Chapter 65 Genetic Diseases
Catecholaminergic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is one of the most malignant inherited arrhythmogenic diseases, with a high incidence of sudden cardiac death among affected individuals. The first report of a patient with this disease was published in 1975, but the first systematic description came in 1978 with the work of Coumel et al and was further refined by the same group in 1995.1–3 In 2001, the authors of this chapter reported for the first time that the autosomal dominant form of the disease was caused by mutations in the gene of the cardiac ryanodine receptor.4 Shortly thereafter, the gene for an autosomal recessive form of CPVT was identified as the gene encoding cardiac calsequestrin5; overall, it became clear that CPVT is caused by abnormal calcium (Ca2+) homeostasis.
Clinical Presentation and Diagnosis
The initial presentation of CPVT is syncope or cardiac arrest occurring during adrenergic stress.3,4 The mean age of onset of the first syncope is 12 years.6–8 In the absence of treatment, the disease is highly lethal: The estimated incidence of sudden death is of 30% before age 40 years.9 Sudden death may be the first manifestation of the disease, thus making prevention of lethal events a difficult task.
Minor, nondiagnostic features are resting sinus bradycardia and prominent U waves, but their diagnostic value has never been systematically addressed.3,10 Furthermore, mild QT prolongation in some CPVT cases has been reported2,3; thus the differential diagnosis for CPVT should include LQTS. Some patients with LQTS do have a mild phenotype (borderline QT interval and no symptoms), but their prognosis is completely different from that of patients with CPVT, which presents a higher incidence of sudden death and a limited response to β-blocker therapy.6,11
In the authors’ series of patients with CPVT, a positive history of sudden death, stress-induced syncope, or seizures was present in 30% of patients, suggesting that careful collection of family history plays an important role in the correct identification of this condition.6
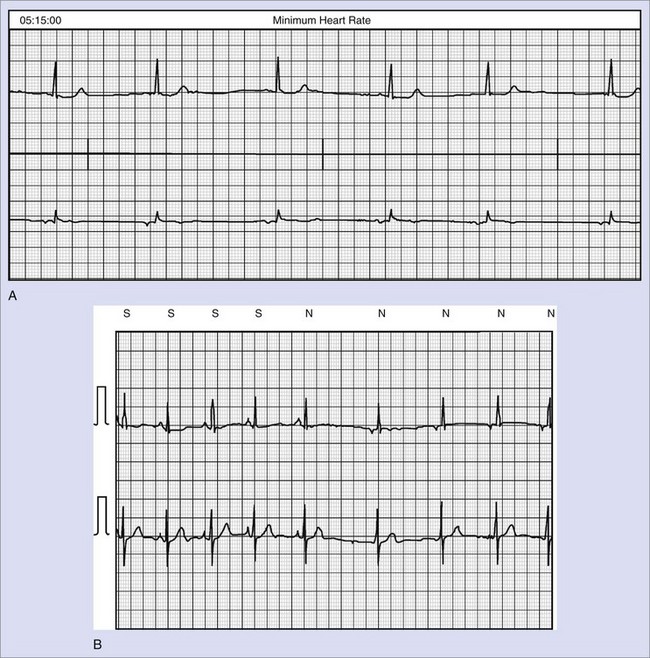
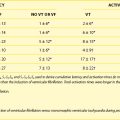
Exercise stress testing is the single most important tool, since CPVT arrhythmias are often reproducible during graded exercise (Figure 65-1). The typical behavior of CPVT arrhythmias is that of a progressive worsening on increase in workload; isolated premature beats or couplets initially appear between 90 and 110 beats per minute (beats/min) followed by runs of nonsustained or sustained ventricular tachycardia (VT) when the heart rate further increases (see Figure 65-1).12 Supraventricular arrhythmias are also a common finding and often precede the onset of ventricular arrhythmias.13 The morphology of VT is a hallmark of the disease: the so-called bidirectional VT, which is characterized by a 180-degree beat-to-beat rotation of the axis of the QRS complexes on the frontal plane.4,6 Although this pattern is recognizable in the majority of patients, it is important to be aware that some patients also present with irregular polymorphic VT. Furthermore, the initial presentation of the disease may also be that of ventricular fibrillation (VF) triggered by sudden adrenergic activation that may be interpreted as idiopathic VF in the absence of documentation of typical CPVT arrhythmias.6 Holter monitoring and implantable loop recorders are helpful for diagnosis, especially in those patients in whom emotional triggers (alone or in combination with exercise) are more arrhythmogenic than exercise alone.14,15
