[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 21
Genetic Defects in Thyroid Hormone Synthesis and Action
Congenital hypothyroidism (CH) is the most frequent endocrine-metabolic disease in infancy, with an incidence of about 1 in 3000 to 4000 newborns.1,2 With the exception of rare cases due to hypothalamic or pituitary defects, CH is characterized by elevated thyroid-stimulating hormone (TSH) in response to reduced thyroid hormone (TH) levels.
Thyroid hormones play critical roles in differentiation, growth, and metabolism. Therefore, THs are required for the normal function of nearly all tissues, with major effects on oxygen consumption and metabolic rate.3
Defects in Thyroid Hormone Synthesis
In the majority of cases (80% to 85%), primary permanent CH is due to alterations occurring during the gland organogenesis, resulting either in a thyroid that is absent (thyroid agenesis, or athyreosis), hypoplastic (thyroid hypoplasia), or located in an unusual position (thyroid ectopy). All these entities are grouped under the term thyroid dysgenesis (TD).4 TD occurs mostly as a sporadic disease, but a genetic cause of the disease has been demonstrated in about 5% of reported cases. Genes associated with TD (Table 21-1) include several thyroid transcription factors expressed in the early phases of thyroid organogenesis (NKX2.1/TITF1, FOXE1/TITF2, PAX8, NKX2.5), as well as genes like the thyrotropin receptor gene (TSHR) expressed later during gland morphogenesis.

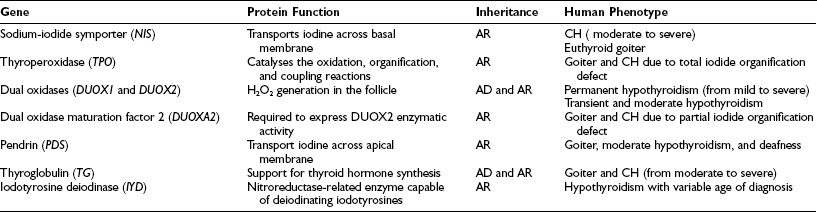
In the remaining 15% of cases, the disease is caused by inborn errors in the molecular steps required for the biosynthesis of thyroid hormones, and generally it is characterized by enlargement of the gland (goiter), presumably due to elevated TSH levels.5 Thyroid dyshormonogenesis shows classical Mendelian recessive inheritance (Table 21-2).
Rarely, CH of central origin is due to hypothalamic and/or pituitary diseases, with reduced production and/or effect of thyrotropin-releasing hormone (TRH) or of the thyrotropin hormone (TSH).6
Hyporesponsiveness to Thyroid-Stimulating Hormone
Defects in the Thyroid-Stimulating Hormone Receptor
The role of the TSHR gene in CH with TSH unresponsiveness and absence of goiter was hypothesized almost 40 years ago. Useful models for studying this autosomal recessive form of CH were offered through (1) identification of hyt/hyt mice that were affected by primary hypothyroidism with elevated TSH and hypoplastic thyroid due to a loss-of-function mutation in the Tshr gene7,8 and (2) the production of Tshr−/− mice.9
TSHR mutations in humans were identified for the first time in three siblings with CH associated with high serum TSH and normal thyroid hormone.10 The siblings were compound heterozygous, carrying a different mutation in each of the two alleles. Since this report, other mutations in the TSHR gene have been identified in several patients with thyroid hypoplasia and increased TSH secretion. All the affected individuals are homozygous or compound heterozygous for loss-of-function mutations, and consistently in the familial forms, the disease is inherited as an autosomal recessive trait. This form of CH is characterized by a “small” thyroid gland in normal position. In the case of total failure of TSHR function, the patient is severely hypothyroid because the complete lack of TSH stimulation almost completely represses the metabolic activity of the thyroid gland.11 When the TSHR has a diminished affinity to its ligand, the effect may largely be compensated for by high plasma TSH concentrations. The high TSH level in these cases does not result in an exaggerated stimulation of thyroid metabolism, and goitrogenesis is not observed.
Abnormalities in the Gs Protein Subunit
Hyporesponsiveness to TSH is found in patients with pseudohypoparathyroidism type 1a (Albright’s hereditary osteodystrophy),12 a variably expressed disorder with autosomal-dominant inheritance. The cause is a defect in the Gsα subunit (gene map locus 20q13). Gsα is involved in the stimulatory pathways of TSH and TRH as well as pathways of other hormones binding to a Gsα-coupled receptor (parathyroid hormone [PTH], gonadotropin-releasing hormone [GnRH], follicle-stimulating hormone [FSH], luteinizing hormone [LH], etc.). Several mutations have been found in these cases.13,14 Patients tend to have only mild manifestations of hypothyroidism, with normal or slightly decreased plasma free thyroxine (FT4) levels and slightly elevated TSH levels. Detection of patients with pseudohypoparathyroidism type 1a by neonatal CH screening has been reported, but it is likely that most affected newborns would be missed because their blood TSH and thyroxine (T4) concentrations would not reach the cutoff levels used in the screening programs. Otherwise, the mild hypothyroidism is just a minor component of the syndrome, and early T4 supplementation therapy is unable to prevent mental and growth retardation.
Other Causes
Hyporesponsiveness to TSH may also be caused by factors other than mutations in the TSHR or G proteins. Many families express the phenotype of resistance to TSH in the absence of a TSHR defect. In many subjects, the inheritance is dominant and the genetic cause has not yet been clarified.15 Possible candidates are factors located downstream of the TSHR/G-protein/cAMP cascade or other thyroid developmental genes.
Thyroid Dysgenesis
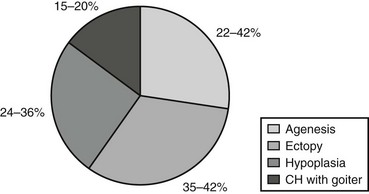
The absence of thyroid follicular cells is called athyreosis or agenesis of the thyroid. The term agenesis should be used to define the absence of the gland due to a defective initiation of thyroid morphogenesis, whereas athyreosis indicates a dysgenesis characterized by the disappearance of the thyroid following any step after the thyroid anlage specification. Athyreosis accounts for 22% to 44% of the cases of primitive permanent CH (Fig. 21-1). So far, the absence of thyroid was reported in patients with CH associated with FOXE1 gene defects (Bamforth-Lazarus syndrome),16,17 in one subject carrying a mutation in PAX818 and in one patient with NKX2-5 mutation.19
The Bamforth-Lazarus syndrome20 is a clinical entity characterized by cleft palate, bilateral choanal atresia, spiky hair, and athyreosis. Two homozygous mutations in the FOXE1 gene have been described in two pairs of siblings affected by this syndrome16,17 and in one patient with syndromic congenital hypothyroidism but not athyreosis.21 All affected members carry homozygous missense mutations in conserved amino acids within the FOXE1 forkhead domain. The mutant proteins were tested in vitro and have shown a reduction in both DNA binding and transcriptional activity.
Ectopic Thyroid
In humans, more than 50% of TD cases are associated with an ectopic thyroid (see Fig. 21-1); however, up to now, only three heterozygous mutations in the NKX2-5 gene have been linked to human ectopic thyroid.19 The functional studies of the mutant NKX2-5 demonstrated a significant functional impairment, with reduction of transactivation properties and a dominant-negative effect. The patients described were all heterozygous, and the mutations were inherited from one of the parents, suggesting that NKX2-5 mutations have variable penetrance and clinical significance.
Hypoplasia
The presence of hypoplastic thyroid has been reported in 24% to 36% of cases of CH (see Fig. 21-1). Thyroid hypoplasia is a genetically heterogeneous disease, since mutations in NKX2-1, PAX8, or TSHR genes have been reported in patients with thyroid hypoplasia.
Patients with NKX2-1 loss-of-function mutations are affected by choreoathetosis, hypothyroidism, and pulmonary alterations, with incomplete penetrance and a variability of the phenotype.22 So far, 22 loss-of-function mutations in the NKX2-1 gene have been identified in patients with this clinical picture.23–40 The unfavorable outcome in the case of impaired NKX2-1 expression, regardless of early T4 supplementation, is most likely caused by defects in the central nervous system rather than fetal hypothyroidism.
The involvement of PAX8 has been described in sporadic and familial cases of CH with TD.18,41–46 In vitro transfection assays demonstrated that the mutated proteins are unable to bind DNA and to drive transcription of the TPO promoter. All affected individuals are heterozygous for the mutations, and in the familial cases, transmission is autosomal dominant with a variable penetrance and expressivity.
Hemiagenesis
Thyroid hemiagenesis is a dysgenesis in which one thyroid lobe fails to develop. The prevalence of this morphologic abnormality ranges from 0.05% to 0.2% in healthy children, with the absence of the left lobe in almost all the cases. In these subjects, thyroid function tests are within the normal range.47
The molecular mechanisms leading to the formation of the two symmetrical thyroid lobes are still unclear, and in humans, candidate genes responsible for hemiagenesis of the thyroid have not yet been described. Indeed, Shh−/− mice embryos can display either a nonlobulated gland48 or hemiagenesis of thyroid.49 Hemiagenesis of the thyroid is also frequent in mice double heterozygous for Titf1+/− and Pax8+/−.50
Dyshormonogenesis
As mentioned before, in about 15% of cases, CH is due to hormonogenesis defects (see Fig. 21-1) caused by mutations in genes involved in thyroid hormone synthesis, secretion, or recycling. These cases are clinically characterized by the presence of goiter, and the molecular mechanisms in most of these forms have been identified.
Defects in any of these steps lead to reduced circulating thyroid hormone, resulting in congenital hypothyroidism and goiter. With the exception of rare cases, all mutations in these genes appear to be inherited in autosomal-recessive fashion (see Table 21-2).
Sodium-Iodide Symporter
The sodium-iodide symporter (NIS) is a member of the sodium/solute symporter family that actively transports iodide across the membrane of the thyroid follicular cells. In 1996, NIS mRNAs from rats51 and humans52 were isolated. The human gene (SLC5A5) maps to chromosome 19p13.2-p12. It has 15 exons and encodes a 643-amino-acid protein expressed primarily in thyroid but also in salivary glands, gastric mucosa, small intestinal mucosa, lacrimal gland, nasopharynx, thymus, skin, lung tissue, choroid plexus, ciliary body, uterus, lactating mammary tissue and mammary carcinoma cells, and placenta.53,54 Only in thyroid cells iodide transport is regulated by TSH.
