Genetic Defects In Vitamin D Metabolism and Action
Vitamin D is a key regulator of mineral homeostasis1,2 and bone development,3 and perturbation of the vitamin D endocrine system leads to rickets and/or osteomalacia (see Chapter 15). In order to gain a complete perspective of the clinical manifestations of genetic anomalies involving vitamin D endocrine function, this chapter will first present a short overview of the salient aspects of the vitamin D metabolic pathway. (An in-depth discussion of vitamin D metabolism and bone development and remodeling can be found in Chapters 3 and 4). The concepts presented here will lay the groundwork for the discussion of the clinical, pathophysiologic, and molecular aspects of hereditary rickets involving the vitamin D endocrine system.
Overview of Vitamin D Metabolism
In humans, a sizeable proportion of vitamin D requirements can be produced endogenously in the skin upon exposure to ultraviolet (UV) light (sunlight). It has been shown, however, that at latitudes where vitamin D synthesis is reduced or absent during winter months, there is a seasonal variation in the photosynthesis of vitamin D.4 People who receive an ample supply of sunlight during the rest of the year are not at risk of developing vitamin D deficiency, because excess cutaneously produced vitamin D is stored in fat and muscle and released at times of need.5 Dietary sources such as fish, plants, and grains can help meet vitamin D requirements. In the absence of any exposure to sunlight, however (such as in elderly people), a daily multivitamin that contains 400 IU of vitamin D is indicated.6,7
Ultraviolet B (UVB) photons penetrate the epidermis and photolize 7-dehydrocholesterol into previtamin D, which rapidly becomes a more thermodynamically stable molecule, vitamin D. Vitamin D then exits the keratinocyte cells and enters the dermal capillary bed, where it becomes bound to the vitamin D binding protein, DBP. Once associated with DBP in the circulation, vitamin D is transported to the liver, where cytochrome P450 25-hydroxylase enzymes (CYP27A1 and/or CYP2R1) add a hydroxyl group on carbon 25 to produce 25-hydroxyvitamin D [25(OH)D]. Early studies using perfused rat liver revealed kinetics of vitamin D metabolism that supported two 25-hydroxylase activities: a microsomal high-affinity, low-capacity enzyme and a mitochondrial low-affinity, high-capacity form.8 The mitochondrial enzyme is the bifunctional CYP27A1 sterol, 27-hydroxylase, that derives its name from its ability to both 27-hydroxylate the side chains of cholesterol-derived intermediates involved in bile acid biosynthesis and 25-hydroxylate vitamin D.9 The microsomal, high-affinity vitamin D 25-hydroxylase was recently identified as CYP2R1, using an elegant expression-based screening strategy.10 Based on specific activity determination, it is estimated that the microsomal enzyme is responsible for about 30% of the total 25-hydroxylation activity, while the mitochondrial enzyme is responsible for the remaining 70%.11
The 25(OH)D metabolite also circulates in the bloodstream bound to DBP. It is an abundant but relatively inactive vitamin D metabolite. Its circulating concentration provides the most readily available evaluation of the vitamin D status in a given individual. 25(OH)D must be further hydroxylated at a different site in the convoluted and straight portions of the proximal kidney tubule to gain hormonal bioactivity. Hydroxylation at position 1α by the mitochondrial cytochrome P450 enzyme 25-hydroxyvitamin D 1α-hydroxylase (CYP27B1) converts 25(OH)D to 1α,25-dihydroxyvitamin D [1,25(OH)2D], the active, hormonal form of vitamin D that plays an essential role in mineral homeostasis, bone growth, and cellular differentiation.12–14
Upon reaching a target tissue, 1,25(OH)2D binds a specific receptor (vitamin D receptor [VDR]) that is a member of the nuclear hormone receptor superfamily.15 The VDR is considered a class II nuclear hormone receptor because it needs to form a heterodimer with the retinoid X receptor (RXR) to bind specific DNA sequence elements with high affinity. These target sequences are termed vitamin D response elements (VDRE), and the best characterized of these binding sites consist of two tandemly repeated hexanucleotide sequences separated by 3 base pairs (bp).15 Transcriptional coactivators and components of the basal transcriptional machinery interact with the liganded, DNA-bound VDR-RXR heterodimer to activate the transcription of vitamin D target genes responsible for carrying out the physiologic actions of 1,25(OH)2D.13,15 Among several target genes, the 1,25(OH)2D hormone induces in target cells the expression of the gene encoding the key effector of its catabolic breakdown: 25-hydroxyvitamin D 24-hydroxylase (CYP24A1).16,17 This insures attenuation of the 1,25(OH)2D biological signal inside target cells and helps regulate vitamin D homeostasis.
Vitamin D–Deficiency Rickets
It is evident from the brief overview presented earlier that vitamin D metabolism can be affected at several levels: inadequate exposure to sunlight; inadequate dietary intake; malabsorption of dietary vitamin D; impaired hepatic 25-hydroxylation; defects in renal 1α-hydroxylation; or defects in receptor function (Fig. 11-1). Reviews on the nonhereditary disorders of the vitamin D endocrine system have been published previously.18 Only defects in renal 1α-hydroxylation or receptor activity have been associated with several specific mutations in genes involved in regulating vitamin D metabolism and action; these will be discussed in this chapter. There is a single report19 of mutations in both alleles of CYP2R1, the microsomal hepatic 25-hydroxylase,10 in a patient with low serum calcium concentrations, low circulating levels of 25(OH)D, and rickets.20 While obviously rare, this finding confirms CYP2R1 as a physiologically relevant vitamin D 25-hydroxylase and demonstrates that selective 25-hydroxylase deficiency can also cause a hereditary defect in vitamin D metabolism.
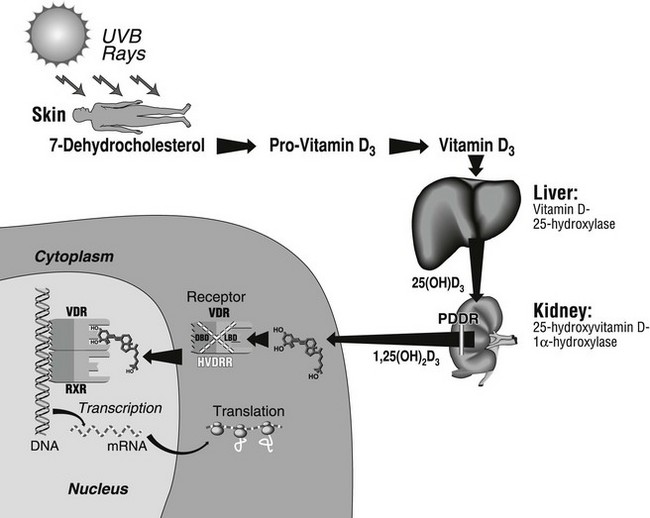
FIGURE 11-1 Schematic representation of the main steps of the vitamin D biosynthetic pathway, where genetic aberrations may lead to rickets and osteomalacia. The renal defect in pseudo–vitamin D–deficiency rickets (PDDR) is indicated by the break in the 1,25(OH)2D3 arrow arising in the kidney. The mutation leads to insufficient synthesis of 1,25(OH)2D. The left part of the figure represents a target cell where schematic coupling of the ligand to its receptor (VDR) takes place in the cytosol or, more likely, in the nucleus. The VDR then heterodimerizes with the RXR receptor. For ease of representation, the RXR ligand (9-cis retinoic acid) is not depicted. The complex then binds to DNA to regulate gene transcription. Various mutations affecting either of the two VDR domains (DBD, DNA-binding domain; LBD, ligand-binding domain), depicted by the stippled X over the receptor complex, cause hereditary vitamin D–resistant rickets (HVDRR).
The prime metabolic consequence of vitamin D deficiency is reduced net intestinal absorption of calcium.1,2 Calcium malabsorption leads to a fall in plasma calcium, secondary hyperparathyroidism, reduced renal tubular reabsorption of phosphate, hypophosphatemia, and thus a reduction in the calcium X phosphate product. Eventually, deposition of mineral in osteoid is impaired because the supply of the relevant ions is reduced. The impaired mineralization triggers the development of the rachitic and/or osteomalacic phenotype.
Clinical Features of Pseudo–Vitamin D–Deficiency Rickets (Vitamin D–Dependent Rickets Type I)
The clinical symptoms of pseudo–vitamin D–deficiency rickets (PDDR), also referred to as vitamin D–dependent rickets type I, are similar to those of common vitamin D–deficiency rickets, including failure to thrive, hypotonia, and growth retardation. Affected babies lie supine because of severe muscle weakness and bone pain. At this age, gross skeletal deformities are rare; however, if diagnosis and treatment are delayed, severe deformities of the spine and long bones occur, together with generalized muscle weakness simulating myopathy. Motor problems translate into regression in head control and ability to stand. In some patients, the initial event is generalized convulsions, tremulations and Bravais-Jacksonian fits, or tetany. Pathologic fractures may occur (Fig. 11-2). The onset in most cases occurs early during the third trimester of life; the patients look healthy at birth.
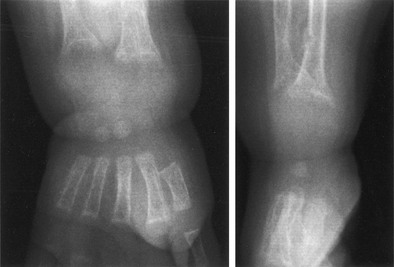
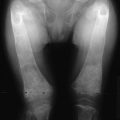
FIGURE 11-2 Radiograph of the wrist of a 19-month-old boy with pseudo–vitamin D–deficiency rickets (PDDR) at diagnosis. Severe rickets and demineralization are evident. Pronounced hypocalcemia and secondary hyperparathyroidism were documented, the latter causing a metaphyseal pseudofracture clearly seen on the lateral view.