Genetic Bases
The majority of familial CPVT cases present an autosomal dominant pattern of inheritance. In 1999, Swan et al identified a significant linkage with some microsatellite markers at locus 1q42-q43.16 On the basis of this information, the authors of this chapter undertook a candidate gene screening in the critical region and identified the cardiac ryanodine receptor (RyR2) as the mutant CPVT gene.4 In the same year, Lahat et al described an autosomal recessive transmission pattern on the chromosomal locus 1p13-21 in a large Bedouin CPVT family.17 Subsequently, they also succeeded in identifying a mutation on the cardiac calsequestrin (CASQ2) gene, encoding for cardiac calsequestrin.5
Autosomal dominant (RyR2) CPVT is, by far, the most frequent variant, and CASQ2 mutations represent 1% to 2% of all genotyped CPVT. More recently, on the basis of the evidence that the patients with Andersen-Tawil syndrome may have bi-directional VT, it has been suggested that some CPVT cases can be explained by KCNJ2 mutations.18,19 A detailed discussion of the link between KCNJ2 mutations and CPVT is beyond the scope of this chapter, but it is relevant to note here that preliminary experimental studies on adenoviral-infected cardiac myocytes with KCNJ2-CPVT mutations show a cellular phenotype very similar to that observed in RyR2-mutant myocytes.20
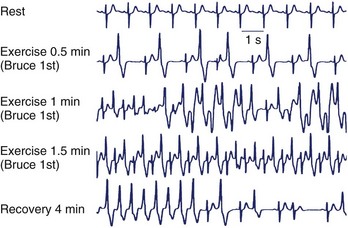
In 2007, a new autosomal recessive form of CPVT mapping on the chromosomal locus 7p14-22 was reported by Bhuiyan et al, but the gene disease has not yet been discovered.21 So far, more than 70 different mutations have been associated with CPVT, and they all are single–base pair substitutions causing the replacement of an amino acid. As expected with regard to autosomal recessive disorders, the number of families with CPVT linked to CASQ2 mutations is fairly small. At present, only seven mutations have been discovered, and they can be inherited in homozygous or compound heterozygous form. A recent analysis from the authors’ group has demonstrated that genetic screening on the RyR2 gene can identify at least 62% of probands who present with a clinical picture suggestive for the disease22; genetic testing helps identify silent mutation carriers and for reproductive counseling (Figure 65-2). Furthermore, RyR2-negative and CASQ2-negative patients should undergo KCNJ2 screening. KCNJ2 mutations are associated with a favorable outcome (sudden death is an exceptional event in this case).
Mechanisms of Arrhythmias in Autosomal Dominant CPVT: From Cellular Studies to Engineered Murine Models
Summary of RyR2 Physiology
Since Ca2+ is not only important for electrical and mechanical activation but also for a wide range of enzymatic and metabolic pathways, it is clear that RyR2 abnormalities may exert additional (and, so far, not thoroughly investigated) consequences on cell physiology.23
RyR2 Mutations and Catecholaminergic Polymorphic Ventricular Tachycardia
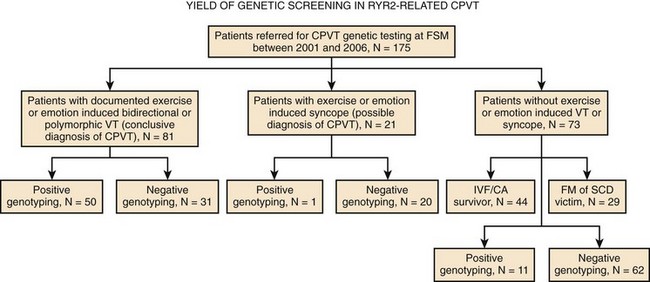
The proposed “primum movens” for RyR2-CPVT mutations to lead to Ca2+ overload is uncontrolled release (leakage) during diastole, which could be detected, according to different authors, only on adrenergic activation (phosphorylation) or already in the unstimulated condition.24–26 Given the complexity of the SR Ca2+ release process, the leakage could, in principle, be caused by several mechanisms.24,25,27 The hypotheses so far advanced are summarized in the following paragraphs (Figure 65-3).
