[level-membership-for-pediatrics-category]
Chapter 428 General Principles of Treatment of Congenital Heart Disease
Bacterial infections should be treated vigorously, but the presence of congenital heart disease is not an appropriate reason to use antibiotics indiscriminately. Prophylaxis against infective endocarditis should be carried out during dental procedures for appropriate patients. The American Heart Association has recently significantly revised these recommendations, with most patients no longer requiring routine prophylaxis (Chapter 431).
Cyanotic patients need to be monitored for a multitude of noncardiac manifestations of oxygen deficiency (Table 428-1). Treatment of iron deficiency anemia is important in cyanotic patients, who will show improved exercise tolerance and general well-being with adequate hemoglobin levels. These patients should also be carefully observed for excessive polycythemia. Cyanotic patients should avoid situations in which dehydration may occur, which leads to increased viscosity and increases the risk of stroke. Diuretics may need to be decreased or temporarily discontinued during episodes of acute gastroenteritis. High altitudes and sudden changes in the thermal environment should also be avoided. Phlebotomy with partial exchange transfusion is carried out only in symptomatic patients with severe polycythemia (usually those hematocrit >65%). Patients with moderate to severe forms of congenital heart disease or a history of rhythm disturbance should be carefully monitored during anesthesia for even routine surgical procedures. Consultation with an anesthesiologist experienced in the care of children with congenital heart disease is encouraged. Women with nonrepaired severe congenital heart disease should be counseled on the risks associated with childbearing and on the use of contraceptives and tubal ligation. Pregnancy may be dangerous for patients with chronic cyanosis or pulmonary arterial hypertension. Women with mild to moderate heart disease and many of those who have had corrective surgery can have normal pregnancies, although those with residual hemodynamic derangements or with systemic right ventricles should optimally be followed by a high-risk perinatologist and a cardiologist with expertise in caring for adults with congenital heart disease.
Table 428-1 EXTRACARDIAC COMPLICATIONS OF CYANOTIC CONGENITAL HEART DISEASE AND EISENMENGER PHYSIOLOGY
| PROBLEM | ETIOLOGY | THERAPY |
|---|---|---|
| Polycythemia | Persistent hypoxia | Phlebotomy |
| Relative anemia | Nutritional deficiency | Iron replacement |
| CNS abscess | Right-to-left shunting | Antibiotics, drainage |
| CNS thromboembolic stroke | Right-to-left shunting or polycythemia | Phlebotomy |
| Low-grade DIC, thrombocytopenia | Polycythemia | None for DIC unless bleeding, then phlebotomy |
| Hemoptysis | Pulmonary infarct, thrombosis, or rupture of pulmonary artery plexiform lesion | Embolization |
| Gum disease | Polycythemia, gingivitis, bleeding | Dental hygiene |
| Gout | Polycythemia, diuretic agent | Allopurinol |
| Arthritis, clubbing | Hypoxic arthropathy | None |
| Pregnancy complications: abortion, fetal growth retardation, prematurity increase, maternal illness | Poor placental perfusion, poor ability to increase cardiac output | Bed rest, pregnancy prevention counseling |
| Infections | Associated asplenia, DiGeorge syndrome, endocarditis | Antibiotics |
| Fatal RSV pneumonia with pulmonary hypertension | Ribavirin; RSV immunoglobulin (prevention) | |
| Failure to thrive | Increased oxygen consumption, decreased nutrient intake | Treat heart failure; correct defect early; increase caloric intake |
| Psychosocial adjustment | Limited activity, cyanotic appearance, chronic disease, multiple hospitalizations | Counseling |
CNS, central nervous system; DIC, disseminated intravascular coagulation; RSV, respiratory syncytial virus.
Postoperative Management
The electrocardiogram should be monitored continuously during the postoperative period. A change in heart rate, even without arrhythmia, may be the first indication of a serious complication such as hemorrhage, hypothermia, hypoventilation, or heart failure. Cardiac rhythm disorders must be diagnosed quickly because a prolonged untreated arrhythmia may add a severe hemodynamic burden to the heart in the critical early postoperative period (Chapter 429). Injury to the heart’s conduction system during surgery can result in postoperative complete heart block. This complication is usually temporary and is treated with surgically placed pacing wires that can later be removed. Occasionally, complete heart block is permanent. If heart block persists beyond 10-14 days postoperatively, insertion of a permanent pacemaker is required. Tachyarrhythmias are a common problem in postoperative patients. Junctional ectopic tachycardia (JET) can be a particularly troublesome rhythm to manage (Chapter 429), although it usually responds to intravenous amiodarone.