The inability of the thyroid gland to accumulate iodine was one of the early known causes of CH, and before the cloning of NIS, a clinical diagnosis of hereditary iodide transport defect had been made on the basis of goitrous hypothyroidism and absent thyroidal radioiodine uptake. To date, several mutations inherited in an autosomal-recessive manner have been described, with a clinical picture characterized by hypothyroidism of variable severity (from severe to fully compensated) and goiter.55,56 Thyroid morphology is heterogeneous in patients with the same NIS mutation.57
In the neonatal period, infants with iodide transport defects are found to have a normal-size or slightly enlarged thyroid gland by ultrasonography and elevated serum thyroglobulin levels.58 Radioactive iodide uptake is absent. Measurement of the saliva-to-plasma 123I ratio is around one. The degree of hypothyroidism is variable and ranges from mild to severe, possibly depending on the amount of iodide in the diet. These children are severely hypothyroid if maintained with a normal iodine diet, but addition of high amounts of iodide to the diet tends to compensate for the iodide transport failure.
Thyroperoxidase
Accumulation of iodine in the thyroid gland reaches a steady state between active influx, protein binding, and efflux, resulting in a relatively low free intracellular iodide concentration in normal conditions, but increased in the presence of TPO defects. The kinetics of iodide uptake and release can be traced by administration of radioiodide, and iodide reuptake can be inhibited by anions of similar molecular size and charge, such as perchlorate or thiocyanate. Radioiodide uptake and perchlorate inhibition give an idea of the intrathyroidal iodide concentration in relation to the circulating iodine. Iodine organification defects can be quantified as total or partial: total iodide organification defects are characterized by discharge of more than 90% of the radioiodide taken up by the gland within 1 hour after administration of sodium perchlorate, usually given 2 hours after radioiodide. A total disappearance of the thyroid image is also observed. Partial iodide organification defects are characterized by discharge of 20% to 90% of the accumulated radioiodine.59
The human TPO gene is located on chromosome 2p25 and spans approximately 150 kb; the coding sequence of 3048 bp is divided over 17 exons60 and encodes for a 933-amino-acid, membrane-bound, glycated, heme-containing protein located on the apical membranes of the thyroid follicular cell.
Defects in the TPO gene have been reported to cause congenital hypothyroidism by a total iodide organification defect, and mutations have been identified in all exons of the TPO gene. Most mutations are found in exons 8, 9, or 10, encoding the active center and heme-binding portion of the enzyme. Nonsense, splice-site, and frameshift mutations have been also described by several groups.56,61,62
DUOX1 and DUOX2
The generation of H2O2 is a crucial step in thyroid hormonogenesis. Recently two new proteins involved in the H2O2 generation in the apical membrane of the follicular thyroid cell have been identified.60 These proteins, initially named THOX1 and THOX2 (for thyroid oxidase), map to chromosome 15q15.3, only 16 kb apart from each other and in opposite transcriptional orientation. In 2001, since these proteins contain two distinct functional domains, it was suggested that they be called DUOX (for dual oxidase).
DUOX1 and DUOX2 are glycoproteins with seven putative transmembrane domains. Their function remained unclear until a factor named DUOXA2, which allows the transition of DUOX2 from the endoplasmic reticulum to the Golgi, was identified.63 The coexpression of this factor with DUOX2 in HeLa cells is able to reconstitute the H2O2 production in vitro. A similar protein (DUOXA1) is necessary for the complete maturation of the DUOX1. Interestingly, both DUOXA genes map in the 16 kb that separate the DUOX1 and DUOX2 genes on chromosome 15.
Several mutations in DUOX genes have been reported in patients with congenital hypothyroidism showing very variable phenotypes.64–67 To produce congenital permanent hypothyroidism, a severe alteration of both alleles of the DUOX2 gene is required. The presence of some residual activity in one of the alleles may produce a less severe phenotype, whereas monoallelic severe inactivation of the DUOX2 gene is associated with transient CH. In addition, the phenotype of monoallelic inactivation seems to be modulated by other factors, including environmental conditions (such as iodine insufficiency) or lifetime events (pregnancy, immediate postnatal life).
So far, no mutation in the DUOX1 gene has been identified in patients with CH. In contrast, very recently a biallelic inactivation in the dual oxidase maturation factor 2 (DUOXA2) gene has been identified in a patient with congenital hypothyroidism.68
Pendrin
In 1896, Vaughan Pendred described a syndrome characterized by congenital neurosensorial deafness and goiter.69 The disease is transmitted as an autosomal-recessive disorder. Patients have a moderately enlarged thyroid gland, are usually euthyroid, and show only a partial discharge of iodide after the administration of thiocyanate or perchlorate. The impaired hearing characteristic of the condition is not constant and is due to a cochlear defect that corresponds to the Mondini’s type of developmental abnormality of the cochlea.
In 1997, the PDS gene was cloned, and the predicted protein of 780 amino acids (86 kD) was called pendrin.70 The PDS gene maps to human chromosome 7q31, contains 21 exons, and is expressed both in the cochlea and in the thyroid. Pendrin has been localized into the apical membrane of thyroid follicular cells.71,72 In thyroid follicular cells and in transfected oocytes, pendrin is able to transport iodide.
A number of mutations in the PDS gene have been described in patients with Pendred’s syndrome.73 Despite the goiter, individuals are likely to be euthyroid and only rarely present congenital hypothyroidism. However, TSH levels are often in the upper limit of the normal range, and hypothyroidism of variable severity may eventually develop.74
Thyroglobulin
Thyroglobulin is a homodimer protein synthesized exclusively in the thyroid. The human gene is located on chromosome 8q24, and the coding sequence, containing 8307 bp,75 is divided into 42 exons.76 Following a signal peptide of 19 amino acids, the polypeptide chain is composed of 2750 amino acids containing 66 tyrosine residues. Thyroglobulin is a dimer with identical 330-kD subunits containing 10% carbohydrate residues.
Patients with disorders of thyroglobulin synthesis are moderately to severely hypothyroid. Usually, plasma thyroglobulin concentration is low, especially in relation to the TSH concentrations, and does not change after T4 treatment or injection of TSH. Patients classified in the category “thyroglobulin synthesis defects” often have abnormal iodoproteins, mainly iodinated plasma albumin, and they excrete iodopeptides of low molecular weight in the urine.77
Several mutations in the thyroglobulin gene have been reported in patients with CH78,79 and in animals, including Afrikander cattle (p.R697X),80 Dutch goats (p.Y296X),81 cog/cog mice (p.L2263P),82 and rdw rats (p.G2300R).83
IYD
In addition to the active transport from the blood due to the NIS, iodine in the thyroid follicular cells derives also from the deiodination of monoiodotyrosine and diiodotyrosine.84 The gene encoding for this enzymatic activity was recently identified and named IYD (or DEHAL1).85,86 The human gene maps to chromosome 6q24-q25 and consists of 6 exons encoding a protein of 293 amino acids, with a nitroreductase-related enzyme capable of deiodinating iodotyrosines.
In the past, it was suggested that IYD mutations could be responsible for congenital hypothyroidism, but only very recently, four patients with three mutations in the IYD gene have been reported.87,88 The disease was transmitted either as an autosomal-recessive87 or an autosomal-dominant pattern of inheritance with incomplete penetration.88 Patients were hypothyroid and goitrous, with a high phenotypic variability, depending on the time of expression of the disease manifestations. The patients born after the introduction of the screening program for CH were not identified by the screening. There is also a variable severity in the clinical picture, and this can derive either from the molecular effects of the mutation (complete absence or partial activity of the protein) or from environmental factors, such as iodine diet content.
Central Congenital Hypothyroidism
Developmental Defects of the Pituitary
Recent studies have identified a variety of mutations in the LHX3 and LHX4 genes in patients with combined pituitary hormone deficiency diseases. These patients have complex and variable syndromes involving short stature, metabolic disorders, reproductive system deficits, and nervous system developmental abnormalities.89
Hesx1 (also called Rpx), a member of the paired-like class of homeobox genes, is one of the earliest markers of the pituitary primordium.90 Extinction of Hesx1 is important for activation of downstream genes such as Prop1, suggesting that both proteins act as opposing transcription factors.91 Targeted disruption of Hesx1 in the mouse revealed a reduction in the prospective forebrain tissue, absent optic vesicles, markedly decreased head size, and severe microphthalmia reminiscent of the syndrome of septo-optic dysplasia (SOD) in humans.
SOD is a rare heterogeneous anomaly hypoplasia of the optic nerves, various types of forebrain defects, and a variety of pituitary hormone deficiencies. Endocrine dysfunction ranges from isolated GH deficiency to complete pituitary hormonal deficiency. The human HESX1 gene maps to chromosome 3p21.1-3p21.2, and its coding region spans 1.7 kb, with a highly conserved genomic organization consisting of 4 coding exons. The first homozygous missense mutation (Arg160Cys) was found in the homeobox of HESX1 in two siblings with SOD.90 Subsequently, several other homozygous and heterozygous mutations have been shown to present with varying phenotypes characterized by pituitary hormone deficiency and SOD.91
Defects in the TRH Gene and the Thyrotropin-Releasing Hormone Receptor
In mice, homozygous deletion of the TRH gene produced a phenotype characterized by hypothyroidism and hyperglycemia.92 Only a few patients with reduced TRH production have been described in the literature,93,94 but no human mutations have been described so far.
Similarly, mice lacking the TRH receptor appear almost normal, with some growth retardation and a considerable decrease in serum T3, T4, and PRL levels but not in serum TSH.95 Thus far, only one family with a compound heterozygous96 and one family with a homozygous97 loss-of-function mutation of the TRH receptor have been described.
Pit1/POU1F1
Pit1 (called POU1F1 in humans) is a pituitary-specific transcription factor belonging to the POU homeodomain family. The human POU1F1 maps to chromosome 3p11 and consists of 6 exons spanning 17 kb encoding for a 291-amino-acid protein. After the initial report,98 several heterozygous, compound heterozygous, and homozygous POU1F1 deletions and missense and nonsense mutations have been reported to cause this type of hereditary CH.99 Deficiency of GH, PRL, and TSH is generally severe in patients harboring mutations in POU1F1.