Subcellular Mechanisms for RyR2 Dysfunction in Catecholaminergic Polymorphic Ventricular Tachycardia
FKBP12.6 Dissociation with RyR2 Mutants
FKBP12.6, also known as calstabin, is thought to play a role as a “stabilizer” of the channel in the closed state during diastole. Hints for the possible role of FKBP12.6 in CPVT were initially collected in experimental studies in heart failure. During congestive heart failure, chronic adrenergic hyperactivation may lead to weaker FKBP12.6 binding that can be the cause of Ca2+ overload-mediated arrhythmogenesis in this common disease.28 The FKBP12.6 hypothesis, originally developed by Andrew Marks and co-workers, was subsequently extended to CPVT pathophysiology; these authors demonstrated that some RyR2 mutants may cause a reduced binding affinity for FKBP12.6, which is further increased by sympathetic stimulation (see Figure 65-3).27 More recently, the same group performed an FKBP12.6 binding assay in a RyR2 knock-in mouse engineered with the R2474S mutation which has a CPVT-like phenotype29; they confirmed their initial hypothesis by demonstrating reduced FKBP12.6 binding in the presence of adrenergic stimulation. In parallel with these studies, Marks’ group also tried to pharmacologically treat the binding defect by using K201, a benzothiazepine derivative that was expected to increase FKBP-RyR2 binding. Long-term administration of this drug (or of its novel derivative, S107) was able to restore binding and to prevent the occurrence of arrhythmias both in the FKBP12.6 knock-out murine model and in the RyR2R2474S/WT knock-in murine model.29,30
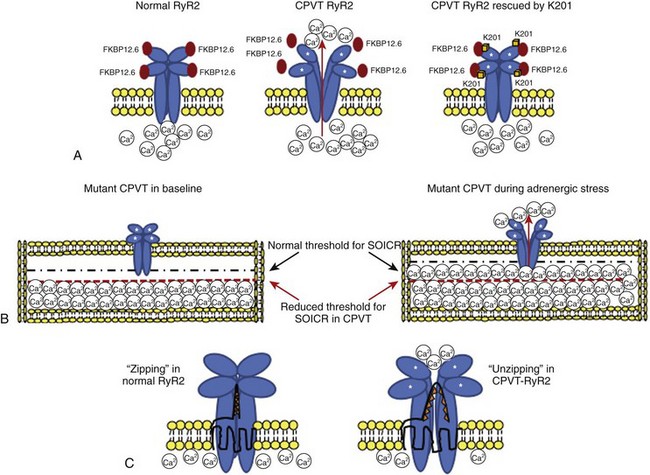
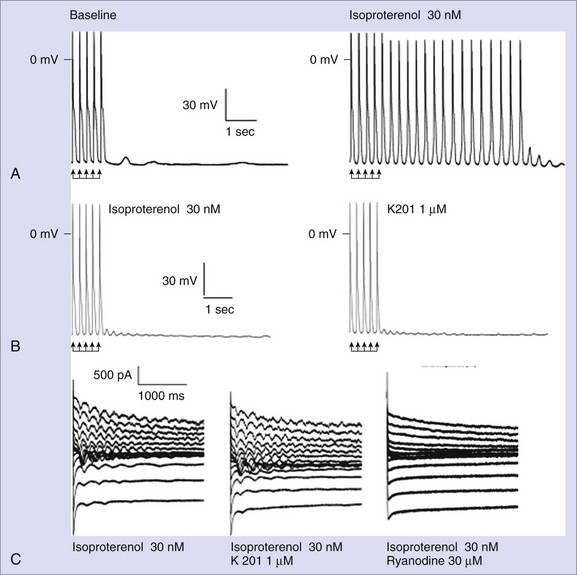
Despite the large amount of evidence provided by the Andrew Marks group, other authors either were not able to replicate their results or provided conflicting evidence when they used alternative approaches. George et al and Jiang et al reported normal FKBP12.6-RyR2 interaction in vitro.24,31 Chronic treatment with K201 did not prevent arrhythmias in vivo or the occurrence of DADs in isolated myocytes in a CPVT knock-in mouse RyR2R4496C/WT in a study by the authors of this chapter (Figure 65-4).32 Furthermore, in this model, Western blot experiments also demonstrated physiological interaction between RyR2 and FKBP12.6.32 Along these lines, Zissimopulos et al demonstrated that FKBP12.6 binding is either unaltered or even increased in the presence of CPVT-RyR2 mutations.33 Another report by Hunt et al provided evidence that K201 may modulate RyR2 independently from FKBP12.6.34 For example, these authors showed that K201 abolished spontaneous Ca2+ release in cardiac myocytes in the presence of FK506 that dissociates FKBP12.6 from RyR2.