Heart failure with poor cardiac output after cardiac surgery may be secondary to respiratory failure, serious arrhythmias, myocardial injury, blood loss, hypovolemia, a significant residual hemodynamic abnormality, or any combination of these factors. Treatment specific to the cause should be instituted. Catecholamines, phosphodiesterase inhibitors, nitroprusside and other afterload-reducing agents, and diuretics are the cardioactive agents most often used in patients with myocardial dysfunction in the early postoperative period (Chapter 436). Postoperative pulmonary hypertension can be managed with hyperventilation and inhaled nitric oxide. In patients who are unresponsive to standard pharmacologic treatment, various ventricular assist devices are available, depending on the patient’s size. If pulmonary function is adequate, a left ventricular assist device (LVAD) may be used. If pulmonary function is inadequate, extracorporeal membrane oxygenation (ECMO) may be used. These extraordinary measures are helpful in maintaining the circulation until cardiac function improves, usually within 2-3 days. They have also been used with moderate success as a bridge to transplantation in patients with severe nonremitting postoperative cardiac failure.
The postpericardiotomy syndrome may occur toward the end of the 1st postoperative week or may sometimes be delayed until weeks or months after surgery. This febrile illness is characterized by fever, decreased appetite, listlessness, nausea, and vomiting. Chest pain is not always present, so a high index of suspicion should be maintained in any recently postoperative patient. Echocardiography is diagnostic. In most instances, the postpericardiotomy syndrome is self-limited; however, when pericardial fluid accumulates rapidly, the potential danger of cardiac tamponade should be recognized (Chapter 434). Rarely, arrhythmias may also occur. Symptomatic patients usually respond to salicylates or indomethacin and bed rest. Occasionally, steroid therapy or pericardiocentesis is required. Late recurrences can occur but are less usual.
Infection is another potentially serious postoperative problem. Patients usually receive a broad-spectrum antibiotic for the initial postoperative period. Potential sites of infection include the lungs (generally related to postoperative atelectasis), the subcutaneous tissues at the incision site, the sternum, and the urinary tract (especially after an indwelling catheter has been in place). Sepsis with infective endocarditis is an infrequent complication, and can be difficult to manage, especially if prosthetic material was placed at the time of surgery (Chapter 431).
Long-Term Management
Patients who have undergone surgery for congenital heart disease can be divided into several major categories: (1) lesions for which total repair has been achieved; (2) lesions for which both anatomic and physiologic correction has been achieved; and (3) lesions for which only palliation, albeit potentially long-term, has been achieved. There is some disagreement among cardiologists as to exactly which categories a particular congenital heart lesion might fall, and to some degree every case should be considered individually. Many argue that only for isolated patent ductus arteriosus is total repair really achieved, with no requirement for long-term follow-up. Patients who are able to undergo anatomic and physiologic correction include many of the left-to-right shunt lesions (atrial and ventricular septal defects) and milder forms of obstructive lesions (e.g., valvar pulmonic stenosis, some forms of valvar aortic stenosis, and coarctation of the aorta), and some forms of cyanotic heart disease, for example, uncomplicated tetralogy of Fallot and simple transposition of the great arteries. These patients usually have achieved total or near-total physiologic correction of their lesion; however, they are still at some risk of long-term sequelae, including late heart failure or arrhythmia, or recurrence of a significant physiologic abnormality (e.g., recoarctation of the aorta, worsening mitral regurgitation in patients with AVSDs, or long-standing pulmonary regurgitation in patients with tetralogy of Fallot repaired with a transannular patch). These patients require regular follow-up with a pediatric cardiologist (and when old enough, with an adult congenital heart disease specialist [Chapter 428.1]), however, their long-term prognosis is generally very good. Patients with more complex lesions, such as those with single ventricle physiology, are at much higher risk of long-term sequelae and require even closer follow-up. These patients, particularly those who have undergone the Fontan procedure, are at risk long-term for arrhythmia, thrombosis, protein losing enteropathy, end-organ (especially hepatic) dysfunction, and heart failure. Some may eventually require cardiac transplantation.