X-ray features include diffuse osteopenia (mild to severe hypomineralization of the skeleton) and classic rachitic metaphyseal changes: fraying, cupping, widening, and fuzziness of the zone of provisional calcification immediately under the growth plate (see Fig. 11-2). These changes are seen better and detected earlier in the most active growth plates—namely, the distal ulna and femur and the proximal and distal tibia. Changes in the diaphyses may not be evident when metaphyseal changes are first detected. However, they will appear a few weeks later as rarefaction, coarse trabeculation, cortical thinning, and subperiosteal erosion. Looser-Milkman’s pseudofractures and curvature of the shafts of long bones may be observed, especially in children older than 1 to 2 years.
Hypocalcemia is the main biochemical feature in PDDR. A rapid decrease in serum calcium concentration may give rise to tetany and convulsions, which may occur prior to any radiologic evidence of rickets. Prolonged hypocalcemia triggers secondary hyperparathyroidism and hyperaminoaciduria.21 Urinary calcium excretion is very low, whereas fecal calcium is high as a consequence of impaired intestinal calcium absorption. Elevated urinary cyclic adenosine monophosphate (cAMP) is not a consistent finding, and normal values have been measured in PDDR patients, despite high circulating parathyroid hormone (PTH) levels.22
Serum phosphate concentrations may be normal or low. When reduced, the hypophosphatemia is usually of a lesser degree than that measured in X-linked hypophosphatemic rickets.23 It results from the combination of impaired intestinal absorption and increased urinary loss induced by the secondary hyperparathyroidism. Serum alkaline phosphatase activity is consistently elevated, and its increase precedes the appearance of clinical symptoms.
Patients with PDDR have normal serum levels of 25(OH)D after exposure to sunlight or oral intake of small doses of vitamin D; the concentrations increase if higher doses are given.22 Circulating levels of 24,25-dihydroxyvitamin D [24,25(OH)2D] are normal and correlate with those of 25(OH)D.24 Serum levels of 1,25(OH)2D are low in untreated patients.22,25 This is evident immediately after birth, months before any clinical evidence of rickets appears. Even when patients are treated with high doses of vitamin D, causing major increases in circulating levels of 25(OH)D, the blood concentration of 1,25(OH)2D does not reach the normal range. These characteristic features of serum vitamin D metabolites have provided key insight into the pathogenesis of PDDR.
Clinical Features of Hereditary Vitamin D–Resistant Rickets (Vitamin D–Dependent Rickets Type II)
Many of the clinical findings in patients with hereditary vitamin D–resistant rickets (HVDRR), also termed vitamin D–dependent rickets type II, are identical to those described for PDDR, including bone pain, muscle weakness, hypotonia, and occasional convulsions.26 Children are often growth retarded, and hypoplasia of the teeth is observed. The radiologic features of rickets are present. A major difference is that many children with HVDRR have sparse body hair, and some have total scalp and body alopecia, sometimes even including eyebrows and eyelashes. Hair loss may be evident at birth or occur during the first months of life. Patients with alopecia generally have more severe resistance to vitamin D. In families with a prior history of the disease, the absence of scalp hair in newborns can provide initial diagnostic clues for HVDRR. A defect in epithelial-mesenchymal communication that is required for normal hair cycling has been shown to be the cause of the alopecia in an animal model of HVDRR.27
Serum biochemistry includes low concentrations of calcium and phosphate and elevated alkaline phosphatase activity. Secondary hyperparathyroidism with elevated circulating PTH is measurable. The key difference concerns circulating levels of vitamin D metabolites. The 25(OH)D values are normal, and in the cases in which it has been measured, 24,25(OH)2D levels have been low. Importantly, serum levels of 1,25(OH)2D are elevated. This clinical feature clearly distinguishes HVDRR from PDDR, where circulating concentrations of 1,25(OH)2D are depressed. Additionally, patients with HVDRR are resistant to supraphysiologic doses of all forms of vitamin D therapy. Table 11-1 outlines the similarities and differences between the two forms of hereditary rickets involving the vitamin D endocrine system.
Table 11-1
Comparison of Pseudo–Vitamin D–Deficiency Rickets (Vitamin D–Dependent Rickets Type I) and Hereditary Vitamin D–Resistant Rickets (Vitamin D–Dependent Rickets Type II)
| Feature | PDDR (or VDDR-I) | HVDRR (or VDDR-II) |
| Mutations | CYP27B1 (25[OH]D-1α-hydroxylase) | VDR (vitamin D receptor) |
| Genetic inheritance | Autosomal recessive | Autosomal recessive |
| Age of onset | Early | Early |
| Rickets | Yes | Yes |
| Hypocalcemia | Yes | Yes |
| Serum alkaline phosphatase | Elevated | Elevated |
| Secondary hyperparathyroidism | Yes | Yes |
| Alopecia | No | Yes |
| Serum 25(OH)D | Normal | Normal |
| Serum 1,25(OH)2D | Low | Elevated |
| Response to 1,25(OH)2D therapy | Yes | No |
Pseudo–Vitamin D–Deficiency Rickets
As previously mentioned, serum levels of 25(OH)D are normal in untreated patients with PDDR and elevated in patients receiving large daily amounts of vitamin D.22 These results indicate that intestinal absorption of vitamin D and its hydroxylation in the liver are not impaired in PDDR. Circulating levels of 24,25(OH)2D are also normal and highly correlated with those of 25(OH)D, indicating a fully functional 25(OH)D-24-hydroxylase enzyme.24 However, serum concentrations of 1,25(OH)2D are low in untreated patients and remain low even when they are treated with high doses of vitamin D.22,25 This clearly identifies defective activity of the 25(OH)D-1α-hydroxylase enzyme (CYP27B1; hereafter referred to as 1α-hydroxylase) as the basic abnormality in PDDR and differentiates it from HVDRR.
PDDR is inherited as a simple autosomal recessive trait.28 No phenotypic abnormalities are observed in heterozygotes.21 By taking advantage of the unusual frequency of PDDR in the French-Canadian population and the availability of sample material from relatively large kindreds, the PDDR locus was mapped to the region of band 14 on the long arm of chromosome 12 (12q13-14).29,30
Tremendous progress has been achieved in the study of the molecular etiology of PDDR through the cloning of the complementary DNA (cDNA) encoding for 1α-hydroxylase.31–34 The human gene has also been cloned, sequenced, and mapped to 12q13.1-13.3 by fluorescence in situ hybridization,33,35,36 consistent with the earlier mapping of the disease to this locus by linkage analysis.
The ultimate proof that mutations in the 1α-hydroxylase gene were responsible for the PDDR phenotype required the identification of such mutations in PDDR patients and carriers of the disease. The first identified mutation was reported by Fu et al.31 in 1997; several additional mutations in various ethnic groups have since been published.35–41 These findings unequivocally establish the molecular genetic basis of PDDR as inactivating mutation(s) in the 1α-hydroxylase gene (CYP27B1). Further proof was provided by developing valid animal models of the disease using targeted inactivation of the cyp27B1 gene in mice.42,43
25-Hydroxyvitamin D 1α-Hydroxylase
The 25(OH)D-1α-hydroxylase (CYP27B1; 1α-hydroxylase) enzyme catalyzes the addition of a hydroxyl group at position 1α of the secosteroid backbone of 25(OH)D. The 1α-hydroxylase is a mitochondrial cytochrome P450 enzyme that requires electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to promote catalysis. These are delivered to the P450 moiety by the flavoprotein NADPH-ferredoxin reductase44 and the nonheme iron protein, ferredoxin.45 The expression of these cofactors is ubiquitous, and their genes were mapped to chromosomes 17 and 11,44,45 respectively, excluding them rapidly in the search for the PDDR mutations, since the PDDR locus was mapped early on to chromosome 12.30
The main site for the 1α-hydroxylation of 25(OH)D is the proximal tubule of the renal cortex.46 In the kidney, the expression of the 1α-hydroxylase gene is subject to complex regulation by PTH, calcitonin, calcium, phosphorus, and 1,25(OH)2D itself.47–49 The 1α-hydroxylase gene exists in a single copy in the human genome and contains nine exons spanning 5 kb of sequence. The ferredoxin-binding domain is encoded by sequences contained in exons 6 and 7, while the heme-binding domain is contained in exon 8.50,51
Mutations
To date, 35 different 1α-hydroxylase mutations have been described in PDDR patients and their parents (Table 11-2). All patients have mutations on both alleles, but a high frequency of compound heterozygosity (a different mutation on each allele) has been observed (23 compound heterozygotes out of 54 cases reported). Splice-site mutations, nucleotide deletions and duplications, and missense and nonsense mutations have been reported (see Table 11-2). The mutations are dispersed throughout the 1α-hydroxylase sequence, affecting all exons (Fig. 11-3).
Table 11-2
CYP27B1 (1α-Hydroxylase) Gene Mutations in Patients with Pseudo–Vitamin D–Deficiency Rickets (Vitamin D–Dependent Rickets Type I)
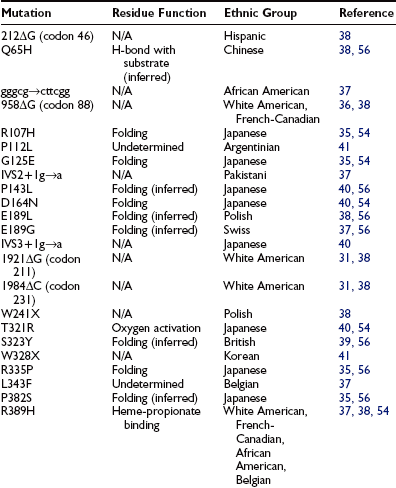

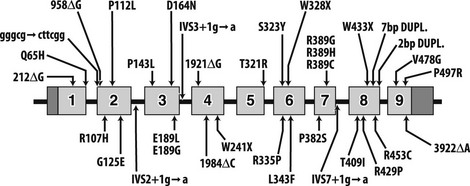
FIGURE 11-3 Mutations in pseudo–vitamin D–deficiency rickets (PDDR) patients. A schematic representation of the CYP27B1 (25[OH]D-1α-hydroxylase) gene is shown. Exons are numbered from 1 to 9 with darker shaded regions representing 5′- and 3′-nontranslated regions. Mutations are presented above and below the gene map. Numbers refer to amino acid residue; the one-letter amino acid code is used. Δ, Deletion; gggcg→cttcg, deletion of gggcg and substitution of cttcgg beginning at nucleotide 897 in exon 2; IVS2, IVS3, or IVS7+1 g→a, splice-site mutation in intron (intervening sequence) 2, 3, or 7; 7bp dupl, 7-base-pairs duplication; 2 bp dupl, 2-base-pairs duplication.