Prop1
Prop1 (Prophet of Pit1) is a pituitary-specific paired-like homeodomain transcription required for the expression of Pit1 and also important in regulating Hesx1 expression. Dwarf mice harboring a homozygous missense mutation in Prop1 exhibit GH, TSH, and PRL deficiency and an anterior pituitary gland reduced in size by about 50%. Additionally, these mice have reduced gonadotropin expression.100
The human PROP1 maps to chromosome 5q. The gene consists of 3 exons encoding for a 226-amino-acid protein. After the first report of mutations in PROP1 in four unrelated pedigrees with GH, TSH, PRL, LH, and FSH deficiencies,101 several distinct mutations have been identified in over 170 patients,91 suggesting that PROP1 mutations account for most cases of familial multiple pituitary hormone deficiency. Affected individuals exhibit recessive inheritance. The timing of initiation and the severity of hormonal deficiency in patients with PROP1 mutations is highly variable; diagnosis of GH deficiency preceded that of TSH deficiency in 80%. Following the deficiencies in GH and TSH, there is a delayed onset of gonadotropin insufficiency. Although most patients fail to enter puberty spontaneously, some start puberty before deficiencies in LH and FSH evolve. ACTH deficiency is a relatively late manifestation of PROP1 mutation, often evolving several decades after birth. The degree of prolactin deficiency and pituitary morphologic alterations are variable.91
Structural Thyroid-Stimulating Hormone Defects
The TSH α subunit has the amino acid sequence in common with LH, FSH, and chorionic gonadotropin. The β subunit is different for each of these hormones and carries specific information for receptor binding and expression of hormonal action. For biological activity, the heterodimer configuration is required.102
Mutations in the TSH-β gene are a rare cause of congenital hypothyroidism, and in all the reported cases, the mutations were homozygous or compound heterozygous. Available data have been recently reviewed by Miyai.103 The phenotype is quite variable, and it may range from a very mild hypothyroidism to severe forms associated with mental retardation in cases of delayed treatment. Patients with mutations in TSHB are characterized by the presence of low levels of circulating TSH that will not be stimulated by TRH. Finally, cases of immunologically reactive but biologically inactive TSH have also been reported.103
Peripheral Congenital Hypothyroidism
Defects in Transmembrane Transport of Thyroid Hormone
Although T3 is the major receptor-active form of thyroid hormone, T4 is the predominant iodothyronine secreted by the thyroid gland under normal conditions. As consequence, target tissues need to convert T4 into T3 by so-called outer-ring deiodination (ORD). Alternatively, T4 is metabolized by inner-ring deiodination (IRD) to receptor-inactive reverse T3 (rT3), and by the same reaction, T3 is inactivated to T2.104 The latter is also produced by ORD from rT3. Three iodothyronine deiodinases (D1–3) are involved in these reactions. These selenoproteins, each with a different catalytic preference and tissue distribution pattern, not only regulate the basic metabolic activity in the various tissues but are also involved in processes that adapt the body to extraordinary conditions such as fasting and illness.
Endemic selenium deficiency may potentially affect iodothyronine activity in large populations, but until now, no clinicopathologic entity that could be ascribed with certainty to genetic defects in any of the iodothyronine deiodinases has been described. Furthermore, mice knockouts for D1 and D2 have a surprisingly mild phenotype, their serum T3 level is normal (probably due to enhanced secretion and formation of T3 from the thyroid gland), and their general health and reproductive capacity are unimpaired.105
Besides secretion of the prohormone T4 and its deiodination to T3, transmembrane transport of thyroid hormone (T4, T3, or both) into the cells of the peripheral tissues is a prerequisite for binding of T3 to its nuclear receptors and the subsequent protein synthesis. It has been thought for a long time that the lipophilic iodothyronines are capable of crossing the plasma membrane by simple diffusion, but it has become increasingly clear that for such crossing, transporters are needed.106
More recently, monocarboxylate transporter 8 (MCT8) has been shown to be an active and specific thyroid hormone transporter.107,108 The gene encoding for MCT8 (named SLC16A2) is located on human chromosome Xq13.2 and consists of 6 exons and 5 introns, the first of which is about 100,000 kb in size.109,110 The mature mRNA is about 4.4 kb large and contains two possible translation start sites (TLSs). Depending on which of these TLSs is used, proteins are generated consisting of 613 or 539 amino acids, respectively. The human MCT8 protein contains 12 putative transmembrane domains, and both the N and C terminus are located intracellularly. The important role of MCT8 in maintaining euthyroidism, especially in brain tissues, is demonstrated by the identification of several male subjects with a particular combination of severe neurologic deficits and abnormal serum thyroid hormone levels.111–125 The neurologic phenotype includes central hypotonia with poor head control; peripheral hypotonia that evolves into spastic quadriplegia; inability to sit, stand, or walk independently; severe mental retardation; and absence of speech.113,126
To date, more than 40 different mutations have been identified in MCT8,127 including large deletions with loss of one or more exons, smaller frame-shift deletions, 3-nucleotide deletions or insertions, and nonsense and missense mutations. Most of these mutations resulted in a complete inactivation of MCT8 function, although some residual activity has been associated with certain MCT8 mutations, ending in a milder clinical phenotype.119,127 Of note, mouse mutants deficient in MCT8 exhibit a marked increase in serum T3 and a decrease in serum T4 and rT3, but in contrast to the human phenotype, Mct8-null animals do not have any overt neurologic abnormalities.128,129
Clinical Manifestations of Congenital Hypothyroidism
TH concentrations are low in the fetus during the first half of pregnancy. During this time, the fetus is entirely dependent on maternal TH. The fetal hypothalamic-pituitary-thyroid axis begins to function by mid-gestation and is mature in term infants at delivery. Despite the critical importance of TH to multiple organ systems, especially the brain, most infants with CH appear normal at birth because of the protective effects of a substantial maternal-fetal transfer of T4130 and in consequence of the increased intracerebral conversion of T4 to T3, resulting in greater local availability of T3 despite its low serum concentration.131,132 The cerebral damage is mainly due to lack of TH after birth. The introduction of screening programs allowing the early identification of affected patients and prompt treatment with l-thyroxine prevent mental and neuromotor damage.
Finally, it should be remembered that infants with CH appear to be at increased risk of other congenital anomalies, mostly cardiac (approximately 10% of infants with CH, compared with 3% in the general population).133
Neonatal Screening and Diagnosis
As mentioned, most newborns with CH have normal appearance and no detectable physical signs. Delayed diagnosis leads to the most severe outcome of CH, such as mental retardation, and this emphasizes the importance of neonatal screening and immediately starting T4 therapy to prevent cerebral damage. Pilot screening programs for CH were developed in Quebec, Canada, and Pittsburgh, Pennsylvania, in 1974 and have now been established in Western Europe, North America, Japan, Australia, and parts of Eastern Europe, Asia, South America, and Central America.134–136 Since then, the apparent overall incidence of CH has increased considerably as a consequence of the detection of mild disorders that previously remained undetected or were not recognized as congenital problems.
International studies show that the incidence of permanent (thyroidal) CH is approximately 1 in 3500 newborns (in iodine-sufficient areas). There is considerable ethnic variation in incidence, ranging from 1 in 30,000 in the African American population in the United States137 to 1 in 900 in Asian populations in the United Kingdom.138 With few exceptions, the international screening programs do not detect patients with permanent central CH.
CH is usually sporadic, with a 2 : 1 female-to-male ratio. Familial cases occur with a frequency that is 15-fold higher than by chance alone139; the genetic basis of these familial cases has been established in some but not all pedigrees.140 On the other hand, more than 90% of monozygotic twin pairs are discordant for thyroid dysgenesis, suggesting that postzygotic events play a major role in the developmental abnormality.141
Certainly, the main objective of screening, the eradication of severe mental retardation after CH, has been achieved. In addition to the great clinical benefit, it has been estimated that the cost of screening for CH is much lower than the cost of diagnosis at an older age. Finally, newborn screening has also shown the prevalence of the various causes of CH (see Fig. 21-1), including transient disorders found predominantly in preterm infants.142
If hypothyroidism is confirmed by laboratory analysis, imaging studies should be performed, but it is not acceptable to delay hormone replacement therapy if imaging studies are not readily available.143
Tests commonly used to determine the underlying cause of congenital hypothyroidism are presented in Table 21-3.
Table 21-3
Tests to Complete the Diagnosis of Congenital Hypothyroidism
In the cases of thyroid dysgenesis, thyroid scintigraphy, with 99mtechnetium or 123I, is the most informative diagnostic procedure.144 Although thyroid ultrasonography is useful in demonstrating enlarged or absent glands, it is less accurate than scintigraphy in showing ectopic glands.145 Large thyroid glands may be due to inborn errors of thyroid hormone synthesis or to diseases produced by maternal TSHR autoantibodies. More specialized tests, such as perchlorate discharge; evaluation of serum, salivary, and urinary radioiodine; and measurement of serum T4 precursors, such as diiodothyronine, may be necessary to delineate specific inborn errors of thyroid hormone biosynthesis (Table 21-4).61
Table 21-4
Diagnostic Features in Congenital Hypothyroidism
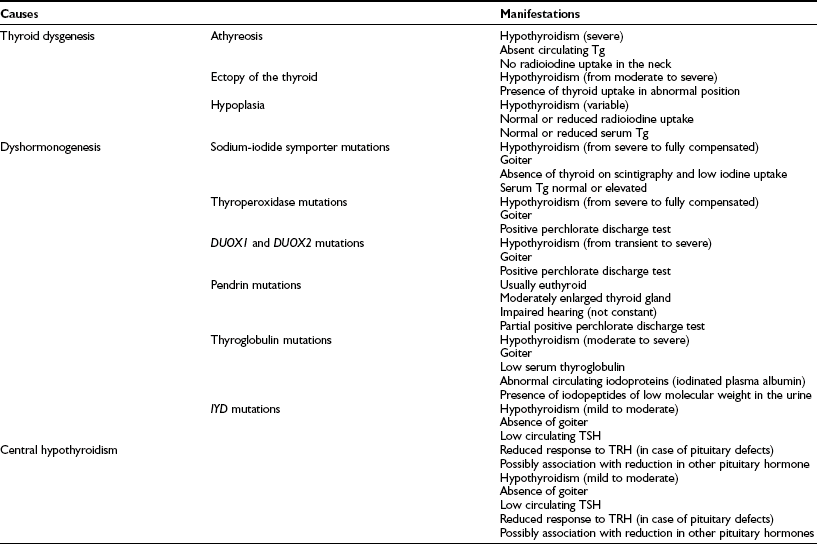
Tg, Thyroglobulin; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
Transient Congenital Hypothyroidism
Transient hypothyroidism may be the consequence of intrauterine exposure to maternal antithyroid drugs, maternal TSHR-blocking antibodies (TSHRBAb), or heterozygous DUOX264 or TSHR germline mutations.146 Endemic iodine deficiency or prenatal or postnatal exposure to iodides excess are also frequent causes of transient CH.147,148
Transplacental passage of potent maternal TSHBAb is a very rare cause of transient CH, but can be suspected in presence of a maternal history of autoimmune thyroid disease. The half-life of maternal TSHBAb in the neonate is approximately 3 to 4 weeks,149 and disappearance from serum lasts from 3 to 6 months of age.
