Chapter 63 Genetic Diseases
Brugada Syndrome
Sudden cardiac death (SCD) is a major cause of mortality in the Western world, with an approximate incidence of 1 per 1000 per year. Coronary artery disease is the most common cause of SCD. In the absence of coronary disease, SCD is commonly caused by a ventricular arrhythmia in patients with some form of structural heart disease. However, in 10% to 20% of cases, no cardiac structural abnormalities are found at autopsy or after extensive medical investigation of the survivors. In 1992, Brugada and Brugada described a new syndrome causing SCD in individuals with normal hearts.1 Today, generally known as Brugada syndrome, this new entity is thought to be responsible for up to 20% of SCDs that occur in individuals with structurally normal hearts.2
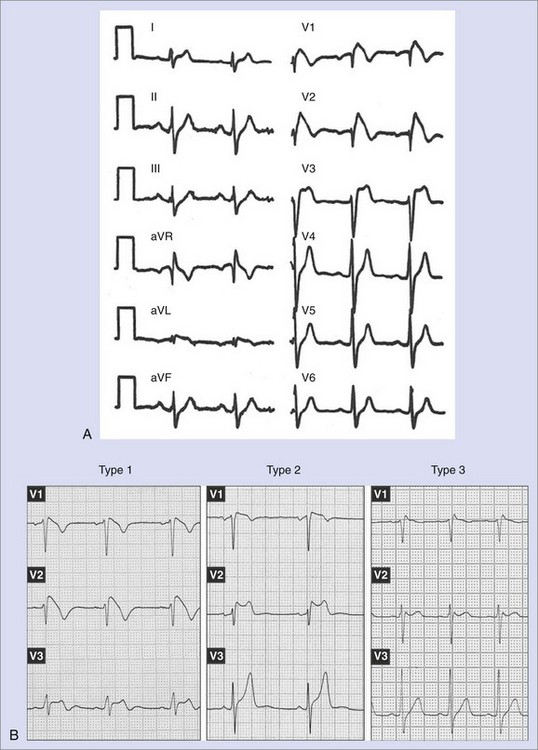
Brugada syndrome was initially described as a clinical syndrome characterized by (1) an electrocardiogram (ECG) resembling a right bundle branch block with a particular morphology of ST-segment elevation in right precordial leads (Figure 63-1, A) and (2) a susceptibility to developing polymorphic ventricular arrhythmias that cause syncope when self-terminating and SCD when long lasting and not terminated by cardiopulmonary resuscitation.1 After the initial description of the first eight patients, numerous works appeared either focusing on clinical characteristics of larger populations or defining the genetic, molecular, and cellular aspects of the disease.3–12 In recent years, major advances in clinical and mechanistic knowledge have provided very valuable information about the disease, but questions still remain, propelling large research activity on the subject. This chapter reviews the current knowledge on clinical, genetic, and molecular features of Brugada syndrome and provides updated information supplied by recent clinical and basic studies.
Definition and Epidemiology
The diagnosis of Brugada syndrome is obvious when the ECG has a certain appearance, as in Figure 63-1, A. However, certain ambiguities appeared in the years following the initial description of the syndrome. Three repolarization patterns were soon identified (Figure 63-1, B)13: (1) type 1 ECG pattern, as described in the initial report in 1992, in which a coved ST-segment elevation 2 mm or greater is followed by a negative T wave, with little or no isoelectric separation, this feature being present in more than one right precordial lead (from V1 to V3); (2) type 2 ECG pattern, also characterized by an ST-segment elevation but followed by a positive or biphasic T wave, which results in a saddle-back configuration; (3) type 3 ECG pattern, a right precordial ST-segment elevation 1 mm or less either with a coved-type or a saddle-back morphology. Although all three patterns can be present in Brugada syndrome patients (see section on Electrocardiography and Modulating Factors), only the type 1 ECG is diagnostic of the syndrome, as it was stated in the first consensus report of the Arrhythmia Working Group of the European Society of Cardiology and subsequently confirmed in the II Consensus Conference published in 2005.2,13 Both documents also held that in order to establish the definite diagnosis of Brugada syndrome, the type 1 ECG pattern should be documented in combination with one of the following clinical criteria: (1) documented ventricular fibrillation (VF), (2) polymorphic ventricular tachycardia (VT), (3) a family history of sudden death (SD) at an age younger than 45 years, (4) the presence of coved-type ECG in family members, (5) inducibility of ventricular arrhythmias with programmed electrical stimulation, (6) syncope, or (7) nocturnal agonal respiration.2,13 However, this definition should be applied with caution, especially when causative mutations have been identified and the disorder can be understood as a disease rather than a syndrome.14,15 In this regard, data from the authors of this chapter confirm that only the presence of the characteristic type 1 ECG pattern, even with no further clinical criteria, may be associated with SD in the follow-up.15 This confirms the need for monitoring all patients, even when an isolated type 1 ECG pattern is found.
The prevalence of Brugada syndrome has been estimated as 5 in 10,000, although this rate may be an underestimation of the real prevalence, as many patients have concealed forms of the disease and remain underdiagnosed. Importantly, significant ethnic and geographic differences have been described.2 For example, the type 1 ECG pattern was observed in 12 of 10,000 in Japan, whereas the few available data on North American and European populations point to a much lower prevalence.16–18 In fact, the syndrome is considered endemic in certain Southeast Asian countries, where it has long been known as sudden unexplained death syndrome (SUDS), also called bangungot (in the Philippines), pokkuri (in Japan), or lai tai (in Thailand); all of these are phenotypically, genetically, and functionally identical to Brugada syndrome.19
Genetics of Brugada Syndrome
Inheritance in Brugada syndrome occurs via an autosomal dominant mode of transmission, although in some cases, the disease can be sporadic, that is, absent in parents and other relatives.2 Thus far, all the mutations that have been linked to Brugada syndrome affect (directly or indirectly) the normal function of specific ion channels participating in the action potential (AP). Therefore, Brugada syndrome is included among the so-called channelopathies, together with other primary electrical disorders such as long QT syndrome (LQTS).
The first mutations related to Brugada syndrome were described in 1998 by Chen and coworkers and were identified in SCN5A, the gene encoding the α-subunit of the cardiac sodium channel (locus 3p21, 28 exons).8 To date, more than 100 other different mutations associated with the syndrome have been found in the same gene.9,12,19–23 For the majority of them, functional studies performed with expression systems have demonstrated a loss of function of the sodium channel that translates into a decrease in sodium current (INa), which is achieved either through a quantitative decrease (failure in their expression) or through a qualitative dysfunction (impaired kinetics) of the channels.9,12,19–23
Although for almost a decade SCN5A has been the only gene linked to Brugada syndrome, mutations in SCN5A are generally found in only 18% to 30% of patients, which suggests a genetic heterogeneity of the disease.2 Accordingly, in the past 2 years, four other genes have been found to be linked to Brugada syndrome. The first of them, glycerol-3-phosphate dehydrogenase 1–like (GPD1-L), was described in 2007 after previous identification of the locus on chromosome 3 (3p22-p24) in 2002.24,25 The A280V mutation in GPD1-L was shown to induce a sodium loss-of-function effect by affecting the trafficking of the cardiac sodium channel to the cell surface.24 Very interestingly, two recent reports demonstrated that mutations in genes other than those involved in the sodium channel function can be responsible for some cases of Brugada syndrome. In 2007, Antzelevitch et al linked loss-of-function mutations in the genes encoding the cardiac calcium channel Cav1.2 (CACNA1c) and its β-subunit CACNB2b to a syndrome overlapping short QT and the Brugada ECG pattern.26 More recently, the same group of investigators described the first family with Brugada syndrome identified to be carrying a mutation (R99H) in the KCNE3 gene, which encodes a β-subunit that is thought to modulate Kv4.3 channels and to be responsible for an increase in transient potassium Ito currents.27 Together, these findings open up new lines of research, where the concept of Brugada syndrome as a pure sodium channelopathy has given way to the concept of the syndrome as an ionic imbalance between inward and outward currents during phase 1 of the AP.