The mutations detected at the highest frequency are 958ΔG, common among French-Canadian patients (owing to a founder effect),29,36,38 and a mutation located at codons 438 to 442 in exon 8. These codons are composed of the duplicated 7-bp sequence 5′-CCCACCC CCCACCC-3′. In 11 families described to date,36,38,39,41 three rather than two copies of the 7-bp sequence are present, which alters the downstream reading frame (Fig. 11-4). Careful analysis of the correlation between ethnic origin, microsatellite haplotyping, and the presence of the 7-bp duplication mutation suggested that the mutation has arisen by several independent de novo events.36,38
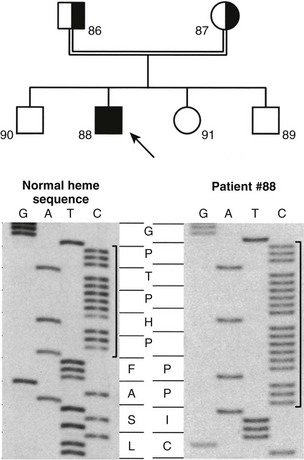
FIGURE 11-4 Identification of the molecular defect in a pseudo–vitamin D–deficiency rickets (PDDR) pedigree. Upper panel, The heterozygote parents are identified by half-filled boxes (male parent, square symbol, no. 86; female parent, round symbol, no. 87). The affected patient (male, no. 88) is identified by a filled square symbol with an arrow. Lower panel, DNA sequence analysis of the mutation within the heme-binding domain of the 25(OH)D-1α-hydroxylase (CYP27B1) gene. The 7-bp duplication is bracketed. The amino acid sequence (one-letter code) is highlighted in the center of the figure. Note the change of reading frame leading to an aberrant protein sequence and premature termination in the affected patient.
Structure/Function Relationships
An important aspect of the identification of mutations in the 1α-hydroxylase gene was to correlate the genotype of the patients with their phenotype—that is, the severity of the disease and the circulating levels of 1,25(OH)2D. In several cases, although 1,25(OH)2D serum levels are low, they are not undetectable,22,25,37,40,52 suggesting some degree of residual 1α-hydroxylase activity. Presumably, some 1α-hydroxylase mutations affect the structural integrity of the enzyme, resulting in a modification of its kinetics. This reasoning cannot apply to the frameshift (deletions, insertions, and duplications) and nonsense mutations described to date. All such mutations eliminate the heme-binding site of the protein and thus completely abolish the 1α-hydroxylase enzymatic activity. The apparent residual 1α-hydroxylase activity observed in some patients could be attributable to missense mutations. Most of these missense mutations were entirely devoid of enzymatic activity in the assays used,38,40 except for the L343F mutation (retained 2.3% of wild-type activity) and the E189G mutation (retained 22% of wild-type activity).37 Thus some missense mutations contribute to the variable phenotype observed in patients with PDDR.
Early modeling efforts16,38 compared the sequence of the 1α-hydroxylase protein (a mitochondrial class I cytochrome P450) with the sequence of bacterial class I cytochrome P450s for which x-ray crystallographic data were available. The tertiary structures of these enzymes show remarkable conservation despite low amino acid sequence identity.53 These sequence alignments yielded improper predictions of the functions of the residues mutated in PDDR patients.54 Further modeling based on the first solved crystal structure of a eukaryotic cytochrome P450,55 combined with extensive structure/function analysis of recombinant 1α-hydroxylase proteins, identified the functions of many residues mutated in PDDR patients (see Table 11-2).54,56,57 Most mutations affect folding and conformation.54,56 Residue T321 is involved in oxygen activation, and amino acid R389 is essential for heme binding.54,56 Spectroscopic analysis of wild-type and mutant 1α-hydroxylase proteins identified residue T409 as critical for binding of the 25(OH)D substrate.57
Treatment
Vitamin D2, at high doses, was initially used to treat PDDR. Under such treatment, circulating levels of 25(OH)D increase sharply, and it is likely that massive concentrations of 25(OH)D can bind to the VDR and induce the response of the target organs to normalize calcium homeostasis. The risk of overdose is high because vitamin D progressively accumulates in fat and muscle, and the therapeutic doses are close to the toxic doses, ultimately placing the patient at risk for nephrocalcinosis and impaired renal function. The use of 25(OH)D3 as a therapeutic agent in PDDR has been reported.58 The mechanism of action is likely to be similar to the one described earlier for vitamin D. The low availability and high cost of the metabolite have not encouraged its widespread use as long-term therapy for PDDR.
The treatment of choice is long-term (lifelong) replacement therapy with 1,25(OH)2D3.22,59 This results in rapid and complete correction of the abnormal phenotype, eliminating hypocalcemia, secondary hyperparathyroidism, and radiographic evidence of rickets. Strikingly, the myopathy disappears within days after initiation of therapy. The restoration of bone mineral content is equally rapid and histologic evidence of healing of the bone structure has been reported.22 Correction of tooth enamel hypoplasia is only partial. An important aspect of treatment is to ensure adequate calcium intake during the bone-healing phase. Needs can be monitored by frequent assessment of urinary calcium excretion. It should be noted that hypercalciuria is common during treatment with 1,25(OH)2D3, particularly during the first year of administration. Its close monitoring is used to adjust the needs in 1,25(OH)2D3. The initial dose will be 1 to 2 µg/d, and the maintenance dose will vary between 0.5 and 1 µg/d. High levels of calcium excretion may amplify the pattern of calcium deposition in the kidney, so frequent renal imaging and assessment of renal function are essential during the course of treatment.
Before 1,25(OH)2D3 became available from commercial sources, several investigators used the monohydroxylated analog, 1α(OH)D3,60 which only requires liver hydroxylation at position 25 to be activated to the hormonally active metabolite. The 25-hydroxylation step is not affected by the PDDR mutation. Response to treatment with 1α(OH)D3 is rapid, with healing of rickets in 7 to 9 weeks, and this compound is still used in several countries. On a weight basis, 1α(OH)D3 is about half as potent as 1,25(OH)2D3, nullifying any possible economic advantage in favor of the monohydroxylated compound.
Hereditary Vitamin D–Resistant Rickets
Patients with HVDRR have normal 25(OH)D and low but measurable 24,25(OH)2D serum values. The circulating levels of 1,25(OH)2D are elevated from 3 to 5 times the normal values. These biochemical findings demonstrate that all the vitamin D metabolic enzymes (25-hydroxylase, 24-hydroxylase, and 1α-hydroxylase) are active in patients with HVDRR. Most patients with the disease are resistant to all forms of vitamin D therapy. This lack of response to vitamin D treatment led Albright et al.61 to introduce the concept of hormonal resistance. The molecular basis of vitamin D end-organ resistance became clearer as the mechanism of action of the hormonal form of vitamin D was elucidated.
Ligand-binding studies first established that the 1,25(OH)2D hormone, like other sex-steroid hormones studied at the time, binds to a high-affinity receptor located in the nucleus.62 It was later discovered that this receptor could bind to DNA.63 This property was utilized to purify sufficient quantities of the VDR to raise antibodies.64,65
The observation that binding sites for 1,25(OH)2D could be detected in many tissues in addition to the classical vitamin D target tissues helped to define the etiology of HVDRR. Investigators began to study the VDR from cultured fibroblasts of patients and relatives, using ligand-binding assays, radioimmunoassays, and DNA-cellulose chromatography. Other fibroblast responses measured included induction of the 24-hydroxylase enzyme activity and vitamin D–mediated growth arrest. These methodologies led to several milestone observations. In the first studies reported, [3H]-1,25(OH)2D3 binding was undetectable in fibroblasts from HVDRR patients, and high doses of 1,25(OH)2D failed to induce the 24-hydroxylase bio-marker.66 The diminished hormone binding provided a clear rationale for the end-organ resistance reported in patients. Subsequent reports continued to describe a lack of response of the patient’s cells to 1,25(OH)2D, but some patients’ fibroblasts exhibited normal [3H]-1,25(OH)2D3 binding.67 From these early reports, it was concluded that at least two classes of HVDRR patients could be recognized: “receptor-negative” patients and “receptor-positive” patients. The development of a sensitive radioimmunoassay for the VDR protein68 demonstrated that these semantic differences were incorrect. Using this assay, it was shown that fibroblasts from so-called receptor-negative patients expressed normal levels of receptor protein. Pike et al.69 hypothesized that the VDR defect in these patients’ cells was due to a structural abnormality in the ligand-binding domain preventing [3H]-1,25(OH)2D3 from binding to the receptor, not from defective synthesis of the VDR protein. The two classes of HVDRR were more adequately described by the terminology ligand-binding positive and ligand-binding negative.
A second type of VDR structural abnormality was identified in cultured cells from patients displaying the ligand-binding positive HVDRR phenotype. The VDR from these cells showed reduced affinity for heterologous DNA as measured by DNA-cellulose chromatography.70 It was suspected that the defect in these patients would likely be a point mutation in the DNA-binding domain of the VDR. Interestingly, measurements of DNA binding affinities of the VDR from parents of ligand-binding positive patients clearly identified two forms of the VDR molecule, one with normal, wild-type affinity for DNA, and the second with reduced, defective DNA binding.70 This was the first clear evidence establishing the heterozygous state of HVDRR parents. Binding and antibody-based assays had failed to reconcile genotype and phenotype of carriers in the past.
Eventually, the purified receptor was used to obtain monoclonal antibodies that led to the cloning of the VDR cDNA from various species.71–73 In turn, the VDR genomic structure was analyzed. Eight exons comprise the entire coding region, spanning approximately 50 kb of genomic DNA.74 The translation start site is contained within exon 2.