Physical limitations are variable, ranging from minimal to none in patients with physiologic correction, to mild to moderate in patients with palliative procedures. The extent to which a patient should be allowed to participate in athletics, both recreational and competitive, can best be determined by the cardiologist, often with the assistance of the data that can be derived from cardiopulmonary exercise testing (Chapter 417.5).
Barbu D, Mert I, Kruger M, et al. Evidence of fetal central nervous system injury in isolated congenital heart defects: microcephaly at birth. Am J Obstet Gynecol. 2009;201:43.e1-43.e7.
Bellinger DC, Newburger JW, Wypij D, et al. Behaviour at eight years in children with surgically corrected transposition: The Boston Circulatory Arrest Trial. Cardiol Young. 2009;19:86-97.
Bjarnason-Wehrens B, Dordel S, Schickendantz S, et al. Motor development in children with congenital cardiac diseases compared to their healthy peers. Cardiol Young. 2007;17:487-498.
Hoffman TM, Bush DM, Wernovsky G, et al. Postoperative junctional ectopic tachycardia in children: incidence, risk factors, and treatment. Ann Thorac Surg. 2002;74:1607-1611.
Hoffman TM, Wernovsky G, Atz AM, et al. Efficacy and safety of milrinone in preventing low cardiac output syndrome in infants and children after corrective surgery for congenital heart disease. Circulation. 2003;107:996-1002.
Karsdorp PA, Everaerd W, Kindt M, et al. Psychological and cognitive functioning in children and adolescents with congenital heart disease: a meta-analysis. J Pediatr Psychol. 2007;32:527-541.
Landolt MA, Valsangiacomo Buechel ER, Latal B. Health-related quality of life in children and adolescents after open-heart surgery. J Pediatr. 2008;152:349-355.
Limperopoulos C, Tworetzky W, McElhinney DB, et al. Brain volume and metabolism in fetuses with congenital heart disease. Circulation. 2010;121:26-33.
Majnemer A, Limperopoulous C, Shevell M, et al. Developmental and functional outcomes at school entry in children with congenital heart disease. J Pediatr. 2008;153:55-60.
Majnemer A, Limperopoulous C, Shevell M, et al. Long-term neuromotor outcome at school entry of infants with congenital heart defects requiring open-heart surgery. J Pediatr. 2006;148:72-77.
Miller SP, McQuillen PS, Hamrick S, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357:1928-1938.
Newburger JW, Bellinger DC. Brain injury in congenital heart disease. Circulation. 2006;113:183-185.
Owens JL, Musa N. Nutrition support after neonatal cardiac surgery. Nutr Clin Pract. 2009;24:242-249.
Parison SM, Mitchell PD, Colan SD, et al. A cross-sectional study of exercise performance during the first 2 decades of life after the Fontan operation. J Am Coll Cardiol. 2008;52:99-107.
Salvin JW, Laussen PC, Thiagarajan RR. Extracorporeal membrane oxygenation for postcardiotomy mechanical cardiovascular support in children with congenital heart disease. Paediatr Anaesth. 2008;18:1157-1162.