Cellular and Ionic Mechanisms
Experimental studies have elucidated cellular and molecular bases for the two main characteristic features of Brugada syndrome: (1) the specific ECG morphology (ST-segment elevation in right precordial leads) and (2) the susceptibility for VF and SD. Figure 63-2, A, represents the normal ventricular myocyte AP and the major ionic currents involved in each one of the phases. Sodium loss-of-function conditions, the most encountered disorder in SCN5A mutations related to Brugada syndrome, create an imbalance between outward and inward positive currents during phase 1, favoring repolarization and the appearance of a particular notch in the AP (dashed line) that is mediated by the outward transient potassium currents (Ito).9,12,19–23 Comparable imbalances can appear either by decrease in ICaL (in calcium channel loss-of-function mutations) or relative increase in Ito (in the recently described KCNE3 mutation).26,27
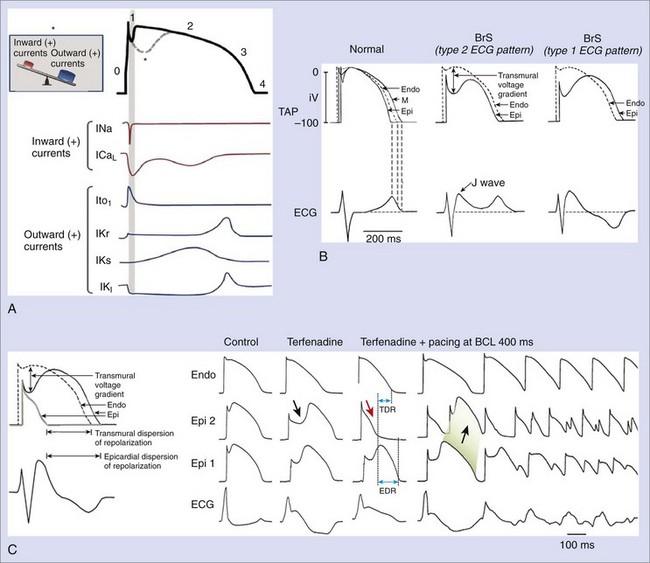
The accentuated notch present in patients with Brugada syndrome, especially in the epicardium, gives rise to a transmural voltage gradient between the epicardium and the endocardium, producing the characteristic ST-segment elevation on ECG (Figure 63-2, B).28 The imbalance between outward and inward positive currents during phase 1 also establishes the basis for the development of ventricular arrhythmias in Brugada syndrome. The proposed mechanism would be a phase 2 re-entry (Figure 63-2, C). When the notch is such that phase 1 reaches approximately −30 mV, all-or-none repolarization can lead to a complete loss of the AP dome. The heterogeneity of the loss of the dome among different sites within the epicardium and between the epicardium and the endocardium results in epicardial and transmural dispersions of repolarization, respectively. This substrate may facilitate the development of premature beats by means of conduction of the AP dome from the sites where it is maintained to the sites where it is lost.11,28
The understanding that the imbalance between inward and outward ionic currents at phase 1 defines the pathologic substrate for Brugada syndrome has multiple implications. First, it has helped in the development of experimental models of the disease, which have been successfully created by the administration of potassium openers (pinacidil), the combination of a sodium channel blocker (flecainide) and acetylcholine, or drugs with combined sodium channel blocker and calcium channel blocker effects.11,29 These interventions create a relative predominance of outward positive currents at the end of phase 1, thus accentuating the notch. The ionic imbalance hypothesis also explains the effects of certain modulators and certain particularities of the syndrome, such as the enhanced phenotypic expression (accompanied by an increased risk of arrhythmias) during vagal situations (acetylcholine inhibits calcium currents, whereas β-adrenergic drugs increase them) or the worse prognosis in men than in women affected with the disease (men could have constitutionally greater Ito density than women).11,30–34 Likewise, it appears that interventions that decrease inward positive currents (as do sodium channel blockers) could be potentially harmful to patients with Brugada syndrome, as they increase ST-segment elevation and the risk of arrhythmic events; however, they could be useful in unmasking concealed forms of the disease.35 In contrast, Ito blockers such as quinidine could be a good therapeutic option, as they reduce the notch at the end of phase 1 (see section on Treatment).36
Clinical Manifestations of Brugada Syndrome
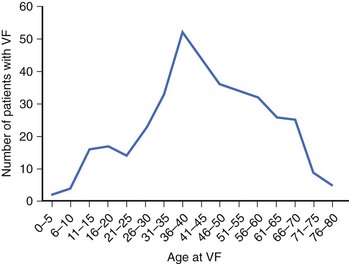
Patients with Brugada syndrome usually remain asymptomatic. However, syncope or cardiac arrest, a consequence of an arrhythmic complication such as polymorphic VT or VF, has been described in up to 17% to 42% of diagnosed individuals.37–40 This rate is probably an overestimation of the real prevalence of symptoms among patients with Brugada syndrome, given that most asymptomatic patients remain underdiagnosed. The age of symptom occurrence (especially cardiac arrest) is consistently around the fourth decade of life in all the series (Figure 63-3), with no definite explanation for this observation thus far.41 Previous syncope may be present in up to 23% of patients who present with cardiac arrest.38 Brugada syndrome should therefore be considered during the workup of patients who have suffered an aborted SD or syncope suspected to be of cardiac origin, especially if no underlying structural heart disease is found (see section on Approach to Patients with Suspected Brugada Syndrome: Family Screening).
Up to 20% of patients with Brugada syndrome may present with supraventricular arrhythmias and thus complain of palpitations, dizziness, or both.42 An increased atrial vulnerability to both spontaneous atrial fibrillation (AF) and induced AF has been reported in patients with Brugada syndrome.43 Other symptoms, such as neurally mediated syncope, have been also recently associated with Brugada syndrome.44,45
As in the case of other sodium channel–related disorders such as type 3 LQTS, ventricular arrhythmias in Brugada syndrome typically occur at rest, especially during night-time or sleep. In a study by Matsuo et al, 26 of 30 episodes of VF documented in implantable cardioverter-defibrillator (ICD) recordings of Brugada syndrome patients appeared during sleep, which suggests that vagal activity may play an important role in the arrhythmogenesis of Brugada syndrome.30 This finding has been confirmed in more recent series.31 As mentioned before, the increase in vagal tone is mediated by acetylcholine decreases in calcium currents, which could favor arrhythmogenesis through a phase 2 re-entry mechanism.11
Sex Differences
It is currently accepted that the clinical phenotype of Brugada syndrome is 8 to 10 times more prevalent in male patients than in female patients.46 Consequently, the main clinical studies published thus far have included 71% to 77% of the male population, which generally appears to be more symptomatic compared with the female population.37–40 The fact that in the case of SUDS, SD mainly occurs in young males during sleep has long been recognized in Southeast Asia, where men from certain small villages dress in women’s bedclothes, as they believe that the syndrome is a female spirit searching for young men at night.
