circulation. 2005;112:1251-1258.
Crotti L, Monti MC, Insolia R, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657-1663.
Crotti L, Spazzolini C, Schwartz PJ, et al. The common long QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: Toward a mutation-specific risk stratification. Circulation. 2007;116:2366-2375.
De Ferrari GM, Schwartz PJ. Long QT syndrome, a purely electrical disease? Not anymore. Eur Heart J. 2009;30:253-255.
Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: Clinical implications. J Intern Med. 2006;259:39-47.
Schwartz PJ. Cutting nerves and saving lives. Heart Rhythm. 2009;6:760-763.
Schwartz PJ, Crotti L. Long QT and short QT syndromes. In Zipes DP, Jalife J, editors: Cardiac electrophysiology: From cell to bedside, ed 5, Philadelphia: Saunders, 2009.
Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381-3386.
Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long QT syndrome. Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD. True or false? Heart Rhythm. 2009;6:113-120.
Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome. Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783-790.
Schwartz PJ, Spazzolini C, Priori SG, et al. Who are the long QT syndrome patients who receive an implantable cardioverter defibrillator and what happens to them? Data from the European LQTS ICD Registry. Circulation. 2010;122(13):1272-1282.
Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761-1767.
Schwartz PJ, Vanoli E, Crotti L, et al. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol. 2008;51:920-929.
Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long QT syndrome type 1: Contribution of non-compliance and QT prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215-221.
1 Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378-390.
2 Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am Heart J. 1957;54:59-68.
3 Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome. Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783-790.
4 Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’età pediatrica. Clin Pediatr. 1963;45:656-683.
5 Ward OC. A new familial cardiac syndrome in children. J Irish Med Assoc. 1964;54:103-106.
6 Schwartz PJ, Crotti L. Long QT and short QT syndromes. In Zipes DP, Jalife J, editors: Cardiac electrophysiology: From cell to bedside, ed 5, Philadelphia: Saunders, 2009.
7 Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569-580.
8 Bennett PB, Yazawa K, Makita N, George ALJr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683-685.
9 Splawski I, Timothy KW, Sharpe LM, et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19-31.
10 Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761-1767.
11 Schwartz PJ, Garson AJr, Paul T, et al. Guidelines for the interpretation of the neonatal electrocardiogram. Eur Heart J. 2002;23:1329-1344.
12 Schwartz PJ. Another role for the sympathetic nervous system in the long QT syndrome? J Cardiovasc Electrophysiol. 2001;12:500-502.
13 Crotti L, Lundquist AL, Insolia R, et al. KCNH2-K897T is a genetic modifier of latent congenital long QT syndrome. Circulation. 2005;112:1251-1258.
14 Schwartz PJ, Vanoli E, Crotti L, et al. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol. 2008;51:920-929.
15 Crotti L, Monti MC, Insolia R, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657-1663.
16 Schwartz PJ, Malliani A. Electrical alternation of the T wave. Clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long QT syndrome. Am Heart J. 1975;89:45-50.
17 Nador F, Beria G, De Ferrari GM, et al. Unsuspected echocardiographic abnormality in the long QT syndrome: Diagnostic, prognostic, and pathogenetic implications. Circulation. 1991;84:1530-1542.
18 Haugaa KH, Edvardsen T, Leren TP, et al. Left ventricular mechanical dispersion by tissue Doppler imaging: A novel approach for identifying high-risk individuals with long QT syndrome. Eur Heart J. 2009;30:330-337.
19 De Ferrari GM, Schwartz PJ. Long QT syndrome, a purely electrical disease? Not anymore. Eur Heart J. 2009;30:253-255.
20 De Ferrari GM, Nador F, Beria G, et al. Effect of calcium channel block on the wall motion abnormality of the idiopathic long QT syndrome. Circulation. 1994;89:2126-2132.
21 Viskin S, Alla SR, Barron HV, et al. The mode of onset of torsades de pointes in the congenital long QT syndrome. J Am Coll Cardiol. 1996;28:1262-1268.
22 Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381-3386.
23 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long QT syndrome. Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
24 Khositseth A, Tester DJ, Will ML, et al. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60-64.
25 Heradien MJ, Goosen A, Crotti L, et al. Does pregnancy increase cardiac risk for LQT1 patients? J Am Coll Cardiol. 2006;48:1410-1415.
26 Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-1874.
27 Etheridge SP, Sanatani S, Cohen MI, et al. Long QT Syndrome in children in the era of implantable defibrillators. J Am Coll Cardiol. 2007;50:1335-1340.
28 Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794-799.
29 Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481-2489.
30 Crotti L, Spazzolini C, Schwartz PJ, et al. The common long QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: Toward a mutation-specific risk stratification. Circulation. 2007;116:2366-2375.
31 Brink PA, Crotti L, Corfield V, et al. Phenotypic variability and unusual clinical severity of congenital long QT syndrome in a founder population. Circulation. 2005;112:2602-2610.
32 Nof E, Cordeiro JM, Pérez GJ, et al. A common single nucleotide polymorphism can exacerbate long QT type 2 syndrome leading to sudden infant death. Circ Cardiovasc Genet. 2010;3(2):199-206.
33 Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644-651.
34 Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109:399-411.
35 Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: An update. Circulation. 1993;88:782-784.
36 Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: Clinical implications. J Intern Med. 2006;259:39-47.
37 Schwartz PJ, Locati E. The idiopathic long QT syndrome. Pathogenetic mechanisms and therapy. Eur Heart J. 1985;6(Suppl D):103-114.
38 Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616-623.
39 Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-1344.
40 Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long QT syndrome type 1: Contribution of non-compliance and QT prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215-221.
41 Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD. True or false? Heart Rhythm. 2009;6:113-120.
42 Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias. The surgical supraclavicular approach to cervico-thoracic sympathectomy. Heart Rhythm. 2010;7(8):1161-1165.
43 Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826-1833.
44 Schwartz PJ, Motolese M, Pollavini G, et althe Italian Sudden Death Prevention Group. Prevention of sudden cardiac death after a first myocardial infarction by pharmacologic or surgical antiadrenergic interventions. J Cardiovasc Electrophysiol. 1992;3:2-16.
45 Wilde AAM, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-2029.
46 Atallah J, Fynn-Thompson F, Cecchin F, et al. Video-assisted thorascopic cardiac denervation: A potential novel therapeutic option for children with intractable ventricular arrhythmias. Ann Thorac Surg. 2008;86:1620-1625.
47 Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752-759.
48 Schwartz PJ. Cutting nerves and saving lives. Heart Rhythm. 2009;6:760-763.
49 Zareba W, Moss AJ, Daubert JP, Hall WJ, et al. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337-341.
50 Schwartz PJ, Spazzolini C, Priori SG, et al. Who are the long QT syndrome patients who receive an implantable cardioverter defibrillator and what happens to them? Data from the European LQTS ICD Registry. 2010;122(13):1272-1282.
51 Wolf MJ, Zeltser IJ, Salerno J, et al. Electrical storm in children with an implantable cardioverter defibrillator: Clinical features and outcome. Heart Rhythm. 2007;4(May Suppl):S43.
52 Wang DW, Crotti L, Shimizu W, et al. Malignant perinatal variant of long QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circulation Arrhythmia Electrophysiol. 2008;1:370-378.