Analysis of the VDR cDNA sequence soon established that the VDR is a member of the nuclear hormone receptor superfamily, and its mechanism of action was subsequently unraveled.15 The ligand-bound receptor forms a heterodimer with the retinoid X receptor (RXR), and the dimer contacts specific sites within the regulatory domains of responsive genes. This results in positive or negative modulation of the transcription of target genes. The VDR is essential to transduce the biological effects of 1,25(OH)2D, such as promoting calcium and phosphate transport across the small intestine and maintaining calcium homeostasis. The underlying molecular basis of the hormone resistance described in patients with HVDRR is mutation in the VDR which renders the receptor nonfunctional or less functional than the wild-type VDR. Several laboratories have independently engineered valid animal models of HVDRR by inactivating the VDR gene through gene targeting.75
Vitamin D Receptor
Structure/function analysis, sequence comparison alignments, and recent crystallographic studies have contributed to our understanding of the domain structure of the members of the nuclear hormone receptor superfamily to which the VDR belongs. The different domains of nuclear receptors have been labeled A to F (Fig. 11-5). Some of these domains exhibit high sequence similarity between individual family members, while others vary considerably or are altogether absent. This diversity is manifested most strongly in domains A/B, D, and F. Domain A/B includes all residues that are N-terminal to the receptor’s DNA-binding domain (DBD). The size of this domain is highly variable, ranging from hundreds of residues in the progesterone receptor, for example, to only 24 amino acids in the VDR. The function of the A/B domain remains somewhat uncertain, but results suggest that polymorphisms in the VDR A/B segment could modulate its transcriptional activity.76 Domain C comprises the highly conserved zinc-finger DBD, the hallmark feature of nuclear receptor family members, and will be discussed in detail later. Region D serves as a flexible hinge domain between the DBD and domain E and is the least conserved among nuclear receptors. Interestingly, because of the splicing in of an additional exon, the D segment of the VDR is longer by 50 amino acids when compared to classical steroid receptors.77 Residues within the D domain of the VDR are subject to posttranslational modification in the form of reversible serine phosphorylation.78,79 This regulation provides an additional degree of control of the VDR activity.15 The E region encodes the ligand-binding domain (LBD) of ligand-activated receptors and also contributes to transactivation and dimerization. The small F domain is not highly conserved between nuclear receptor family members.
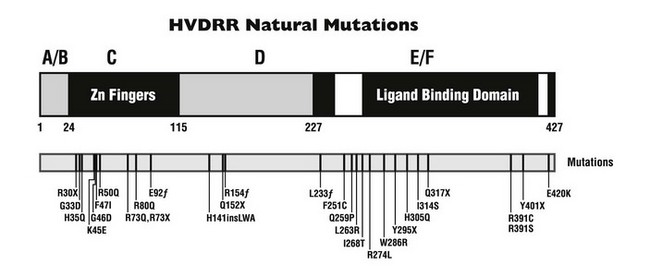
FIGURE 11-5 Natural mutations in hereditary vitamin D–resistant rickets (HVDRR). A schematic view of the vitamin D receptor is presented. The 427-amino-acid protein can be separated into domains, designated A/B, C, D, and E/F. The white regions represent helices within the E/F domain that are important for transcriptional activation. Point mutations in human patients with the HVDRR syndrome are indicated below (one-letter amino acid code). f, Frameshift mutation; ins, insertion; X, premature stop codon.
Structure/Function
The Vitamin D Receptor DNA-Binding Domain: The DNA-binding domain of the VDR consists of two zinc-finger motifs located between residues 24 and 90. The zinc fingers are of the C2C2 type, with two zinc atoms tetrahedrally coordinated through four cysteine residues, each of which serves to stabilize the finger structure itself. The α-helical motifs residing on the C-terminal side of each zinc finger constitute the DNA-recognition and phosphate backbone–binding helices, respectively.15 The region immediately C-terminal to the second zinc finger, covering residues 91 to 115, also forms an α-helical structure that could contribute to DNA contacts. The functional importance of those helical domains is confirmed by the analysis of the HVDRR mutations described in the Mutations section.
The VDR exhibits a unique characteristic when compared to other nuclear receptors: a cluster of five basic amino acids is located at residues 49 to 55 in the intervening sequence between the two zinc fingers. This domain is predicted to make DNA contact80 and regulate the nuclear localization of the receptor.81 Interestingly, this cluster contains residue serine-51, a site of posttranslational modification through phosphorylation by protein kinase C.82,83
Aside from their well-characterized role in the binding of the DNA response element, residues within the zinc-finger motifs of the VDR also contribute to association with the partner receptor, RXR, to form the functional heterodimer (see Fig. 11-1).84,85 The α-helical domain immediately C-terminal to the second zinc finger also provides interactions with partner proteins.85 These specific contact sites in the DBD of the VDR facilitate weak heterodimerization between VDR and RXR, while the stronger, ligand-dependent heterodimerization interactions are provided by residues located within the LBD.
The Vitamin D Receptor Ligand-Binding Domain: The E/F region of the VDR (see Fig. 11-5) represents a complex multifunctional domain involved in binding of the 1,25(OH)2D ligand, heterodimerization with RXR, and transactivation. The structure of the LBD of the VDR has been modeled following x-ray crystallographic analysis.86 It consists of 13 α-helices and several short β-strands organized as a “sandwich” around a lipophilic hormone-binding pocket. The LBD of the VDR is bordered by helices H3, H5, H7, H11, and residues Ser275 (loop H5-β), Trp286 (β-1), and Leu233 (helix H3). Once the ligand enters the pocket, a “lid” formed by H12 closes over the pocket. Several of the mutations characterized in HVDRR coincide with hormone contact sites.
It is hypothesized that ligand binding leads to conformational changes that expose, enhance, or produce novel dimerization and/or transactivation interfaces. Proteolytic digestion assays have provided indirect evidence that binding of 1,25(OH)2D induces changes in the conformation of the VDR.87,88 Interestingly, different vitamin D analogs induce different conformational changes.86,87,89
Two regions within the LBD of the VDR are involved in strong, ligand-dependent heterodimerization with RXR. These have been identified following mutagenesis experiments and by analogy to the RXR homodimer crystal.90 These two subdomains consist of residues 244 to 26391–93 and amino acids 317 to 395,93,94 corresponding to portions of helices 3 to 4 and 7 to 10, respectively. Thus the ligand-binding and heterodimerization functions of the VDR are interrelated within the context of the tertiary structure of the molecule, presumably through allosteric effects that ultimately generate an active receptor conformation.
Transactivation
As previously mentioned, ligand binding probably serves to induce conformational changes that allow the VDR to contact pertinent partners. Mutagenesis experiments have identified some of the key residues involved in these contacts. Two regions of the receptor are required exclusively for transcriptional activation. One of these regions is located between residues 244 and 263, a domain also involved in heterodimerization with RXR,91–93 but residue lysine-246 is not involved in contacts with the RXR partner, and its alteration severely compromises transactivation.92 This residue, highly conserved among nuclear receptors, must form part of the binding interface with transcriptional coactivators.92,95
The second region is known as activation function 2, or AF-2, and corresponds to helix 12 (residues 416 to 422).96–98 Alteration of residues leucine-417 and glutamic acid-420 does not affect hormone binding or heterodimeric DNA binding but completely abrogates transactivation.97,98 These residues also function to stimulate transcription by a mechanism involving coactivators. Some of the proteins interacting with the VDR to allow ligand-activated transcription have been identified. Within the basal transcriptional machinery, the VDR interacts with transcription factor TFIIB.99,100 This contact involves the AF-2 domain of the VDR but also requires the wild-type residue arginine-391, located N-terminal to the AF-2 region within helix 10/11.98
Three categories of multicomponent transcriptional coactivator complexes that are involved in nuclear receptor–mediated transcription have been identified: those involved in ATP-dependent chromatin remodeling, those that physically interact with general transcription factors and RNA polymerase II, and complexes that modify histone tails covalently.101 For VDR-mediated activation, the WINAC chromatin remodeling complex has been biochemically characterized.102 The DRIP/TRAP complex that physically interacts with components of the general transcriptional machinery has been shown to be involved in ligand-dependent transcription by the VDR.103,104 Finally, complexes that include histone acetyltransferase enzymes of the p160 family have been shown to coactivate the VDR.105,106 Structure/function analysis of the AF-2 domain of the VDR identified residues that abolished interaction with p160 family members and DRIP components but retained ligand-dependant transcriptional activation. This suggests that yet uncharacterized coactivator complexes may participate in VDR-mediated transcription.107 All of those categories of complexes transiently associate with the VDR, and their recruitment is thought to be cyclic and highly regulated.105
Mutations
Our understanding of the structural and functional consequences of HVDRR-causing mutations in the VDR has followed the introduction of molecular biological techniques as routine detection methods. For example, the amplification of mRNA by RT-PCR (reverse transcription-coupled polymerase chain reaction) has been of tremendous help for analysis of mutations (Fig. 11-6). Similarly, the functional consequences of particular mutations, first analyzed using ligand-binding assays and nonspecific binding to calf thymus DNA, can now be analyzed routinely using transient transfection assays on bona fide vitamin D–responsive promoter elements. Some laboratories have begun to test VDR protein-protein interactions. Finally, the availability of crystal structures for related nuclear receptors allows us to understand the consequences of particular mutations at the tertiary structure level.
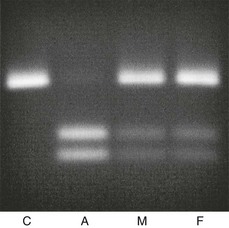
FIGURE 11-6 Prenatal diagnosis in a family where a first child had hereditary vitamin D–resistant rickets (HVDRR) caused by an R30X mutation. This mutation in the vitamin D receptor gene (C-to-T substitution in exon 2) has introduced a recognition site for the restriction endonuclease DdeI. Using primers internal to the affected exon, an 89-bp DNA fragment was amplified using PCR with 100 ng of genomic DNA prepared from amniotic cells (A), or from whole blood of an unrelated control (C), the mother (M), or the father (F). An aliquot of each amplimer was incubated with the restriction endonuclease DdeI. The digested fragments were visualized on a 2.5% agarose gel. The data show unambiguous homozygosity for the R30X mutation in the fetus, with both parents carriers for the mutation.