428.1 Congenital Heart Disease in Adults
Long-Term Medical Considerations
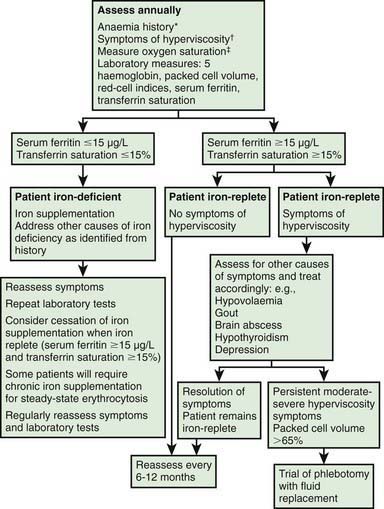
About 25% of adults with congenital heart disease have a mild form that has allowed them to survive into adulthood without surgical or interventional cardiac catheterization. The most common lesions in this category include mild aortic valve stenosis (usually in setting of bicuspid aortic valve), small restrictive ventricular septal defects, mild pulmonary valve stenosis, and mitral valve prolapse (Table 428-2). These patients need less frequent follow-up to assess for progression of disease and to identify associated complications. The majority of adults with congenital heart disease living in the USA are patients who have had previous intervention (Table 428-3). Although the majority of children who undergo surgical intervention will survive to adulthood, with few exceptions, “total correction” is not the rule. The few exceptions include patent ductus arteriosus, ventricular septal defects, and atrial septal defects; this is true only if they are closed early before the development of irreversible pulmonary vascular changes and no residual lesions exist. Because adult patients with congenital heart disease are surviving longer than ever, it is becoming increasingly apparent that even the simplest lesions can be associated with long-term complications. These long-term complications include both cardiac and noncardiac problems (Tables 428-4 and 428-5, Fig. 428-1). Cardiac complications include arrhythmias and conduction defects, ventricular dysfunction, residual shunts, valvular lesions (regurgitation and stenosis), hypertension, and aneurysms. Noncardiac sequelae include developmental abnormalities such as developmental delay, somatic abnormalities such as facial dysmorphism (cleft palate/lip), central nervous abnormalities such as seizure disorders from previous thromboembolic events or cerebrovascular accidents, disturbances of the senses such as hearing loss or vision loss, and pulmonary sequelae such as both restrictive and obstructive lung disease. Psychosocial problems involving employment, life and health insurance, participation in sports, sexual activity, and contraception are common. As result of these long-term complications, the majority of adults with congenital heart disease need lifelong follow-up.
Table 428-2 CONGENITAL HEART DEFECTS ASSOCIATED WITH SURVIVAL INTO ADULTHOOD WITHOUT SURGERY OR INTERVENTIONAL CARDIAC CATHETERIZATION
Table 428-3 MOST COMMON CONGENITAL HEART DEFECTS SURVIVING TO ADULTHOOD AFTER SURGERY OR INTERVENTIONAL CATHETERIZATION
Table 428-4 RISKS IN ADULTS WHO HAVE CONGENITAL HEART DISEASE
ASD, atrial septal defect; PDA, patent ductus arteriosus; SBE, subacute bacterial endocarditis; VSD, ventricular septal defect.
Table 428-5 LESION SPECIFIC RISKS OF MATERNAL AND NEONATAL COMPLICATIONS OF PREGNANCY
| No additional risk | Small septal defects |
| Surgically closed ASD, VSD, PDA | |
| Mild to moderate aortic regurgitation | |
| Mild to moderate pulmonary stenosis | |
| Slightly increased risk | Postoperative repair of tetralogy of Fallot |
| Transposition of the great arteries, s/p arterial switch procedure | |
| Moderate risk | Transposition of the great arteries, s/p atrial switch procedure |
| Congenitally corrected transposition of the great arteries | |
| Single ventricle physiology, s/p Fontan procedure | |
| Severe risk | Cyanotic congenital heart disease, unoperated or palliated |
| Marfan syndrome | |
| Prosthetic valves | |
| Obstructive lesions including coarctation | |
| Pregnancy contraindicated | Severe pulmonary hypertension |
| Severe obstructive lesions | |
| Marfan syndrome, aortic root >40 mm |
ASD, atrial septal defect; PDA, patent ductus arteriosus; s/p, status post (after); VSD, ventricular septal defect.
Specific Lesions
Left-to-Right Shunts
If the shunt is large and nonrestrictive (allowing transmission of near systemic pressure to the pulmonary arteries), irreversible pulmonary vascular changes can occur, resulting in pulmonary hypertension at systemic levels with reversed or bidirectional shunting at the level of the defect (Eisenmenger syndrome) (Chapter 427.2).
Atrial Septal Defects (ASDs) (Chapter 420.1)
Pregnancy and Congenital Heart Disease
Despite these hemodynamic changes, the outcome of pregnancy is favorable in most women with CHD provided that functional class and systemic ventricular function are good (see Table 428-5). Pulmonary artery hypertension presents a serious risk during pregnancy, particularly when the pulmonary pressure exceeds 70% of systemic pressure, regardless of functional class. Other contraindications to pregnancy include severe obstructive left-sided lesions (coarctation of the aorta, aortic valve stenosis, mitral valve stenosis, hypertrophic cardiomyopathy), Marfan syndrome with coexisting dilated ascending aorta (defined as >4.0 cm), persistent cyanosis, and systemic ventricular dysfunction (ejection fraction of ≤40%). The need for full anticoagulation during pregnancy, although not a contraindication, poses an increased risk to both mother and fetus. The relative risks and benefits of the different anticoagulant approaches need to be discussed fully with the prospective mother.