Defective Interdomain Interaction
RyR2 is a large molecule with a complex tertiary and quaternary structure, in which interactions between domains are important for channel function. Indeed, domain-domain interactions ensure proper folding during the opening and closing of the channel.35
Several RyR2-CPVT mutations occur in the N-terminal (residue 70-466) and the central domains (residues 2246-2543).36 The N-terminal and central domains interact, and their relative conformational change from zipped (tight interaction) to unzipped (loose interaction) is involved in the channel gating process.35 Recent data have highlighted how some CPVT-related mutants may affect this mechanism by inducing a shift toward a mostly unzipped interaction that destabilizes the channel (see Figure 65-3). To test this hypothesis, Oda et al generated a synthetic peptide, DPc10, encompassing the Gly2460-Pro2495 region of RyR2 that mimics an RyR2 mutant: The peptide destabilizes the N-terminal–central region interaction through competitive activity and reproduces the biophysical effect of CPVT mutations.37 The same group engineered several other peptides, mimicking mutations located either in the N-terminal or in the central region of the protein, demonstrating that the mutations caused Ca2+ leakage through the disruption of interdomain interaction.38 Independent support to the hypothesis of unzipping has been provided by George et al by means of high-resolution confocal microscopy and fluorescence resonance energy transfer analysis.39
Store Overload–Induced Calcium Release
The open-state probability of RyR2 increases as the SR Ca2+ increases. If SR Ca2+ reaches a critical threshold, spontaneous Ca2+ leakage may occur even in the absence of membrane depolarization (i.e., calcium-induced calcium release). Chen et al proposed the hypothesis that some RyR2 mutants increase channel sensitivity to luminal Ca2+ (in other words, reduce the threshold for spontaneous Ca2+ release) (see Figure 65-3).25,26,31 This phenomenon was termed store overload–induced calcium release (SOICR). A reduced SOCIR threshold was shown for nine RyR2 mutations. Interestingly, while luminal (i.e., inside the SR) Ca2+ sensitivity was increased, no evidence for an increased sensitivity to cytosolic Ca2+ was observed. On the contrary, Thomas et al reported increased sensitivity of the receptor to cytosolic Ca2+ in L433P and R176Q/T2504M RyR2 mutants.40 While the role of cytosolic Ca2+ sensitivity has still to be determined, it appears evident that a reduced threshold for release has an important role, since, as shown by Venetucci et al, increasing ryanodine receptor open probability alone does not produce arrhythmogenic diastolic Ca2+ release because of the accompanying decrease of SR Ca2+content (and reduced driving force).41,42
Insights from RyR2-CPVT Murine Models
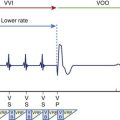
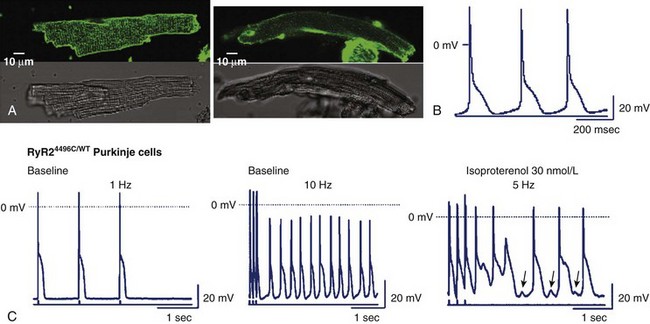
Knock-in murine models have been pivotal to the understanding of the cellular and whole-heart pathophysiology of CPVT.29,43,44 In 2005, the chapter authors’ group provided initial evidence that by engineering a RyR2-CPVT mutation in the mouse genome (at variance with what observed for several other murine models of inherited arrhythmias), it is possible to reproduce the phenotype observed in the clinical setting. By homologous recombination, a conditional knock-in mouse harboring the R4496C mutation (a frequent