The authors of this chapter recently conducted a study aimed at analyzing gender differences in a large population of patients with Brugada syndrome.33 The study population (n = 384) included 272 men (70.8%) and 112 women (29.2%). General demographic characteristics were similar between both groups (mean age, 45.8 years), but at diagnosis, men presented more frequently with symptoms (syncope in 18%, previous aborted SD in 6%) than did women (14% and 1%, respectively; P = .04). Men also had higher rates of spontaneous type 1 ECG (47% vs. 23%; P = .0001) and inducibility of VF during the electrophysiological study (32% vs. 12%; P = .0001) (Figure 63-4, A).33 The prognosis also differed between men and women. Cardiac events (defined as SD or documented VF) appeared in 31 men (11.6%) and in 3 women (2.8%) during a mean follow-up period of 58 ± 48 months (log-rank test, P = .007). The Kaplan-Meier estimate of cardiac event–free survival according to gender is given in Figure 63-4, B. In accordance with these results, in a recent meta-analysis pooling data from 30 studies and including more than 1500 patients, male gender appeared as an independent predictor of cardiac events defined as SD, syncope, or internal defibrillator shock, with a relative risk (RR) of 3.47 (95% confidence interval [CI], 1.58 to 7.63) compared with women.47
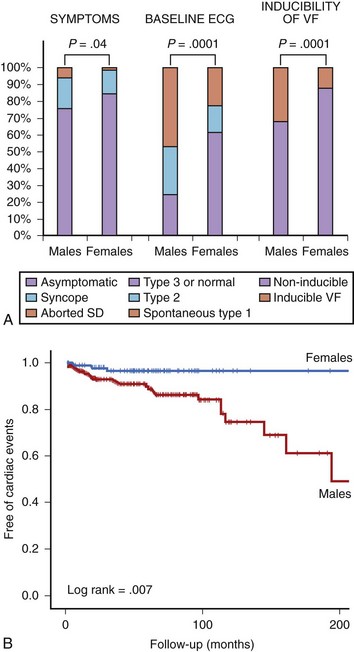
Two main hypotheses have been proposed for the gender distinction, which perhaps interact with each other. First, according to some experimental models, it appears that men could have constitutionally greater Ito density in the right ventricular epicardium than do women, which enhances the ionic imbalance. Second, sex hormones could play a role. Regression of the typical ECG features has been reported in castrated men, and levels of testosterone seem to be higher in male patients with Brugada syndrome compared with those in controls.48,49 Because of this hypothesis, the few data available thus far regarding Brugada syndrome in children have not shown a difference in phenotypic presentation between boys and girls before age 16.50
Children
Although 3 of the 8 patients reported in the first description of the disease were in the pediatric age group, little information has been available on the behavior of Brugada syndrome during childhood.1 Probst et al recently provided data from a multicenter study including 30 patients with Brugada syndrome aged younger than 16 years (mean age, 8 ± 5).50 More than half (n = 17) had been diagnosed during family screening, but symptoms were present in 11 patients (1 aborted SD and 10 syncope). Interestingly, 10 of the 11 symptomatic patients displayed spontaneous type 1 ECG, and in 5 of them, symptoms were precipitated by fever. Five patients received an ICD, and 4 were treated with hydroquinidine.50 During a mean follow-up period of 37 ± 23 months, 3 patients (10% of the population) experienced SD (n = 1) or an appropriate shock from an ICD (n = 2). Importantly, all the 3 patients had presented with syncope at the time of diagnosis and displayed spontaneous type 1 ECG. The 4 patients receiving quinidine remained asymptomatic during 28 ± 24 months of follow-up.50
The results of the chapter authors’ study on 58 pediatric patients with Brugada syndrome are in line with the ones from Probst et al and provide further information on prognosis markers during childhood.51 In the authors’ population (mean age, 11.8 ± 4), up to 22 patients (38%) were symptomatic (11 with previous syncope and 11 with aborted SD), and 36 had been diagnosed during family screening. Spontaneous type 1 ECG was present in 18 patients (31%), and the electrophysiological study (EPS), which was performed in 31 patients, induced VF in 7 of them. An ICD was indicated for 14 patients. Cardiac events appeared in 6 patients (2 SD and 4 appropriate ICD shocks) during a mean follow-up of 48.8 ± 48 months, this rate of 2.5% per year being somewhat lower than the 3.2% per year reported by Probst et al.50 Cardiac events occurred more frequently among patients with spontaneous type 1 ECG and among those with inducible VF at the EPS, but in the authors’ series, symptoms at diagnosis was the strongest variable to predict events during follow-up.51
Electrocardiogram and Modulating Factors
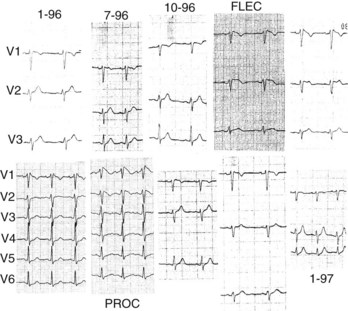
As mentioned above, three types of repolarization have been described in patients with Brugada syndrome, but only the coved-type ST-segment elevation (type 1 ECG pattern) is diagnostic of the syndrome (see Figure 63-1). However, it must be stressed that the ECG pattern typically fluctuates over time in patients with Brugada syndrome, and thus can change from type 1 to type 2 or type 3 or even be transiently normal (Figure 63-5). A large variety of conditions influence the electrocardiographic aspect (see below). The prevalence of spontaneous ECG fluctuations was assessed in a work by Veltmann et al, which included 310 ECGs on 43 patients followed up for 17.7 months.52 Among 15 patients with initial diagnostic ECGs, 14 revealed at least one nondiagnostic ECG in a median period of 12 days, while 8 of 28 patients with nondiagnostic ECGs developed a type 1 ECG pattern in a median period of 16 days. On the basis of these results, it appears that repetitive ECG recordings may be mandatory in patients with the syndrome; however, the role of the basal ECG as a risk marker should be understood with some caution (see section on Prognosis and Risk Stratification).52,53
It is worth noting that some factors can account for an ECG abnormality that can closely resemble the Brugada ECG (Table 63-1). Importantly, some of them are conditions different from the syndrome and should be carefully excluded during differential diagnosis, and some others may induce ST-segment elevation probably because an underlying genetic predisposition is present.54
Table 63-1 Electrocardiogram Abnormalities That Can Lead to ST-Segment Elevation in Leads V1 to V3
| DIFFERENTIAL DIAGNOSIS | GENETIC PREDISPOSITION? |
|---|---|
| Atypical right bundle branch block Acute MI, especially of the RV Acute pericarditis/myopericarditis Hemopericardium Pulmonary embolism Dissecting aortic aneurysm Central and autonomic nervous system disorders Duchenne muscular dystrophy Friedreich ataxia LV hypertrophy Arrhythmogenic RV cardiomyopathy Mechanical compression of the RV outflow tract: Mediastinal tumor Pectus excavatum After electrical cardioversion Early repolarization, especially in athletes Hypothermia |
Hyperkalemia Hypercalcemia Cocaine intoxication/alcohol intoxication Treatment with: I.Antiarrhythmic drugs: Sodium channel blockers (class IC, class IA) Calcium channel blockers β-blockers II.Antianginal drugs: Calcium channel blockers Nitrates III. Psychotropic drugs: Tricyclic antidepressants Tetracyclic antidepressants Phenothiazines Selective serotonin reuptake inhibitors Lithium |
MI, Myocardial infarction; RV, right ventricle; LV, left ventricle.
From Benito B, Brugada R, Brugada J, Brugada P: Brugada syndrome, Prog Cardiovasc Dis 51(1):1–22, 2008.
Modulating factors play a major role in the dynamic nature of the ECG and also may be responsible for the ST-segment elevation in genetically predisposed patients (see Table 63-1). As mentioned before, sympathovagal balance, hormones, metabolic factors, and pharmacologic agents, by means of specific effects on transmembrane ionic currents, are thought to not only modulate the ECG morphology but also explain the development of ventricular arrhythmias under certain conditions (see section on Cellular and Ionic Mechanisms).11,30,31,48,49,55,56 Temperature may be an important modulator in some patients with Brugada syndrome. Premature inactivation of the sodium channel has been shown to be accentuated at higher temperatures in some SCN5A mutations.10 Accordingly, several case reports in which fever precipitates the syndrome or an arrhythmic complication have been published recently.57,58 It seems that fever would be a particularly important trigger factor among pediatric patients.50
Numerous studies have analyzed the ECG of Brugada syndrome aiming to identify new electrocardiographic hallmarks or risk markers. Pitzalis and coworkers described a prolongation of the corrected Q-T interval (QTc) in right precordial leads, but not in left precordial leads, after administration of flecainide to patients with Brugada syndrome and nondiagnostic basal ECG.59 Subsequently, other groups have correlated a QTc 460 ms or greater in V2 to the occurrence of life-threatening arrhythmias.60 More recently, the aVR sign (an R wave ≥3 mm or an R/q ratio ≥0.75 in lead aVR; Figure 63-6, A) has also been defined as a risk marker of cardiac events in Brugada syndrome, the prominent R wave possibly reflecting some degree of right ventricular conduction delay and consequently more electrical heterogeneity.61 In addition, T-wave alternans (Figure 63-6, B), an indicator of transmural dispersion of repolarization, has been reported after administration of sodium blockers to patients with Brugada syndrome and is thought to be associated with an increased risk for the development of VF.62 Indeed, in a recent study conducted in 77 patients with Brugada syndrome, who were undergoing a pharmacologic test, the presence of T-wave alternans after administration of a sodium blocker identified a subgroup of patients with higher risk of spontaneous VF (52.9% vs. 8.3%, P < .001).63