Adolescent Transition
It is well recognized that, as part of the process of obtaining independence, adolescents or young adults must develop a forward-looking, independent approach to their medical care. For children with heart disease, the transition process must begin during early adolescence and should be encouraged by both the primary care provider and the pediatric cardiologist, who must identify an appropriate adult congenital heart program to which transition and transfer will be made at an appropriate time (Table 428-6).
Table 428-6 ISSUES THAT REQUIRE COORDINATION OF CARE BETWEEN THE CARDIOLOGIST AND THE PRIMARY CARE PHYSICIAN
A successful transition program includes the following elements:
Betranon EG, Blackstone EH, Hazelrig JB, et al. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol. 1978;42:458-466.
Cantor WJ, Harrison DA, Moussadji JS, et al. Determinants of survival and length of survival in adults with Eisenmenger’s syndrome. Am J Cardiol. 1999;84:677-681.
Cohen M, Foster V, Steele PM, et al. Coarctation of the aorta: long-term follow-up and prediction of outcome after surgical correction. Circulation. 1989;80:840-845.
Earing MG, Connolly HM, Dearani JA, et al. Long-term follow-up of patients after surgical treatment of isolated pulmonary valve stenosis. Mayo Clin Proc. 2005 Jul;80(7):871-876.
Earing MG, Webb G. Congenital heart disease and pregnancy: maternal and fetal risks. Clin Perinatol. 2005;32:913-919.
Keane JF, Driscoll DJ, Gersony WM, et al. Second natural history study of congenital heart defects: results of treatment of patients with aortic valve stenosis. Circulation. 1993;87:I-21-I-26.
Losay J, Touchot S, Serraf A, et al. Late outcome after arterial switch operation for transposition of the great arteries. Circulation. 2001;104:I121-I126.
Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births: incidence and natural history. Circulation. 1971;43:323-332.
Nollert G, Fischlein T, Bouterwek S, et al. Long-term survival in patients with repair of tetralogy of Fallot: 36-year follow-up of 490 survivors of the first year after surgical repair. J Am Coll Cardiol. 1997;30:1374-1383.
Perloff JK. Congenital heart disease after childhood: an expanding patient population: 22nd Bethesda Conference, Maryland, October 18–19, 1990. J Am Coll Cardiol. 1991;18:311-342.
Perloff JK, Warnes CA. Challenges posed by adults with repaired congenital heart disease. Circulation. 2001;103:2637-2643.
Spence MS, Balaratnam MS, Gatzoulis MA. Clinical update: cyanotic adult congenital heart disease. Lancet. 2007;370:1530-1532.
Therrien J, Dore A, Gersony W, et al. CCS consensus conference 2001 update: recommendations for the management of adults with congenital heart disease, Part I. Can J Cardiol. 2001;17:940-959.
Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on management of adults with congenital heart disease). Circulation. 2008;118:e714-e833.
Wilson NJ, Clarkson PM, Barratt-Boyes BG, et al. Long-term outcome after the mustard repair for simple transposition of the great arteries: 28 year follow-up. J Am Coll Cardiol. 1998;32:758-765.
Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736-1754.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 428 General Principles of Treatment of Congenital Heart Disease
Bacterial infections should be treated vigorously, but the presence of congenital heart disease is not an appropriate reason to use antibiotics indiscriminately. Prophylaxis against infective endocarditis should be carried out during dental procedures for appropriate patients. The American Heart Association has recently significantly revised these recommendations, with most patients no longer requiring routine prophylaxis (Chapter 431).
Cyanotic patients need to be monitored for a multitude of noncardiac manifestations of oxygen deficiency (Table 428-1). Treatment of iron deficiency anemia is important in cyanotic patients, who will show improved exercise tolerance and general well-being with adequate hemoglobin levels. These patients should also be carefully observed for excessive polycythemia. Cyanotic patients should avoid situations in which dehydration may occur, which leads to increased viscosity and increases the risk of stroke. Diuretics may need to be decreased or temporarily discontinued during episodes of acute gastroenteritis. High altitudes and sudden changes in the thermal environment should also be avoided. Phlebotomy with partial exchange transfusion is carried out only in symptomatic patients with severe polycythemia (usually those hematocrit >65%). Patients with moderate to severe forms of congenital heart disease or a history of rhythm disturbance should be carefully monitored during anesthesia for even routine surgical procedures. Consultation with an anesthesiologist experienced in the care of children with congenital heart disease is encouraged. Women with nonrepaired severe congenital heart disease should be counseled on the risks associated with childbearing and on the use of contraceptives and tubal ligation. Pregnancy may be dangerous for patients with chronic cyanosis or pulmonary arterial hypertension. Women with mild to moderate heart disease and many of those who have had corrective surgery can have normal pregnancies, although those with residual hemodynamic derangements or with systemic right ventricles should optimally be followed by a high-risk perinatologist and a cardiologist with expertise in caring for adults with congenital heart disease.