mutation found in CPVT families) was created.43 The typical CPVT bi-directional VT can be generated in this mouse with adrenergic stimulation in the absence of structural abnormalities.43 Furthermore, it was demonstrated that the presence of adrenergic-dependent DADs increased NCX-transient inward current (Iti) and triggered activity as the cellular mechanisms for CPVT (see Figure 65-4).32 In a subsequent study, the same group observed the onset of abnormal Ca2+ waves during diastole, which paralleled the occurrence of DAD development both at baseline and during isoproterenol superfusion.45 Increased propensity to DAD development in RyR2-R4496C mice was demonstrated also in isolated Purkinje cells by Cerrone et al (Figure 65-5).46
Finally, additional data supporting the concept that DADs are the arrhythmogenic mechanisms in CPVT were provided in an elegant study by Paavola et al in patients with CPVT; they used monophasic action potential recordings that showed DAD-like humps originating from the right ventricular septal wall and preceding the occurrence of ventricular arrhythmias.47
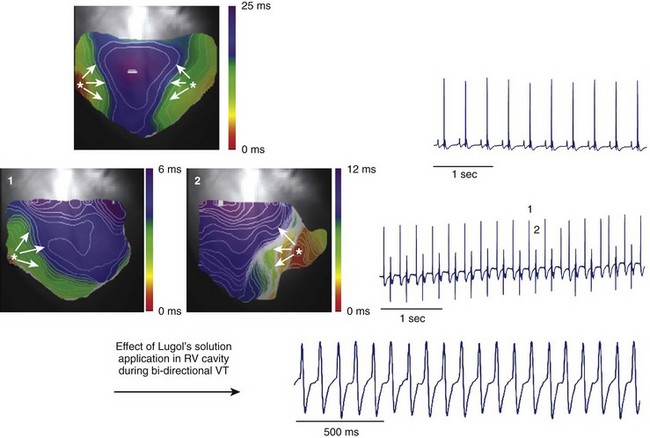
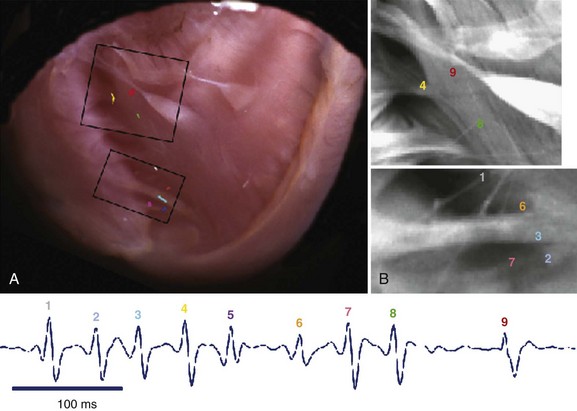
Transgenic models of CPVT have also provided information on the mechanisms of CPVT arrhythmias at the whole-heart level in studies using the optical mapping technique. In collaboration with José Jalife, the authors of this chapter showed that the arrhythmic beats of both polymorphic and bi-directional VT have a focal origin.46 Epicardial optical mapping demonstrated that during bi-directional VT, the beats alternately originate from the right ventricle and from the left ventricle and arise from an area coincident with the anatomic insertion of the major bundle branches of the conduction system (Figure 65-6).46 Additionally, administration of Lugol’s solution to chemically ablate the Purkinje network in the right ventricle converts bi-directional VT to monomorphic left-sided VT (see Figure 65-6).46 Endocardial optical maps have also shown that during polymorphic VT, the site of origin of the beats mapped on the endocardial right ventricular wall corresponded to free-running Purkinje fibers (Figure 65-7).46 Overall, these experiments support the relevant role of the Purkinje network in the pathogenesis of CPVT arrhythmias.
Mechanisms of Altered Ca2+ Homeostasis in CPVT Linked to CASQ2 Mutations
Summary of CASQ2 Physiology
CASQ2 has been initially described as a Ca2+ buffering protein resident in the SR lumen. It exists in monomeric and polymeric form; polymerization occurs at high Ca2+ and the polymerized protein presents higher Ca2+ buffering capacity. When luminal Ca2+ is low, CASQ2 binds to junctin and triadin and inhibits RyR2. In the presence of a rise in luminal SR Ca2+, the binding among CASQ2, triadin, and junctin is weakened by the replacement of the binding sites by Ca2+, and this process gradually increases the open probability for the RyR2 channel.48 Thus, CASQ2 is both a Ca2+ buffer molecule and a RyR2 modulator.
Subcellular Mechanisms for CASQ2 Dysfunction in Catecholaminergic Polymorphic Ventricular Tachycardia
Terentyev et al suggested that a reduction or absence of CASQ2, as it happens with the truncation mutants, leads to a decrease of the time necessary to re-establish Ca2+ storage, thus facilitating a premature activation of the RyR2 and, as a consequence, diastolic Ca2+ leakage.49
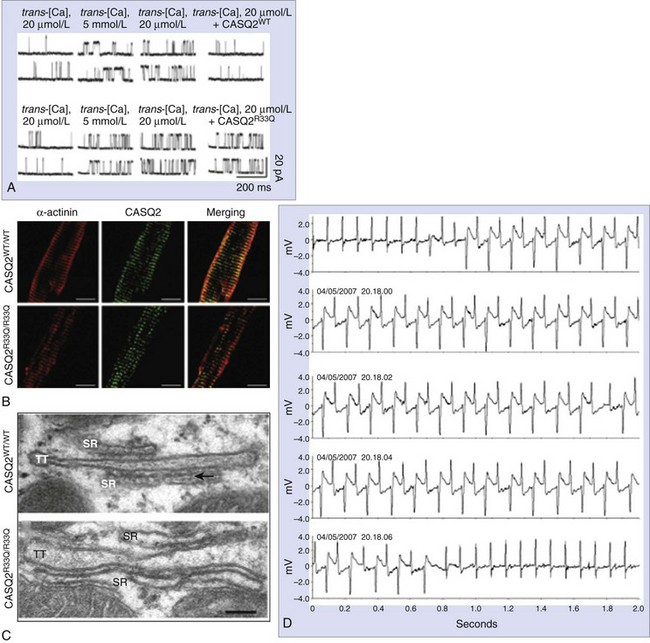