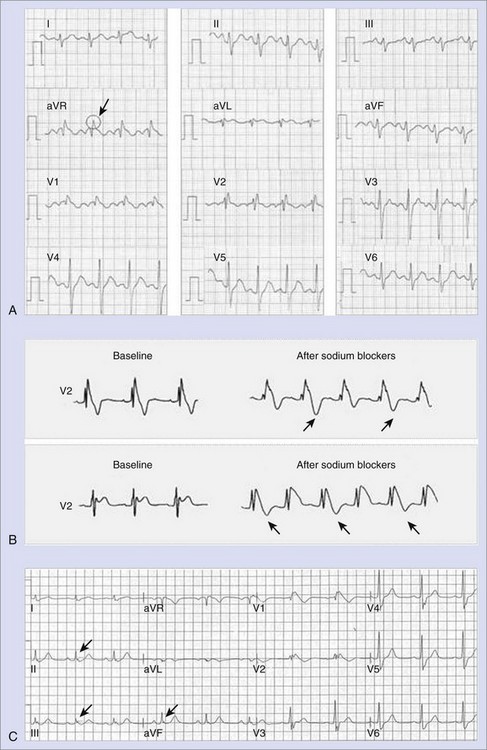
Following the description of several sporadic cases, a recent study has confirmed that up to 11% of patients with Brugada syndrome have inferior-lateral spontaneous repolarization pattern, defined as 1 mm or greater J-wave elevation or slurring in leads other than V1 to V3 (usually inferior, lateral, or both leads; Figure 63-6, C).64,65 Almost always, this pattern appears together with the typical coved-type ST-segment elevation in leads V1 to V3, which defines Brugada syndrome, either spontaneously or after administration of sodium channel blockers (see section on Diagnostic Tools: Drug Challenge). These patients present with the same clinical characteristics as those of patients with the typical form of Brugada syndrome, although they seem to develop more arrhythmic complications in their lifetime.65 Interestingly, the early repolarization pattern in inferior and lateral leads—found isolated and in the absence of ECG features typical of Brugada syndrome—has been associated with an increased susceptibility to VF and cardiac death in population studies, which suggests that this could be an independent syndrome responsible for some cases of SD in individuals with a normal heart.66,67 Although some similarities between Brugada syndrome and the new early repolarization disorder (high rate of arrhythmic complications at night, relative effectiveness of quinidine and isoproterenol in the treatment of arrhythmias) have been described, recent data suggest that both entities have distinct particularities, at least with regard to the mode of initiation of arrhythmic complications.66,68 On the basis of this, Brugada syndrome should always be carefully excluded in patients with inferior-lateral early repolarization pattern and idiopathic VF, even when the spontaneous ECG in leads V1 to V3 is normal. To date, the main difference between early repolarization disorder and Brugada syndrome seems to be the appearance of coved-type ST-segment elevation in response to sodium channel blockers in Brugada syndrome (see section on Diagnostic Tools: Drug Challenge). Therefore, it seems appropriate to recommend a pharmacologic test with sodium blockers for all individuals with early repolarization pattern and idiopathic VF in order to establish the definite diagnosis and unmask possible atypical forms of Brugada syndrome.65
Cardiac conduction disturbances may be present in patients with Brugada syndrome. Both phenotypes (Brugada syndrome and cardiac conduction disorders) can be explained by a reduction in the sodium current and have been described within the same family carrying a mutation on the SCN5A gene.69 Consequently, conduction parameters (specifically P-Q interval, QRS duration, and H-V interval) seem to be longer among those patients with Brugada syndrome who are SCN5A gene carriers (and do have a mutation in the sodium channel) compared with non-SCN5A gene carriers, in whom the underlying mechanism or mutation is not identified.70 These differences have been recently shown to become progressively accentuated during follow-up.71 In a recent study by this chapter authors’ group, it was observed that some conduction parameters such as QRS duration are increased among symptomatic patients. Indeed, in a population of 200 patients with Brugada syndrome, of whom 66 (33%) were symptomatic, the optimized cutoff point of QRS in lead V2 was 120 ms or greater, with an odds ratio (OR) of 2.5 (95% CI, 1.4 to 4.6; P = .003) for being symptomatic.72
Although sinus rhythm is the most common, supraventricular arrhythmias, especially AF, can be found in up to one third of patients with Brugada syndrome.42,43 Recently, it has been shown that patients with Brugada syndrome who present with spontaneous AF have more severe clinical phenotype.73 Other rhythm disorders such as bradycardia secondary to sick sinus syndrome or atrial standstill have also been reported in association with Brugada syndrome.28
Diagnostic Tools: Drug Challenge
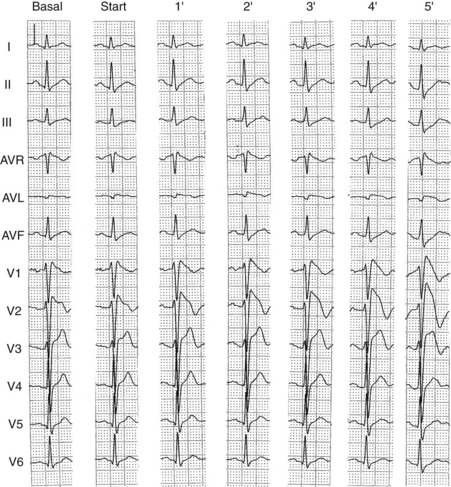
The diagnosis of Brugada syndrome can be established when a type 1 (coved-type) ECG pattern is found in right precordial leads in the absence of confounding factors and other causes of ST-segment elevation (see Table 63-1). However, because the ECG in Brugada syndrome is dynamic in nature and can even be transiently normal in affected patients, pharmacologic provocative tests have been used in an attempt to unmask concealed forms of the disease. Sodium channel blockers, which increase the ionic imbalance at the end of phase 1 of the AP by decreasing sodium currents, appear as the most attractive option.35 Ajmaline, flecainide, procainamide, pilsicainide, disopyramide, and propafenone have been used.2 The recommended dose regimens for the most commonly used drugs are listed in Table 63-2. Brugada syndrome is diagnosed if a coved-type (type 1 ECG) pattern appears after administration of a sodium channel blocker (Figure 63-7). According to the Second Consensus Committee, the pharmacologic test should be monitored under continuous ECG recording and should be terminated when (1) the diagnostic test is positive, (2) premature ventricular beats or other arrhythmias develop, or (3) QRS widens to 130% or more of baseline.2 The last criterion has recently been questioned. In a retrospective study, Batchvarov et al evaluated QRS duration and the occurrence of ventricular arrhythmias in 148 patients during ajmaline challenge and found that QRS prolonged to 130% or more of baseline in more than 50% of cases.74 The incidence of ventricular arrhythmias did not differ between patients with QRS prolongation and those without QRS prolongation, and no sustained arrhythmias were documented after drug administration. More importantly, in 40% of the positive tests, prolongation of QRS 130% or more occurred more than 1 minute earlier than the diagnostic Brugada ECG changes developed. Therefore, it seems that early termination of the test could possibly have resulted in false-negative results.74 On the basis of these results, these authors have proposed that the criteria for test termination be redefined according to baseline QRS duration (>150% in patients with normal QRS duration and >125% in patients with major intraventricular conduction disturbances).74
| DRUG | DOSAGE | ADMINISTRATION |
|---|---|---|
| Ajmaline | 1 mg/kg over 5 min | IV |
| Flecainide | 2 mg/kg over 10 min | IV |
| 400 mg | PO | |
| Procainamide | 10 mg/kg over 10 min | IV |
| Pilsicainide | 1 mg/kg over 10 min | IV |
IV, Intravenous; PO, oral.
From Antzelevitch C, Brugada P, Borggrefe M, et al: Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association, Circulation 111(5):659–670, 2005; and Benito B, Brugada R, Brugada J, Brugada P: Brugada syndrome, Prog Cardiovasc Dis 51(1):1–22, 2008.
Current data indicate that ajmaline is probably the best drug available to unmask Brugada syndrome, whereas procainamide, the only drug available in the United States, seems to be the weakest to uncover ECG abnormalities. In a study with 147 individuals from four large families with identified SCN5A mutations, ajmaline provided a sensitivity of 80%, a specificity of 94.4%, a positive predictive value of 93.3%, and a negative predictive value of 82.9% for the diagnosis of Brugada syndrome.75 The penetrance of the disease phenotype increased from 32.7% to 78.6% with the use of a sodium channel blocker.75 These results show values considerably higher than those obtained for flecainide in another study with 110 genotyped patients, in which the sensitivity, the specificity, and the positive and the negative predictive values for the diagnosis were 77%, 80%, 96%, and 36%, respectively.76 It is important to note the low negative predictive value, which should be taken into account when using flecainide, especially during genetic screening. Ajmaline and flecainide were compared in a study with 22 patients with confirmed Brugada syndrome who were subjected to both pharmacologic tests. Although the test was positive in all 22 patients after ajmaline administration, only 15 patients showed a positive response to flecainide.77 Experimental studies revealed that flecainide inhibits not only sodium currents but also Ito currents, which explains its lesser effectiveness in enhancing the ionic imbalance at phase 1.77
Given the limitations of drug tests and the restricted availability of genetic analysis, new strategies have been proposed to help in the clinical diagnosis of Brugada syndrome. Placement of the right precordial leads in an upper position (second or third intercostal spaces) can increase the sensitivity of the ECG to detect the Brugada phenotype, in the presence or absence of a drug challenge, although it is still uncertain whether the greater sensitivity is at the expense of a lower specificity.78,79 Recent data demonstrate that the presence of a type 1 ECG pattern recorded at higher intercostal spaces, even when the standard ECG is normal, can identify a subgroup of patients with a prognosis similar to those with spontaneous type 1 ECG pattern at standard leads (Figure 63-8).80 Therefore, this strategy seems to allow the identification of a subset of patients at risk who would otherwise be underdiagnosed.