Because the transient nature of the hypothyroidism will not be recognized clinically or through laboratory tests, initial treatment will be similar to that of the infant with permanent CH; however, at a later age, interruption of therapy allows transient hypothyroidism to be distinguished from permanent hypothyroidism.142
Treatment
All infants with hypothyroidism, with or without goiter, should be rendered euthyroid as promptly as possible by replacement therapy with TH.150–153 An optimal cognitive outcome depends on both the adequacy and timing of postnatal therapy, particularly in severe cases of CH. The goal of therapy is to normalize T4 within 2 weeks and TSH within 1 month. An initial dosage of 10 to 15 µg/kg of l-thyroxine (LT4) has been recommended.142
Growth rate and adult height are normal in children with CH who received adequate treatment.154–156 The best reported outcome occurred with l-thyroxine therapy started within 2 weeks of age with 9.5 µg/kg or more per day.142,153
After 3 years of age, LT4 administration should be discontinued for 30 to 45 days if no definite cause of CH was found by scan or if there was no TSH increase after the newborn period.157 After this withdrawal period, serum FT4 and TSH should be measured. Low FT4 and elevated TSH values indicate permanent hypothyroidism, and TH therapy should be started again. If the FT4 and TSH concentrations remain within the normal range, the diagnosis of transient hypothyroidism is confirmed. Nevertheless, these children need to continue follow-up to detect a later increase in TSH.
Patients with CH receiving early treatment with TH show only minor differences in intelligence, school achievement, and neuropsychological tests when compared with classmates and siblings.158–162 If treatment is started within the first 2 months of life, although physical recovery is good and stature is normal,156 patients may still have a low to normal IQ.163 Similarly, although more than 80% of infants receiving replacement therapy before 3 months of age have an IQ greater than 85, 77% show some signs of minimal brain damage, including impairment of arithmetic ability, speech, or fine motor coordination. Even in early-treated patients with CH, auditory brainstem evoked potentials were abnormal in 25% of the children studied. The reason for this is unknown, but it may suggest that prenatal maternal T4 production does not provide complete protection to alterations in the developing central nervous system.164
Transplacental transfer of maternal T4 in the first trimester may protect the brain during early development.165 For the same reason, maternal hypothyroidism during fetal development can have persistent effects on the child’s neural development.165–167
Finally, in the case of severe CH due to genetic defects diagnosed before birth, starting intrauterine treatment with l-thyroxine should be considered. This may offer definite short-term and possible long-term beneficial effects for the patient.168–173
References
1. Toublanc, JE. Comparison of epidemiological data on congenital hypothyroidism in Europe with those of other parts in the world. Horm Res. 1992;38:230–235.
2. Klett, M. Epidemiology of congenital hypothyroidism. Exp Clin Endocrinol Diabetes. 1997;105(Suppl 4):19–23.
3. Oppenheimer, JH, Schwartz, HL, Mariash, CN, et al. Advances in our understanding of thyroid hormone action at the cellular level. Endocr Rev. 1987;8:288–308.
4. Fisher, DA, Klein, A. Thyroid development and disorders of thyroid function in the newborn. New Engl J Med. 1981;304:702–712.
5. Madeiros-Neto, G, Stanbury, JB. Inherited Disorders of the Thyroid System. Boca Raton, FL: CRC Press; 1994.
6. Grüters, A, Krude, H, Biebermann, H. Molecular genetic defects in congenital hypothyroidism. Eur J Endocrinol. 2004;151(Suppl 3):U39–U44.
7. Stuart, A, Oates, E, Hall, C, et al. Identification of a point mutation in the thyrotropin receptor of the hyt/hyt hypothyroid mouse. Mol Endocrinol. 1994;8:129–138.
8. Beamer, WJ, Cresswell, LA. Defective thyroid ontogenesis in fetal hypothyroid (hyt/hyt) mice. Anat Rec. 1982;202:387–393.
9. Marians, RC, Ng, L, Blair, HC, et al. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781.
10. Sunthornthepvarakui, T, Gottschalk, M, Hayashi, Y, Refetoff, S. Brief report: resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med. 1995;332:155–160.
11. Biebermann, H, Schoneberg, T, Krude, H, et al. Mutations of the human thyrotropin receptor gene causing thyroid hypoplasia and persistent congenital hypothyroidism. J Clin Endocrinol Metab. 1997;82:3471–3480.
12. Albright, F, Burnett, C, Smith, P, Parson, W. Pseudohypoparathyroidism: An example of “Seabright-Bantam syndrome”: Report of three cases. Endocrinology. 1942;30:922.
13. Spiegel, AM, Shenker, A, Weinstein, LS. Receptor-effector coupling by G proteins: implications for normal and abnormal signal transduction. Endocr Rev. 1992;13:536–565.
14. Weinstein, LS, Liu, J, Sakamoto, A, et al. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–5464.
15. Refetoff, S. Resistance to thyrotropin. J Endocrinol Invest. 2003;26:770–779.
16. Clifton-Bligh, RJ, Wentworth, JM, Heinz, P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399–401.
17. Castanet, M, Park, SM, Smith, A, et al. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051–2059.
18. Macchia, PE, Lapi, P, Krude, H, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. 1998;19:83–86.
19. Dentice, M, Cordeddu, V, Rosica, A, et al. Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91:1428–1433.
20. Bamforth, JS, Hughes, IA, Lazarus, JH, et al. Congenital hypothyroidism, spiky hair, and cleft palate. J Med Genet. 1989;26:49–60.
21. Baris, I, Arisoy, AE, Smith, A, et al. A novel missense mutation in human TTF-2 (FKHL15) gene associated with congenital hypothyroidism but not athyreosis. J Clin Endocrinol Metab. 2006;91:4183–4187.
22. De Felice, M, Di Lauro, R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev. 2004;25:722–746.
23. Devriendt, K, Vanhole, C, Matthijs, G, de Zegher, F. Deletion of Thyroid Transcription Factor-1 gene in an infant with neonatal thyroid dysfunction and respiratory failure. N Engl J Med. 1998;338:1317–1318.
24. Iwatani, N, Mabe, H, Devriendt, K, et al. Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J Pediatr. 2000;137:272–276.
25. de Vries, BB, Arts, WF, Breedveld, GJ, et al. Benign hereditary chorea of early onset maps to chromosome 14q. Am J Hum Genet. 2000;66:136–142.
26. Guala, A, Nocita, G, Di Maria, E, et al. Benign hereditary chorea: a rare cause of disability. Riv Ital Pediatr. 2001;27:150–152.
27. Pohlenz, J, Dumitrescu, A, Zundel, D, et al. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest. 2002;109:469–473.
28. Krude, H, Schutz, B, Biebermann, H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109:475–480.
29. Breedveld, GJ, Percy, AK, MacDonald, ME, et al. Clinical and genetic heterogeneity in benign hereditary chorea. Neurology. 2002;59:579–584.
30. Kleiner-Fisman, G, Rogaeva, E, Halliday, W, et al. Benign hereditary chorea: clinical, genetic, and pathological findings. Ann Neurol. 2003;54:244–247.
31. Doyle, DA, Gonzalez, I, Thomas, B, Scavina, M. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX2-1. J Pediatr. 2004;145:190–193.
32. Asmus, F, Horber, V, Pohlenz, J, et al. A novel TITF-1 mutation causes benign hereditary chorea with response to levodopa. Neurology. 2005;64:1952–1954.
33. do Carmo Costa, M, Costa, C, Silva, AP, et al. Nonsense mutation in TITF1 in a Portuguese family with benign hereditary chorea. Neurogenetics. 2005;6:209–215.
34. Willemsen, MA, Breedveld, GJ, Wouda, S, et al. Brain-thyroid-lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr. 2005;164:28–30.
35. Kleiner-Fisman, G, Calingasan, NY, Putt, M, et al. Alterations of striatal neurons in benign hereditary chorea. Mov Disord. 2005;20:1353–1357.
36. Devos, D, Vuillaume, I, de Becdelievre, A, et al. New syndromic form of benign hereditary chorea is associated with a deletion of TITF-1 and PAX-9 contiguous genes. Mov Disord. 2006;21:2237–2240.
37. Moya, CM, Perez de Nanclares, G, Castano, L, et al. Functional study of a novel single deletion in the TITF1/NKX2.1 homeobox gene that produces congenital hypothyroidism and benign chorea but not pulmonary distress. J Clin Endocrinol Metab. 2006;91:1832–1841.
38. Provenzano, C, Veneziano, L, Appleton, R, et al. Functional characterization of a novel mutation in TITF-1 in a patient with benign hereditary chorea. J Neurol Sci. 2008;264:56–62.
39. Nagasaki, K, Narumi, S, Asami, T, et al. Mutation of a gene for thyroid transcription factor-1 (TITF1) in a patient with clinical features of resistance to thyrotropin. Endocr J. 2008;55:875–888.
40. Ferrara, AM, De Michele, G, Salvatore, E, et al. A novel NKX2.1 mutation in a family with hypothyroidism and benign hereditary chorea. Thyroid. 2008;18:1005–1009.