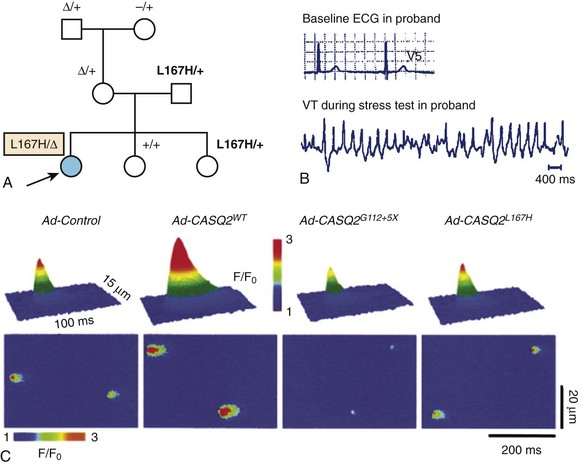
The D307H was the first missense mutation to be linked to the disease.5 In in vitro studies, the mutant protein was not able to form properly oriented dimers, and this affected the Ca2+ binding capacity.50 Adenoviral-mediated overexpression of D307H in rat cardiomyocytes showed that Ca2+ binding capacity is impaired despite the presence of endogenous native protein (dominant-negative effect)51,52; similar results have been obtained in CASQ2D307H myocytes from a transgenic murine model.53 In contrast, CASQ2-R33Q increases diastolic SR Ca2+ release, without affecting Ca2+ storing capacity, and it may cause CPVT through a loss of CASQ2-inhibiting effect on RyR2 (Figure 65-8).54,55 The L167H mutation was identified in a patient with compound heterozygosity (Figure 65-9), and it appears to cause a complex phenotype including abnormal (increased) Ca2+ buffering properties, impaired polymerization, and loss of RyR2 inhibitory properties.55,56
Insights from CASQ2-CPVT Murine Models
Knollmann et al created a CASQ2 knock-out murine model, in which VT and VF could be induced by β-adrenergic stimulation (isoproterenol).57 In isolated CASQ2 null myocytes, increased diastolic Ca2+ leakage leading to DADs and triggered activity was observed, thus proving the similarities between RyR2-CVPT and CASQ2-CVPT. Interestingly, analysis of this model proved that even in the absence of CASQ2, the SR Ca2+ storage capacity was not changed. The authors of this study pointed to the ultrastructural abnormalities involving the increase of the SR volume, which may represent a compensatory phenomenon to preserve SR Ca2+ storing capacity.57
More recently, the chapter authors’ group developed the CASQ2R33Q/R33Q knock-in model (R33Q was found in a severe case of recessive CPVT), which reproduces the CPVT phenotype.58 In contrast to the RyR2-R4496C model, in this model arrhythmias occur even spontaneously in the presence of mild stressors (i.e., loud sudden noise) (see Figure 65-8). CASQ2R33Q/R33Q cardiomyocytes showed DADs and triggered activity not only during adrenergic stimulation but also in resting conditions.58 In contrast to the CASQ2 knock-out model, SR Ca2+ is reduced (see Figure 65-8). Rather unexpectedly for a missense mutation, this model is also characterized by reduction of CASQ2 protein (both in the total cell homogenate and in the microsomal fraction) in the presence of normal expression at the mRNA level. Further investigations showed that the mutant R33Q protein, compared with the wild type, is significantly more prone to proteolysis.58
Song et al studied two additional murine models, one carrying a null mutation (knock-out) and the other with the D307H mutation.53 No significant phenotypic differences between the two models, including increased levels of RyR2 and calreticulin, were detected.53 Calreticulin is a fetal protein that shows Ca2+ binding capacity but is not present in adult hearts; the authors thus inferred that its increase in their model should be interpreted as a compensatory mechanism to balance the lack of CASQ2.
Neither the CASQ2 knock-out model generated by Knollmann nor the CASQ2R33Q/R33Q mouse showed altered expression of RyR2 or calreticulin.57,58 Therefore, until further data are available, it remains unclear why these two different models present a common compensatory mechanism that is not evident in other models, in particular in the knock-out model.