Table 428-1 EXTRACARDIAC COMPLICATIONS OF CYANOTIC CONGENITAL HEART DISEASE AND EISENMENGER PHYSIOLOGY
| PROBLEM | ETIOLOGY | THERAPY |
|---|---|---|
| Polycythemia | Persistent hypoxia | Phlebotomy |
| Relative anemia | Nutritional deficiency | Iron replacement |
| CNS abscess | Right-to-left shunting | Antibiotics, drainage |
| CNS thromboembolic stroke | Right-to-left shunting or polycythemia | Phlebotomy |
| Low-grade DIC, thrombocytopenia | Polycythemia | None for DIC unless bleeding, then phlebotomy |
| Hemoptysis | Pulmonary infarct, thrombosis, or rupture of pulmonary artery plexiform lesion | Embolization |
| Gum disease | Polycythemia, gingivitis, bleeding | Dental hygiene |
| Gout | Polycythemia, diuretic agent | Allopurinol |
| Arthritis, clubbing | Hypoxic arthropathy | None |
| Pregnancy complications: abortion, fetal growth retardation, prematurity increase, maternal illness | Poor placental perfusion, poor ability to increase cardiac output | Bed rest, pregnancy prevention counseling |
| Infections | Associated asplenia, DiGeorge syndrome, endocarditis | Antibiotics |
| Fatal RSV pneumonia with pulmonary hypertension | Ribavirin; RSV immunoglobulin (prevention) | |
| Failure to thrive | Increased oxygen consumption, decreased nutrient intake | Treat heart failure; correct defect early; increase caloric intake |
| Psychosocial adjustment | Limited activity, cyanotic appearance, chronic disease, multiple hospitalizations | Counseling |
CNS, central nervous system; DIC, disseminated intravascular coagulation; RSV, respiratory syncytial virus.
Postoperative Management
The electrocardiogram should be monitored continuously during the postoperative period. A change in heart rate, even without arrhythmia, may be the first indication of a serious complication such as hemorrhage, hypothermia, hypoventilation, or heart failure. Cardiac rhythm disorders must be diagnosed quickly because a prolonged untreated arrhythmia may add a severe hemodynamic burden to the heart in the critical early postoperative period (Chapter 429). Injury to the heart’s conduction system during surgery can result in postoperative complete heart block. This complication is usually temporary and is treated with surgically placed pacing wires that can later be removed. Occasionally, complete heart block is permanent. If heart block persists beyond 10-14 days postoperatively, insertion of a permanent pacemaker is required. Tachyarrhythmias are a common problem in postoperative patients. Junctional ectopic tachycardia (JET) can be a particularly troublesome rhythm to manage (Chapter 429), although it usually responds to intravenous amiodarone.
Heart failure with poor cardiac output after cardiac surgery may be secondary to respiratory failure, serious arrhythmias, myocardial injury, blood loss, hypovolemia, a significant residual hemodynamic abnormality, or any combination of these factors. Treatment specific to the cause should be instituted. Catecholamines, phosphodiesterase inhibitors, nitroprusside and other afterload-reducing agents, and diuretics are the cardioactive agents most often used in patients with myocardial dysfunction in the early postoperative period (Chapter 436). Postoperative pulmonary hypertension can be managed with hyperventilation and inhaled nitric oxide. In patients who are unresponsive to standard pharmacologic treatment, various ventricular assist devices are available, depending on the patient’s size. If pulmonary function is adequate, a left ventricular assist device (LVAD) may be used. If pulmonary function is inadequate, extracorporeal membrane oxygenation (ECMO) may be used. These extraordinary measures are helpful in maintaining the circulation until cardiac function improves, usually within 2-3 days. They have also been used with moderate success as a bridge to transplantation in patients with severe nonremitting postoperative cardiac failure.