Close to 30 mutations in the VDR have been described in HVDRR patients (see Fig. 11-5 and Table 11-3). Nine of these genetic alterations are nonsense (X) or frameshift (fs) mutations that introduce premature stop codons in the receptor: R30X (see Fig. 11-6), R73X, E92fs, Q152X, R154fs, L233fs, Y295X, Q317X, and Y401X. The premature translation termination codons lead to truncated VDRs that lack the DBD (R30X and R73X), the LBD (E92fs, Q152X, R154fs, L233fs, Y295X, and Q317X), or the AF-2 activation domain (Y401X); in most cases, the mRNAs for these truncated receptors are unstable.108 The mutations involving amino acid substitutions (missense mutations) have been more revealing in terms of structure/function relationships.
Table 11-3
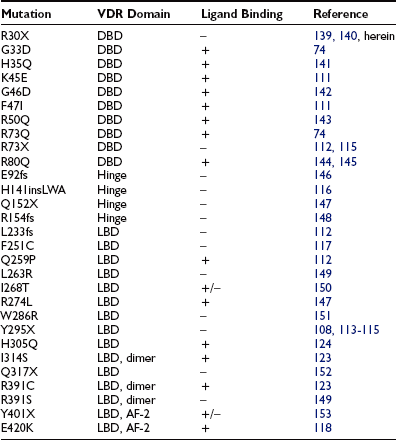
Vitamin D–Receptor Mutations in Patients With Hereditary Vitamin D–Resistant Rickets (Vitamin D–Dependent Rickets Type II)
DNA-Binding Domain Mutations: The first VDR mutation ever reported was described by Hughes et al.74 and affected the DBD of the receptor. The mutation substitutes a polar uncharged glutamine for a positively charged arginine at position 73 (R73Q) within the second zinc finger of the DBD. The VDR mutation identified by Hughes et al.74 was the first natural disease-causing mutation reported for the entire steroid-thyroid-retinoid receptor gene superfamily.
Several additional missense mutations affecting the DBD have been reported since (see Table 11-3). Based on the crystal structure of the related GR, RXR, and TR molecules,90,109,110 the H35Q, K45E, R50Q, R73Q, and R80Q mutations are thought to affect residues that contact DNA.15,26,111 The conversion of residue glycine-46 to aspartic acid (G46D) introduces a bulky, charged amino acid that would create unfavorable electrostatic interactions with the negatively charged phosphate backbone of the DNA helix.112 The G33D mutation would be expected to have the same effect.111 The F47I mutation is a relatively conserved substitution, but the loss of the phenylalanine ring structure may disrupt the hydrophobic core of the DBD and affect the proposed α-helical structure at the base of the first zinc finger.111
Ligand-Binding Domain Mutations: The first identified mutation affecting the LBD was the Y295X mutation reported by Ritchie et al.113 This mutation truncates 132 amino acids from the carboxy-terminus of the VDR, which results in the deletion of a major portion of the LBD. This mutation has been described in seven families forming a large kindred in which consanguineous marriages occurred.108 The mutation was also identified in three additional families unrelated to the extended kindred.114,115 As previously mentioned, the mutant mRNA is unstable, and the truncated VDR is undetectable using immunology-based assays.108
The VDR crystallographic data86 can be used to understand the structural consequences of the missense mutations in the VDR LBD described in HVDRR patients. The Q259P mutation (helix H4), the L263R and I268T (H4/5), the R274L mutation (H5), the W286R mutation (loop H5-H6), the H305Q mutation (loop H6-H7), the I314S mutation (H7), and the R391C or R391S mutations (positioned within H10) should perturb ligand binding and/or dimerization. Mutation H141insLWA consists of a unique 5-bp deletion combined with an 8-bp insertion in exon 4, leading to deletion of residues H141 and T142 and their substitution by mutant residues L141, W142, and A143 in helix H1.116 The resulting mutant VDR does not bind ligand.116
Mutation F251C is located in the E1 region (amino acids 244 to 263) that overlaps helices 3 and 4 and loop 3 to 4 between them.117 The mutation of residue F251 thus likely disrupts the ligand-binding pocket and perturbs optimal function of the VDR. Mutation E420K modifies a residue in helix H12 that provides coactivator dimerization interfaces, and thus it results in loss of transactivation.118
Treatment
Therapies with pharmacologic doses of vitamin D metabolites, including vitamin D itself, 25(OH)D, 1α(OH)D, and 1,25(OH)2D, have been used to attempt to overcome the target-organ resistance to vitamin D associated with HVDRR. Patients with HVDRR without alopecia are generally more responsive to treatment with high doses of vitamin D preparations than patients with alopecia.119 Reported effective doses range from 5000 to 40,000 IU/day for vitamin D, 20 to 200 µg/day for 25(OH)D, and 17 to 20 µg/day for 1,25(OH)2D.120–122 The efficacy of treatment with high doses of vitamin D metabolites can be reconciled with the molecular cause of HVDRR. When the resistance to vitamin D is caused by mutations in the VDR that moderately decrease the affinity of the receptor for its ligand, such as the H305Q or I314S mutations,123,124 high doses of the hormone can apparently overcome the low-affinity binding defect and achieve adequate VDR occupancy to mediate normal 1,25(OH)2D responses. A few patients with HVDRR with alopecia have also been treated successfully using vitamin D metabolites.67,74,125–129
When vitamin D therapy is proven ineffective, intensive calcium therapy is the alternative treatment of choice. Some success has been achieved with high-dose oral calcium (Fig. 11-7).130 To bypass the calcium absorption defect in the intestine caused by the mutant VDR, long-term intravenous (IV) calcium infusions should be considered (see Fig. 11-7). High doses of IV calcium are infused during the night over a several-month period. Rapid disappearance of bone pains has been documented, and gradual improvement of calcemia and parathyroid function, followed by improvement of rickets (Fig. 11-8), and weight and height gains ensue.131–134 The syndrome can recur when the IV infusions are discontinued.131 After the serum calcium normalizes, and radiologic control of the rickets has been achieved with IV calcium infusions, high-dose oral calcium therapy is effective in maintaining normocalcemia.133 For HVDRR patients who do not respond to high doses of vitamin D therapy, the two-step calcium protocol (IV infusions followed by high oral doses) appears to be the preferred therapeutic approach.
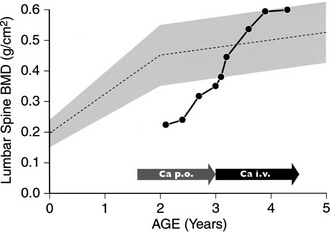
FIGURE 11-7 Bone mineral density (BMD) response in a patient with hereditary vitamin D–resistant rickets (HVDRR) treated with oral and intravenous (IV) calcium. BMD of the lumbar spine was measured by dual-energy x-ray absorptiometry (DXA). Patient received oral calcium, 2 g/d (Ca p.o.), for 17 months before treatment with IV calcium (Ca i.v.). The shaded area represents the reference values for the corresponding ages. Note the rapid correction of the BMD upon initiation of IV calcium therapy.
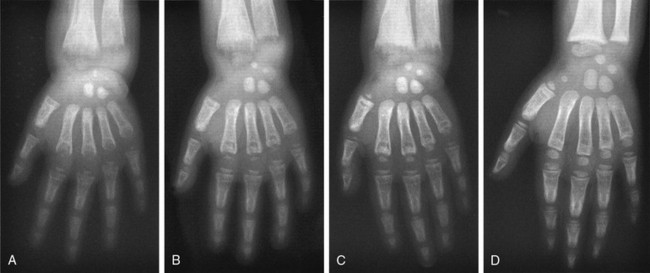
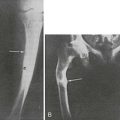
FIGURE 11-8 Radiographic analysis of response to calcium therapy in a patient with hereditary vitamin D–resistant rickets (HVDRR). A, At referral, the patient had been treated for 3 months with 1,25(OH)2D3 (15 µg/d). Rickets is still active, and the bone is demineralized. B, After 6 months on 1,25(OH)2D3 (30 µg/d) and oral calcium (2 g/d), no significant improvement. C, After 2 months of continuous intravenous calcium (without 1,25[OH]2D3), mineralization is improved. D, Following 11 months of continuous infusion, mineralization is adequate, but the growth plate remains irregular. Clinical status is satisfactory and maintained with oral calcium (2 g/d).
It is interesting to note several reports of spontaneous improvement in the disease of HVDRR patients.67,135,136 This spontaneous healing of rickets usually happens between 7 and 15 years of age and has not been consistently associated with the onset of puberty. Spontaneous recovery does not appear to be related to treatment, since therapy was often ineffective, and improvement occurred after the treatment was discontinued. The patients appear to remain normocalcemic without therapy and show no evidence of rickets or osteomalacia. Spontaneous improvement has been reported for both ligand-binding-positive and ligand-binding-negative HVDRR patients.67,135,136 The alopecia persists despite healing of the rickets.67,135,136 It is not uncommon for children to “outgrow” genetic diseases, and the organism appears able to compensate for the loss of VDR function after skeletal growth has been completed. This apparent functional redundancy does not apply to the hair follicle, however, a situation analogous to what has been described in the VDR-ablated animal model of the disease.137
Perspectives
Conceptually, because the signal transduction pathway of 1,25(OH)2D involves accessory molecules such as RXR, the coactivators, and components of the basic transcriptional machinery, it is possible that target-organ resistance to 1,25(OH)2D may be due to mutations in other proteins involved in the transactivation process. When the proteins interact with numerous partners, like RXR, the phenotype associated with mutations would be expected to be broader than the clinical symptoms of HVDRR. The recent identification of transcriptional coactivators with specificity towards the VDR widens the range of putative targets for mutations that could cause HVDRR. It is interesting to note that Hewison et al.138 described a case of HVDRR in an English patient in which a mutation could not be found in the VDR. The patient exhibited all of the clinical features of HVDRR, including alopecia. The patient’s fibroblasts expressed wild-type VDR mRNA, and the receptor bound ligand with normal affinity. Ligand-dependent transcriptional activation by the VDR was deficient in the patient’s cells but normal when the patient’s VDR was expressed in heterologous cells. These results clearly demonstrate that the patient’s VDR was normal and suggest that the vitamin D resistance is the result of a mutation affecting an accessory protein specific for 1,25(OH)2D-dependent transcriptional activation. The characterization of the molecular cause of these types of clinical manifestations will further enhance our understanding of vitamin D biology.