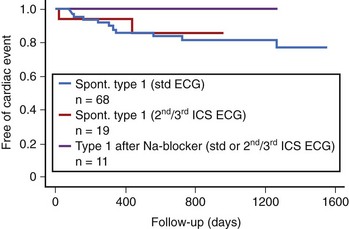
Pharmacologic challenge with sodium channel blockers should be performed on patients with syncope, aborted SD of unknown origin, or both to unmask possible concealed forms of Brugada syndrome. Other indications of pharmacologic testing include asymptomatic patients with a suspicious ECG (spontaneous type 2 or type 3 ECG) and family members of a confirmed index case during family screening if they do not have a spontaneous type 1 ECG (see section on Approach to Patients with Suspected Brugada Syndrome: Family Screening).
Prognosis and Risk Stratification
In the authors’ most recent study population with Brugada syndrome coming from the international registry, the percentage of patients who experienced SCD or VF throughout their lifetime was 25% (177 of 724 patients).81 The mean age at cardiac events was 42 ± 15 years. Of course, such a high rate might have been influenced by a baseline high-risk population referred to the international registry and included in this analysis. In fact, the authors’ reported annual rate of events has decreased from the first patients included in the registry to the most recent published series; the change probably reflects the inherent bias during the first years after the description of a novel disease, in which particularly severe forms of the disease are most likely to be diagnosed.4,37,39,81 It is important to note that in the global series, the lifetime probability of having a cardiac event during varied widely (from 3% to 45%), depending on the baseline characteristics of the individuals. Thus, a careful risk stratification of every individual is mandatory.
Several clinical variables have been demonstrated to predict a poor outcome in patients with Brugada syndrome. In all the analyses of the authors’ series over time, the presence of symptoms before diagnosis, a spontaneous type 1 ECG at baseline, the inducibility of ventricular arrhythmias at the EPS, and male gender have consistently been related to the occurrence of cardiac events during follow-up.4,33,37,39,81
Little controversy exists on the value of a previous cardiac arrest as a risk marker for future events. The authors’ data show that up to 62% of patients recovered from an aborted SCD are at risk of a new arrhythmic event within the following 54 months.37 Thus, these patients should be protected with an ICD irrespective of the presence of other risk factors (indication class I).2,82 Because a general agreement on the best approach toward patients who have never developed VF is lacking, the authors conducted a prospective study including 547 individuals with Brugada syndrome and no previous cardiac arrest.39 Of them (mean age 41 ± 45 years, 408 men), 124 had presented with syncope (22.7%) and 423 (77.3%) were asymptomatic and had been diagnosed during routine ECG or family screening. The baseline ECG showed a type 1 ECG pattern spontaneously in 391 patients (71.5%) and after sodium channel blocker challenge in 156 (28.5%). During a mean follow-up of 24 ± 32 months, 45 individuals (8.2%) developed a first major cardiac event (documented VF or SD).39 By univariable analysis, a previous history of syncope (heart rate [HR], 2.79; 95% CI, 1.5 to 5.1; P = .002), a spontaneous type 1 ECG (HR, 7.69; 95% CI, 1.9 to 33.3; P = .0001), male gender (HR, 5.26; 95% CI, 1.6 to 16.6; P = .001), and inducibility of ventricular arrhythmias at the EPS (HR, 8.33; 95% CI, 2.8 to 25; P = .0001) were significantly related to VF or SCD in follow-up. Multivariable analysis identified previous syncope and inducibility of VF as the only independent risk factors for the occurrence of events during follow-up (Figure 63-9, A).39 Logistic regression analysis allowed the definition of eight categories of risk, of which asymptomatic patients with normal ECG at baseline and non-inducible VF at the EPS would represent the population at lowest risk; and patients with syncope, spontaneous type 1 ECG, and inducibility at EPS would have the worst outcome (Figure 63-9, B). Further analysis indicated that the EPS was particularly useful in predicting cardiac events among asymptomatic patients with no family history of SCD (named fortuitous cases, n = 167).81 Indeed, 11 (6%) of 167 patients presented with VF during follow-up, and the only independent predictor was inducibility at the EPS. In contrast, not performing an EPS in this subgroup of patients with the aim of identifying those at risk was shown to be predictive of effective SD (P = .002).81
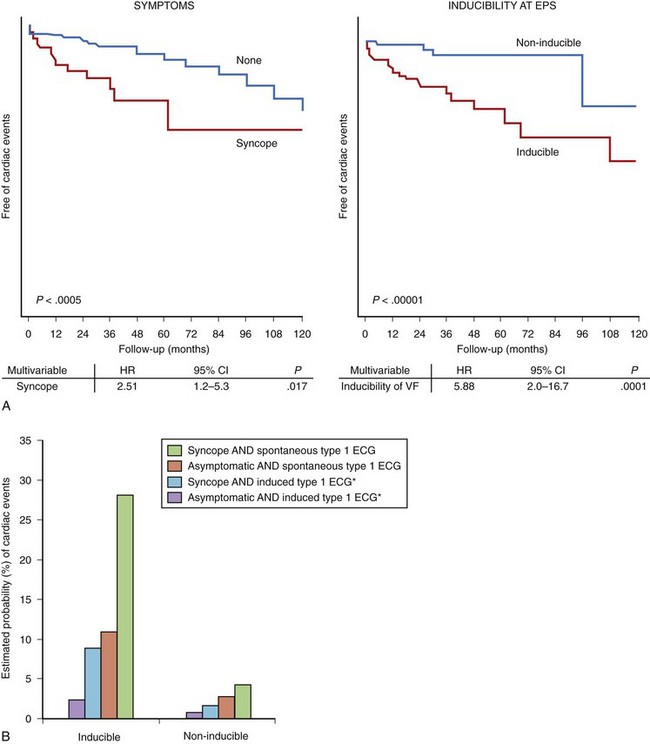
Other groups agree that previous symptoms and a spontaneous type 1 ECG are risk factors, although they have found a much lower incidence of arrhythmic events for the whole population (6.5% in 34 ± 44 months of follow-up in the work of Priori et al and 4.2% in 40 ± 50 months of follow-up in that of Eckardt et al).38,40 The worse outcome in the authors’ series may probably reflect a more severely ill baseline population.40 The other large registries also agree that EPS inducibility is greatest among patients with previous SCD or syncope, but failed to demonstrate the value of the EPS in predicting outcome.38,40 Several reasons could explain this discrepancy: (1) the use of multiple testing centers with nonstandardized stimulation protocols; (2) the inclusion of patients with type 2 and type 3 ST-segment elevation (and not type 1) in some series, which suggests that they may contain individuals who do not have the syndrome; and (3) the lack of events during follow-up in the other registries.81 The latter might change when longer follow-ups are available because events can only increase in follow-up and so will the positive predictive value.81
Males have consistently shown a trend to experience more arrhythmic events in all the studies, and male sex even has been defined as an independent predictor for a worse outcome in a recent meta-analysis.47 A very recent study by the authors’ group indicates that men with Brugada syndrome display a higher risk profile than do women and thus have a worse prognosis during follow-up (see section on Sex Differences).33 This study also suggests that although the classical risk markers (symptoms, spontaneous type 1 ECG, and inducibility of VF) are useful in identifying male patients at risk, the female population with worse outcome is usually characterized by more severe conduction disturbances.33 Spontaneous AF, which can appear in 10% to 53% of cases, has been recently shown to have prognostic significance.43,73 Kusano et al, in a series of 73 patients with Brugada syndrome, observed that spontaneous AF was associated with a higher incidence of syncopal episodes (60.0% vs. 22.2%; P < .03) and documented VF (40.0% vs. 14.3%; P < .05).73 In the same line, the authors’ results demonstrate that AF is more frequently observed in patients who experience SCD or VF during follow-up compared with asymptomatic patients, both in the male and female populations.33 Multiple ECG parameters have been assessed in the search for new risk markers, of which a prolonged QTc in V2, the aVR sign, the presence of T-wave alternans, the early repolarization pattern in inferior or lateral leads, and probably a wide QRS complex seem to be the most important (see section on Electrocardiogram and Modulating Factors).60–63,65,72 Interestingly, a positive family history of SD or the presence of an SCN5A mutation have not been proven to be risk markers in any of the large studies conducted thus far.38,40,47
However, recent data suggest that other genetic findings might have prognostic implications. In a recent study with 147 patients with Brugada syndrome or progressive cardiac conduction disease carrying 32 different mutations in the SCN5A gene, Meregalli et al found that those with a mutation leading to a premature stop codon (and thus a truncated protein) had a higher rate of syncope than did patients with other types of mutation (25.3% vs. 5.7%, respectively; P = .03).83 Nevertheless, these authors could not find differences in the rate of major arrhythmic complications (SCD or VF) according to the type of mutation.83 The chapter authors’ data on 188 patients (all with Brugada syndrome) carrying 69 different mutations in SCN5A demonstrated that the presence of a mutation leading to a premature stop codon is related to a greater rate of major cardiac events defined as SCD or documented VF.84 Moreover, in the authors’ series, the presence of a mutation leading to a truncated protein was confirmed as an independent predictor of cardiac events (HR, 2.9; 95% CI, 1.2 to 7.2; P = .02), together with the classic clinical risk factors reported in previous series.84 From these data, it can be concluded that genetic testing could be useful in the risk stratification of patients with Brugada syndrome who are carriers of an SCN5A mutation. This finding is particularly important because, in contrast to previously defined clinical variables, genetic information is constitutional and thus invariable over time within the same individual.