The postpericardiotomy syndrome may occur toward the end of the 1st postoperative week or may sometimes be delayed until weeks or months after surgery. This febrile illness is characterized by fever, decreased appetite, listlessness, nausea, and vomiting. Chest pain is not always present, so a high index of suspicion should be maintained in any recently postoperative patient. Echocardiography is diagnostic. In most instances, the postpericardiotomy syndrome is self-limited; however, when pericardial fluid accumulates rapidly, the potential danger of cardiac tamponade should be recognized (Chapter 434). Rarely, arrhythmias may also occur. Symptomatic patients usually respond to salicylates or indomethacin and bed rest. Occasionally, steroid therapy or pericardiocentesis is required. Late recurrences can occur but are less usual.
Infection is another potentially serious postoperative problem. Patients usually receive a broad-spectrum antibiotic for the initial postoperative period. Potential sites of infection include the lungs (generally related to postoperative atelectasis), the subcutaneous tissues at the incision site, the sternum, and the urinary tract (especially after an indwelling catheter has been in place). Sepsis with infective endocarditis is an infrequent complication, and can be difficult to manage, especially if prosthetic material was placed at the time of surgery (Chapter 431).
Long-Term Management
Patients who have undergone surgery for congenital heart disease can be divided into several major categories: (1) lesions for which total repair has been achieved; (2) lesions for which both anatomic and physiologic correction has been achieved; and (3) lesions for which only palliation, albeit potentially long-term, has been achieved. There is some disagreement among cardiologists as to exactly which categories a particular congenital heart lesion might fall, and to some degree every case should be considered individually. Many argue that only for isolated patent ductus arteriosus is total repair really achieved, with no requirement for long-term follow-up. Patients who are able to undergo anatomic and physiologic correction include many of the left-to-right shunt lesions (atrial and ventricular septal defects) and milder forms of obstructive lesions (e.g., valvar pulmonic stenosis, some forms of valvar aortic stenosis, and coarctation of the aorta), and some forms of cyanotic heart disease, for example, uncomplicated tetralogy of Fallot and simple transposition of the great arteries. These patients usually have achieved total or near-total physiologic correction of their lesion; however, they are still at some risk of long-term sequelae, including late heart failure or arrhythmia, or recurrence of a significant physiologic abnormality (e.g., recoarctation of the aorta, worsening mitral regurgitation in patients with AVSDs, or long-standing pulmonary regurgitation in patients with tetralogy of Fallot repaired with a transannular patch). These patients require regular follow-up with a pediatric cardiologist (and when old enough, with an adult congenital heart disease specialist [Chapter 428.1]), however, their long-term prognosis is generally very good. Patients with more complex lesions, such as those with single ventricle physiology, are at much higher risk of long-term sequelae and require even closer follow-up. These patients, particularly those who have undergone the Fontan procedure, are at risk long-term for arrhythmia, thrombosis, protein losing enteropathy, end-organ (especially hepatic) dysfunction, and heart failure. Some may eventually require cardiac transplantation.
Physical limitations are variable, ranging from minimal to none in patients with physiologic correction, to mild to moderate in patients with palliative procedures. The extent to which a patient should be allowed to participate in athletics, both recreational and competitive, can best be determined by the cardiologist, often with the assistance of the data that can be derived from cardiopulmonary exercise testing (Chapter 417.5).
Barbu D, Mert I, Kruger M, et al. Evidence of fetal central nervous system injury in isolated congenital heart defects: microcephaly at birth. Am J Obstet Gynecol. 2009;201:43.e1-43.e7.
Bellinger DC, Newburger JW, Wypij D, et al. Behaviour at eight years in children with surgically corrected transposition: The Boston Circulatory Arrest Trial. Cardiol Young. 2009;19:86-97.
Bjarnason-Wehrens B, Dordel S, Schickendantz S, et al. Motor development in children with congenital cardiac diseases compared to their healthy peers. Cardiol Young. 2007;17:487-498.
Hoffman TM, Bush DM, Wernovsky G, et al. Postoperative junctional ectopic tachycardia in children: incidence, risk factors, and treatment. Ann Thorac Surg. 2002;74:1607-1611.
[/not-level-membership-for-pediatrics-category]