41. Vilain, C, Rydlewski, C, Duprez, L, et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J Clin Endocrinol Metab. 2001;86:234–238.
42. Congdon, T, Nguyen, LQ, Nogueira, CR, et al. A novel mutation (Q40P) in PAX8 associated with congenital hypothyroidism and thyroid hypoplasia: evidence for phenotypic variability in mother and child. J Clin Endocrinol Metab. 2001;86:3962–3967.
43. Komatsu, M, Takahashi, T, Takahashi, I, et al. Thyroid dysgenesis caused by PAX8 mutation: the hypermutability with CpG dinucleotides at codon 31. J Pediatr. 2001;139:597–599.
44. de Sanctis, L, Corrias, A, Romagnolo, D, et al. Familial PAX8 small deletion (c.989_992delACCC) associated with extreme phenotype variability. J Clin Endocrinol Metab. 2004;89:5669–5674.
45. Al Taji, E, Biebermann, H, Limanova, Z, et al. Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur J Endocrinol. 2007;156:521–529.
46. Esperante, SA, Rivolta, CM, Miravalle, L, et al. Identification and characterization of four PAX8 rare sequence variants (p.T225M, p.L233L, p.G336S and p.A439A) in patients with congenital hypothyroidism and dysgenetic thyroid glands. Clin Endocrinol (Oxf). 2008;68:828–835.
47. Maiorana, R, Carta, A, Floriddia, G, et al. Thyroid hemiagenesis: prevalence in normal children and effect on thyroid function. J Clin Endocrinol Metab. 2003;88:1534–1536.
48. Alt, B, Elsalini, OA, Schrumpf, P, et al. Arteries define the position of the thyroid gland during its developmental relocalisation. Development. 2006;133:3797–3804.
49. Fagman, H, Grande, M, Gritli-Linde, A, Nilsson, M. Genetic deletion of sonic hedgehog causes hemiagenesis and ectopic development of the thyroid in mouse. Am J Pathol. 2004;164:1865–1872.
50. Amendola, E, De Luca, P, Macchia, PE, et al. A mouse model demonstrates a multigenic origin of congenital hypothyroidism. Endocrinology. 2005;146:5038–5047.
51. Dai, G, Levy, O, Carrasco, N. Cloning and characterization of the thyroid iodide symporter. Nature. 1996;379:458–460.
52. Smanik, PA, Ryu, KY, Theil, KS, et al. Expression, exon-intron organization, and chromosome mapping of the human sodium iodide symporter. Endocrinology. 1997;138:3555–3558.
53. Caillou, B, Troalen, F, Baudin, E, et al. Na+/I− symporter distribution in human thyroid tissues: an immunohistochemical study. J Clin Endocrinol Metab. 1998;83:4102–4106.
54. Lazar, V, Bidart, J, Caillou, B, et al. Expression of the Na+/I− symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab. 1999;84:3228–3234.
55. Dohan, O, De la Vieja, A, Paroder, V, et al. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24:48–77.
56. Park, SM, Chatterjee, VK. Genetics of congenital hypothyroidism. J Med Genet. 2005;42:379–389.
57. De la Vieja, A, Dohan, O, Levy, O, Carrasco, N. Molecular analysis of the sodium/iodide symporter: Impact on thyroid and extrathyroid pathophysiology. Physiol Rev. 2000;80:1083–1105.
58. Vulsma, T, Rammeloo, J, Gons, M, de Vijlder, J. The role of serum thyroglobulin concentration and thyroid ultrasound imaging in the detection of iodide transport defects in infants. Acta Endocrinol (Copenh). 1991;124:405–410.
59. de Vijlder, J. Primary congenital hypothyroidism: Defects in iodine pathways. Eur J Endocrinol. 2003;149:247–256.
60. De Deken, X, Wang, D, Many, M, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233.
61. LaFranchi, S. Congenital hypothyroidism: etiologies, diagnosis, and management. Thyroid. 1999;9:735–740.
62. Bakker, B, Bikker, H, Vulsma, T, et al. Two decades of screening for congenital hypothyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update). J Clin Endocrinol Metab. 2000;85:3708–3712.
63. Grasberger, H, Refetoff, S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272.
64. Moreno, JC, Visser, TJ. New phenotypes in thyroid dyshormonogenesis: hypothyroidism due to DUOX2 mutations. Endocr Dev. 2007;10:99–117.
65. Johnson, KR, Marden, CC, Ward-Bailey, P, et al. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol Endocrinol. 2007;21:1593–1602.
66. Ohye, H, Fukata, S, Hishinuma, A, et al. A novel homozygous missense mutation of the dual oxidase 2 (DUOX2) gene in an adult patient with large goiter. Thyroid. 2008;18:561–566.
67. Maruo, Y, Takahashi, H, Soeda, I, et al. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab. 2008;93:4261–4267.
68. Zamproni, I, Grasberger, H, Cortinovis, F, et al. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab. 2008;93:605–610.
69. Pendred, V. Deaf mutism and goitre. Lancet. 1896;2:532.
70. Gausden, E, Coyle, B, Armour, JA, et al. Pendred syndrome: evidence for genetic homogeneity and further refinement of linkage. J Med Genet. 1997;34:126–129.
71. Royaux, I, Suzuki, K, Mori, A, et al. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–845.
72. Yoshida, A, Taniguchi, S, Hisatome, I, et al. Pendrin is an iodide-specific apical porter responsible for iodide efflux from thyroid cells. J Clin Endocrinol Metab. 2002;87:3356–3361.
73. Kopp, P. Pendred’s syndrome and genetic defects in thyroid hormone synthesis. Rev Endocr Metab Disord. 2000;1:109–121.
74. Glaser, B. Pendred syndrome. Pediatr Endocrinol Rev. 2003;1(Suppl 2):199–204.
75. Van de Graaf, S, Pauws, E, de Vijlder, J, Ris-Stalpers, C. The revised 8307 base pair coding sequence of human thyroglobulin transiently expressed in eukaryotic cells. Eur J Endocrinol. 1997;136:508–515.
76. Mendive, F, Rivolta, C, Vassart, G, Targovnik, H. Genomic organization of the 3′ region of the human thyroglobulin gene. Thyroid. 1999;9:903–912.
77. Gons, M, Kok, J, Tegelaers, W, de Vijlder, J. Concentration of plasma thyroglobulin and urinary excretion of iodinated material in the diagnosis of thyroid disorders in congenital hypothyroidism. Acta Endocrinol (Copenh). 1983;104:27–34.
78. Rivolta, CM, Targovnik, HM. Molecular advances in thyroglobulin disorders. Clin Chim Acta. 2006;374:8–24.
79. Caputo, M, Rivolta, CM, Esperante, SA, et al. Congenital hypothyroidism with goitre caused by new mutations in the thyroglobulin gene. Clin Endocrinol (Oxf). 2007;67:351–357.
80. Ricketts, MH, Simons, MJ, Parma, J, et al. A nonsense mutation causes hereditary goiter in the Afrikander cattle and unmasks alternative splicing of thyroglobulin transcripts. Proc Natl Acad Sci U S A. 1987;84:3181–3184.
81. Veenboer, G, de Vijlder, J. Molecular basis of the thyroglobulin synthesis defect in Dutch goats. Endocrinology. 1993;132:377–381.
82. Kim, PS, Hossain, SA, Park, YN, et al. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc Natl Acad Sci U S A. 1998;95:9909–9913.
83. Hishinuma, A, Furudate, S, Oh-Ishi, M, et al. A novel missense mutation (G2320R) in thyroglobulin causes hypothyroidism in rdw rats. Endocrinology. 2000;141:4050–4055.
84. Roche, J, Michel, R, Michel, O, Lissitzky, S. Enzymatic dehalogenation of iodotyrosine by thyroid tissue; on its physiological role. Biochim Biophys Acta. 1952;9:161–169.
85. Moreno, JC, Pauws, E, van Kampen, AH, et al. Cloning of tissue-specific genes using serial analysis of gene expression and a novel computational subtraction approach. Genomics. 2001;75:70–76.
86. Moreno, JC. Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res. 2003;60(Suppl 3):96–102.
87. Moreno, JC, Klootwijk, W, van Toor, H, et al. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med. 2008;358:1811–1818.
88. Afink, G, Kulik, W, Overmars, H, et al. Molecular characterization of iodotyrosine dehalogenase deficiency in patients with hypothyroidism. J Clin Endocrinol Metab. 2008;93:4894–4901.
89. Colvin, SC, Mullen, RD, Pfaeffle, RW, Rhodes, SJ. LHX3 and LHX4 transcription factors in pituitary development and disease. Pediatr Endocrinol Rev. 2009;6(Suppl 2):283–290.
90. Dattani, MT, Martinez-Barbera, JP, Thomas, PQ, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125–133.
91. Mehta, A, Dattani, MT. Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism. Best Pract Res Clin Endocrinol Metab. 2008;22:191–206.
92. Yamada, M, Saga, Y, Shibusawa, N, et al. Tertiary hypothyroidism and hyperglycemia in mice with targeted disruption of the thyrotropin-releasing hormone gene. Proc Natl Acad Sci U S A. 1997;94:10862–10867.
93. Niimi, H, Inomata, H, Sasaki, N, Nakajima, H. Congenital isolated thyrotrophin releasing hormone deficiency. Arch Dis Child. 1982;57:877–878.
94. Katakami, H, Kato, Y, Inada, M, Imura, H. Hypothalamic hypothyroidism due to isolated thyrotropin-releasing hormone (TRH) deficiency. J Endocrinol Invest. 1984;7:231–233.
95. Rabeler, R, Mittag, J, Geffers, L, et al. Generation of thyrotropin-releasing hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol Endocrinol. 2004;18:1450–1460.
96. Collu, R, Tang, J, Castagne, J, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561–1565.
97. Bonomi, M, Busnelli, M, Beck-Peccoz, P, et al. A family with complete resistance to thyrotropin-releasing hormone. N Engl J Med. 2009;360:731–734.