Current Therapy and Future Directions
Catecholaminergic Polymorphic Ventricular Tachycardia Therapy in the Clinical Setting
On the basis of the clear evidence of the critical role of adrenergic stimulation as a trigger for arrhythmias in CPVT, β-blockers were proposed as a reasonable therapeutic approach following the initial reports on this disease.2,3 β-Blockers should be started immediately when CPVT is diagnosed; agreement in the scientific community seems to indicate nadolol as the first choice among the available options—for its once-daily dosing and nonselective inhibition of adrenergic stimuli. Asymptomatic bradycardia in these patients should not be used as a reason to reduce the dosage of β-blocker therapy. In fact, the demonstration that DAD-induced arrhythmias are facilitated by faster heart rates provides the rationale to consider the bradycardic effect of β-blockers as an additional antiarrhythmic benefit, along with their inhibitory effect on the sympathetic drive.59
More recently, larger epidemiologic studies showed that β-blockers may fail to provide protection from life-threatening events in a relevant proportion of patients.6,8 In the Italian CPVT Registry, the incidence of recurrent arrhythmia while on therapy is as high as 30%.6 In case of recurrences of syncopal episodes or VT while on therapy, an implantable cardioverter-defibrillator (ICD) should be considered. Obviously, after ICD implantation, β-blocker treatment should be maintained to minimize the risk of device interventions. Indeed, in the series by the authors of this chapter, 50% of implanted patients received an appropriate ICD therapy during a 2-year follow-up.9
The incomplete protection afforded by β-blockers calls for the need of identifying adjunctive effective therapies. Calcium channel blockers, in particular verapamil, have been studied in a limited patient series as a possible alternative to β-blocker therapy by Swan et al and Sumitomo et al.60,61 The study of Rosso et al evaluated the efficacy of a combined association between β-blockers and verapamil. In their series, the combination of therapies reduced or even suppressed the recurrences of exercise-induced arrhythmias, ICD shocks, or both.62
Preliminary data on the possible efficacy of flecainide have been recently published.63 Flecainide has been shown to prevent stress-induced arrhythmias in CASQ2 knock-out mice and spontaneous SR Ca2+ release in isolated myocytes. It was also tested in two patients with CPVT who were implanted with an ICD and were refractory to conventional therapy, and it prevented arrhythmias during exercise test. Although presented only in an anecdotal report, these data are opening encouraging paths to be explored in the attempt to find new therapeutic strategies for treating CPVT.
Wilde et al provided preliminary evidence for the long-term effectiveness of left sympathetic cardiac denervation (LCSD) in three patients with CPVT. However, patients on β-blockers and LCSD who still experience recurrences of sustained VT and syncope do also exist (Priori SG, personal communication).64 Thus, caution is needed before considering this approach as an effective therapy for high-risk CPVT patients.
Experimental Therapies for Catecholaminergic Polymorphic Ventricular Tachycardia
Another interesting approach is that of inhibiting the effects of β-adrenergic stimulation by acting on the downstream targets of RyR2 phosphorylation. The pharmacologic inhibition of CAMKII (which phosphorylates RyR2 during adrenergic activation) is a promising approach. CAMKII phosphorylates RyR2 at different sites. Moreover, it is known that CAMKII inhibition reduces diastolic Ca2+ leakage and the transient inward Iti current (the transmembrane current generating DADs).65 Preliminary observations from the chapter authors’ group in the RyRR4496C/WT murine model suggest that a specific CAMKII inhibitor, KN93, could prevent arrhythmias both in vitro and in vivo, which provides encouraging support for novel therapeutic strategies involving this pathway.66
Key References
Cerrone M, Colombi B, Santoro M, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77-e82.
Cerrone M, Noujaim SF, Tolkacheva EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039-1048.
George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531-540.
Jiang D, Wang R, Xiao B, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173-1181.
Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004;101:13062-13067.
Knollmann BC, Chopra N, Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510-2520.
Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378-1384.
Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512-1519.
Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292-298.
Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196-200.
Rizzi N, Liu N, Napolitano C, et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008;103:298-306.
Tateishi H, Yano M, Mochizuki M, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536-545.
Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759-11764.
Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829-840.
1 Reid DS, Tynan M, Braidwood L, Fitzgerald GR. Bidirectional tachycardia in a child. A study using His bundle electrography. Br Heart J. 1975;37:339-344.
2 Coumel P, Fidelle J, Lucet V, et al. Catecholamine-induced severe ventricular arrhythmias with Adam-Stokes in children: Report of four cases. Br Heart J. 1978;40(Suppl):28-37.
3 Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512-1519.
4 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
5 Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378-1384.
6 Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
7 Rossenbacker T, Bloise R, De Giuli L, et al. Catecholaminergic polymorphic ventricular tachycardia: Genetics, natural history and response to therapy. Circulation. 2007;116(Suppl II):179.
8 Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426-2434.
9 Cerrone M, Colombi B, Bloise R, et al. Clinical and molecular characterization of a large cohort of patients affected with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2004;110(Suppl):552.
10 Postma AV, Denjoy I, Kamblock J, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42:863-870.
11 Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-1344.
12 Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51:23-30.
13 Monteforte N, Raytcheva-Buono E, Bloise R, et al. Electrocardiographic analysis of arrhythmias developing during exercise in patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2007;116(II):492.
14 Ormaetxe JM, Saez R, Arkotxa MF, Martinez-Alday JD. Catecholaminergic polymorphic ventricular tachycardia detected by an insertable loop recorder in a pediatric patient with exercise syncopal episodes. Pediatr Cardiol. 2004;25:693-695.
15 Hasdemir C, Priori SG, Overholt E, Lazzara R. Catecholaminergic polymorphic ventricular tachycardia, recurrent syncope, and implantable loop recorder. J Cardiovasc Electrophysiol. 2004;15:729.