References
1. Canadillas, S, Rodriguez-Benot, A, Rodriguez, M. More on the bone-kidney axis—lessons from hypophosphataemia. Nephrol Dial Transplant. 2007;22:1521–1523.
2. DeLuca, HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80:1689S–1696S.
3. St-Arnaud, R. The direct role of vitamin D on bone homeostasis. Arch Biochem Biophys. 2008;473:225–230.
4. Webb, AR, Kline, L, Holick, MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. J Clin Endocrinol Metab. 1988;67:373–378.
5. Mawer, EB, Backhouse, J, Holman, CA, et al. The distribution and storage of vitamin D and its metabolites in human tissues. Clin Sci. 1972;43:413–431.
6. Chapuy, M-C, Meunier, PJ. Vitamin D insufficiency in adults and the elderly. In: Feldman D, Glorieux FH, Pike JW, eds. Vitamin D. San Diego: Academic Press; 1997:679–693.
7. Holick, MF. Photobiology of vitamin D. In: Feldman D, Glorieux FH, Pike JW, eds. Vitamin D. San Diego: Academic Press; 1997:33–39.
8. Guo, YD, Strugnell, S, Back, DW, et al. Transfected human liver cytochrome P-450 hydroxylates vitamin D analogs at different side-chain positions. Proc Natl Acad Sci U S A. 1993;90:8668–8672.
9. Okuda, KI, Usui, E, Ohyama, Y. Recent progress in enzymology and molecular biology of enzymes involved in vitamin D metabolism. J Lipid Res. 1995;36:1641–1652.
10. Cheng, JB, Motola, DL, Mangelsdorf, DJ, et al. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxylase. J Biol Chem. 2003;278:38084–38093.
11. Bjorkhem, I, Holmberg, I. Assay and properties of a mitochondrial 25-hydroxylase active on vitamine D3. J Biol Chem. 1978;253:842–849.
12. Bikle, DD. What is new in vitamin D: 2006–2007. Curr Opin Rheumatol. 2007;19:383–388.
13. Christakos, S, Dhawan, P, Liu, Y, et al. New insights into the mechanisms of vitamin D action. J Cell Biochem. 2003;88:695–705.
14. Sutton, AL, MacDonald, PN. Vitamin D: More than a “bone-a-fide” hormone. Mol Endocrinol. 2003;17:777–791.
15. Haussler, MR, Whitfield, GK, Haussler, CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349.
16. Omdahl, JL, Bobrovnikova, EA, Choe, S, et al. Overview of regulatory cytochrome P450 enzymes of the vitamin D pathway. Steroids. 2001;66:381–389.
17. Makin, G, Lohnes, D, Byford, V, et al. Target cell metabolism of 1,25-dihydroxyvitamin D3 to calcitroic acid. Evidence for a pathway in kidney and bone involving 24-oxidation. Biochem J. 1989;262:173–180.
18. Glorieux, FH, Pettifor, JM, Juppner, H. Pediatric Bone: Biology and Diseases. San Diego: Academic Press; 2003.
19. Cheng, JB, Levine, MA, Bell, NH, et al. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101:7711–7715.
20. Casella, SJ, Reiner, BJ, Chen, TC, et al. A possible genetic defect in 25-hydroxylation as a cause of rickets. J Pediatr. 1994;124:929–932.
21. Arnaud, C, Maijer, R, Reade, T, et al. Vitamin D dependency: An inherited postnatal syndrome with secondary hyperparathyroidism. Pediatrics. 1970;46:871–880.
22. Delvin, EE, Glorieux, FH, Marie, PJ, et al. Vitamin D dependency: replacement therapy with calcitriol. J Pediatr. 1981;99:26–34.
23. Feldman, D, Glorieux, FH, Pike, JW. Vitamin D. San Diego: Academic Press; 1997.
24. Mandla, S, Jones, G, Tenenhouse, HS. Normal 24-hydroxylation of vitamin D metabolites in patients with vitamin D-dependency rickets type I, Structural implications for the vitamin D hydroxylases. J Clin Endocrinol Metab. 1992;74:814–820.
25. Scriver, CR, Reade, TM, DeLuca, HF, et al. Serum 1,25-dihydroxyvitamin D levels in normal subjects and in patients with hereditary rickets or bone disease. N Engl J Med. 1978;299:976–979.
26. Malloy, PJ, Pike, JW, Feldman, D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D–resistant rickets. Endocr Rev. 1999;20:156–188.
27. Sakai, Y, Kishimoto, J, Demay, MB. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J Clin Invest. 2001;107:961–966.
28. Scriver, CR. Vitamin D dependency. Pediatrics. 1970;45:361–363.
29. Labuda, M, Labuda, D, Korab-Laskowska, M, et al. Linkage disequilibrium analysis in young populations: pseudo–vitamin D–deficiency rickets and the founder effect in French Canadians. Am J Hum Genet. 1996;59:633–643.
30. Labuda, M, Morgan, K, Glorieux, FH. Mapping autosomal recessive vitamin D–dependency type I to chromosome 12q14 by linkage analysis. Am J Hum Genet. 1990;47:28–36.
31. Fu, GK, Lin, D, Zhang, MY, et al. Cloning of human 25-hydroxyvitamin D 1-alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11:1961–1970.
32. Shinki, T, Shimada, H, Wakino, S, et al. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci U S A. 1997;94:12920–12925.
33. St-Arnaud, R, Messerlian, S, Moir, JM, et al. The 25-hydroxyvitamin D 1alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res. 1997;12:1552–1559.
34. Takeyama, K, Kitanaka, S, Sato, T, et al. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277:1827–1830.
35. Kitanaka, S, Takeyama, K, Murayama, A, et al. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338:653–661.
36. Yoshida, T, Monkawa, T, Tenenhouse, HS, et al. Two novel 1alpha-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type I. Kidney Int. 1998;54:1437–1443.
37. Wang, X, Zhang, MY, Miller, WL, et al. Novel gene mutations in patients with 1alpha-hydroxylase deficiency that confer partial enzyme activity in vitro. J Clin Endocrinol Metab. 2002;87:2424–2430.
38. Wang, JT, Lin, CJ, Burridge, SM, et al. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63:1694–1702.
39. Smith, SJ, Rucka, AK, Berry, JL, et al. Novel mutations in the 1alpha-hydroxylase (P450c1) gene in three families with pseudovitamin D-deficiency rickets resulting in loss of functional enzyme activity in blood-derived macrophages. J Bone Miner Res. 1999;14:730–739.
40. Kitanaka, S, Murayama, A, Sakaki, T, et al. No enzyme activity of 25-hydroxyvitamin D3 1alpha-hydroxylase gene product in pseudovitamin D deficiency rickets, including that with mild clinical manifestation. J Clin Endocrinol Metab. 1999;84:4111–4117.
41. Kim, CJ, Kaplan, LE, Perwad, F, et al. Vitamin D 1alpha-hydroxylase gene mutations in patients with 1alpha-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:3177–3182.
42. Dardenne, O, Prud’homme, J, Arabian, A, et al. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)- hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D- deficiency rickets. Endocrinology. 2001;142:3135–3141.
43. Panda, DK, Miao, D, Tremblay, ML, et al. Targeted ablation of the 25-hydroxyvitamin D 1alpha-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498–7503.
44. Solish, SB, Picado-Leonard, J, Morel, Y, et al. Human adrenodoxin reductase: two mRNAs encoded by a single gene on chromosome 17cen–q25 are expressed in steroidogenic tissues. Proc Natl Acad Sci U S A. 1988;85:7104–7108.
45. Chang, CY, Wu, DA, Mohandas, TK, et al. Structure, sequence, chromosomal location, and evolution of the human ferredoxin gene family. DNA Cell Biol. 1990;9:205–212.
46. Brunette, MG, Chan, M, Ferriere, C, et al. Site of 1,25(OH)2 vitamin D3 synthesis in the kidney. Nature. 1978;276:287–289.
47. Murayama, A, Takeyama, K, Kitanaka, S, et al. The promoter of the human 25-hydroxyvitamin D3 1 alpha-hydroxylase gene confers positive and negative responsiveness to PTH, calcitonin, and 1 alpha,25(OH)2D3. Biochem Biophys Res Commun. 1998;249:11–16.
48. Murayama, A, Takeyama, K, Kitanaka, S, et al. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology. 1999;140:2224–2231.
49. Yoshida, T, Yoshida, N, Monkawa, T, et al. Dietary phosphorus deprivation induces 25-hydroxyvitamin D(3) 1alpha-hydroxylase gene expression. Endocrinology. 2001;142:1720–1726.
50. Fu, GK, Portale, AA, Miller, WL. Complete structure of the human gene for the vitamin D 1alpha-hydroxylase, P450c1alpha. DNA Cell Biol. 1997;16:1499–1507.
51. Monkawa, T, Yoshida, T, Wakino, S, et al. Molecular cloning of cDNA and genomic DNA for human 25-hydroxyvitamin D3 1 alpha-hydroxylase. Biochem Biophys Res Commun. 1997;239:527–533.
52. Rosen, JF, Finberg, L. Vitamin D-dependent rickets: actions of parathyroid hormone and 25- hydroxycholecalciferol. Pediatr Res. 1972;6:552–562.
53. Hasemann, CA, Kurumbail, RG, Boddupalli, SS, et al. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3:41–62.
54. Sawada, N, Sakaki, T, Kitanaka, S, et al. Structure-function analysis of CYP27B1 and CYP27A1. Studies on mutants from patients with vitamin D-dependent rickets type I (VDDR-I) and cerebrotendinous xanthomatosis (CTX). Eur J Biochem. 2001;268:6607–6615.