Treatment
Implantable Cardioverter Defibrillator
The implantable cardioverter defibrillator (ICD) has been the only proven effective treatment so far for Brugada syndrome. On the basis of available clinical and basic science data, a second Consensus Conference was held in September 2003, focusing on risk stratification schemes and approaches to therapy.2 The recommendations for ICD implantation stated by this consensus are summarized in Figure 63-10, although not all of them have been endorsed in the most recent ACC/AHA/ESC 2006 Guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD (Table 63-3).82 Symptomatic patients should always receive an ICD. The EPS could be performed in these patients to better assess the sensitivity and specificity of the test to predict the outcome and also for the study of supraventricular arrhythmias. Asymptomatic patients may benefit from the EPS for risk stratification, especially if they have a spontaneous type 1 ECG: An ICD should be implanted in those with inducible VF. Indications for the EPS and ICD implantation in asymptomatic patients with sodium channel blocker–induced ECG are, however, less established. Although the second Consensus Conference proposed performing an EPS for risk stratification in these patients and ICD implantation in those who had inducible arrhythmias (indication class IIb), this recommendation has not been included in the most updated guidelines.2,82 According to the latter, asymptomatic patients who develop a type 1 ECG only after sodium channel blockade should probably be closely followed up, whether or not they have a family history of SCD (see Figure 63-10).82
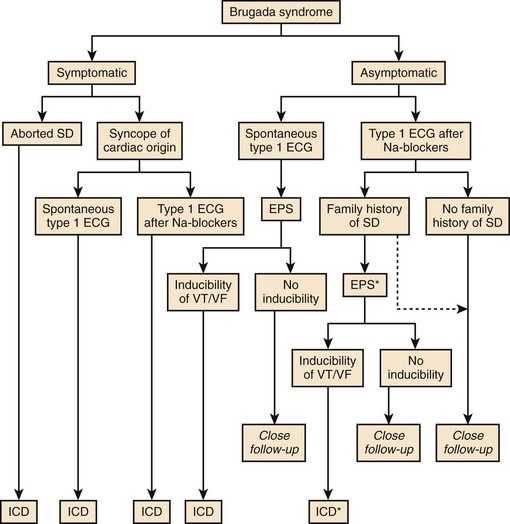
Table 63-3 Recommendations from the ACC/AHA/ESC 2006 Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Brugada Syndrome
| Class I benefit > risk (“should be done”) | An ICD is indicated for patients with Brugada syndrome who have had a previous cardiac arrest and who have reasonable expectation of survival with a good functional status for more than 1 year. |
| Class IIa benefit ≅ risk (“it is reasonable”) | An ICD is reasonable for patients with Brugada syndrome who have spontaneous elevation in V1, V2, or V3, who have had syncope with or without causative mutations demonstrated, and who have reasonable expectation of survival with a good functional status for more than 1 year. |
| An ICD is reasonable for patients with Brugada syndrome who have documented VT that has not resulted in cardiac arrest and who have reasonable expectation of survival with a good functional status for more than 1 year. | |
| Clinical monitoring for the development of a spontaneous ST-segment elevation pattern is reasonable for the management of patients with ST-segment elevation induced only with provocative pharmacologic challenge in the presence or absence of symptoms. | |
| Isoproterenol can be useful in treating an electrical storm in patients with Brugada syndrome. | |
| Class IIb benefit ≥ risk (“may be considered”) | EPS testing may be considered for risk stratification in asymptomatic patients with Brugada syndrome who have spontaneous ST-segment elevation with or without a mutation identified. |
| Quinidine might be reasonable for the treatment of electrical storm in patients with Brugada syndrome. |
ICD, Inplantable cardioverter-defibrillator; VT, ventricular tachycardia.
Modified from Zipes DP, Camm AJ, Borggrefe M, et al: ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society, Europace 8(9):746–837, 2006.
Single- or dual-chamber ICDs can be implanted in patients with Brugada syndrome, and the decision usually depends on a history of supraventricular arrhythmias, concomitant pacing indication, or both. In the routine protocol employed by the authors of this chapter, a single VF zone is programmed, with a detection rate between 180 and 220 beats/min (individualized for all patients) and backup pacing at a rate of 35 to 50 beats/min.85
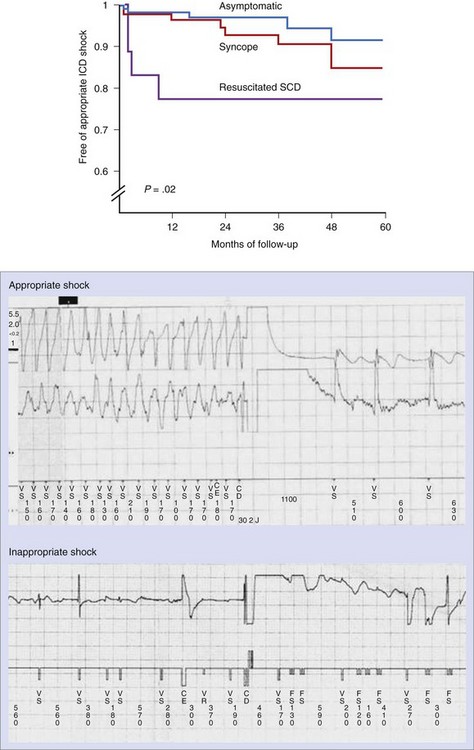
From the two main retrospective studies conducted on patients with Brugada syndrome who had received ICD, it can be concluded that ICD is an effective therapy for patients at risk, which can have an annual rate of appropriate shocks of up to 3.7%.85,86 It is important to note that this rate is not only comparable with ICD trials dealing with other cardiac diseases, but it also affects young and otherwise healthy people, whose life expectancy could be more than 30 years.87,88 Therefore, should this rate remain constant in time, most patients would be likely to receive appropriate ICD therapy in their lifetime. The rate of appropriate shocks is significantly higher among patients who were symptomatic prior to ICD implantation (22% in patients with previous resuscitated SCD and 10% in patients with previous syncope during a mean follow-up of 38 ± 27 months) than in previously asymptomatic patients (4% during comparable follow-up; P = .025), symptoms prior to ICD implantation being the only predictor of subsequent appropriate ICD therapies (Figure 63-11).86 However, a noteworthy rate of inappropriate shocks from the device has also been reported (see Figure 63-11). In the study by Sacher et al, 45 (20%) of 220 patients had inappropriate shocks in a follow-up.86 In the chapter authors’ series, the rate was even higher (36%).85 The reasons for inappropriate therapies were mainly sinus tachycardia, supraventricular arrhythmias, T-wave oversensing, and lead failure in both studies.85,86 In fact, a previous history of supraventricular arrhythmias, the presence of T-wave oversensing, and a low R-wave amplitude (<5 mV) at the time of implantation have all been found to be predictors of inappropriate therapies in follow-up. On the basis of these results and because ICD is not affordable worldwide, efforts to find pharmacologic approaches to help treat the disease are growing.
Pharmacologic Options
With the aim of rebalancing the ion channel currents active during phase 1 of the AP, so as to reduce the magnitude of the notch (see Figure 63-2, A), two main pharmacologic approaches have been assessed (Table 63-4): (1) drugs that decrease outward positive currents, such as Ito inhibitors; and (2) drugs that increase inward positive currents (ICa, INa).