16 Swan H, Piippo K, Viitasalo M, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035-2042.
17 Lahat H, Eldar M, Levy-Nissenbaum E, et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: Clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation. 2001;103:2822-2827.
18 Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511-519.
19 Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;110:381-388.
20 Ruan Y, Boveri L, Rossenbacker T, et al. Arrhythmogenesis in mutant KCNJ2 associated catecholaminergic polymorphic ventricular tachycardia. Circulation. 2008;118:525.
21 Bhuiyan ZA, Hamdan MA, Shamsi ET, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18:1060-1066.
22 Rong B, Napolitano C, Bloise R, et al. Yield of genetic screening in inherited cardiac channelopathies: How to prioritize access to genetic testing. Circ Arrhythmia Electrophysiol. 2009;2:6-15.
23 Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198-205.
24 George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531-540.
25 Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218-225.
26 Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004;101:13062-13067.
27 Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829-840.
28 Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365-376.
29 Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230-2245.
30 Wehrens XH, Lehnart SE, Reiken SR, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292-296.
31 Jiang D, Wang R, Xiao B, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173-1181.
32 Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292-298.
33 Zissimopoulos S, Thomas NL, Jamaluddin WW, Lai FA. FKBP12.6 binding of ryanodine receptors carrying mutations associated with arrhythmogenic cardiac disease. Biochem J. 2009;419:273-278.
34 Hunt DJ, Jones PP, Wang R, et al. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J. 2007;404:431-438.
35 Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671-d683.
36 George CH, Jundi H, Thomas NL, Fry DL, Lai FA. Ryanodine receptors and ventricular arrhythmias: Emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol. 2007;42:34-50.
37 Oda T, Yano M, Yamamoto T, et al. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400-3410.
38 Tateishi H, Yano M, Mochizuki M, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536-545.
39 George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:88-97.
40 Thomas NL, George CH, Lai FA. Functional heterogeneity of ryanodine receptor mutations associated with sudden cardiac death. Cardiovasc Res. 2004;64:52-60.
41 Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285-292.
42 Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: Threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105-111.
43 Cerrone M, Colombi B, Santoro M, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77-e82.
44 Kannankeril PJ, Mitchell BM, Goonasekera SA, et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:12179-12184.
45 Fernandez-Velasco M, Rueda A, Rizzi N, et al. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201-209.
46 Cerrone M, Noujaim SF, Tolkacheva EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039-1048.
47 Paavola J, Viitasalo M, Laitinen-Forsblom PJ, et al. Mutant ryanodine receptors in catecholaminergic polymorphic ventricular tachycardia generate delayed afterdepolarizations due to increased propensity to Ca2+ waves. Eur Heart J. 2007;28:1135-1142.
48 Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417-429.
49 Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759-11764.
50 Kim E, Youn B, Kemper L, et al. Characterization of human cardiac calsequestrin and its deleterious mutants. J Mol Biol. 2007;373:1047-1057.
51 Dirksen WP, Lacombe VA, Chi M, et al. A mutation in calsequestrin, CASQ2D307H, impairs sarcoplasmic reticulum Ca2+ handling and causes complex ventricular arrhythmias in mice. Cardiovasc Res. 2007;75:69-78.
52 Viatchenko-Karpinski S, Terentyev D, Gyorke I, et al. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471-477.
53 Song L, Alcalai R, Arad M, et al. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007;117:1814-1823.
54 Terentyev D, Nori A, Santoro M, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006;98:1151-1158.
55 Qin J, Valle G, Nani A, et al. Luminal Ca2+ regulation of single cardiac ryanodine receptors: Insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325-334.
56 di Barletta MR, Viatchenko-Karpinski S, Nori A, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012-1019.
57 Knollmann BC, Chopra N, Hlaing T, et al. CASQ2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510-2520.
58 Rizzi N, Liu N, Napolitano C, et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008;103:298-306.
59 Rosen MR, Danilo PJr. Effects of tetrodotoxin, lidocaine, verapamil, and AHR-2666 on ouabain-induced delayed afterdepolarizations in canine Purkinje fibers. Circ Res. 1980;46:117-124.
60 Swan H, Laitinen P, Kontula K, Toivonen L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005;16:162-166.
61 Sumitomo N, Harada K, Nagashima M, et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart (British Cardiac Society). 2003;89:66-70.
62 Rosso R, Kalman JM, Rogowski O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149-1154.
63 Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380-383.
64 Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-2029.
65 Wu Y, Roden DM, Anderson ME. Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res. 1999;84:906-912.
66 Liu N, Colombi B, Napolitano C, Priori S. Ca2+/calmodulin kinase II inhibitor abolishes and prevents triggered activity in myocytes from RyR2 R4496C+/- knock-in mice. Heart Rhythm. 2007;4(Suppl):S107-S108.