98. Tatsumi, K, Miyai, K, Notomi, T, et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. 1992;1:56–58.
99. Cohen, LE, Radovick, S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev. 2002;23:431–442.
100. Ward, RD, Raetzman, LT, Suh, H, et al. Role of PROP1 in pituitary gland growth. Mol Endocrinol. 2005;19:698–710.
101. Wu, W, Cogan, JD, Pfaffle, RW, et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18:147–149.
102. Cohen, R, Weintraub, B, Wondisford, F. Thyrotropin: Chemistry and biosynthesis of thyrotropin. In: Braverman L, Utiger R, eds. Werner and Ingbar’s the Thyroid. Philadelphia: Lippincott Williams & Wilkins; 2000:202–218.
103. Miyai, K. Congenital thyrotropin deficiency–from discovery to molecular biology, postgenome and preventive medicine. Endocr J. 2007;54:191–203.
104. Gereben, B, Zavacki, AM, Ribich, S, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938.
105. Galton, VA, Schneider, MJ, Clark, AS, St Germain, DL. Life without thyroxine to 3,5,3′-triiodothyronine conversion: Studies in mice devoid of the 5′-deiodinases. Endocrinology. 2009;150:2502–2504.
106. Hennemann, G, Docter, R, Friesema, EC, et al. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001;22:451–476.
107. Friesema, EC, Ganguly, S, Abdalla, A, et al. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135.
108. Friesema, EC, Kuiper, GG, Jansen, J, et al. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772.
109. Lafreniere, RG, Carrel, L, Willard, HF. Lafreniere RG, Carrel L, Willard HF: A novel transmembrane transporter encoded by the XPCT gene in Xq13.2. Hum Mol Genet. 1994;3:1133–1139.
110. Halestrap, AP, Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628.
111. Dumitrescu, AM, Liao, XH, Best, TB, et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175.
112. Biebermann, H, Ambrugger, P, Tarnow, P, et al. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005;153:359–366.
113. Brockmann, K, Dumitrescu, AM, Best, TT, et al. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663–666.
114. Friesema, EC, Grueters, A, Biebermann, H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437.
115. Friesema, EC, Jansen, J, Heuer, H, et al. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2:512–523.
116. Frints, SG, Lenzner, S, Bauters, M, et al. MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet. 2008;16:1029–1037.
117. Fuchs, O, Pfarr, N, Pohlenz, J, Schmidt, H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009;51:240–244.
118. Herzovich, V, Vaiani, E, Marino, R, et al. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2007;67:1–6.
119. Jansen, J, Friesema, EC, Kester, MH, et al. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92:2378–2381.
120. Jansen, J, Friesema, EC, Kester, MH, et al. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. 2008;149:2184–2190.
121. Maranduba, CM, Friesema, EC, Kok, F, et al. Decreased cellular uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J Med Genet. 2006;43:457–460.
122. Namba, N, Etani, Y, Kitaoka, T, et al. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. 2008;167:785–791.
123. Vaurs-Barriere, C, Deville, M, Sarret, C, et al. Pelizaeus-Merzbacher-like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65:114–118.
124. Visser, WE, Friesema, EC, Jansen, J, Visser, TJ. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab. 2008;19:50–56.
125. Wemeau, JL, Pigeyre, M, Proust-Lemoine, E, et al. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084–2088.
126. Holden, KR, Zuniga, OF, May, MM, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005;20:852–857.
127. Heuer, H, Visser, TJ. Pathophysiological importance of thyroid hormone transport. Endocrinology. 2009;150:1078–1083.
128. Dumitrescu, AM, Liao, XH, Weiss, RE, et al. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147:4036–4043.
129. Trajkovic, M, Visser, TJ, Mittag, J, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635.
130. Vulsma, T, Gons, MH, de Vijlder, JJ. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N Engl J Med. 1989;321:13–16.
131. Ruiz de Ona, C, Obregon, MJ, Escobar del Rey, F, Morreale de Escobar, G. Developmental changes in rat brain 5′-deiodinase and thyroid hormones during the fetal period: the effects of fetal hypothyroidism and maternal thyroid hormones. Pediatr Res. 1988:24.
132. Kester, MH, Martinez de Mena, R, Obregon, MJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab. 2004;89:3117–3128.
133. Olivieri, A, Stazi, MA, Mastroiacovo, P, et al. A population-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: Data from the Italian Registry for Congenital Hypothyroidism, 1991–1998. J Clin Endocrinol Metab. 2002;87:557–562.
134. Dussault, JH, Coulombe, P, Laberge, C, et al. Preliminary report on a mass screening program for neonatal hypothyroidism. J Pediatr. 1975;86:670–674.
135. Revised guidelines for neonatal screening programmes for primary congenital hypothyroidism, Working Group on Neonatal Screening of the European Society for Paediatric Endocrinology. Horm Res. 1999;52:49–52.
136. LaFranchi, SH, Snyder, DB, Sesser, DE, et al. Follow-up of newborns with elevated screening T4 concentrations. J Pediatr. 2003;143:296–301.
137. Brown, AL, Fernhoff, PM, Milner, J, et al. Racial differences in the incidence of congenital hypothyroidism. J Pediatr. 1981;99:934–936.
138. Rosenthal, M, Addison, GM, Price, DA. Congenital hypothyroidism: increased incidence in Asian families. Arch Dis Child. 1988;63:790–793.
139. Castanet, M, Polak, M, Bonaiti-Pellie, C, et al. Nineteen years of national screening for congenital hypothyroidism: familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J Clin Endocrinol Metab. 2001;86:2009–2014.
140. Castanet, M, Sura Trueba, S, Chauty, A, et al. Linkage and mutational analysis of familial thyroid dysgenesis suggest genetic heterogeneity. Eur J Hum Genet. 2005;13:232–239.
141. Perry, R, Heinrichs, C, Bourdoux, P, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab. 2002;87:4072–4077.
142. Rose, SR, Brown, RS, Foley, T, et al. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117:2290–2303.
143. Gruters, A, Krude, H. Update on the management of congenital hypothyroidism. Horm Res. 2007;68(Suppl 5):107–111.
144. Muir, A, Daneman, D, Daneman, A, Ehrlich, R. Thyroid scanning, ultrasound, and serum thyroglobulin in determining the origin of congenital hypothyroidism. Am J Dis Child. 1988;142:214–216.
145. Takashima, S, Nomura, N, Tanaka, H, et al. Congenital hypothyroidism: assessment with ultrasound. AJNR Am J Neuroradiol. 1995;16:1117–1123.
146. Calaciura, F, Motta, RM, Miscio, G, et al. Subclinical hypothyroidism in early childhood: a frequent outcome of transient neonatal hyperthyrotropinemia. J Clin Endocrinol Metab. 2002;87:3209–3214.
147. Glinoer, D. Pregnancy and iodine. Thyroid. 2001;11:471–481.
148. Bartalena, L, Bogazzi, F, Braverman, LE, Martino, E. Effects of amiodarone administration during pregnancy on neonatal thyroid function and subsequent neurodevelopment. J Endocrinol Invest. 2001;24:116–130.
149. van Der Zwet, WC, Vandenbroucke-Grauls, CM, van Elburg, RM, et al. Neonatal antibody titers against varicella-zoster virus in relation to gestational age, birth weight, and maternal titer. Pediatrics. 2002;109:79–85.
150. de Escobar, GM, Obregon, MJ, del Rey, FE. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract Res Clin Endocrinol Metab. 2004;18:225–248.
151. Bakker, B, Kempers, MJ, De Vijlder, JJ, et al. Dynamics of the plasma concentrations of TSH, FT4 and T3 following thyroxine supplementation in congenital hypothyroidism. Clin Endocrinol (Oxf). 2002;57:529–537.
152. Cassio, A, Cacciari, E, Cicognani, A, et al. Treatment for congenital hypothyroidism: thyroxine alone or thyroxine plus triiodothyronine? Pediatrics. 2003;111:1055–1060.
153. Bongers-Schokking, JJ, Koot, HM, Wiersma, D, et al. Influence of timing and dose of thyroid hormone replacement on development in infants with congenital hypothyroidism. J Pediatr. 2000;136:292–297.
154. Ohnishi, H, Inomata, H, Watanabe, T, et al. Clinical utility of thyroid ultrasonography in the diagnosis of congenital hypothyroidism. Endocr J. 2002;49:293–297.
155. Salerno, M, Micillo, M, Di Maio, S, et al. Longitudinal growth, sexual maturation and final height in patients with congenital hypothyroidism detected by neonatal screening. Eur J Endocrinol. 2001;145:377–383.
156. Morin, A, Guimarey, L, Apezteguia, M, et al. Linear growth in children with congenital hypothyroidism detected by neonatal screening and treated early: a longitudinal study. J Pediatr Endocrinol Metab. 2002;15:973–977.
157. Eugster, EA, LeMay, D, Zerin, JM, Pescovitz, OH. Definitive diagnosis in children with congenital hypothyroidism. J Pediatr. 2004;144:643–647.
158. Bongers-Schokking, JJ. Pre- and postnatal brain development in neonates with congenital hypothyroidism. J Pediatr Endocrinol Metab. 2001;14(Suppl 6):1463–1468.
159. Gruters, A, Jenner, A, Krude, H. Long-term consequences of congenital hypothyroidism in the era of screening programmes. Best Pract Res Clin Endocrinol Metab. 2002;16:369–382.
160. Oerbeck, B, Sundet, K, Kase, BF, Heyerdahl, S. Congenital hypothyroidism: influence of disease severity and l-thyroxine treatment on intellectual, motor, and school-associated outcomes in young adults. Pediatrics. 2003;112:923–930.
161. Rovet, J, Daneman, D. Congenital hypothyroidism: a review of current diagnostic and treatment practices in relation to neuropsychologic outcome. Paediatr Drugs. 2003;5:141–149.
162. Rovet, JF. Congenital hypothyroidism: an analysis of persisting deficits and associated factors. Child Neuropsychol. 2002;8:150–162.
163. Mirabella, G, Feig, D, Astzalos, E, et al. The effect of abnormal intrauterine thyroid hormone economies on infant cognitive abilities. J Pediatr Endocrinol Metab. 2000;13:191–194.
164. Chou, YH, Wang, PJ. Auditory brainstem evoked potentials in early-treated congenital hypothyroidism. J Child Neurol. 2002;17:510–514.