Table 63-4 Pharmacologic Approach to Therapy in Brugada Syndrome
| ACTION | PROVED ON |
|---|---|
| Ito BLOCKERS | |
| 4-Aminopyridine |
See references 28, 36, 50, 89, and 90 to 93.
ICD, Implantable cardioverter-defibrillator; VF, ventricular fibrillation.
* Fish JM, Welchons DR, Kim YS, et al: Dimethyl lithospermate B, an extract of Danshen, suppresses arrhythmogenesis associated with the Brugada syndrome, Circulation 113(11):1393–1400, 2006.
From Benito B, Brugada R, Brugada J, Brugada P: Brugada syndrome, Prog Cardiovasc Dis 51(1):1–22, 2008.
Quinidine, a drug with Ito-blocking and IKr-blocking properties, has been the most assayed drug in clinical studies. In a work by Belhassen et al, 25 patients with inducible VF were treated with quinidine (1483 ± 240 mg orally).36 After treatment, 22 (88%) of 25 patients were no longer inducible at the EPS, and none of the 19 patients with ongoing medical therapy with oral quinidine developed arrhythmias during follow-up (56 ± 67 months).36 However, 36% of the patients had transient side effects that led to drug discontinuation. Preliminary data have also proven quinidine as a good adjunctive therapy in patients with ICD and multiple shocks and as an effective treatment of electrical storms associated with Brugada syndrome.89,90 More recently, quinidine has been proposed as a good alternative to ICD implantation in children with the syndrome and at high risk for malignant arrhythmias.50
β-Adrenergic agents, through an increase in ICa currents, have been shown to decrease transmural dispersion of repolarization and epicardial dispersion of repolarization in experimental models.11 Clinically, they have proven effectiveness in the treatment of electrical storms associated with Brugada syndrome.91 Recently, phosphodiesterase III inhibitors have appeared as a new appealing option because they would increase ICa and decrease Ito. Indeed, cilostazol was reported to be effective in preventing ICD shocks in a patient with recurrent episodes of VF.92 However, a recent publication reports the failure of this drug in another patient with multiple ICD discharges despite sustained therapy.93
Approach to Patients with Suspected Brugada Syndrome: Family Screening
The diagnosis of Brugada syndrome strongly depends on the degree of suspicion on the part of the physician. Patients with syncope, aborted SD of unknown origin, or both and normal hearts should be recommended to have a standard ECG to rule out a spontaneous diagnostic type 1 ECG pattern. If the ECG is found to be normal, a modified ECG with recording from upper precordial leads and a pharmacologic challenge with a sodium channel blocker should be performed to detect possible concealed forms of Brugada syndrome. If a type 1 ECG is documented, the diagnosis of Brugada syndrome can be established (Figure 63-12).
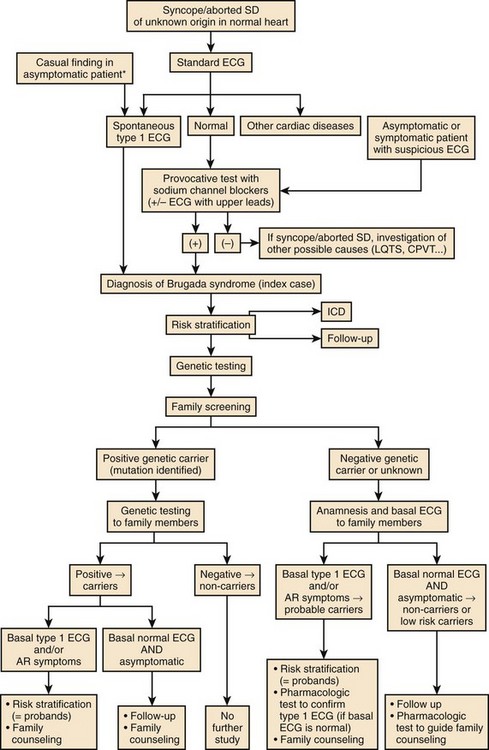
The first step after identification of a proband with Brugada syndrome is to evaluate his or her individual risk of SD and recommend ICD if necessary (see section on Prognosis and Risk Stratification). Currently, genetic testing is recommended because it helps confirm the disease in patients with borderline phenotype, may provide information on arrhythmic risk during lifetime (see above), and can be helpful in family screening.
Given that Brugada syndrome is commonly an inherited disorder, family screening should always be performed to identify possible relatives who are unaware of the risk of SD (see Figure 65-12). It is important to remember that hereditary forms of Brugada syndrome are autosomal dominant and not linked to sex and that each affected patient has a 50% probability of transmitting the disease to his or her offspring.
Key References
Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart rhythm Association. Circulation. 2005;111(5):659-670.
Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442-449.
Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110(13):1731-1737.
Benito B, Sarkozy A, Mont L, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52:1567-1573.
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101(5):510-515.
Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108(25):3092-3096.
Brugada P, Brugada R, Brugada J, et al. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292.
Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-296.
Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1(3):209-218.
Eckardt L, Probst V, Smits JPP, et al. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation. 2005;111(3):257-263.
London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1–like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116(20):2260-2268.
Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation. 2002;105(11):1342-1347.
Probst V, Denjoy I, MeregalIi PG, et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation. 2007;115(15):2042-2048.
Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-1666.
1 Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
2 Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659-670.
3 Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac death. J Cardiovasc Electrophysiol. 1997;8(3):325-331.
4 Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: A marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97(5):457-460.
5 Alings M, Wilde A. “Brugada” syndrome: Clinical data and suggested pathophysiological mechanism. Circulation. 1999;99(5):666-673.
6 Brugada P, Brugada R, Brugada J. Sudden death in patients and relatives with the syndrome of right bundle branch block, ST segment elevation in the precordial leads V1to V3 and sudden death. Eur Heart J. 2000;21(4):321-326.
7 Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102(20):2509-2515.
8 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-296.
9 Rook MB, Bezzina Alshinawi C, Groenewegen WA, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44(3):507-517.
10 Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85(9):803-809.
11 Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-1666.
12 Deschênes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46(1):55-65.
13 Wilde AAM, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome. Eur Heart J. 2002;23(21):1648-1654.
14 van den Berg MP, de Boer RA, van Tintelen JP. Brugada syndrome or Brugada electrocardiogram? J Am Coll Cardiol. 2009;53(17):1569.
15 Benito B, Brugada J, Brugada R, Brugada P. Brugada syndrome or Brugada electrocardiogram? Authors’ reply. J Am Coll Cardiol. 2009;53(17):1569-1570.
16 Miyasaka Y, Tsuji H, Yamada K, et al. Prevalence and mortality of the Brugada-type electrocardiogram in one city in Japan. J Am Coll Cardiol. 2001;38(3):771-774.
17 Hermida JS, Lemoine JL, Aoun FB, Jarry G, Rey JL, Quiret JC. Prevalence of the Brugada syndrome in an apparently healthy population. Am J Cardiol. 2000;86(1):91-94.
18 Donohue D, Tehrani F, Jamehdor R, Lam C, Movahed MR. The prevalence of Brugada ECG in adult patients in a large university hospital in the Western United States. Am Heart Hosp J. 2008;6(1):48-50.
19 Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11(3):337-345.
20 Vatta M, Dumaine R, Antzelevitch C, et al. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol Genet Metab. 2002;75(4):317-324.
21 Cordeiro JM, Barajas-Martinez H, Hong K, et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006;114(19):2026-2033.
22 Casini S, Tan HL, Bhuiyan ZA, et al. Characterization of a novel SCN5A mutation associated with Brugada syndrome reveals involvement of DIIIS4-S5 linker in slow inactivation. Cardiovasc Res. 2007;76(3):418-429.
23 Pfahnl AE, Viswanathan PC, Weiss R, et al. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm. 2007;4(1):46-53.
24 London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1–like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116(20):2260-2268.
25 Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105(6):707-713.
26 Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442-449.
27 Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1(3):209-218.
28 Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29(10):1130-1159.
29 Fish J, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1(2):210-217.
30 Matsuo K, Kurita T, Inagaki M, et al. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. Eur Heart J. 1999;20(6):465-470.
31 Takigawa M, Noda T, Shimizu W, et al. Seasonal and circadian distributions of ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm. 2008;5(11):1523-1527.
32 Litovsky SH, Antzelevitch C. Differences in the electrophysiological response of canine ventricular subendocardium and subepicardium to acetylcholine and isoproterenol. A direct effect of acetylcholine in ventricular myocardium. Circ Res. 1990;67(3):615-627.