55. Williams, PA, Cosme, J, Sridhar, V, et al. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131.
56. Yamamoto, K, Masuno, H, Sawada, N, et al. Homology modeling of human 25-hydroxyvitamin D3 1alpha-hydroxylase (CYP27B1) based on the crystal structure of rabbit CYP2C5. J Steroid Biochem Mol Biol. 2004;89–90:167–171.
57. Yamamoto, K, Uchida, E, Urushino, N, et al. Identification of the amino acid residue of CYP27B1 responsible for binding of 25-hydroxyvitamin D3 whose mutation causes vitamin D-dependent rickets type 1. J Biol Chem. 2005;280:30511–30516.
58. Balsan, S, Garabedian, M, Lieberherr, M, et al. Serum 1,25-dihydroxyvitamin D concentrations in two different types of pseudo-deficiency rickets. In: Norman AW, Schaefer K, von Herrath D, et al, eds. Vitamin D: Basic Research and Its Clinical Application. Berlin: Walter de Gruyter; 1979:1143–1149.
59. Glorieux, FH. Calcitriol treatment in vitamin D-dependent and vitamin D-resistant rickets. Metabolism. 1990;39(suppl):10–12.
60. Reade, TM, Scriver, CR, Glorieux, FH, et al. Response to crystalline 1alpha-hydroxyvitamin D3 in vitamin D dependency. Pediatr Res. 1975;9:593–599.
61. Albright, F, Buttler, A, Bloomberg, E. Rickets resistant to vitamin D therapy. Am J Dis Child. 1937;54:529–547.
62. Haussler, MR, Norman, AW. Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci U S A. 1969;62:155–162.
63. Pike, JW, Haussler, MR. Purification of chicken intestinal receptor for 1,25-dihydroxyvitamin D. Proc Natl Acad Sci U S A. 1979;76:5485–5489.
64. Pike, JW, Marion, SL, Donaldson, CA, et al. Serum and monoclonal antibodies against the chick intestinal receptor for 1,25-dihydroxyvitamin D3, Generation by a preparation enriched in a 64,000-dalton protein. J Biol Chem. 1983;258:1289–1296.
65. Dame, MC, Pierce, EA, Prahl, JM, et al. Monoclonal antibodies to the porcine intestinal receptor for 1,25-dihydroxyvitamin D3: interaction with distinct receptor domains. Biochemistry. 1986;25:4523–4534.
66. Feldman, D, Chen, T, Cone, C, et al. Vitamin D resistant rickets with alopecia: cultured skin fibroblasts exhibit defective cytoplasmic receptors and unresponsiveness to 1,25(OH)2D3. J Clin Endocrinol Metab. 1982;55:1020–1022.
67. Hirst, MA, Hochman, HI, Feldman, D. Vitamin D resistance and alopecia: a kindred with normal 1,25-dihydroxyvitamin D binding, but decreased receptor affinity for deoxyribonucleic acid. J Clin Endocrinol Metab. 1985;60:490–495.
68. Dokoh, S, Haussler, MR, Pike, JW. Development of a radioligand immunoassay for 1,25-dihydroxycholecalciferol receptors utilizing monoclonal antibody. Biochem J. 1984;221:129–136.
69. Pike, JW, Dokoh, S, Haussler, MR, et al. Vitamin D3–resistant fibroblasts have immunoassayable 1,25-dihydroxyvitamin D3 receptors. Science. 1984;224:879–881.
70. Malloy, PJ, Hochberg, Z, Pike, JW, et al. Abnormal binding of vitamin D receptors to deoxyribonucleic acid in a kindred with vitamin D-dependent rickets, type II. J Clin Endocrinol Metab. 1989;68:263–269.
71. Baker, AR, McDonnell, DP, Hughes, M, et al. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85:3294–3298.
72. Burmester, JK, Maeda, N, DeLuca, HF. Isolation and expression of rat 1,25-dihydroxyvitamin D3 receptor cDNA. Proc Natl Acad Sci U S A. 1988;85:1005–1009.
73. McDonnell, DP, Mangelsdorf, DJ, Pike, JW, et al. Molecular cloning of complementary DNA encoding the avian receptor for vitamin D. Science. 1987;235:1214–1217.
74. Hughes, MR, Malloy, PJ, Kieback, DG, et al. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242:1702–1705.
75. Bouillon, R, Van Cromphaut, S, Carmeliet, G. Intestinal calcium absorption: molecular vitamin D mediated mechanisms. J Cell Biochem. 2003;88:332–339.
76. Arai, H, Miyamoto, K, Taketani, Y, et al. A vitamin D receptor gene polymorphism in the translation initiation codon: effect on protein activity and relation to bone mineral density in Japanese women. J Bone Miner Res. 1997;12:915–921.
77. Miyamoto, K, Kesterson, RA, Yamamoto, H, et al. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol Endocrinol. 1997;11:1165–1179.
78. Jurutka, PW, Hsieh, JC, MacDonald, PN, et al. Phosphorylation of serine 208 in the human vitamin D receptor. The predominant amino acid phosphorylated by casein kinase II, in vitro, and identification as a significant phosphorylation site in intact cells. J Biol Chem. 1993;268:6791–6799.
79. Hilliard, GMt, Cook, RG, Weigel, NL, et al. 1,25-dihydroxyvitamin D3 modulates phosphorylation of serine 205 in the human vitamin D receptor: site-directed mutagenesis of this residue promotes alternative phosphorylation. Biochemistry. 1994;33:4300–4311.
80. Rastinejad, F, Perlmann, T, Evans, RM, et al. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–211.
81. Hsieh, JC, Shimizu, Y, Minoshima, S, et al. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J Cell Biochem. 1998;70:94–109.
82. Hsieh, JC, Jurutka, PW, Galligan, MA, et al. Human vitamin D receptor is selectively phosphorylated by protein kinase C on serine 51, a residue crucial to its trans-activation function. Proc Natl Acad Sci U S A. 1991;88:9315–9319.
83. Hsieh, JC, Jurutka, PW, Nakajima, S, et al. Phosphorylation of the human vitamin D receptor by protein kinase C. Biochemical and functional evaluation of the serine 51 recognition site. J Biol Chem. 1993;268:15118–15126.
84. Nishikawa, J, Kitaura, M, Imagawa, M, et al. Vitamin D receptor contains multiple dimerization interfaces that are functionally different. Nucleic Acids Res. 1995;23:606–611.
85. Hsieh, JC, Jurutka, PW, Selznick, SH, et al. The T-box near the zinc fingers of the human vitamin D receptor is required for heterodimeric DNA binding and transactivation. Biochem Biophys Res Commun. 1995;215:1–7.
86. Rochel, N, Wurtz, JM, Mitschler, A, et al. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179.
87. Peleg, S, Sastry, M, Collins, ED, et al. Distinct conformational changes induced by 20-epi analogues of 1 alpha,25-dihydroxyvitamin D3 are associated with enhanced activation of the vitamin D receptor. J Biol Chem. 1995;270:10551–10558.
88. Allegretto, EA, Pike, JW, Haussler, MR. Immunochemical detection of unique proteolytic fragments of the chick 1,25-dihydroxyvitamin D3 receptor. Distinct 20-kDa DNA-binding and 45-kDa hormone-binding species. J Biol Chem. 1987;262:1312–1319.
89. Gardezi, SA, Nguyen, C, Malloy, PJ, et al. A rationale for treatment of hereditary vitamin D-resistant rickets with analogs of 1 alpha,25-dihydroxyvitamin D(3). J Biol Chem. 2001;276:29148–29156.
90. Bourguet, W, Ruff, M, Chambon, P, et al. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–382.
91. Rosen, ED, Beninghof, EG, Koenig, RJ. Dimerization interfaces of thyroid hormone, retinoic acid, vitamin D, and retinoid X receptors. J Biol Chem. 1993;268:11534–11541.
92. Whitfield, GK, Hsieh, JC, Nakajima, S, et al. A highly conserved region in the hormone-binding domain of the human vitamin D receptor contains residues vital for heterodimerization with retinoid X receptor and for transcriptional activation. Mol Endocrinol. 1995;9:1166–1179.
93. Jin, CH, Kerner, SA, Hong, MH, et al. Transcriptional activation and dimerization functions in the human vitamin D receptor. Mol Endocrinol. 1996;10:945–957.
94. Nakajima, S, Hsieh, JC, MacDonald, PN, et al. The C-terminal region of the vitamin D receptor is essential to form a complex with a receptor auxiliary factor required for high-affinity binding to the vitamin D-responsive element. Mol Endocrinol. 1994;8:159–172.
95. Henttu, PM, Kalkhoven, E, Parker, MG. AF-2 activity and recruitment of steroid receptor coactivator 1 to the estrogen receptor depend on a lysine residue conserved in nuclear receptors. Mol Cell Biol. 1997;17:1832–1839.
96. Danielian, PS, White, R, Lees, JA, et al. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11:1025–1033.
97. Jurutka, PW, Hsieh, JC, Remus, LS, et al. Mutations in the 1,25-dihydroxyvitamin D3 receptor identifying C-terminal amino acids required for transcriptional activation that are functionally dissociated from hormone binding, heterodimeric DNA binding, and interaction with basal transcription factor IIB, in vitro. J Biol Chem. 1997;272:14592–14599.
98. Masuyama, H, Brownfield, CM, St-Arnaud, R, et al. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol Endocrinol. 1997;11:1507–1517.
99. Blanco, JC, Wang, IM, Tsai, SY, et al. Transcription factor TFIIB and the vitamin D receptor cooperatively activate ligand-dependent transcription. Proc Natl Acad Sci U S A. 1995;92:1535–1539.
100. MacDonald, PN, Sherman, DR, Dowd, DR, et al. The vitamin D receptor interacts with general transcription factor IIB. J Biol Chem. 1995;270:4748–4752.