165. Haddow, JE, Palomaki, GE, Allan, WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341:549–555.
166. Lavado-Autric, R, Auso, E, Garcia-Velasco, JV, et al. Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J Clin Invest. 2003;111:1073–1082.
167. Morreale de Escobar, G, Obregon, MJ, Escobar del Rey, F. Is neuropsychological development related to maternal hypothyroidism or to maternal hypothyroxinemia? J Clin Endocrinol Metab. 2000;85:3975–3987.
168. Perelman, AH, Johnson, RL, Clemons, RD, et al. Intrauterine diagnosis and treatment of fetal goitrous hypothyroidism. J Clin Endocrinol Metab. 1990;71:618–621.
169. Abuhamad, AZ, Fisher, DA, Warsof, SL, et al. Antenatal diagnosis and treatment of fetal goitrous hypothyroidism: case report and review of the literature. Ultrasound Obstet Gynecol. 1995;6:368–371.
170. Caron, P, Moya, CM, Malet, D, et al. Compound heterozygous mutations in the thyroglobulin gene (1143delC and 6725G→A [R2223H]) resulting in fetal goitrous hypothyroidism. J Clin Endocrinol Metab. 2003;88:3546–3553.
171. Gruner, C, Kollert, A, Wildt, L, et al. Intrauterine treatment of fetal goitrous hypothyroidism controlled by determination of thyroid-stimulating hormone in fetal serum. A case report and review of the literature. Fetal Diagn Ther. 2001;16:47–51.
172. Borgel, K, Pohlenz, J, Holzgreve, W, Bramswig, JH. Intrauterine therapy of goitrous hypothyroidism in a boy with a new compound heterozygous mutation (Y453D and C800R) in the thyroid peroxidase gene. A long-term follow-up. Am J Obstet Gynecol. 2005;193:857–858.
173. Miyata, I, Abe-Gotyo, N, Tajima, A, et al. Successful intrauterine therapy for fetal goitrous hypothyroidism during late gestation. Endocr J. 2007;54:813–817.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 21
Genetic Defects in Thyroid Hormone Synthesis and Action
Congenital hypothyroidism (CH) is the most frequent endocrine-metabolic disease in infancy, with an incidence of about 1 in 3000 to 4000 newborns.1,2 With the exception of rare cases due to hypothalamic or pituitary defects, CH is characterized by elevated thyroid-stimulating hormone (TSH) in response to reduced thyroid hormone (TH) levels.
Thyroid hormones play critical roles in differentiation, growth, and metabolism. Therefore, THs are required for the normal function of nearly all tissues, with major effects on oxygen consumption and metabolic rate.3
Defects in Thyroid Hormone Synthesis
In the majority of cases (80% to 85%), primary permanent CH is due to alterations occurring during the gland organogenesis, resulting either in a thyroid that is absent (thyroid agenesis, or athyreosis), hypoplastic (thyroid hypoplasia), or located in an unusual position (thyroid ectopy). All these entities are grouped under the term thyroid dysgenesis (TD).4 TD occurs mostly as a sporadic disease, but a genetic cause of the disease has been demonstrated in about 5% of reported cases. Genes associated with TD (Table 21-1) include several thyroid transcription factors expressed in the early phases of thyroid organogenesis (NKX2.1/TITF1, FOXE1/TITF2, PAX8, NKX2.5), as well as genes like the thyrotropin receptor gene (TSHR) expressed later during gland morphogenesis.
In the remaining 15% of cases, the disease is caused by inborn errors in the molecular steps required for the biosynthesis of thyroid hormones, and generally it is characterized by enlargement of the gland (goiter), presumably due to elevated TSH levels.5 Thyroid dyshormonogenesis shows classical Mendelian recessive inheritance (Table 21-2).
Rarely, CH of central origin is due to hypothalamic and/or pituitary diseases, with reduced production and/or effect of thyrotropin-releasing hormone (TRH) or of the thyrotropin hormone (TSH).6
Hyporesponsiveness to Thyroid-Stimulating Hormone
Defects in the Thyroid-Stimulating Hormone Receptor
The role of the TSHR gene in CH with TSH unresponsiveness and absence of goiter was hypothesized almost 40 years ago. Useful models for studying this autosomal recessive form of CH were offered through (1) identification of hyt/hyt mice that were affected by primary hypothyroidism with elevated TSH and hypoplastic thyroid due to a loss-of-function mutation in the Tshr gene7,8 and (2) the production of Tshr−/− mice.9
TSHR mutations in humans were identified for the first time in three siblings with CH associated with high serum TSH and normal thyroid hormone.10 The siblings were compound heterozygous, carrying a different mutation in each of the two alleles. Since this report, other mutations in the TSHR gene have been identified in several patients with thyroid hypoplasia and increased TSH secretion. All the affected individuals are homozygous or compound heterozygous for loss-of-function mutations, and consistently in the familial forms, the disease is inherited as an autosomal recessive trait. This form of CH is characterized by a “small” thyroid gland in normal position. In the case of total failure of TSHR function, the patient is severely hypothyroid because the complete lack of TSH stimulation almost completely represses the metabolic activity of the thyroid gland.11 When the TSHR has a diminished affinity to its ligand, the effect may largely be compensated for by high plasma TSH concentrations. The high TSH level in these cases does not result in an exaggerated stimulation of thyroid metabolism, and goitrogenesis is not observed.
Abnormalities in the Gs Protein Subunit
Hyporesponsiveness to TSH is found in patients with pseudohypoparathyroidism type 1a (Albright’s hereditary osteodystrophy),12 a variably expressed disorder with autosomal-dominant inheritance. The cause is a defect in the Gsα subunit (gene map locus 20q13). Gsα is involved in the stimulatory pathways of TSH and TRH as well as pathways of other hormones binding to a Gsα-coupled receptor (parathyroid hormone [PTH], gonadotropin-releasing hormone [GnRH], follicle-stimulating hormone [FSH], luteinizing hormone [LH], etc.). Several mutations have been found in these cases.13,14 Patients tend to have only mild manifestations of hypothyroidism, with normal or slightly decreased plasma free thyroxine (FT4) levels and slightly elevated TSH levels. Detection of patients with pseudohypoparathyroidism type 1a by neonatal CH screening has been reported, but it is likely that most affected newborns would be missed because their blood TSH and thyroxine (T4) concentrations would not reach the cutoff levels used in the screening programs. Otherwise, the mild hypothyroidism is just a minor component of the syndrome, and early T4 supplementation therapy is unable to prevent mental and growth retardation.
Other Causes
Hyporesponsiveness to TSH may also be caused by factors other than mutations in the TSHR or G proteins. Many families express the phenotype of resistance to TSH in the absence of a TSHR defect. In many subjects, the inheritance is dominant and the genetic cause has not yet been clarified.15 Possible candidates are factors located downstream of the TSHR/G-protein/cAMP cascade or other thyroid developmental genes.
Thyroid Dysgenesis
The absence of thyroid follicular cells is called athyreosis or agenesis of the thyroid. The term agenesis should be used to define the absence of the gland due to a defective initiation of thyroid morphogenesis, whereas athyreosis indicates a dysgenesis characterized by the disappearance of the thyroid following any step after the thyroid anlage specification. Athyreosis accounts for 22% to 44% of the cases of primitive permanent CH (Fig. 21-1). So far, the absence of thyroid was reported in patients with CH associated with FOXE1 gene defects (Bamforth-Lazarus syndrome),16,17 in one subject carrying a mutation in PAX818 and in one patient with NKX2-5 mutation.19
The Bamforth-Lazarus syndrome20 is a clinical entity characterized by cleft palate, bilateral choanal atresia, spiky hair, and athyreosis. Two homozygous mutations in the FOXE1 gene have been described in two pairs of siblings affected by this syndrome16,17 and in one patient with syndromic congenital hypothyroidism but not athyreosis.21 All affected members carry homozygous missense mutations in conserved amino acids within the FOXE1 forkhead domain. The mutant proteins were tested in vitro and have shown a reduction in both DNA binding and transcriptional activity.
Ectopic Thyroid
In humans, more than 50% of TD cases are associated with an ectopic thyroid (see Fig. 21-1); however, up to now, only three heterozygous mutations in the NKX2-5 gene have been linked to human ectopic thyroid.19 The functional studies of the mutant NKX2-5 demonstrated a significant functional impairment, with reduction of transactivation properties and a dominant-negative effect. The patients described were all heterozygous, and the mutations were inherited from one of the parents, suggesting that NKX2-5 mutations have variable penetrance and clinical significance.
Hypoplasia
The presence of hypoplastic thyroid has been reported in 24% to 36% of cases of CH (see Fig. 21-1). Thyroid hypoplasia is a genetically heterogeneous disease, since mutations in NKX2-1, PAX8, or TSHR genes have been reported in patients with thyroid hypoplasia.
Patients with NKX2-1 loss-of-function mutations are affected by choreoathetosis, hypothyroidism, and pulmonary alterations, with incomplete penetrance and a variability of the phenotype.22 So far, 22 loss-of-function mutations in the NKX2-1 gene have been identified in patients with this clinical picture.23–40 The unfavorable outcome in the case of impaired NKX2-1 expression, regardless of early T4 supplementation, is most likely caused by defects in the central nervous system rather than fetal hypothyroidism.
The involvement of PAX8 has been described in sporadic and familial cases of CH with TD.18,41–46 In vitro transfection assays demonstrated that the mutated proteins are unable to bind DNA and to drive transcription of the TPO promoter. All affected individuals are heterozygous for the mutations, and in the familial cases, transmission is autosomal dominant with a variable penetrance and expressivity.
Hemiagenesis
Thyroid hemiagenesis is a dysgenesis in which one thyroid lobe fails to develop. The prevalence of this morphologic abnormality ranges from 0.05% to 0.2% in healthy children, with the absence of the left lobe in almost all the cases. In these subjects, thyroid function tests are within the normal range.47
The molecular mechanisms leading to the formation of the two symmetrical thyroid lobes are still unclear, and in humans, candidate genes responsible for hemiagenesis of the thyroid have not yet been described. Indeed, Shh−/− mice embryos can display either a nonlobulated gland48 or hemiagenesis of thyroid.49 Hemiagenesis of the thyroid is also frequent in mice double heterozygous for Titf1+/− and Pax8+/−.50
Dyshormonogenesis
As mentioned before, in about 15% of cases, CH is due to hormonogenesis defects (see Fig. 21-1) caused by mutations in genes involved in thyroid hormone synthesis, secretion, or recycling. These cases are clinically characterized by the presence of goiter, and the molecular mechanisms in most of these forms have been identified.