33 Benito B, Sarkozy A, Mont L, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52:1567-1573.
34 Di Diego JM, Cordeiro JM, Goodrow RJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106(15):2004-2011.
35 Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101(5):510-515.
36 Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110(13):1731-1737.
37 Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105(1):73-78.
38 Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation. 2002;105(11):1342-1347.
39 Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108(25):3092-3096.
40 Eckardt L, Probst V, Smits JPP, et al. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation. 2005;111(3):257-263.
41 Benito B, Arzamendi D, Porres J, et al. Seguimiento a largo plazo de los pacientes con sindrome de Brugada. Estudio multicentrico de los factores de mal pronostico [translation, abstract]. Rev Esp Cardiol. 2007;60(Suppl 2):116.
42 Eckardt L, Kirchhof P, Loh P, et al. Brugada syndrome and supraventricular tachyarrhythmias: A novel association? J Cardiovasc Electrophysiol. 2001;12(6):680-685.
43 Morita H, Kusano-Fukushima K, Nagase S, et al. Atrial fibrillation and atrial vulnerability in patients with Brugada syndrome. J Am Coll Cardiol. 2002;40(8):1437-1444.
44 Benito B, Brugada J. Recurrent syncope: An unusual presentation of Brugada syndrome. Nat Clin Pract Cardiovasc Med. 2006;3(10):573-577.
45 Makita N, Sumitomo N, Watanabe I, Tsutsui H. Novel SCN5A mutation (Q55X) associated with age-dependent expression of Brugada syndrome presenting as neurally mediated syncope. Heart Rhythm. 2007;4(4):516-519.
46 Eckardt L. Gender differences in Brugada syndrome. J Cardiovasc Electrophysiol. 2007;18(4):422-424.
47 Gehi A, Duong T, Metz L, Gomes J, Mehta D. Risk stratification of individuals with the Brugada electrocardiogram: A meta-analysis. J Cardiovasc Electrophysiol. 2006;17(6):577-583.
48 Matsuo K, Akahoshi M, Seto S, Yan K. Disappearance of the Brugada-type electrocardiogram after surgical castration: A role for testosterone and an explanation for the male preponderance. Pacing Clin Electrophysiol. 2003;26:1551-1553.
49 Shimizu W, Matsuo K, Kokubo Y, et al. Sex hormone and gender difference—role of testosterone on male predominance in Brugada syndrome. J Cardiovas Electrophysiol. 2007;18(4):415-421.
50 Probst V, Denjoy I, MeregalIi PG, et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation. 2007;115(15):2042-2048.
51 Benito B, Sarkozy A, Berne P, et al. [Características clínicas y pronóstico del síndrome de Brugada en la población pediátrica [abstract]. Rev Esp Cardiol. 2007;60(Suppl 2):70.
52 Veltmann C, Schimpf R, Echternach C, et al. A prospective study on spontaneous fluctuations between diagnostic and non-diagnostic ECGs in Brugada syndrome: Implications for correct phenotyping and risk stratification. Eur Heart J. 2006;27(21):2544-2552.
53 Wilde AA. Spontaneous electrocardiographic fluctuations in Brugada syndrome: Does it matter? Eur Heart J. 2006;27(21):2493-2494.
54 Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008;51(1):1-22.
55 Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27(5):1061-1070.
56 Tatsumi H, Takagi M, Nakagawa E, Yamashita H, Yoshiyama M. Risk stratification in patients with Brugada syndrome: Analysis of daily fluctuations in 12-lead electrocardiogram (ECG) and signal-averaged electrocardiogram (SAECG). J Cardiovasc Electrophysiol. 2006;17(7):705-711.
57 Gonzalez Rebollo JM, Hernandez MA, Garcia A, Garcia de CA, Mejias A, Moro C. Recurrent ventricular fibrillation during a febrile illness in a patient with the Brugada syndrome [translation]. Rev Esp Cardiol. 2000;53(5):755-757.
58 Porres JM, Brugada J, Urbistondo V, Garcia F, Reviejo K, Marco P. Fever unmasking the Brugada syndrome. Pacing Clin Electrophysiol. 2002;25(11):1646-1648.
59 Pitzalis MV, Anaclerio M, Iacoviello M, et al. QT-interval prolongation in right precordial leads: An additional electrocardiographic hallmark of Brugada syndrome. J Am Coll Cardiol. 2003;42(9):1632-1637.
60 Castro Hevia J, Antzelevitch C, Tornés Bárzaga F, et al. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol. 2006;47(9):1828-1834.
61 Babai Bigi MA, Aslani A, Shahrzad S. aVR sign as a risk factor for life-threatening arrhythmic events in patients with Brugada syndrome. Heart Rhythm. 2007;4(8):1009-1012.
62 Fish JM, Antzelevitch C. Cellular mechanism and arrhythmogenic potential of T-wave alternans in the Brugada syndrome. J Cardiovasc Electrophysiol. 2008;19(3):301-308.
63 Tada T, Kusano KF, Nagase S, et al. The relationship between the magnitude of T wave alternans and amplitude of the corresponding T wave in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2008;19(1):56-61.
64 Ogawa M, Kumagai K, Yamanouchi Y, Saku K. Spontaneous onset of ventricular fibrillation in Brugada syndrome with J wave and ST-segment elevation in the inferior leads. Heart Rhythm. 2005;2(1):97-99.
65 Sarkozy A, Chierchia GB, Paparella G, et al. Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome. Circ Arrhythmia Electrophysiol. 2009;2(2):154-161.
66 Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016-2023.
67 Tikkanen JT, Anttonen O, Junttila MJ, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361(26):2529-2537.
68 Nam GB, Ko KH, Kim J, et al. Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J. 2009;31(3):330-339.
69 Kyndt F, Probst V, Potet F, et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104(25):3081-3086.
70 Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: Electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40(2):350-356.
71 Yokokawa M, Noda T, Okamura H, et al. Comparison of long-term follow-up of electrocardiographic features in Brugada syndrome between the SCN5A-positive probands and the SCN5A-negative probands. Am J Cardiol. 2007;100(4):649-655.
72 Junttila MJ, Brugada P, Hong K, et al. Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol. 2007;19(4):380-383.
73 Kusano KF, Taniyama M, Nakamura K, et al. Atrial fibrillation in patients with Brugada syndrome relationships of gene mutation, electrophysiology, and clinical backgrounds. J Am Coll Cardiol. 2008;51(12):1169-1175.
74 Batchvarov VN, Govindan M, Camm AJ, Behr ER. Significance of QRS prolongation during diagnostic ajmaline test in patients with suspected Brugada syndrome. Heart Rhythm. 2009;6(5):625-631.
75 Hong K, Brugada J, Oliva A, et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation. 2004;110(19):3023-3027.
76 Meregalli PG, Ruijter JM, Hofman N, Bezzina CR, Wilde AA, Tan HL. Diagnostic value of flecainide testing in unmasking SCN5A-related Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17(8):857-864.
77 Wolpert C, Echternach C, Veltmann C, et al. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2(3):254-260.
78 Shimizu W, Matsuo K, Takagi M, et al. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: Clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms. J Cardiovasc Electrophysiol. 2000;11(4):396-404.
79 Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001;22(24):2290-2296.
80 Miyamoto K, Yokokawa M, Tanaka K, et al. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol. 2007;99(1):53-57.
81 Brugada P, Brugada R, Brugada J, et al. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292.
82 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8(9):746-837.
83 Meregalli PG, Tan HL, Probst V, et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 2009;6(3):341-348.
84 Benito B, Campuzano O, Ishac R, et al. Role of genetic testing in risk stratification of Brugada syndrome [abstract]. Heart Rhythm. 2009;6:S102.
85 Sarkozy A, Boussy T, Kourgiannides G, et al. Long-term follow-up of primary prophylactic implantable cardioverter-defibrillator therapy in Brugada syndrome. Eur Heart J. 2007;28(3):334-344.
86 Sacher F, Probst V, Iesaka Y, et al. Outcome after implantation of a cardioverter-defibrillator in patients with Brugada syndrome: A multicenter study. Circulation. 2006;114(22):2317-2324.
87 Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342(6):365-373.
88 Bardy GH, Lee KL, Mark DB, et al. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005;352(3):225-237.
89 Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol. 2004;43(10):1853-1860.
90 Mok NS, Chan NY, Chiu AC. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol. 2004;27(6 Pt 1):821-823.
91 Ohgo T, Okamura H, Noda T, et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm. 2007;4(6):695-700.
92 Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13(7):698-701.
93 Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17(2):210-212.