101. Glass, CK, Rosenfeld, MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141.
102. Kitagawa, H, Fujiki, R, Yoshimura, K, et al. The chromatin-remodeling complex WINAC targets a nuclear receptor to promoters and is impaired in Williams syndrome. Cell. 2003;113:905–917.
103. Gu, W, Malik, S, Ito, M, et al. A novel human SRB/MED-containing cofactor complex, SMCC, involved in transcription regulation. Mol Cell. 1999;3:97–108.
104. Rachez, C, Suldan, Z, Ward, J, et al. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 1998;12:1787–1800.
105. Kim, S, Shevde, NK, Pike, JW. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J Bone Miner Res. 2005;20:305–317.
106. Takeyama, K, Masuhiro, Y, Fuse, H, et al. Selective interaction of vitamin D receptor with transcriptional coactivators by a vitamin D analog. Mol Cell Biol. 1999;19:1049–1055.
107. Yamaoka, K, Shindo, M, Iwasaki, K, et al. Multiple co-activator complexes support ligand-induced transactivation function of VDR. Arch Biochem Biophys. 2007;460:166–171.
108. Malloy, PJ, Hochberg, Z, Tiosano, D, et al. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest. 1990;86:2071–2079.
109. Renaud, JP, Rochel, N, Ruff, M, et al. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689.
110. Wagner, RL, Apriletti, JW, McGrath, ME, et al. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697.
111. Rut, AR, Hewison, M, Kristjansson, K, et al. Two mutations causing vitamin D resistant rickets: modelling on the basis of steroid hormone receptor DNA-binding domain crystal structures. Clin Endocrinol (Oxf). 1994;41:581–590.
112. Cockerill, FJ, Hawa, NS, Yousaf, N, et al. Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab. 1997;82:3156–3160.
113. Ritchie, HH, Hughes, MR, Thompson, ET, et al. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proc Natl Acad Sci U S A. 1989;86:9783–9787.
114. Malloy, PJ, Hughes, MR, Pike, JW, et al. Vitamin D receptor mutations and hereditary 1,25-dihydroxyvitamin D resistant rickets. In: Norman AW, Bouillon R, Thomasset M, eds. Vitamin D: Gene Regulation, Structure-Function Analysis, and Clinical Application. Eighth Workshop on Vitamin D. New York: Walter de Gruyter; 1991:116–124.
115. Wiese, RJ, Goto, H, Prahl, JM, et al. Vitamin D-dependency rickets type II: Truncated vitamin D receptor in three kindreds. Mol Cell Endocrinol. 1993;90:197–201.
116. Malloy, PJ, Xu, R, Cattani, A, et al. A unique insertion/substitution in helix H1 of the vitamin D receptor ligand binding domain in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Bone Miner Res. 2004;19:1018–1024.
117. Malloy, PJ, Zhu, W, Zhao, XY, et al. A novel inborn error in the ligand-binding domain of the vitamin D receptor causes hereditary vitamin D-resistant rickets. Mol Genet Metab. 2001;73:138–148.
118. Malloy, PJ, Xu, R, Peng, L, et al. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol. 2002;16:2538–2546.
119. Marx, SJ, Bliziotes, MM, Nanes, M. Analysis of the relation between alopecia and resistance to 1,25-dihydroxyvitamin D. Clin Endocrinol (Oxf). 1986;25:373–381.
120. Brooks, MH, Bell, NH, Love, L, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–999.
121. Marx, SJ, Spiegel, AM, Brown, EM, et al. A familial syndrome of decrease in sensitivity to 1,25-dihydroxyvitamin D. J Clin Endocrinol Metab. 1978;47:1303–1310.
122. Zerwekh, JE, Glass, K, Jowsey, J, et al. An unique form of osteomalacia associated with end organ refractoriness to 1,25-dihydroxyvitamin D and apparent defective synthesis of 25-hydroxyvitamin D. J Clin Endocrinol Metab. 1979;49:171–175.
123. Whitfield, GK, Selznick, SH, Haussler, CA, et al. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol. 1996;10:1617–1631.
124. Malloy, PJ, Eccleshall, TR, Gross, C, et al. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304.
125. Kudoh, T, Kumagai, T, Uetsuji, N, et al. Vitamin D dependent rickets: decreased sensitivity to 1,25-dihydroxyvitamin D. Eur J Pediatr. 1981;137:307–311.
126. Balsan, S, Garabedian, M, Liberman, UA, et al. Rickets and alopecia with resistance to 1,25-dihydroxyvitamin D: two different clinical courses with two different cellular defects. J Clin Endocrinol Metab. 1983;57:803–811.
127. Tsuchiya, Y, Matsuo, N, Cho, H, et al. An unusual form of vitamin D-dependent rickets in a child: alopecia and marked end-organ hyposensitivity to biologically active vitamin D. J Clin Endocrinol Metab. 1980;51:685–690.
128. Castells, S, Greig, F, Fusi, MA, et al. Severely deficient binding of 1,25-dihydroxyvitamin D to its receptors in a patient responsive to high doses of this hormone. J Clin Endocrinol Metab. 1986;63:252–256.
129. Takeda, E, Kuroda, Y, Saijo, T, et al. 1 alpha-hydroxyvitamin D3 treatment of three patients with 1,25-dihydroxyvitamin D-receptor-defect rickets and alopecia. Pediatrics. 1987;80:97–101.
130. Sakati, N, Woodhouse, NJ, Niles, N, et al. Hereditary resistance to 1,25-dihydroxyvitamin D: clinical and radiological improvement during high-dose oral calcium therapy. Horm Res. 1986;24:280–287.
131. Balsan, S, Garabedian, M, Larchet, M, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–1667.
132. Weisman, Y, Bab, I, Gazit, D, et al. Long-term intracaval calcium infusion therapy in end-organ resistance to 1,25-dihydroxyvitamin D. Am J Med. 1987;83:984–990.
133. Hochberg, Z, Tiosano, D, Even, L. Calcium therapy for calcitriol-resistant rickets. J Pediatr. 1992;121:803–808.
134. Bliziotes, M, Yergey, AL, Nanes, MS, et al. Absent intestinal response to calciferols in hereditary resistance to 1,25-dihydroxyvitamin D: documentation and effective therapy with high dose intravenous calcium infusions. J Clin Endocrinol Metab. 1988;66:294–300.
135. Hochberg, Z, Benderli, A, Levy, J, et al. 1,25-Dihydroxyvitamin D resistance, rickets, and alopecia. Am J Med. 1984;77:805–811.
136. Chen, TL, Hirst, MA, Cone, CM, et al. 1,25-Dihydroxyvitamin D resistance, rickets, and alopecia: analysis of receptors and bioresponse in cultured fibroblasts from patients and parents. J Clin Endocrinol Metab. 1984;59:383–388.
137. Li, YC, Amling, M, Pirro, AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–4396.
138. Hewison, M, Rut, AR, Kristjansson, K, et al. Tissue resistance to 1,25-dihydroxyvitamin D without a mutation of the vitamin D receptor gene. Clin Endocrinol (Oxf). 1993;39:663–670.
139. Zhu, W, Malloy, PJ, Delvin, E, et al. Hereditary 1,25-dihydroxyvitamin D-resistant rickets due to an opal mutation causing premature termination of the vitamin D receptor. J Bone Miner Res. 1998;13:259–264.
140. Mechica, JB, Leite, MO, Mendonca, BB, et al. A novel nonsense mutation in the first zinc finger of the vitamin D receptor causing hereditary 1,25-dihydroxyvitamin D3-resistant rickets. J Clin Endocrinol Metab. 1997;82:3892–3894.
141. Yagi, H, Ozono, K, Miyake, H, et al. A new point mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor in a kindred with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Clin Endocrinol Metab. 1993;76:509–512.
142. Lin, NU, Malloy, PJ, Sakati, N, et al. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Clin Endocrinol Metab. 1996;81:2564–2569.
143. Saijo, T, Ito, M, Takeda, E, et al. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Am J Hum Genet. 1991;49:668–673.
144. Malloy, PJ, Weisman, Y, Feldman, D. Hereditary 1 alpha,25-dihydroxyvitamin D-resistant rickets resulting from a mutation in the vitamin D receptor deoxyribonucleic acid-binding domain. J Clin Endocrinol Metab. 1994;78:313–316.
145. Sone, T, Marx, SJ, Liberman, UA, et al. A unique point mutation in the human vitamin D receptor chromosomal gene confers hereditary resistance to 1,25-dihydroxyvitamin D3. Mol Endocrinol. 1990;4:623–631.
146. Hawa, NS, Cockerill, FJ, Vadher, S, et al. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin Endocrinol (Oxf). 1996;45:85–92.
147. Kristjansson, K, Rut, AR, Hewison, M, et al. Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1,25 dihydroxyvitamin D3. J Clin Invest. 1993;92:12–16.
148. Katavetin, P, Katavetin, P, Wacharasindhu, S, et al. A girl with a novel splice site mutation in VDR supports the role of a ligand-independent VDR function on hair cycling. Horm Res. 2006;66:273–276.
149. Nguyen, M, d’Alesio, A, Pascussi, JM, et al. Vitamin D-resistant rickets and type 1 diabetes in a child with compound heterozygous mutations of the vitamin D receptor (L263R and R391S): dissociated responses of the CYP-24 and rel-B promoters to 1,25-dihydroxyvitamin D3. J Bone Miner Res. 2006;21:886–894.
150. Malloy, PJ, Xu, R, Peng, L, et al. Hereditary 1,25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology. 2004;145:5106–5114.
151. Nguyen, TM, Adiceam, P, Kottler, ML, et al. Tryptophan missense mutation in the ligand-binding domain of the vitamin D receptor causes severe resistance to 1,25-dihydroxyvitamin D. J Bone Miner Res. 2002;17:1728–1737.
152. Malloy, PJ, Zhu, W, Bouillon, R, et al. A novel nonsense mutation in the ligand binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab. 2002;77:314–318.
153. Malloy, PJ, Wang, J, Peng, L, et al. A unique insertion/duplication in the VDR gene that truncates the VDR causing hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Arch Biochem Biophys. 2007;460:285–292.