Defects in any of these steps lead to reduced circulating thyroid hormone, resulting in congenital hypothyroidism and goiter. With the exception of rare cases, all mutations in these genes appear to be inherited in autosomal-recessive fashion (see Table 21-2).
Sodium-Iodide Symporter
The sodium-iodide symporter (NIS) is a member of the sodium/solute symporter family that actively transports iodide across the membrane of the thyroid follicular cells. In 1996, NIS mRNAs from rats51 and humans52 were isolated. The human gene (SLC5A5) maps to chromosome 19p13.2-p12. It has 15 exons and encodes a 643-amino-acid protein expressed primarily in thyroid but also in salivary glands, gastric mucosa, small intestinal mucosa, lacrimal gland, nasopharynx, thymus, skin, lung tissue, choroid plexus, ciliary body, uterus, lactating mammary tissue and mammary carcinoma cells, and placenta.53,54 Only in thyroid cells iodide transport is regulated by TSH.
The inability of the thyroid gland to accumulate iodine was one of the early known causes of CH, and before the cloning of NIS, a clinical diagnosis of hereditary iodide transport defect had been made on the basis of goitrous hypothyroidism and absent thyroidal radioiodine uptake. To date, several mutations inherited in an autosomal-recessive manner have been described, with a clinical picture characterized by hypothyroidism of variable severity (from severe to fully compensated) and goiter.55,56 Thyroid morphology is heterogeneous in patients with the same NIS mutation.57
In the neonatal period, infants with iodide transport defects are found to have a normal-size or slightly enlarged thyroid gland by ultrasonography and elevated serum thyroglobulin levels.58 Radioactive iodide uptake is absent. Measurement of the saliva-to-plasma 123I ratio is around one. The degree of hypothyroidism is variable and ranges from mild to severe, possibly depending on the amount of iodide in the diet. These children are severely hypothyroid if maintained with a normal iodine diet, but addition of high amounts of iodide to the diet tends to compensate for the iodide transport failure.
Thyroperoxidase
Accumulation of iodine in the thyroid gland reaches a steady state between active influx, protein binding, and efflux, resulting in a relatively low free intracellular iodide concentration in normal conditions, but increased in the presence of TPO defects. The kinetics of iodide uptake and release can be traced by administration of radioiodide, and iodide reuptake can be inhibited by anions of similar molecular size and charge, such as perchlorate or thiocyanate. Radioiodide uptake and perchlorate inhibition give an idea of the intrathyroidal iodide concentration in relation to the circulating iodine. Iodine organification defects can be quantified as total or partial: total iodide organification defects are characterized by discharge of more than 90% of the radioiodide taken up by the gland within 1 hour after administration of sodium perchlorate, usually given 2 hours after radioiodide. A total disappearance of the thyroid image is also observed. Partial iodide organification defects are characterized by discharge of 20% to 90% of the accumulated radioiodine.59
The human TPO gene is located on chromosome 2p25 and spans approximately 150 kb; the coding sequence of 3048 bp is divided over 17 exons60 and encodes for a 933-amino-acid, membrane-bound, glycated, heme-containing protein located on the apical membranes of the thyroid follicular cell.
Defects in the TPO gene have been reported to cause congenital hypothyroidism by a total iodide organification defect, and mutations have been identified in all exons of the TPO gene. Most mutations are found in exons 8, 9, or 10, encoding the active center and heme-binding portion of the enzyme. Nonsense, splice-site, and frameshift mutations have been also described by several groups.56,61,62
DUOX1 and DUOX2
The generation of H2O2 is a crucial step in thyroid hormonogenesis. Recently two new proteins involved in the H2O2 generation in the apical membrane of the follicular thyroid cell have been identified.60 These proteins, initially named THOX1 and THOX2 (for thyroid oxidase), map to chromosome 15q15.3, only 16 kb apart from each other and in opposite transcriptional orientation. In 2001, since these proteins contain two distinct functional domains, it was suggested that they be called DUOX (for dual oxidase).
DUOX1 and DUOX2 are glycoproteins with seven putative transmembrane domains. Their function remained unclear until a factor named DUOXA2, which allows the transition of DUOX2 from the endoplasmic reticulum to the Golgi, was identified.63 The coexpression of this factor with DUOX2 in HeLa cells is able to reconstitute the H2O2 production in vitro. A similar protein (DUOXA1) is necessary for the complete maturation of the DUOX1. Interestingly, both DUOXA genes map in the 16 kb that separate the DUOX1 and DUOX2 genes on chromosome 15.
Several mutations in DUOX genes have been reported in patients with congenital hypothyroidism showing very variable phenotypes.64–67 To produce congenital permanent hypothyroidism, a severe alteration of both alleles of the DUOX2 gene is required. The presence of some residual activity in one of the alleles may produce a less severe phenotype, whereas monoallelic severe inactivation of the DUOX2 gene is associated with transient CH. In addition, the phenotype of monoallelic inactivation seems to be modulated by other factors, including environmental conditions (such as iodine insufficiency) or lifetime events (pregnancy, immediate postnatal life).
So far, no mutation in the DUOX1 gene has been identified in patients with CH. In contrast, very recently a biallelic inactivation in the dual oxidase maturation factor 2 (DUOXA2) gene has been identified in a patient with congenital hypothyroidism.68
Pendrin
In 1896, Vaughan Pendred described a syndrome characterized by congenital neurosensorial deafness and goiter.69 The disease is transmitted as an autosomal-recessive disorder. Patients have a moderately enlarged thyroid gland, are usually euthyroid, and show only a partial discharge of iodide after the administration of thiocyanate or perchlorate. The impaired hearing characteristic of the condition is not constant and is due to a cochlear defect that corresponds to the Mondini’s type of developmental abnormality of the cochlea.
In 1997, the PDS gene was cloned, and the predicted protein of 780 amino acids (86 kD) was called pendrin.70 The PDS gene maps to human chromosome 7q31, contains 21 exons, and is expressed both in the cochlea and in the thyroid. Pendrin has been localized into the apical membrane of thyroid follicular cells.71,72 In thyroid follicular cells and in transfected oocytes, pendrin is able to transport iodide.
A number of mutations in the PDS gene have been described in patients with Pendred’s syndrome.73 Despite the goiter, individuals are likely to be euthyroid and only rarely present congenital hypothyroidism. However, TSH levels are often in the upper limit of the normal range, and hypothyroidism of variable severity may eventually develop.74
Thyroglobulin
Thyroglobulin is a homodimer protein synthesized exclusively in the thyroid. The human gene is located on chromosome 8q24, and the coding sequence, containing 8307 bp,75 is divided into 42 exons.76 Following a signal peptide of 19 amino acids, the polypeptide chain is composed of 2750 amino acids containing 66 tyrosine residues. Thyroglobulin is a dimer with identical 330-kD subunits containing 10% carbohydrate residues.
Patients with disorders of thyroglobulin synthesis are moderately to severely hypothyroid. Usually, plasma thyroglobulin concentration is low, especially in relation to the TSH concentrations, and does not change after T4 treatment or injection of TSH. Patients classified in the category “thyroglobulin synthesis defects” often have abnormal iodoproteins, mainly iodinated plasma albumin, and they excrete iodopeptides of low molecular weight in the urine.77
Several mutations in the thyroglobulin gene have been reported in patients with CH78,79 and in animals, including Afrikander cattle (p.R697X),80 Dutch goats (p.Y296X),81 cog/cog mice (p.L2263P),82 and rdw rats (p.G2300R).83
IYD
In addition to the active transport from the blood due to the NIS, iodine in the thyroid follicular cells derives also from the deiodination of monoiodotyrosine and diiodotyrosine.84 The gene encoding for this enzymatic activity was recently identified and named IYD (or DEHAL1).85,86 The human gene maps to chromosome 6q24-q25 and consists of 6 exons encoding a protein of 293 amino acids, with a nitroreductase-related enzyme capable of deiodinating iodotyrosines.
In the past, it was suggested that IYD mutations could be responsible for congenital hypothyroidism, but only very recently, four patients with three mutations in the IYD gene have been reported.87,88 The disease was transmitted either as an autosomal-recessive87 or an autosomal-dominant pattern of inheritance with incomplete penetration.88 Patients were hypothyroid and goitrous, with a high phenotypic variability, depending on the time of expression of the disease manifestations. The patients born after the introduction of the screening program for CH were not identified by the screening. There is also a variable severity in the clinical picture, and this can derive either from the molecular effects of the mutation (complete absence or partial activity of the protein) or from environmental factors, such as iodine diet content.
Central Congenital Hypothyroidism
Developmental Defects of the Pituitary
Recent studies have identified a variety of mutations in the LHX3 and LHX4 genes in patients with combined pituitary hormone deficiency diseases. These patients have complex and variable syndromes involving short stature, metabolic disorders, reproductive system deficits, and nervous system developmental abnormalities.89
Hesx1 (also called Rpx), a member of the paired-like class of homeobox genes, is one of the earliest markers of the pituitary primordium.90 Extinction of Hesx1 is important for activation of downstream genes such as Prop1, suggesting that both proteins act as opposing transcription factors.91 Targeted disruption of Hesx1 in the mouse revealed a reduction in the prospective forebrain tissue, absent optic vesicles, markedly decreased head size, and severe microphthalmia reminiscent of the syndrome of septo-optic dysplasia (SOD) in humans.
SOD is a rare heterogeneous anomaly hypoplasia of the optic nerves, various types of forebrain defects, and a variety of pituitary hormone deficiencies. Endocrine dysfunction ranges from isolated GH deficiency to complete pituitary hormonal deficiency. The human HESX1 gene maps to chromosome 3p21.1-3p21.2, and its coding region spans 1.7 kb, with a highly conserved genomic organization consisting of 4 coding exons. The first homozygous missense mutation (Arg160Cys) was found in the homeobox of HESX1 in two siblings with SOD.90
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
